
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A solid xantphos macroligand based on porous organic polymers for the catalytic hydrogenation of CO2†
Arne
Nisters
 a,
Torsten
Gutmann
b,
Sun-Myung
Kim
c,
Jan Philipp
Hofmann
c and
Marcus
Rose
*a
a,
Torsten
Gutmann
b,
Sun-Myung
Kim
c,
Jan Philipp
Hofmann
c and
Marcus
Rose
*a
aTechnical University of Darmstadt, Ernst-Berl-Institute of Technical and Macromolecular Chemistry, Peter-Grünberg-Straße 8, 64287 Darmstadt, Germany. E-mail: marcus.rose@tu-darmstadt.de
bTechnical University of Darmstadt, Eduard-Zintl-Institute of Inorganic and Physical Chemistry, Peter-Grünberg-Straße 8, 64287 Darmstadt, Germany
cSurface Science Laboratory, Department of Materials and Earth Sciences, Technical University of Darmstadt, Otto-Berndt-Straße 3, 64287 Darmstadt, Germany
First published on 25th June 2024
Abstract
Porous organic polymers enable a novel approach to incorporate xantphos into a solid macroligand. Immobilizing a ruthenium complex on the xantphos framework results in an excellent catalyst for the hydrogenation of CO2 to formic acid. Recycling experiments indicate a minor partial degradation of the heterogenous catalyst after a certain induction period, which is referred to its structural changes.
Sustainability spotlightIntending the defossilization of the chemical industry, alternative carbon feedstocks need to be implemented into the chemical value chain. Beside biomass, CO2 offers a promising carbon source as prospectively captured and abundant C1 building block. However, its kinetic inertness requires the development of appropriate catalysts. This works aims an advanced understanding of this key technology, focusing on a heterogenized catalyst for the hydrogenation of CO2 to formic acid, which is discussed as a storage molecule for hydrogen as well as carbon monoxide and is an important chemical intermediate. Overall, we hope to contribute with the present research to UN SDGs No. 9 (Industry, Innovation and Infrastructure) and No. 12 (Responsible Consumption and Production). |
In the context of the ecological transformation of today's economy, the concept of catalysis plays a pivotal role. It is considered to be a “key technology for rapid introduction of renewable energy”,1,2 a “workhorse”3 for the defossilization of the energy system, a “primary tool for achieving all of the 12 principles of green chemistry”4 and a “pillar of chemical industry to reach the sustainability of the society”.5 Beyond the usual classification between heterogeneous and homogenous catalysts, there is growing recognition of the vital link between these two fields of research.6–8 The evolvement of the latter one, the catalytic application of transition metal complexes, is strongly accompanied by the evolution of diverse ligand types and structures. In particular, soft and strongly ligating phosphines are the donor atoms of choice.9 The early work by Reppe established triphenylphosphine as the prototypical phosphine and initiated the exploration of the ligand sphere, crafting the microenvironment of the metal center.10,11 A remarkable example of this design process is xantphos, a bidentate diphosphine ligand with a rigid backbone and a wide bite angle, which has been addressed by recent reviews.11–13 While xantphos was originally applied for hydroformylation catalysis,14 its superior ligand properties also occur in hydrocyanation,15 cross-coupling reactions16 and C–H activation reactions forming C–C and C–X bonds.17 Due to its versatility, xantphos derivatives have been the subject of various immobilization approaches, which aim to transfer the molecular moiety into an easily separable catalyst. These attempts include encapsulation,18 anchoring on a silica surface19–21 or polymer matrix,22 immobilization on a supported ionic liquid phase23 or the radical polymerization of vinyl functionalized xantphos species.24–26 However, all these approaches require several complex preparation steps, increasing the cost of immobilization.27 In our work, we developed a simple and scalable design strategy for a heterogenized xantphos-based ruthenium catalyst, where the arising aluminum waste is less harmful and easily recyclable. Based on the concept of hyper-crosslinked polymers (HCPs),28 we incorporated xantphos into a porous organic polymer and investigated the ruthenium impregnated catalyst in the direct hydrogenation of CO2 to formic acid (FA). Herein, we present the synthesis and characterization of the novel catalyst and highlight its catalytic behavior, focusing on the stability and its alterations during the catalytic process.
