
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile synthesis of polycarbonates from biomass-based eugenol: catalyst optimization for selective copolymerization of CO2 and eugenol to achieve polycarbonates†
Mani
Sengoden
a,
Gulzar A.
Bhat
 *b,
Tristan
Roland
a,
Chia-Min
Hsieh
a and
Donald J.
Darensbourg
*a
*b,
Tristan
Roland
a,
Chia-Min
Hsieh
a and
Donald J.
Darensbourg
*a
aDepartment of Chemistry, Texas A&M University College Station, Texas 77843, USA. E-mail: djdarens@chem.tamu.edu
bCentre for Interdisciplinary Research and Innovations University of Kashmir, Srinagar, Jammu and Kashmir 190006, India. E-mail: gulzarbhat@uok.edu.in
First published on 16th April 2024
Abstract
In recent years, the quest for achieving polycarbonates in a sustainable, atom economical, and green manner has achieved significant momentum. Much focus has been given to epoxide monomers that are derived from renewable feedstocks. Renewable feedstocks are attractive green alternatives to established petroleum-based materials for reducing the dependency of the polymer industry on fossil resources. Herein, we have employed a series of Co and Cr(salen)-based complexes as selective catalysts for the copolymerization of CO2 and eugenol epoxide. We have observed that varying both diamine backbone as well as the substituents on the phenolate rings of the salen ligand, thereby altering the electron density around the metal center, has notable effects on the reactivity and selectivity of polymer formation. It was also observed that a decrease in CO2 pressure led to the formation of cyclic carbonates. Thermal gravimetric analysis (TGA), and differential scanning calorimetry (DSC) studies of the polymers reveal that these polymers are quite stable up to 250 °C and have relatively high glass transition (Tg) temperatures (83 °C). These findings present an easy strategy to prepare polycarbonates by using biobased eugenol monomers, and provide examples for potential sustainable polymer design and synthesis.
Sustainability spotlightCarbon dioxide is one of the greenhouse gases emitted in large quantities, therefore its emission must be reduced for environmental concerns. Solutions to this issue include capture/storage, along with recycling into chemicals. A very promising method for valorization of CO2 involves the preparation of aliphatic polycarbonates from epoxides and CO2. This process not only consumes CO2, but does so in a catalytic process involving low energy and atom economical pathways, consistent with the principles of green chemistry. Herein, we present studies aimed at defining the best catalyst system for selectively producing aliphatic polycarbonates based not only from CO2, but where the epoxide is derived from an inexpensive, non-food renewable resource, eugenol, thereby avoiding petroleum acquired monomers. Nonetheless, we have not at this stage attempted to avoid all petroleum-based reagents in this strategy. The use of easily available, safe, biobased eugenol epoxides, along with carbon dioxide, affords a process for producing useful, high-valued polymeric materials partially/potentially in a sustainable manner. |
Introduction
The necessity for replacing petroleum-based non-biodegradable polymers with natural biomass-derived biodegradable materials (bioplastics) is of much concern in order to reduce the environmental problem of plastic pollution.1 In an effort to assist in this endeavour, the utilization of carbon dioxide as a feedstock for polymer synthesis represents a partial solution, as well as a contribution to reducing CO2 emissions. It is important to note that the utilization of CO2 for the synthesis of chemicals or polymers is only indirectly related to climate change because of the negligible quantity of CO2 emissions used in the absence of the synthesis of fuels from CO2.2 Nevertheless, the production of polymers or chemicals from CO2 can offset the expenses resulting from its capture and storage. A pathway for CO2 usage that has garnered much attention over the last two decades is the copolymerization of CO2 with three- or four-membered cyclic ethers, namely, oxiranes or oxetanes.3–20 In attempts to enhance the sustainability of these processes, it is necessary to employ cyclic ether monomers from natural non-food resources.21Biomass feedstocks can represent environmentally benign substitutes for petroleum derived chemicals, especially when utilized as monomers for the preparation of biobased polymers. A promising phenolic compound is the low viscosity liquid eugenol, which is readily obtained from various sources such as clove oil.22 Of further importance, the U.S. Food and Drug Administration (FDA) considers eugenol as “generally recognized as safe” (GRAS). Indeed, it has numerous applications involving human subjects, e.g., in antimicrobial, antioxidants, anticancer, cosmetics and perfumes, and dental composites.23–27 Important to this report, eugenol has a chemical structure capable of leading to a variety of bio-based monomers for producing polymers with a broad range of thermal and mechanical properties.28 That is, the eugenol molecule contains several reaction sites, including phenolic hydroxyl and methoxy groups, and an allylic double bond. Particular to our needs, eugenol can be easily converted to an epoxide or the substituent to an epoxide via reactions at the allylic or hydroxy groups (Scheme 1).29,30
 | ||
| Scheme 1 Synthetic procedures for two bio-based monomers from eugenol. | ||
Currently, the literature contains several references illustrating the use of eugenol in the synthesis of polymeric materials. Included are the synthesis of methylacrylate polymers,31,32 self-healing polymers,33 and hydroxyethylmethacrylate34 based polymers. Unlike the methacrylate polymers, the latter eugenol derived hydroxyethyl derivative demonstrated significant antioxidant and antimicrobial properties. Previous studies from our laboratory have revealed that in the presence of (salen)MX (M = Co or Cr, X = CH3CO2 or Cl)29 and organo-boron phosphonium salt catalysts,35 coupling of the protected eugenol monomer (A) with CO2 quantitatively produces the corresponding cyclic carbonate. On the other hand, utilizing the chromium catalyst system and the more reactive COS monomer at ambient or 40 °C provided poly(monothiocarbonate) with tail-to-head linkages and no oxygen/sulfur exchange reactions. Similar polymer production was obtained with COS and the unprotected version of the eugenol monomer. It is important to note that in the synthesis of monomer B, epichlorohydrin is available from natural resources.21,30,36
An interesting change in product selectivity in the coupling reaction of EuO and CO2 was noted when the reaction was carried out in the presence of propylene oxide.37 That is, the terpolymerization of PO/EuO/CO2 performed at various ratios of EuO/PO less than 3 provided extensive eugenol carbonate linkages in the polymer chain. These studies also demonstrated via a Fineman-Ross analysis that the reactivity of EuO entering the polymer chain was greater than that of PO. Furthermore, a sequential addition of PO followed by EuO using the (salen)CoX/PPNX (X = CF3CO2) cocatalyst system at ambient temperature and 3.0 MPa of CO2 provided well-defined diblock polymers of poly(propylenecarbonate-b-eugenol carbonate). The origin of this reversal in product selectivity was attributed to the back-biting process leading to cyclic carbonate for EuO/CO2 being retarded once EuO is enchained in the polymer backbone.
It is well-known that product selectivity resulting from the coupling of CO2 and epoxides is highly dependent on the catalyst system utilized in the process, or in other words, “no catalyst system is effective for the selective copolymer formation indiscriminately with all epoxides”38 In this report we have examined a series of metal catalysts for the coupling of CO2 and EuO in an effort to establish the best catalyst system for affording copolymer compared to its cycloaddition product.
