
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photopolymerization of lichen derived usnic acid†
Ruby R.
Zhou
 a,
Jack L.
Vargo
a,
Bella G.
Andjelkovic
a,
Maya G.
Vermeer
a,
Spencer J.
Goyette
b and
Bassil M.
El-Zaatari
*a
a,
Jack L.
Vargo
a,
Bella G.
Andjelkovic
a,
Maya G.
Vermeer
a,
Spencer J.
Goyette
b and
Bassil M.
El-Zaatari
*a
aDavidson College, Department of Chemistry, Box 5000, Davidson, North Carolina 28035, USA. E-mail: baelzaatari@davidson.edu
bBeaty Biodiversity Museum, The University of British Columbia, 2212 Main Mall, Vancouver, British Columbia V6T 1Z4, Canada
First published on 27th February 2024
Abstract
Exploration of natural sources for polymer synthesis is critical. We report the use of a lichen secondary metabolite, (+)-usnic acid, as a precursor for polymer network formation. The networks are synthesized via the use of visible light thiol–ene photo click chemistry, proceed under high conversions and possess glass transition temperatures above room temperature.
Sustainability spotlightCross-linked polymer networks are ubiquitous in everyday life. However, the predominant method for producing these materials has been historically centered around petroleum-based resources. As we are faced with the challenge of climate change along with a plastic accumulation problem, we are prompted to investigate naturally occurring sources for polymer synthesis. As such, we probe the use of lichens as a natural source for polymer network synthesis. Specifically, (+)-usnic acid, a common and easily extractable secondary metabolite of many lichen species, has been modified into a monomer through a simple one step esterification process and subsequently polymerized into a cross-linked polymer network. The work aligns with the UN sustainable development goals: industry, innovation, and infrastructure (SDG9), and responsible consumption and production (SDG12). |
The current production of polymeric materials relies predominantly on petroleum-based chemistry. As we grapple with climate change, and a global-scale plastics accumulation crisis, there is a growing need to shift towards utilizing naturally-sourced polymers and polymer precursors. The synthesis of these materials requires use of naturally occurring building blocks that can be employed as is or transformed into monomers. As such, researchers have investigated synthesizing polymers derived from various bio-based systems such as plant,1–3 vegetal,4,5 and fungal sources.6 Notably, the synthesis of polymers from poly(hydroxyalkanote),7 cellulose,8 lignin,9–13 terpenes or terpenoids,14–17 and vegetable oils18–21 has gained traction over the past decade. The continual exploration of natural sources for the synthesis of diverse multifunctional monomers, polymer precursors, and macromolecular structures promotes the development of sustainable chemistry and green materials.
Lichens are stable symbiotic associations between a fungus and a photosynthesizing partner such as an algae or cyanobacteria, though additional yeasts22,23 and bacteria24 have also been identified as possible core symbiotic partners.25 Sensitive to pollution, lichens are found in habitats across the planet and are colonizers of different substrates such as rocks, trees, and plant leaves.26 One interesting aspect of lichen biology is their ability to produce secondary metabolites.27 Secondary metabolites are compounds not directly involved in the primary metabolism of an organism but instead are chemical defences which help that organism survive in their environment by limiting predation, reducing damage by ultraviolet light, and providing antimicrobial protection.28,29 There are hundreds of secondary metabolites which have been extracted and characterized from lichens, and most of these compounds are only known from these symbiotic assemblages.30 (+)-Usnic acid (UA) (Fig. 1a) – a dibenzofuran – is one of the most studied lichen secondary metabolites31 and has potential industrial use in the development of sunscreens,32–35 antibiotics36–39 and chemotherapies.40–42 To our knowledge, lichen secondary metabolites have yet to be utilized as a source for the synthesis of polymers. In this paper, we investigate the use of lichen secondary metabolites, specifically UA, as a naturally sourced precursor for polymerization. Though UA has been widely studied and synthetic derivatives are available for experimentation,43–45 little work has been performed investigating its use as a building block for polymeric biomaterials. Several examples exist in the literature in which UA is physically incorporated into polymers mostly in polymer-drug conjugate applications.46–49 Here, we demonstrate the functionalization and subsequent polymerization of UA into a cross-linked network through photoinduced thiol–ene ‘click’ chemistry.