The Xantphos-based HCP (X-HCP) was prepared, following an external knitting strategy (Fig. 1a).28 In accordance with Yang et al., we employed AlCl3 as strong Lewis catalyst for the activation of the less reactive phosphine monomer.29 The successful crosslinking in the X-HCP skeleton was demonstrated by Fourier-transform infrared spectroscopy (FTIR) (Fig. 1b), where the characteristic band at 2911 cm−1 shows the stretching vibration of the formed methylene bridges. The vanishing of the C–Cl vibration at 643 cm−1 indicates the substitution of chloride, whereas the broad band at 1611 cm−1 is assigned to the vibrations of the present aromatic units. The presence of xantphos is clearly revealed by the preservation of the bands at 2952 cm−1 (CH3), 1433 cm−1 (C–P) and 1232 cm−1 (C–O–C). The band at 1178 cm−1 suggests oxidation processes of the phosphorus atoms during the synthesis. X-ray fluorescence spectroscopy (XRF) confirms the presence of phosphorus, with additional small residues of aluminum and chloride remaining inside the network (Table S1†). In comparison to an ideal HCP with entirely crosslinked monomers, only one third of the monomeric xantphos is incorporated in the final X-HCP. This difference is driven by the ability of the external crosslinker to interconnect with itself, without involving Xantphos.30 XRF confirms the uptake of ruthenium (0.31 wt%) in X-HCP-Ru after the ligand exchange (Fig. 1a).31 The molar ratio of phosphorus to ruthenium of 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 indicates that the donor centers of the macroligand are either not all capable to coordinate or only partially accessible for the metal species. This imbalance may be caused by its topological conformation, which is characterized by a combination of HCP-typical amorphous as well as crystalline domains (Fig. S1†).32 Beside a non-ordered structure, the sharp reflexes of X-HCP in X-ray powder diffraction measurements coincide with those of the monomeric xantphos. Thus, it seems that xantphos units in the X-HCP framework arrange in a stacked manner, probably diminishing their ligating ability. The X-HCP exhibits a porosity with a specific surface area (BET) of 222 m2 g−1 (Fig. 1c). Nitrogen physisorption reveals the presence of micropores and a broad distribution of mesopores. The pronounced hysteresis, which does not close, is known for non-rigid materials and is attributed to capillary condensation, pore blocking and swelling effects.33–35 Water vapor sorption measurements refer to the hydrophilic characteristics of the X-HCP. The gradual uptake is related to a moderate hydrophobicity, but demonstrates a wettability, which is of importance for catalytic application in the aqueous phase (Fig. 1d). Thermogravimetric analysis (TGA) indicates a mass loss of 7 wt% up to 350 °C, followed by a one-step decomposition at 450 °C (Fig. S2†). Furthermore, the morphology of the polydisperse particles was investigated by scanning electron microscopy (SEM) (Fig. S3†) and indicates uniform particles with sizes between 200 and 500 μm. The absence of larger ruthenium nanoparticle clusters was proven by transmission electron microscopy (TEM) (Fig. S4†). Quantitative 31P solid-state nuclear magnetic resonance spectroscopy (31P-MAS-NMR) of X-HCP-Ru (Fig. 1e) reveals the presence of oxidized phosphorus atoms (δ = 22.4 ppm) accounting for 39% of all phosphines. The majority of the phosphines (δ = −17.3 ppm) remain non-oxidized (61%), with the peak consisting of a sharp one from the ordered domains and a broader part from the less ordered domains. X-ray photoelectron spectroscopy (XPS) emphasizes the partial oxidation of the phosphines and suggests that 29% of the phosphines are oxidized. This quantification is roughly in agreement with the NMR results, considering that XPS measurements overrepresent near-surface regions and not the entire bulk phase (Fig. 1f and S5b†). Due to the low metal loading and the overlap with the dominant C1s peak, XPS does not allow an unambiguous electronic specification of the ruthenium species (Fig. S5†).