Results and discussion
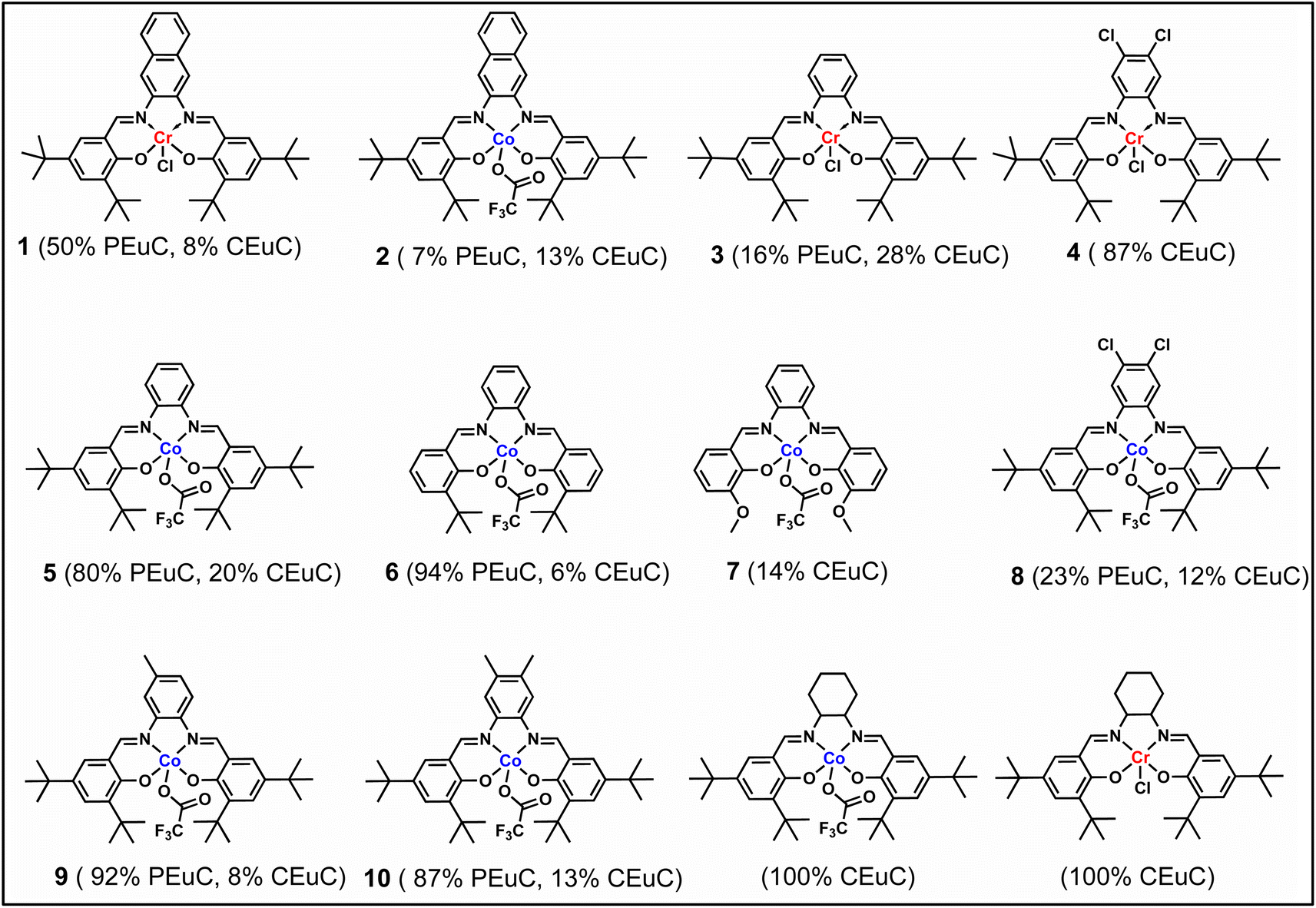
In continuation of our significant interest in selectively producing polycarbonates from the natural product-based eugenol epoxide (EuO), herein, we have synthesized various closely related metal complexes and have tested and compared their effectiveness in catalyzing the copolymerization of CO2 and EuO. As reported recently by our group, we have successfully established the multigram synthesis of biomass-based eugenol-epoxide.29,37 We further modified it by protecting the phenol group (OH) to mitigate the chain-transfer process during this immortal copolymerization.39 This modification, in turn, prevented potential inhibition of polymer production, as previously observed. Fig. 1 lists the M(Salophen)X (M = Co, Cr) complexes employed in this study. After thoroughly characterizing these metal complexes by different spectroscopic and analytical methods (see ESI; Fig. S1–S17†), we have explored their catalytic activity in coupling naturally obtained eugenol epoxides with CO2.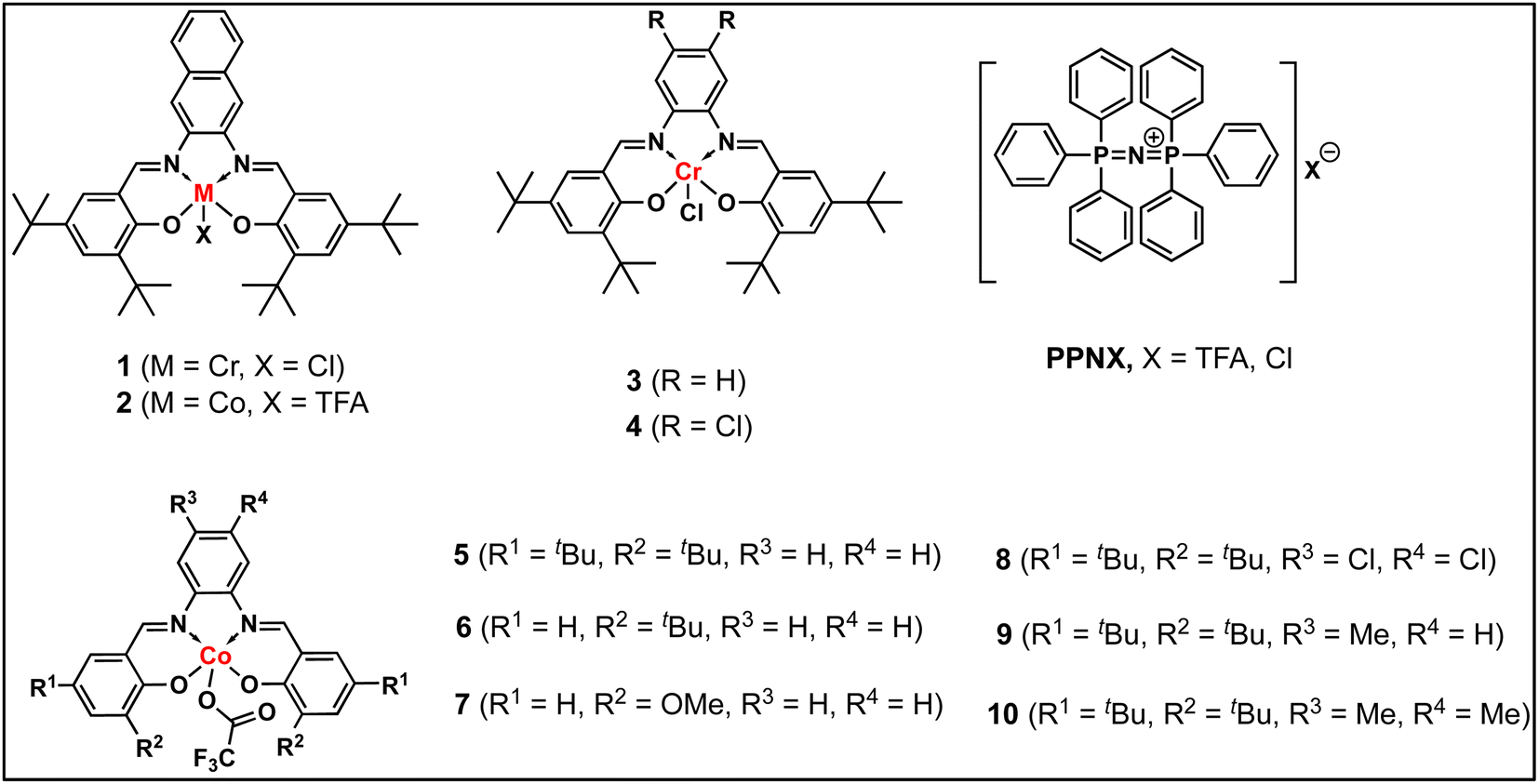 | ||
| Fig. 1 Different M(Salophen)X (M = Co, Cr) metal complexes. | ||
Table 1 summarizes the results of the investigation of utilizing M(Salophen)X/PPNX (M = Co, Cr; X = Cl, CF3CO2) catalytic systems for the coupling of CO2 and EuO at ambient temperature and 3.0 MPa pressure in dichloromethane/toluene over a two day reaction period. As apparent in Table 1, the best results for selective copolymer production are provided by catalyst 6 and 9, similarly catalysts 5 and 10 are also highly selective for copolymer whereas catalysts 4 and 7 provided cyclic carbonates exclusively. Further in-depth studies utilizing complex 9 along with PPNTFA showed that the selectivity for copolymer formation decreased with decreasing CO2 pressure, with only a trace (2% conversion) of cyclic product being provided at atmospheric pressure of CO2. Under identical reaction conditions, these ten closely related catalyst systems displayed a wide range of reactivity and product selectivity. An extended reaction time of two days was employed for comparing catalysts activities, shorter completion times are required for the more reactive catalysts. This highlights our earlier investigations regarding the reactivity and selectivity of the copolymerization reaction being highly sensitive to the catalyst system and nature of epoxide. We have also performed the time dependent kinetic experiments using three catalysts (5, 8 and 10) by stirring the reaction mixture at room temperature for the designated duration and confirming the conversions via1H NMR spectroscopy (Fig. 2 and Table S2†).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Co-catalyst | CO2 (MPa) | Conv. (%) | Selectivity | Carbonate linkages | M n [kg mol−1] | Đ | |
| PEuC | CEuC | ||||||||
| 1 | 1 | PPNCl | 3.0 | 58 | 50 | 8 | 99 | 17.7 | 1.31 |
| 2 | 2 | PPNTFA | 3.0 | 20 | 7 | 13 | — | — | — |
| 3 | 3 | PPNCl | 3.0 | 44 | 16 | 28 | 99 | — | — |
| 4 | 4 | PPNCl | 3.0 | 87 | — | 87 | — | — | — |
| 5 | 5 | PPNTFA | 3.0 | 100 | 80 | 20 | 99 | 27.1 | 1.15 |
| 6 | 6 | PPNTFA | 3.0 | 100 | 94 | 6 | 99 | 48.6 | 1.13 |
| 7 | 7 | PPNTFA | 3.0 | 14 | — | 14 | — | — | — |
| 8 | 8 | PPNTFA | 3.0 | 35 | 23 | 12 | 99 | — | — |
| 9 | 9 | PPNTFA | 3.0 | 100 | 92 | 8 | 99 | 45.8 | 1.13 |
| 10 | 9 | PPNTFA | 1.5 | 100 | 81 | 19 | 99 | 39.7 | 1.09 |
| 11 | 9 | PPNTFA | 1.0 | 100 | 81 | 19 | 99 | 34.7 | 1.13 |
| 12 | 9 | PPNTFA | 0.5 | 100 | 77 | 23 | 99 | 28.2 | 1.15 |
| 13 | 10 | PPNTFA | 3.0 | 100 | 87 | 13 | 99 | 37.1 | 1.12 |
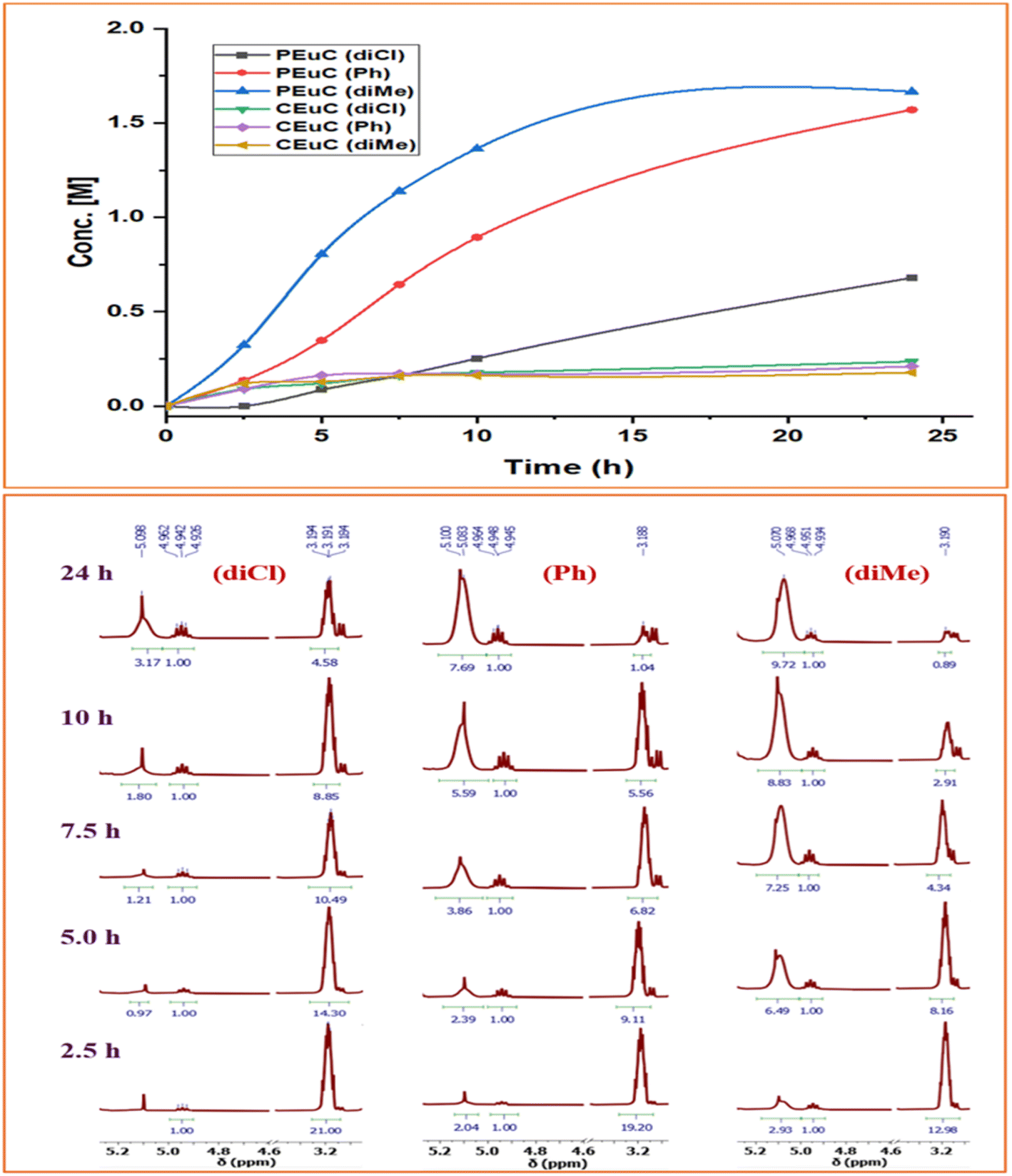 | ||
| Fig. 2 Time dependent plots illustrating the kinetic behaviour shown by these Co(III) catalyst systems having different backbone structures exhibiting different product selectivity. | ||
The copolymers produced were characterized by different spectroscopic techniques, i.e., 1H NMR, 13C NMR, and FT-IR analysis which strongly advocates their formation (Fig. S18–21†). For example, in the FT-IR spectrum an intense band centered around 1751 cm−1 is observed which is the characteristic peak for carbonate linkages (Fig. S18†). In 1H NMR spectrum peaks at δ = 5.04 ppm and δ = 4.26 ppm corresponds to methine and methylene protons of the polycarbonate linkages in the polymer backbone (Fig. 3a). Additionally, the presence of the carbonate linkages are confirmed by the characteristic carbonate carbonyl peak at δ = 154 ppm in the 13C NMR spectrum, along with other distinctive peaks (Fig. 3b). To further understand the structural features of these polycarbonates, the copolymers obtained by using 1,2 propanediol as chain transfer agent were subjected to MALDI-ToF analysis (Fig. 4a). The resulting MALDI-ToF spectrum reveals two distinctive sets of peak, with a monomodal molecular weight distribution, consistent with the observations from gel permeation chromatography (GPC) traces (Fig. 4b). Each series exhibited a consistent separation of 308 m/z aligning with the mass of repeating unit of protected eugenol epoxide and CO2 within the polycarbonate chain. The series denoted by green triangles is attributed to copolymer initiated by the chain transfer agent (1, 2 propanediol) and its mass corresponds to the potassium adduct of the alternating copolymer. The other series (denoted by red circles) is assigned to the protic adduct of the alternating copolymer, also initiated by the chain transfer agent. The GPC further revealed that high molecular weight polymers with Mn up to 84.8 kg mol−1 and polydispersity (Mw/Mn) of 1.13 can be obtained when higher equivalents of eugenol epoxides are used (Fig. S23†). The effect of CO2 pressure on molecular weight of these copolymers was probed by GPC (Fig. S22†), which revealed that on increasing the CO2 pressure from 0.5 MPa to 3 MPa the molecular weight (Mn) of polymers increases from 28.2 to 45.8 kg mol−1.