:1 indicates that the donor centers of the macroligand are either not all capable to coordinate or only partially accessible for the metal species. This imbalance may be caused by its topological conformation, which is characterized by a combination of HCP-typical amorphous as well as crystalline domains (Fig. S1†).32 Beside a non-ordered structure, the sharp reflexes of X-HCP in X-ray powder diffraction measurements coincide with those of the monomeric xantphos. Thus, it seems that xantphos units in the X-HCP framework arrange in a stacked manner, probably diminishing their ligating ability. The X-HCP exhibits a porosity with a specific surface area (BET) of 222 m2 g−1 (Fig. 1c). Nitrogen physisorption reveals the presence of micropores and a broad distribution of mesopores. The pronounced hysteresis, which does not close, is known for non-rigid materials and is attributed to capillary condensation, pore blocking and swelling effects.33–35 Water vapor sorption measurements refer to the hydrophilic characteristics of the X-HCP. The gradual uptake is related to a moderate hydrophobicity, but demonstrates a wettability, which is of importance for catalytic application in the aqueous phase (Fig. 1d). Thermogravimetric analysis (TGA) indicates a mass loss of 7 wt% up to 350 °C, followed by a one-step decomposition at 450 °C (Fig. S2†). Furthermore, the morphology of the polydisperse particles was investigated by scanning electron microscopy (SEM) (Fig. S3†) and indicates uniform particles with sizes between 200 and 500 μm. The absence of larger ruthenium nanoparticle clusters was proven by transmission electron microscopy (TEM) (Fig. S4†). Quantitative 31P solid-state nuclear magnetic resonance spectroscopy (31P-MAS-NMR) of X-HCP-Ru (Fig. 1e) reveals the presence of oxidized phosphorus atoms (δ = 22.4 ppm) accounting for 39% of all phosphines. The majority of the phosphines (δ = −17.3 ppm) remain non-oxidized (61%), with the peak consisting of a sharp one from the ordered domains and a broader part from the less ordered domains. X-ray photoelectron spectroscopy (XPS) emphasizes the partial oxidation of the phosphines and suggests that 29% of the phosphines are oxidized. This quantification is roughly in agreement with the NMR results, considering that XPS measurements overrepresent near-surface regions and not the entire bulk phase (Fig. 1f and S5b†). Due to the low metal loading and the overlap with the dominant C1s peak, XPS does not allow an unambiguous electronic specification of the ruthenium species (Fig. S5†).
 | ||
| Fig. 1 (a) Synthetic route of X-HCP-Ru, consisting of a Friedel-Crafts-alkylation and a subsequent ligand exchange. L corresponds to unspecified, coordinating ligands, remaining from the Ru precursor (in particular Cl− ligands). (b) ATR-FTIR-spectra of the monomer xantphos (orange), the crosslinker BCMBP (grey) and the X-HCP (blue). The intensities are normalized to the height of the most intense peak. (c) Nitrogen physisorption of X-HCP at 77 K. Filled blue diamonds denote the adsorption, empty blue diamonds denote the desorption. (d) Water vapor sorption of X-HCP at 293 K. Filled blue diamonds denote the adsorption, empty blue diamonds denote the desorption. (e) 31P-MAS-NMR-spectrum of X-HCP-Ru. Spinning side bands are marked with an asterisk. (f) P2p XPS spectrum of X-HCP-Ru. | ||
The X-HCP-Ru catalyst was applied in the base-mediated CO2 hydrogenation reaction to formic acid, where an initial rate (TOF) of 1249 molFA molRu h−1 was obtained. In order to put the catalytic performance in context, reference experiments were carried out (Fig. 2a). The (Ru-)metal-free X-HCP was inactive. Additionally, a comparable HCP framework without phosphine units, which contains solely crosslinked BCMBP units, was synthesized and subsequently impregnated with ruthenium by ligand exchange (BCMBP-HCP-Ru). BCMBP-HCP-Ru showed a negligible activity, which reveals the involvement of both species, the phosphine microenvironment