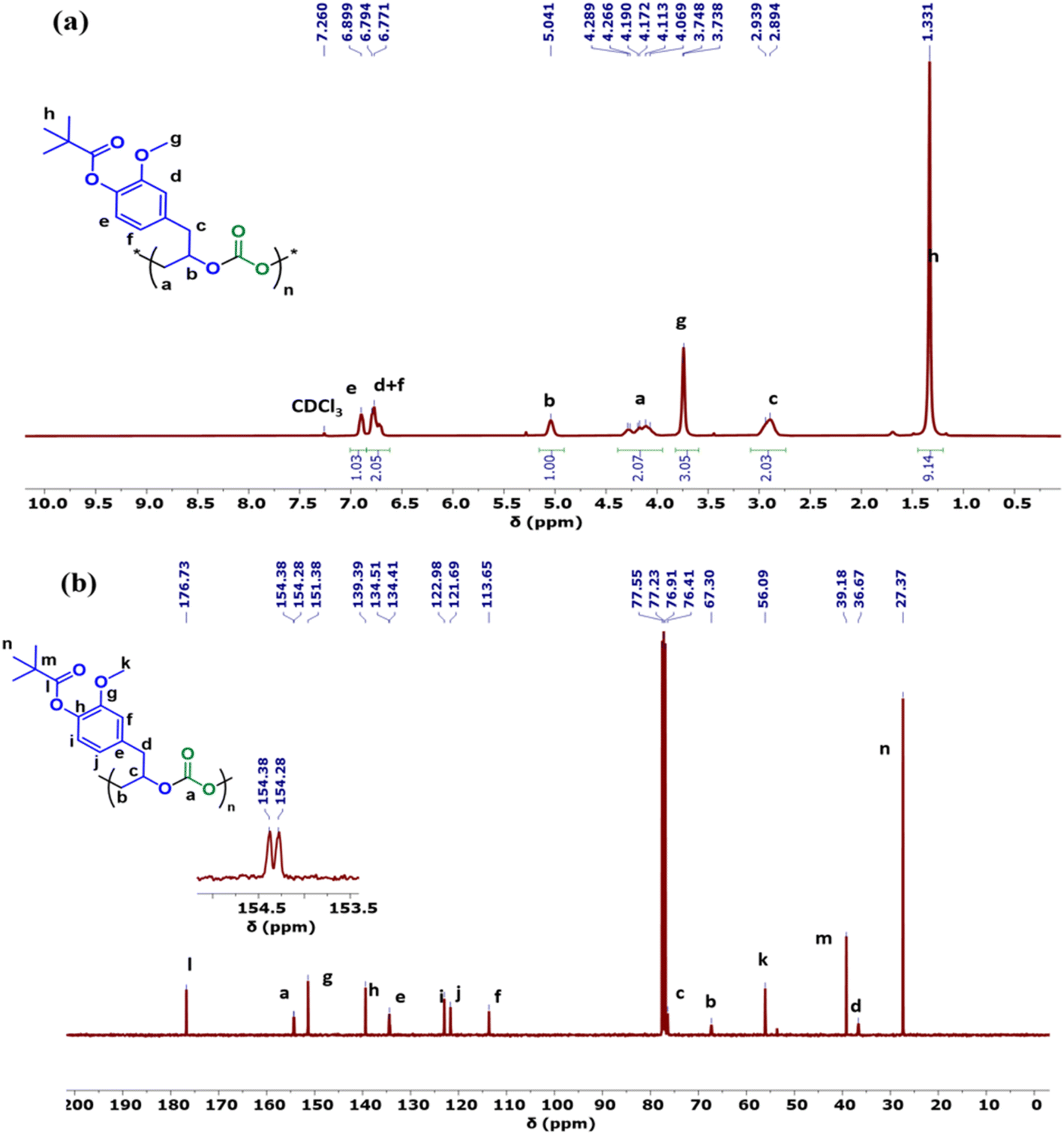 | ||
| Fig. 3 (a) 1H and (b) 13C NMR spectra of polycarbonate polymer. | ||
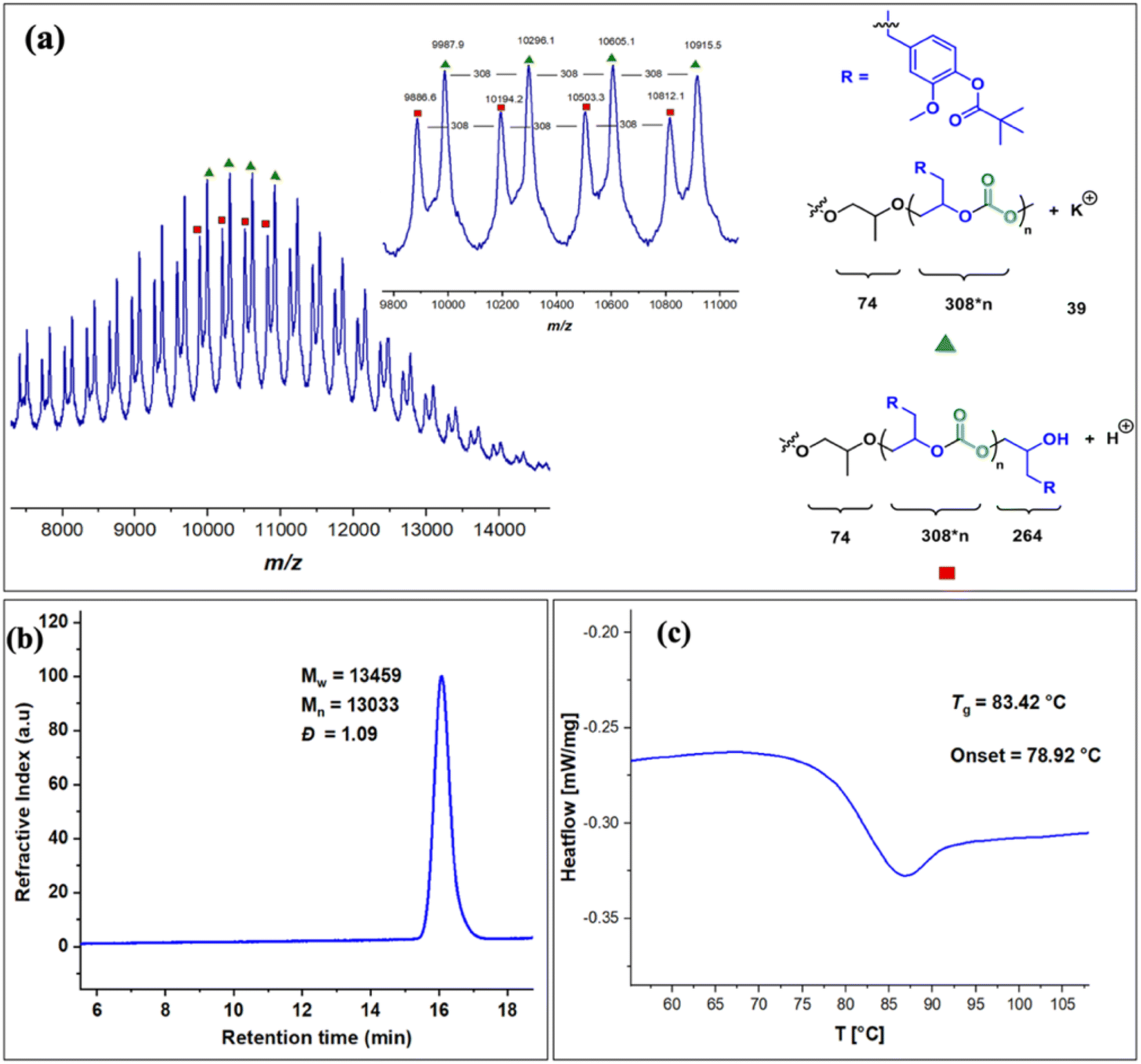 | ||
| Fig. 4 (a). MALDI-ToF spectrum of EuO/CO2 copolymer using 1,2 propanediol as chain transfer agent. (b) GPC traces for copolymer synthesized by using 5 equivalents of 1,2 propanediol as chain transfer agent during EuO/CO2 copolymerization (c) DSC curve for the polycarbonate of EuO with CO2. | ||
The thermal stability of these copolymers was established by performing the thermogravimetric analysis (TGA) within the temperature range of 30–500 °C, under a dry nitrogen gas flow. The TGA traces of these copolymers revealed that the polymers are stable up to 250 °C (Fig. S24†). The copolymer exhibited a moderately high glass-transition temperature (Tg) of approximately 83.4 °C, similar to our recently reported EuO/COS copolymer (75.5 °C) as revealed by DSC (Fig. 4c). This notable Tg is comparable to the copolymers derived from petroleum-based counterparts involving styrene oxide (76 °C) or 2-benzyloxirane (78 °C), positions our polycarbonate as an intriguing candidate for diverse applications. We have further also demonstrated that it is possible to engineer the Tg of these copolymers by varying the EuO content which can be pivotal for achieving different functions (Fig. S25†).
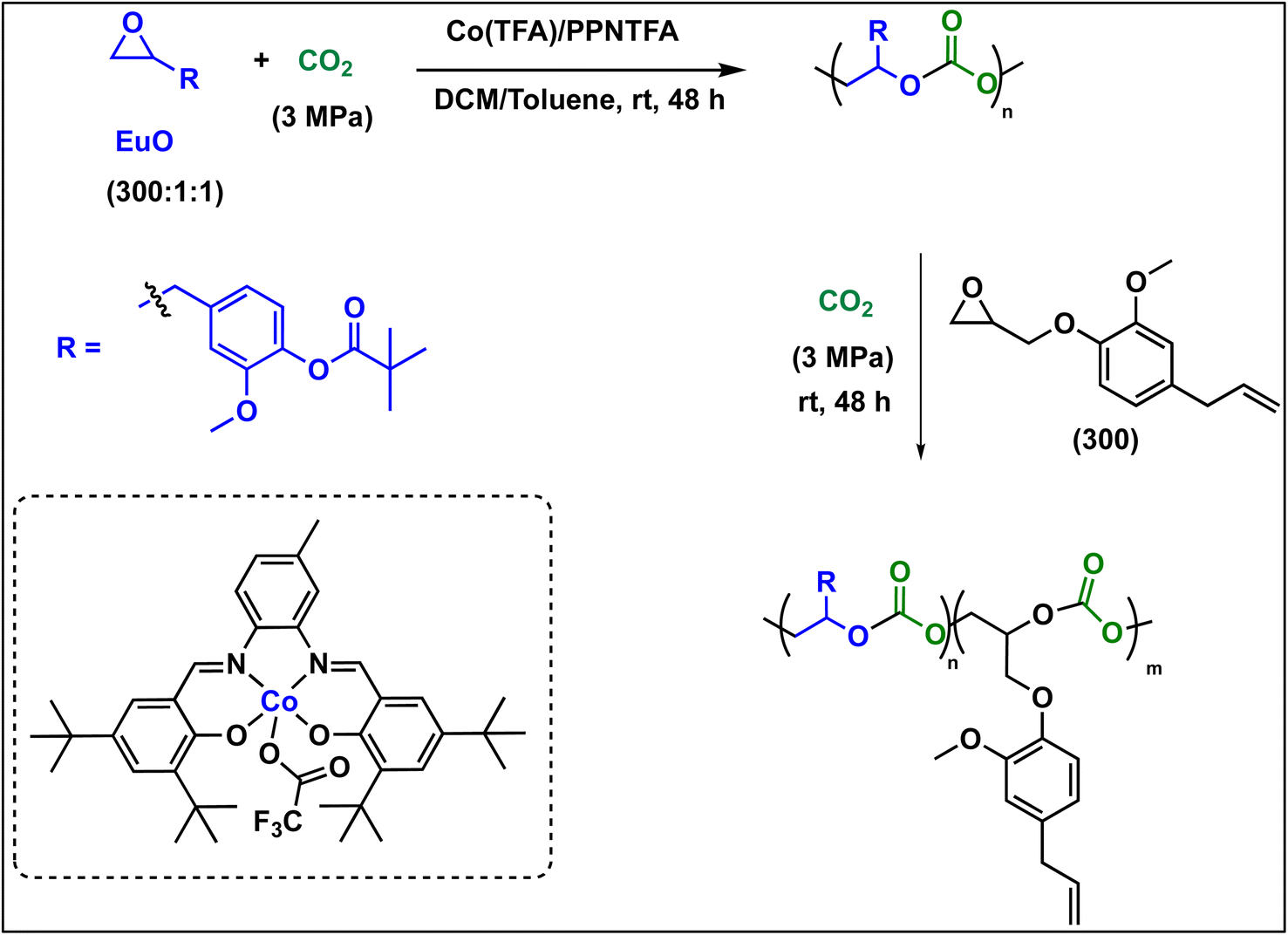
To further introduce the post polymerization functionalization prospectus in these eugenol-based copolymers we have synthesized the double bond functional group containing eugenol glycidyl ether epoxide (EGE) (B) in a one-pot synthesis by employing a previously reported procedure (Scheme S3†).40 After thoroughly establishing the purity of this monomer by different spectroscopic and analytical techniques (see ESI Fig. S26–S29†), we have explored its copolymerization reaction with CO2 using above binary M(Salophen)X/PPNX catalytic systems (Scheme 2). The copolymerization process revealed that among the ten catalysts some selectivity led to the formation of cyclic (CEGC) products (1–3 and 8), whereas others led to the formation of both cyclic or polymeric products (5, 6, 9 and 10), with catalysts 4 and 7 showing no results (Table S1†). The catalyst structure of our standard cyclohexane diamine backbone-based catalysts along with their % selectivity towards cyclic or polymeric products are shown in Scheme 3.