and the metal center, in the catalytically active species of X-HCP-Ru. A low activity was observed for the commercial Ru@C as an example of a heterogenous catalyst and the homogeneous Ru(NO)(NO3)3 complex. Since the xantphos ligand is poorly soluble in the aqueous phase, the reference reaction of the xantphos ruthenium complex was carried out in DMSO with NEt3 as a base. The resulting initial rate (TOF) of 577 molFA molRu h−1 lies in a comparable range to other reported homogeneous diphosphine ruthenium complexes.36,37 These reference experiments emphasize the remarkable performance of X-HCP-Ru. The catalytic stability of X-HCP-Ru was investigated by recycling experiments (Fig. 2b), where the reaction solution was exchanged after each recycling step. During the first and second runs, a continuous deactivation of the X-HCP-Ru is observed, resulting in a significant drop in catalytic productivity after 5.5 hours. Since the hydrogenation reaction is far away from thermodynamic limitations, it can be associated to a gradual degradation of the catalytic species. Starting from the third run, a nearly linear slope with a slight decrease in productivity occurs. Thus, after a certain induction period, the X-HCP-Ru forms a stable catalyst, which coincides with findings about comparable catalytic systems for the CO2 hydrogenation reaction to formic acid.38 In addition, the influence of storing the prepared catalyst on the catalytic performance was examined (Fig. S6†). In comparison to the freshly prepared X-HCP-Ru, the aged catalyst (stored for eight days after preparation) exhibits lower productivities in the first run (75% after 5.5 h). Subsequently, the productivities of the fresh and aged catalysts coincide in the following runs and form a similar steady-state activity. In order to investigate the initial degradation, the retained X-HCP-Ru after four catalytic runs was further analyzed and compared to the pristine X-HCP-Ru. 31P-MAS-NMR (Fig. S7†) shows no significant increase in the proportion of oxidized phosphines (from 39 to 43%). ATR-IR does not indicate any structural changes of the network (Fig. S8†). In TEM images (Fig. S9†) no larger agglomeration of the dispersed metal species was observed.39 However, the formation of highly dispersed nanoparticles cannot be fully excluded, as the decomposition of single source precursors in the presence of hydrogen is a known synthesis approach for very small ruthenium nanoparticles.40 Since these nanoparticles are active catalysts for CO2 methanation,41 an occurring agglomeration may imply a change in selectivity from formic acid to methane in the recycling runs, which is subject to further investigation. In contrast to previous works, leaching of active species into solution could be excluded as the origin of the catalyst degradation.42–44 While the filtrate contains negligible amounts of ruthenium (0.2 wt-ppm) and phosphorus (0.5 wt-ppm) (Table S2†), the hot leaching experiment clearly indicates no catalytic conversion after the removal of X-HCP-Ru, proving its heterogeneous nature (Fig. 2c). Kann et al. proposed the formation of ligating carbon monoxide as a reason for the catalytic degradation, whose retarding influence was mentioned by Jessop et al. and confirmed in this work by catalytic experiments of the CO-exposed X-HCP-Ru (Fig. 2a).38,45 Neither CO nor CH4, pointing towards the formation of Ru nanoparticles, could be detected in the gas phase (Fig. S10†) nor observed in the ATR-IR spectrum (Fig. S8†), which may also be caused by the low density of metal sites. Regarding the topological changes, nitrogen physisorption reveals the loss of pore volume under the catalytic conditions (from 0.18 to 0.046 cm3 g−1), which can affect the accessibility of catalytically active sites (Fig. S11 and Table S3†). However, the dynamic properties of porous polymers and their ability to swell do not allow a definitive conclusion about its porous state under solvated, catalytic conditions.