 | ||
| Scheme 2 Summary of product selectivity of EuO/CO2 copolymerization reaction towards cyclic or polymeric products catalysed by different M(Salophen)X/PPNX (M = Co, Cr; X = Cl, CF3CO2) (1–10) catalysts. | ||
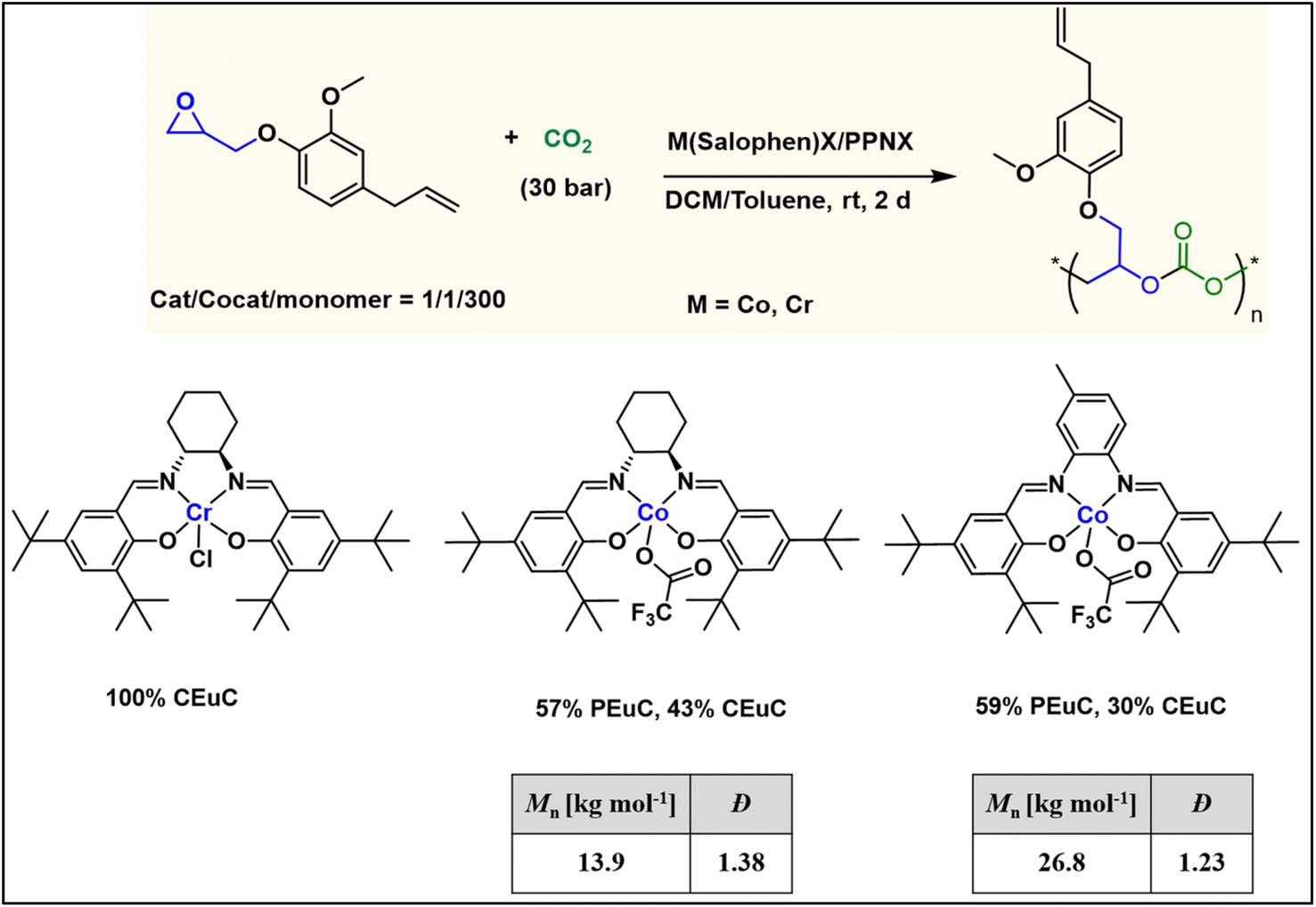 | ||
| Scheme 3 (above) Copolymerization of eugenol glycidyl ether (EGE) with CO2 to form cyclic or polymeric products. (below) Summary of product selectivity of EGE/CO2 copolymerization reaction towards cyclic or polymeric products catalysed by our standard salen catalysts. | ||
The relative proportion of cyclic and polymeric products were established using FT-IR and NMR spectroscopy. The copolymer PEGC showed characteristic carbonate peaks in FT-IR around 1756 cm−1, whereas the cyclic carbonate product (CEuC) peak appears around 1805 cm−1 (Fig. 5a). The copolymers were further thoroughly characterized by spectroscopic techniques including 1H NMR, 13C NMR, 13C (DEPT-135) NMR, which strongly supported their formation. For example, in the 1H NMR spectrum besides showing the characteristic peaks for polymer formation, a quintet peak at δ = 5.9 ppm and merged doublet around δ = 5.2 ppm corresponds to the presence of double bonds in these copolymers (Fig. 5b). The 13C NMR spectrum, along with showing characteristic carbonate carbon peak at δ = 154 ppm, also shows peaks at δ = 115 and 138 ppm for double bond carbons and other distinctive peaks (Fig. 5c). The 13C NMR spectrum obtained through DEPT-135 analysis of these copolymers shows distinctive peaks in both positive and negative directions reflecting diverse carbon environments due to the presence of methyl, methylene and methine carbons (Fig. 5d).
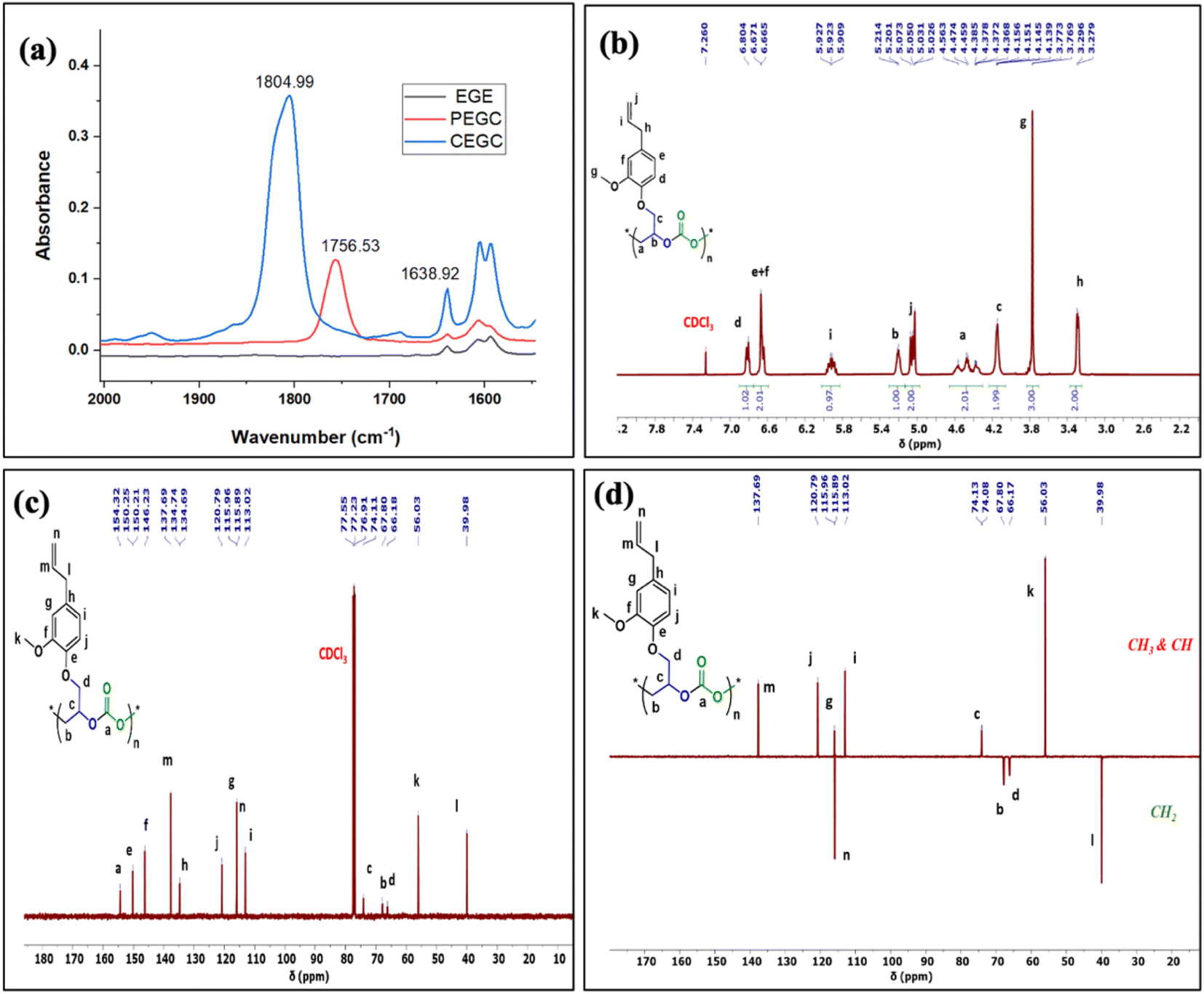 | ||
| Fig. 5 (a) Comparison of ATR FT-IR spectrum of EGE, cyclic carbonate (CEGC), and polycarbonate (PEGC) in CH2Cl2. (b) 1H and (c) 13C NMR and (d) 13C (DEPT-135) spectra of polycarbonate polymers of EGE/CO2 in CDCl3. | ||
The molecular weight distributions of these copolymers were confirmed by using gel permeation chromatography which revealed that copolymers with Mn up to 27 kg mol−1 and polydispersity (Mw/Mn) of 1.26 can be achieved which are smaller compared to EuO (A) under similar conditions. GPC also revealed that lower molecular weight polymers (Mn = 6.2 kg mol−1) are obtained by using terephthalic acid as a CTA (Fig. 6b and S34†). Structural characterization using MALDI-ToF analysis shows two distinct series with a monomodal molecular weight distribution as also supported by GPC trace (Fig. 6a). Each series exhibited a consistent separation of 264 m/z aligning with the mass of repeating unit of EGE epoxide and CO2 within the polycarbonate chain. Both the series are initiated by the CTA and their masses corresponds to the protic adducts. The glass transition temperature of these copolymers as revealed by DSC is around 24 °C which is much lower than EuO/CO2 or EuO/COS copolymers (Fig. 6c).
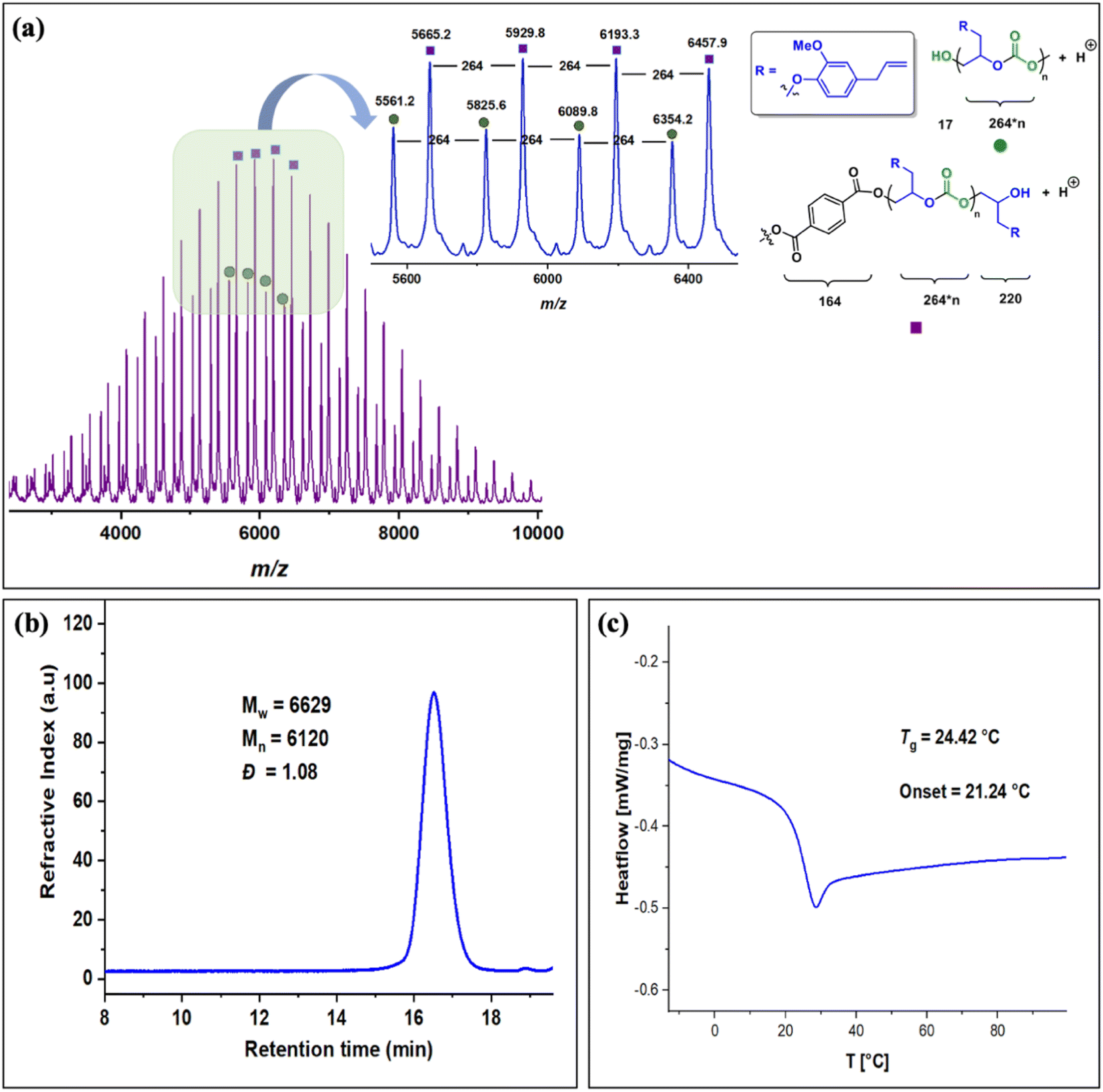 | ||
| Fig. 6 (a). MALDI-ToF spectrum of EGE/CO2 copolymer (obtained using terephthalic acid as chain transfer agent) (b) GPC traces for copolymer synthesized by using 5 equivalents of terephthalic acid as CTA during EGE/CO2 coupling (c) DSC curve for the polycarbonate of EGE with CO2. | ||
We have initially employed a sequential one pot-two step protocol for achieving the block copolymers between EuO/EGE/CO2 using Co(salen)TFA/PPNTFA binary catalyst systems at ambient temperature and 3 MPa CO2 pressure (Scheme 4). These block polymers we have characterized by 1H-NMR (Fig. S35†) and increase in Mn was observed from 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 to 52800 during the addition of EGE/CO2 block to EuO/CO2 (Fig. S36†). The diffusion ordered spectroscopy (DOSY) map of the PEuC–PEGC block polymer in CDCl3 is shown in Fig. 7. Signals corresponding to the PEuC block (δ = 6.89, 5.03, 4.13, 2.89 and 1.32 ppm) and PEGC block (δ = 6.71, 5.19, 4.26, and 3.28 ppm) were observed in the 1H NMR spectrum. Conspicuously, both the PEuC and PEGC blocks exhibited identical diffusion coefficients (D = 1.017 × 10−10 m2 s−1), providing strong evidence for the formation of a block copolymer.
900 to 52800 during the addition of EGE/CO2 block to EuO/CO2 (Fig. S36†). The diffusion ordered spectroscopy (DOSY) map of the PEuC–PEGC block polymer in CDCl3 is shown in Fig. 7. Signals corresponding to the PEuC block (δ = 6.89, 5.03, 4.13, 2.89 and 1.32 ppm) and PEGC block (δ = 6.71, 5.19, 4.26, and 3.28 ppm) were observed in the 1H NMR spectrum. Conspicuously, both the PEuC and PEGC blocks exhibited identical diffusion coefficients (D = 1.017 × 10−10 m2 s−1), providing strong evidence for the formation of a block copolymer.
 | ||
| Scheme 4 Sequential block polymer formation between (EuO/EGE)/CO2. | ||
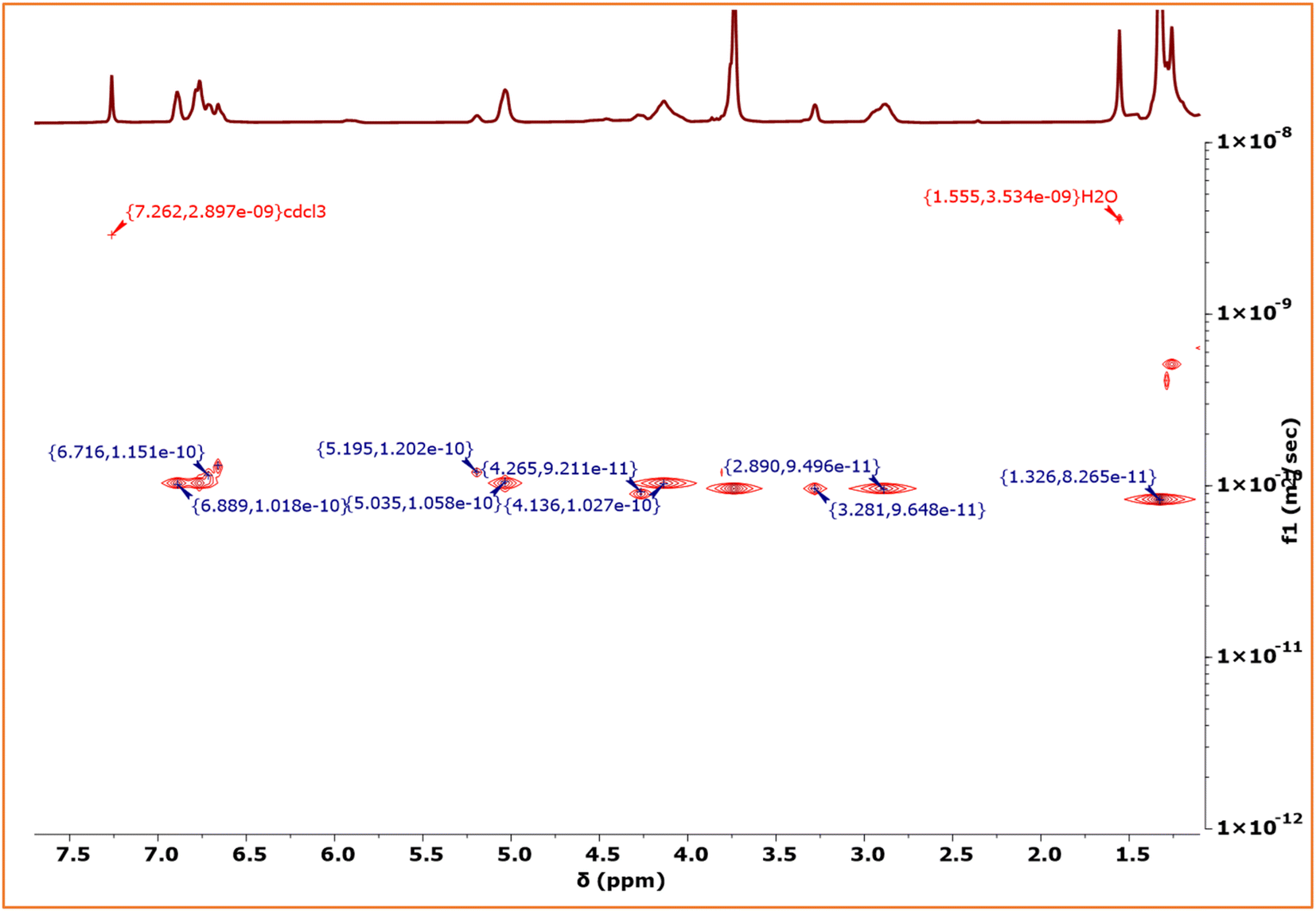 | ||
| Fig. 7 DOSY NMR spectrum of diblock polymer (CDCl3, 500 MHz). | ||
In order to compare with diblock polymers derived from petroleum-based monomers with those from these biomass monomers, we have initiated studies employing diblock polymer produced from EuO/CO2 and EGE/CO2 for 3D printing. As shown earlier, diblock polymers of these type not only allow for tuning of the thermal and mechanical properties of the polymeric materials, but also contain pendant groups for crosslinking or further modification.41 That is, these polymers were examined for 3D printing by preparing a resin by dissolving the diblock polymer in propylene carbonate and ethyl acetate, followed by the addition of reactive diluents (polyethylene glycol diacrylate and triallyl isocyanurate), crosslinker (pentaerythritol tetrakis(3-mercaptopropionate)), photoinitiator (phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide), and radical inhibitor (pyrogallol) (Fig. 8a). The resin was then centrifuged at 2000 rpm to remove air bubbles. The tensile tests of the printed tensile bars were performed to evaluate the mechanical properties of the polymer. The printed polymer exhibited an average Young's modulus of 13.5 MPa with high replicability (Fig. 8b). Research efforts in this area are a subject of ongoing interests.