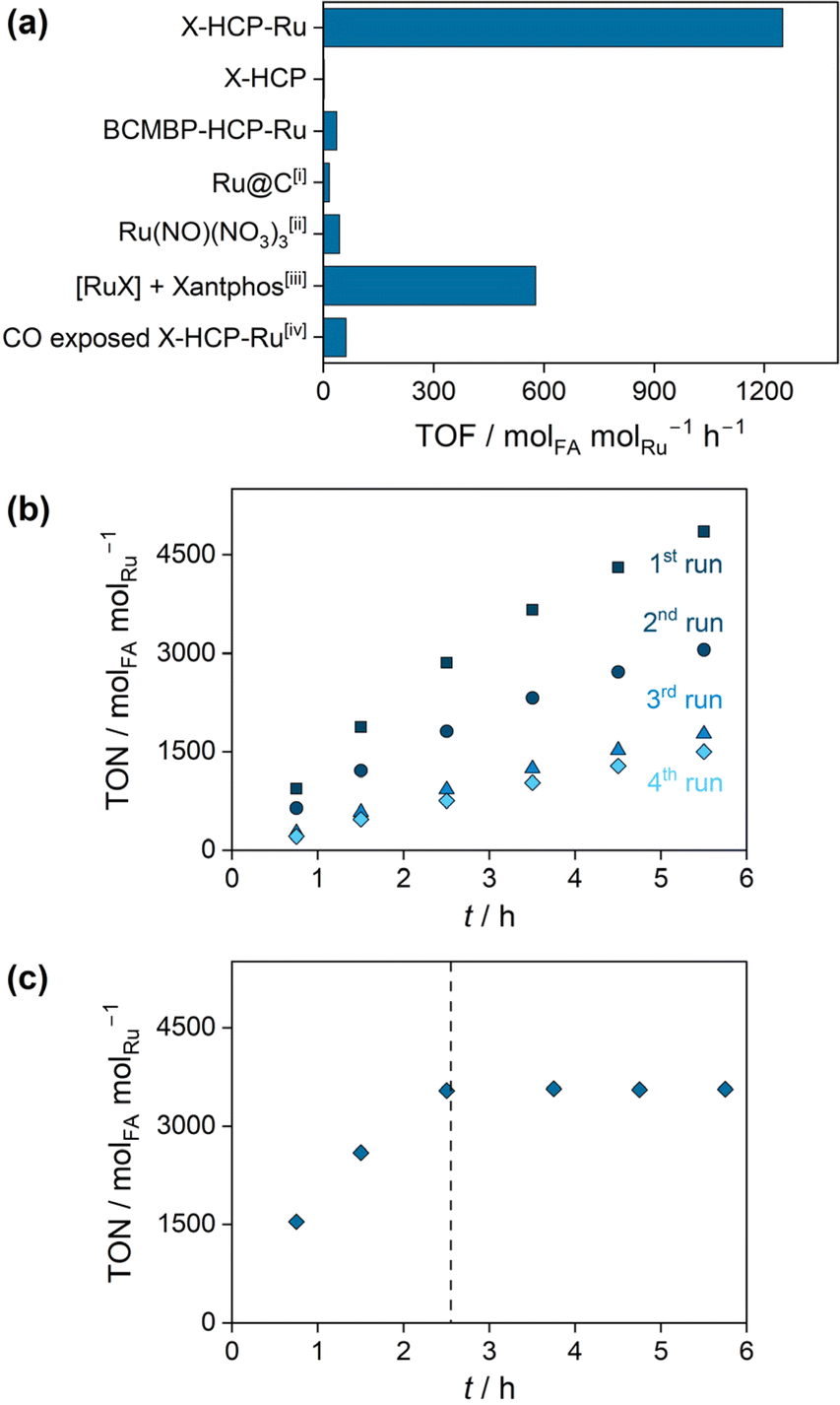 | ||
| Fig. 2 (a) Catalytic evaluation of X-HCP-Ru and reference experiments for the CO2 hydrogenation to formic acid. (b) Catalytic recycling of X-HCP-Ru over four recycling steps. Kinetic curves of the 1st run (squares), 2nd run (circles), 3rd run (triangles) and 4th run (diamonds). (c) Hot leaching experiments of X-HCP-Ru. The catalyst was filtered off after 2.5 h, the remaining filtrate was pressurized and heated up to initial reaction conditions. Reaction conditions: 300 mL Hastelloy autoclave, V(H2O) = 150 mL, n(NaHCO3) = 150 mmol, m(cat.) = 200 mg, p(CO2/H2) = 60 bar (1:1), T = 110 °C, t = 5.5 h, 700 rpm. [i] m(Ru@C) = 50 mg (5 wt%) [ii] n(Ru(NO)(NO3)3) = 15 μmol [iii] V(DMSO) = 150 mL, n(NEt3) = 150 mmol, n([Ru(p-cymene)Cl2]2) = 0.5 μmol, n(Xantphos) = 0.5 μmol. [iv] Exposure of X-HCP-Ru to CO (5 bar), which is released from the autoclave before the catalysis. | ||
In summary, we developed a novel heterogenized xantphos macroligand, following a simple crosslinking approach. Molecular ruthenium species were immobilized on the porous framework and the catalyst was evaluated in the aqueous CO2 hydrogenation to formic acid. Reference experiments reveal its heterogeneous nature and highlight its excellent activity. The investigation of its recyclability suggests an initial deactivation of the catalyst, leading to a slight decrease in catalytic productivity after a certain induction period. This alteration can be presumably referred to changes in the topology of the porous system and the metal coordination sphere.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
A. N. thanks the German Academic Scholarship Foundation (Studienstiftung des deutschen Volkes) for the financial support. M. R. acknowledges funding by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG), project number 450360023. The collaboration was supported by the DFG within the CRC 1487, “Iron, upgraded!”, project number 443703006.Notes and references
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 RSC.
- G. Centi and S. Perathoner, Catal. Today, 2023, 423, 113935 CrossRef CAS.
- R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed.
- P. T. Anastas, L. B. Bartlett, M. M. Kirchhoff and T. C. Williamson, Catal. Today, 2000, 55, 11–22 CrossRef CAS.
- G. Centi and S. Perathoner, Top. Catal., 2009, 52, 948–961 CrossRef CAS.
- J. C. Bailar, Catal. Rev., 1974, 10, 17–36 CrossRef CAS.
- M. Rose, ChemCatChem, 2014, 6, 1166–1182 CrossRef CAS.
- F. Poovan, V. G. Chandrashekhar, K. Natte and R. V. Jagadeesh, Catal. Sci. Technol., 2022, 12, 6623–6649 RSC.
- J. A. Gillespie, E. Zuidema, P. W. N. M. van Leeuwen and P. C. J. Kamer, in Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, John Wiley & Sons, Chichester, 1st edn, 2012, Chapter I, pp. 1–26 Search PubMed.
- W. Reppe and W. J. Schweckendiek, Liebigs Ann. Chem., 1948, 560, 104–116 CrossRef CAS.
- P. W. N. M. van Leeuwen and P. C. J. Kamer, Catal. Sci. Technol., 2018, 8, 26–113 RSC.
- M. W. Haenel, D. Jakubik, E. Rothenberger and G. Schroth, Chem. Ber., 1991, 124, 1705–1710 CrossRef CAS.
- L. Veth and P. Dydio, Nat. Chem., 2022, 14, 1088–1091 CrossRef CAS PubMed.
- M. Kranenburg, Y. E. M. van Der Burgt, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz and J. Fraanje, Organometallics, 1995, 14, 3081–3089 CrossRef CAS.
- W. Goertz, W. Keim, D. Vogt, U. Englert, M. D. K. Boele, L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 1998, 18, 2981–2988 RSC.
- R. Takahashi, K. Kubota and H. Ito, Chem. Commun., 2020, 56, 407–410 RSC.
- J. B. Diccianni, C. T. Hu and T. Diao, Angew. Chem., 2019, 131, 14003–14006 CrossRef.
- K. Dong, Q. Sun, Y. Tang, C. Shan, B. Aguila, S. Wang, X. Meng, S. Ma and F. Xiao, Nat. Commun., 2019, 10, 3059 CrossRef PubMed.
- F. Marras, J. Wang, M. Coppens and J. N. H. Reek, Chem. Commun., 2010, 46, 6587 RSC.