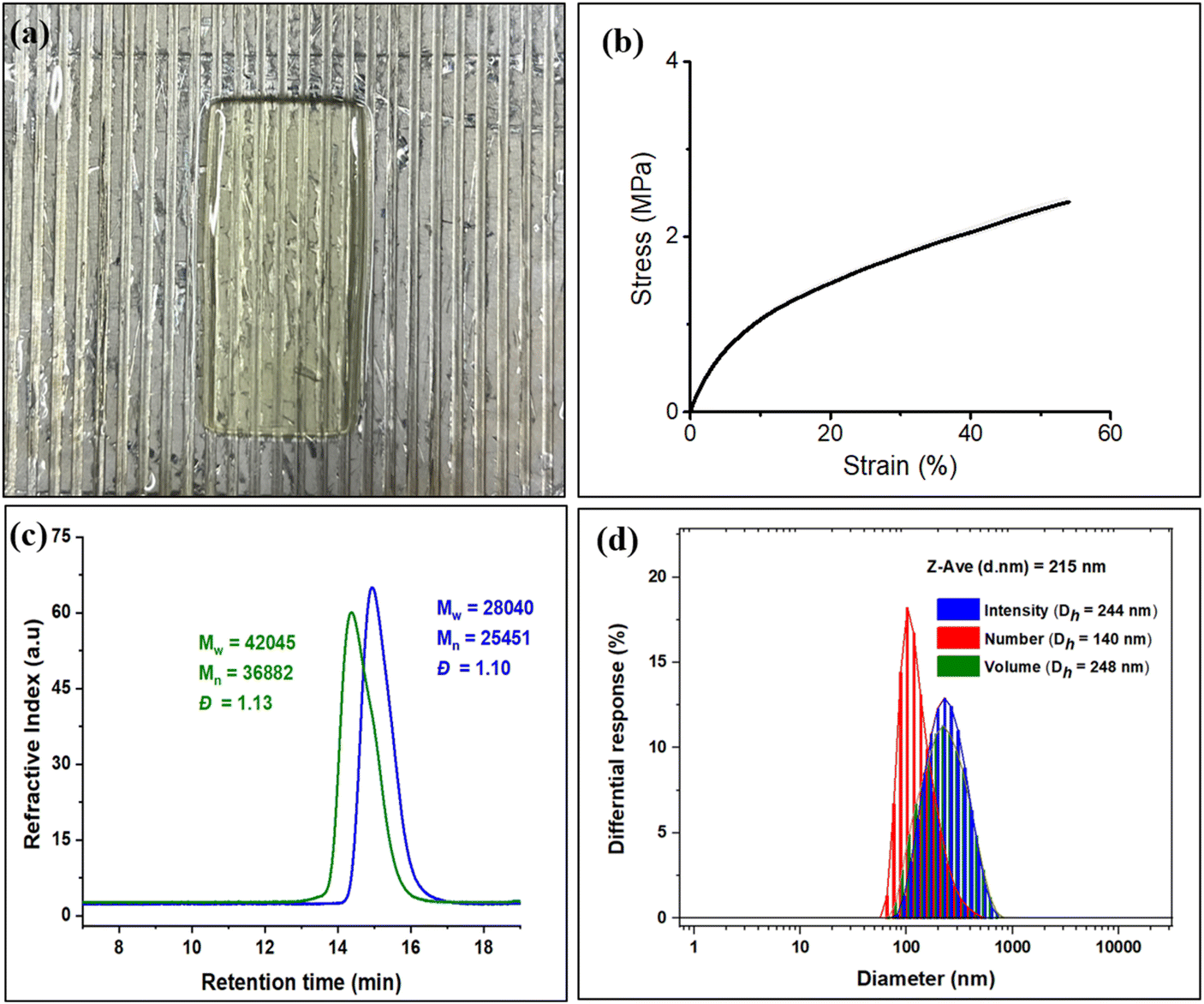 | ||
| Fig. 8 (a) Resin formed from the polymer after removing the air bubbles. (b) Stress–Strain curve of the printed bars. (c) GPC traces for triblock copolymer synthesized by using EuO, EGE with CO2 (green color) and GPC of copolymer of EuO with CO2 (blue color). (d) DLS data of polymer solution (5 mg in 10 mL of deionized water). | ||
To make this approach more versatile, we have later extended this block polymer approach for achieving the ABA triblock copolymers from EuO/CO2 and EGE/CO2 using 1,4 dicarboxylic acid as chain transfer agent at ambient temperature and 3 MPa CO2 pressure (Scheme 5). The ABA triblock synthesis involves coupling of EuO/CO2 using 9/PPNTFA binary catalyst system, yielding dihydroxy end-capped poly(eugenol carbonate), or HO–PEuC–OH homopolymer. After full conversion of EuO, the unreacted CO2 was carefully vented, then EGE was added to the reactor, and it was recharged with CO2. Thus, HO–PEuC–OH served as a telechelic macro chain transfer agent for the growth of poly(eugenol glycidyl ether carbonate) (PEGE) blocks, resulting in the formation of PEGC-b-PEuC-b-PEGC. Confirmation of block polymer synthesis was achieved through various spectroscopic techniques, such as NMR and FTIR, (Fig. S38–S41†) and further validated by GPC, where an increase in molecular weight (Mn) from 25 K to 36 K was observed (Fig. 8c). Subsequent thiol–ene click chemistry of the ABA triblock polymers with mercaptoacetic acid, followed by deprotonation with NH4OH, led to the self-assembly of the terpolymer into nano-micelles in water, representing a unique strategy utilizing sustainable natural resources. The deprotonated polymer dispersion in deionized water was achieved through sonication at ambient temperature. The morphology of these micelles was characterized using dynamic light scattering (DLS) and transmission electron microscopy (TEM) studies. DLS measurements of the negatively charged triblock amphiphilic polymers, as shown in Fig. 8d, demonstrated that the amphiphilic polymer self-assembles in deionized water, forming stable nanoparticles with uniform size distribution, with an intensity-averaged hydrodynamic diameter of 215 nm which is also corroborated by the TEM measurements (Fig. S42†). Again, this study was undertaken to illustrate the use of these bio-based eugenol monomers as replacements for petroleum-based monomers in the preparation of amphiphilic polymers for applications in micellar catalysis and biomedicine.42–46
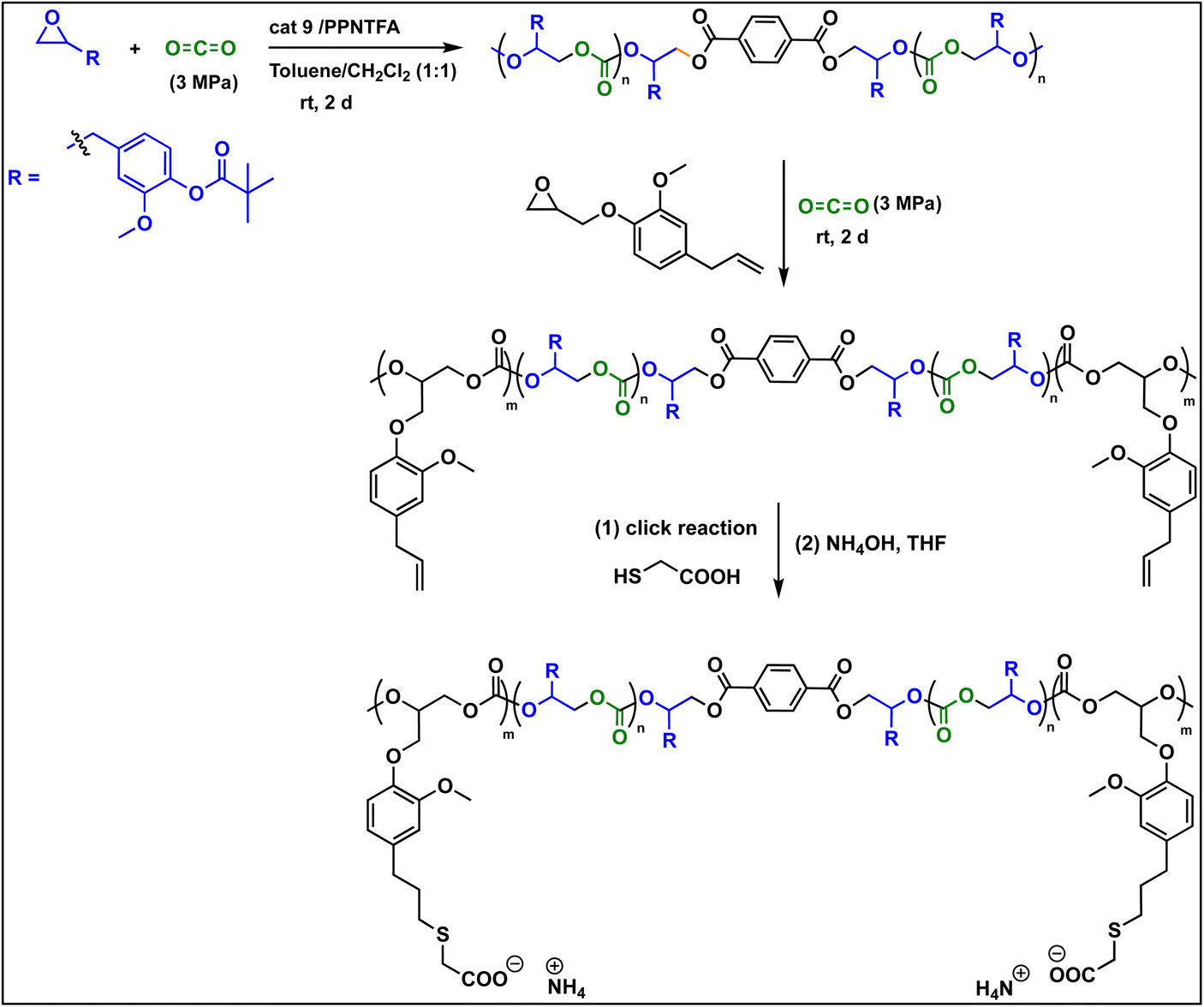 | ||
| Scheme 5 Sequential block polymer formation between (EuO/EGE)/CO2. | ||
Conclusions
Herein, we have described several closely related metal complexes as being highly reactive and selective catalysts in the presence of onium salts for the coupling of epoxide monomers derived from the natural product eugenol and carbon dioxide. Nevertheless, we acknowledge the fact that we have not at this stage designed a strategy for making this process truly sustainable by avoiding all petroleum-based reagents.47 Several of these complexes afford high levels of copolymers or cyclic carbonates depending on the electron density at the metal center. Included in these studies is the synthesis of a triblock polymer of PEGC-b-PEuC-b-PEGC which upon undergoing thiol–ene click chemistry with mercaptoacetic acid and deprotonation with NH4OH, provides nano-micelles in water. This represents an excellent strategy for the incorporation of metal catalysts via CTAs for performing various reactions in aqueous medium as we have previously described.42,43,45,46 However, in this instance the polymeric material is derived completely from renewable resources. Some of the important lessons gleamed from these studies that are consistent with the reactivity and selectivity trends noted by these catalyst systems are: phenylene moieties of M(salophen)X complexes are less steric hindering at the metal center than their cyclohexylene analogs consistent with solid-state structures,29,48 this is especially important for epoxides containing bulky substituents. The reactivity and selectivity for copolymer production of the series of Co(salophen)X catalysts increased with increasing electron density at the Co(III) center, with complexes 6 ≈ 9 > 10 > 5 ≫ 4 and 7.49 Cyclic carbonate production via back-biting from anionic alkoxide chain ends is faster than that of anionic carbonate ends, which accounts for an increase in cyclic carbonate formation with a decrease in CO2 pressure.50,51 Finally, COS insertion is faster and much less reversible and less impacted by steric hinderance at the metal center than CO2 insertion resulting in greater selectivity for copolymer compared to CO2, where the anionic alkoxide chain end has a longer lifetime.52,53Conflicts of interest
The authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
DJD gratefully acknowledges the financial support from Robert A. Welch Foundation (A-0923), and GAB gratefully acknowledges DST-SERB for Ramanujan fellowship Grant (RJF 2020/000116) and core research Grant (CRG/2022/003558). The authors also thank Krista Schoonover, Department of Chemistry, Texas A&M University for helping in DOSY NMR experiment. The use of Texas A&M University Microscopy and Imaging Center Core Facility (RRID:SCR_022128) is acknowledged. Triston Roland was an undergraduate summer researcher from Prairie View A&M University.References
- R. Mori, RSC Sustainability, 2023, 1, 179–212 RSC.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC.