- T. T. Dang, S. P. Shan, B. Ramalingam and A. M. Seayad, RSC Adv., 2015, 5, 42399–42406 RSC.
- P. W. N. M. van Leeuwen, A. J. Sandee, J. N. H. Reek and P. C. J. Kamer, J. Mol. Catal. A: Chem., 2002, 182–183, 107–123 CrossRef CAS.
- S. Ricken, P. W. Osinski, P. Eilbracht and R. Haag, J. Mol. Catal. A: Chem., 2006, 257, 78–88 CrossRef CAS.
- A. Riisager, P. Wasserscheid, R. van Hal and R. Fehrmann, J. Catal., 2003, 219, 452–455 CrossRef CAS.
- C. Li, K. Sun, W. Wang, L. Yan, X. Sun, Y. Wang, K. Xiong, Z. Zhan, Z. Jiang and Y. Ding, J. Catal., 2017, 353, 123–132 CrossRef CAS.
- W. Li, C. Li, Y. Li, H. Tang, H. Wang, Y. Pan and Y. Ding, Chem. Commun., 2018, 54, 8446–8449 RSC.
- Y. Zhou, Z. Liu, X. Fan, R. Li, G. Zhang, L. Chen, Y. Pan, H. Tang, J. Zeng and Z. Zhan, Org. Lett., 2018, 20, 7748–7752 CrossRef CAS.
- S. Hübner, J. G. de Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3–25 CrossRef.
- B. Li, R. Gong, W. Wang, X. Huang, W. Zhang, H. Li, C. Hu and B. Tan, Macromolecules, 2011, 44, 2410–2414 CrossRef CAS.
- Y. Yang, T. Wang, X. Jing and G. Zhu, J. Mater. Chem. A, 2019, 7, 10004–10009 RSC.
- K. Schute and M. Rose, ChemSusChem, 2015, 8, 3419–3423 CrossRef CAS PubMed.
- A. B. Chaplin, C. Fellay, G. Laurenczy and P. J. Dyson, Organometallics, 2007, 26, 586–593 CrossRef CAS.
- L. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- J. Weber, M. Antonietti and A. Thomas, Macromolecules, 2008, 41, 2880–2885 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. SiemieniewskaI, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- C. Federsel, R. Jackstell, A. Boddien, G. Laurenczy and M. Beller, ChemSusChem, 2010, 3, 1048–1050 CrossRef CAS PubMed.
- Y. Gao, J. K. Kuncheria, H. A. Jenkins, R. J. Puddephatt and G. P. A. Yap, J. Chem. Soc., Dalton Trans., 2000, 18, 3212–3217 RSC.
- A. Kann, H. Hartmann, A. Besmehn, P. J. C. Hausoul and R. Palkovits, ChemSusChem, 2018, 11, 1857–1865 CrossRef CAS PubMed.
- N. D. McNamara and J. C. Hicks, ChemSusChem, 2014, 7, 1114–1124 CrossRef CAS PubMed.
- P. Lara, K. Philippot and B. Chaudret, ChemCatChem, 2013, 5, 28–45 CrossRef CAS.
- S. Navarro-Jaén, J. C. Navarro, L. F. Bobadilla, M. A. Centeno, O. H. Laguna and J. A. Odriozola, Appl. Surf. Sci., 2019, 483, 750–761 CrossRef.
- G. Gunasekar, D. Hyun, P. Natarajan, K. Jung and S. Yoon, Catal. Today, 2016, 265, 52–55 CrossRef.
- K. Park, G. H. Gunasekar, N. Prakash, K. Jung and S. Yoon, ChemSusChem, 2015, 8, 3410–3413 CrossRef CAS PubMed.
- G. H. Gunasekar, K. Park, V. Ganesan, K. Lee, N. Kim, K. Jung and S. Yoon, Chem. Mater., 2017, 29, 6740–6748 CrossRef CAS.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00164h. All data presented in this publication are available from the open access repository Zenodo (https://zenodo.org/doi/10.5281/zenodo.10805184). |
| This journal is © The Royal Society of Chemistry 2024 |