- D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef CAS.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed.
- H. Sugimoto and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5561–5573 CrossRef CAS.
- D. J. Darensbourg, R. M. Mackiewicz, A. L. Phelps and D. R. Billodeaux, Acc. Chem. Res., 2004, 37, 836–844 CrossRef CAS.
- M. H. Chisholm and Z. Zhou, J. Mater. Chem., 2004, 14, 3081–3092 RSC.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- X.-B. Lu, W.-M. Ren and G.-P. Wu, Acc. Chem. Res., 2012, 45, 1721–1735 CrossRef CAS PubMed.
- D. J. Darensbourg and S. J. Wilson, Green Chem., 2012, 14, 2665–2671 RSC.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- G. Trott, P. Saini and C. Williams, Philos. Trans. R. Soc., A, 2016, 374, 20150085 CrossRef.
- C. M. Kozak, K. Ambrose and T. S. Anderson, Coord. Chem. Rev., 2018, 376, 565–587 CrossRef CAS.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- J. Huang, J. C. Worch, A. P. Dove and O. Coulembier, ChemSusChem, 2020, 13, 469–487 CrossRef CAS.
- Y.-Y. Zhang, G.-P. Wu and D. J. Darensbourg, Trends Chem., 2020, 2, 750–763 CrossRef CAS.
- G. A. Bhat, M. Luo and D. J. Darensbourg, Green Chem., 2020, 22, 7707–7724 RSC.
- S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990–5011 RSC.
- A. A. Khalil, U. u. Rahman, M. R. Khan, A. Sahar, T. Mehmood and M. Khan, RSC Adv., 2017, 7, 32669–32681 RSC.
- A. E. Kaplan, M. Picca, M. I. Gonzalez, R. L. Macchi and S. L. Molgatini, Dent. Traumatol., 1999, 15, 42–45 CrossRef CAS PubMed.
- L. Rojo, B. Vázquez, S. Deb and J. San Román, Acta Biomater., 2009, 5, 1616–1625 CrossRef CAS PubMed.
- A. L. Carbone-Howell, N. D. Stebbins and K. E. Uhrich, Biomacromolecules, 2014, 15, 1889–1895 CrossRef CAS PubMed.
- L. Rojo, J. M. Barcenilla, B. Vázquez, R. González and J. San Román, Biomacromolecules, 2008, 9, 2530–2535 CrossRef CAS PubMed.
- S. Woranuch and R. Yoksan, Carbohydr. Polym., 2013, 96, 586–592 CrossRef CAS.
- R. Morales-Cerrada, S. Molina-Gutierrez, P. Lacroix-Desmazes and S. Caillol, Biomacromolecules, 2021, 22, 3625–3648 CrossRef CAS PubMed.
- M. Sengoden, G. A. Bhat and D. J. Darensbourg, Green Chem., 2022, 24, 2535–2541 RSC.
- R. Morodo, R. Gérardy, G. Petit and J.-C. M. Monbaliu, Green Chem., 2019, 21, 4422–4433 RSC.
- A.-B. Al-Odayni, W. S. Saeed, A. Y. B. H. Ahmed, A. Alrahlah, A. Al-Kahtani and T. Aouak, Polymers, 2020, 12, 160–188 CrossRef CAS PubMed.
- S. Molina-Gutiérrez, S. Dalle Vacche, A. Vitale, V. Ladmiral, S. Caillol, R. Bongiovanni and P. Lacroix-Desmazes, Molecules, 2020, 25, 3444–3461 CrossRef PubMed.
- C. Cheng, X. Zhang, X. Chen, J. Li, Q. Huang, Z. Hu and Y. Tu, J. Polym. Res., 2016, 23, 110–122 CrossRef.
- M. Di Consiglio, E. Sturabotti, B. Brugnoli, A. Piozzi, L. M. Migneco and I. Francolini, Polym. Chem., 2023, 14, 432–442 RSC.
- M. Sengoden, G. A. Bhat and D. J. Darensbourg, RSC Adv., 2022, 12, 32440–32447 RSC.
- G. M. Lari, G. Pastore, M. Haus, Y. Ding, S. Papadokonstantakis, C. Mondelli and J. Pérez-Ramírez, Energy Environ. Sci., 2018, 11, 1012–1029 RSC.
- M. Sengoden, G. A. Bhat and D. J. Darensbourg, Macromolecules, 2023, 56, 2362–2369 CrossRef CAS.
- G. A. Bhat and D. J. Darensbourg, Green Chem., 2022, 24, 5007–5034 RSC.
- D. J. Darensbourg, Green Chem., 2019, 21, 2214–2223 RSC.
- S. Le Luyer, B. Quienne, M. Bouzaid, P. Guégan, S. Caillol and N. Illy, Green Chem., 2021, 23, 7743–7750 RSC.
- P. Wei, G. A. Bhat, C. E. Cipriani, H. Mohammad, K. Schoonover, E. B. Pentzer and D. J. Darensbourg, Angew. Chem., Int. Ed., 2022, 61, e202208355 CrossRef CAS PubMed.
- T. M. Folsom, G. A. Bhat, A. Z. Rashad and D. J. Darensbourg, Macromolecules, 2019, 52, 5217–5222 CrossRef CAS.
- G. A. Bhat, A. Z. Rashad, T. M. Folsom and D. J. Darensbourg, Organometallics, 2020, 39, 1612–1618 CrossRef CAS.
- Y. Li, S. Liu, X. Zhao, Y. Wang, J. Liu, X. Wang and L. Lu, Theranostics, 2017, 7, 4689 CrossRef CAS PubMed.
- F. Y. Ren, K. Chen, L. Q. Qiu, J. M. Chen, D. J. Darensbourg and L. N. He, Angew. Chem., 2022, 134, e202200751 CrossRef.
- M. Sengoden, G. A. Bhat, R. J. Rutledge, S. Rashid, A. A. Dar and D. J. Darensbourg, Proc. Natl. Acad. Sci. U.S.A., 2023, 120, e2312907120 CrossRef CAS PubMed.
- T. Theerathanagorn, T. Kessaratikoon, H. U. Rehman, V. D'Elia and D. Crespy, Chin. J. Chem., 2024, 42, 652–685 CrossRef CAS.
- D. J. Darensbourg, R. M. Mackiewicz, J. L. Rodgers, C. C. Fang, D. R. Billodeaux and J. H. Reibenspies, Inorg. Chem., 2004, 43, 6024–6034 CrossRef CAS PubMed.
- W. Lindeboom, A. C. Deacy, A. Phanopoulos, A. Buchard and C. K. Williams, Angew. Chem., Int. Ed., 2023, 62, e202308378 CrossRef CAS PubMed.
- D. J. Darensbourg and A. D. Yeung, Polym. Chem., 2014, 5, 3949–3962 RSC.
- A. C. Deacy, A. Phanopoulos, W. Lindeboom, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2022, 144, 17929–17938 CrossRef CAS PubMed.
- D. J. Darensbourg, B. L. Mueller, C. J. Bischoff, S. S. Chojnacki and J. H. Reibenspies, Inorg. Chem., 1991, 30, 2418–2424 CrossRef CAS.
- D. J. Darensbourg, W.-Z. Lee, A. L. Phelps and E. Guidry, Organometallics, 2003, 22, 5585–5588 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00145a |
| This journal is © The Royal Society of Chemistry 2024 |