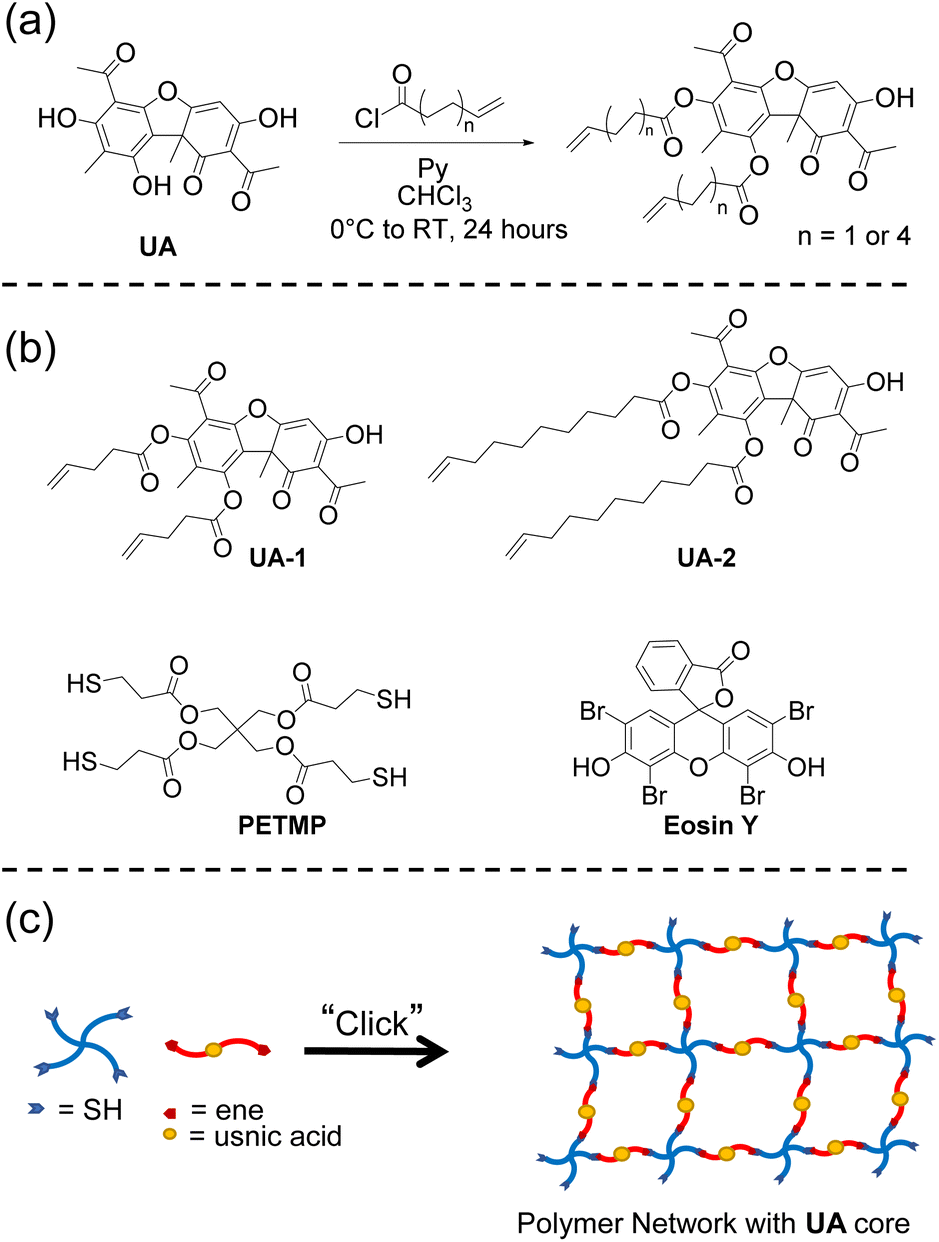 | ||
| Fig. 1 (a) General synthetic procedure for the usnic acid difunctional alkenes used in this study. (b) Structures of the different monomers and photoinitiator used. (c) General polymerization approach depicting an A4 + B2 step growth approach where a tetrafunctional thiol is reacted with a difunctional alkene. Here the tetrafunctional thiol is PETMP and the difunctional ene is the usnic acid derivatives (UA-1 and UA-2). | ||
In order to enable the polymerization of UA, we sought to synthesize multifunctional alkene derivatives of UA. Inspired by the synthetic approach of Erba et al.,50 difunctional alkene UA derivatives were synthesized through an esterification process achieved by reacting the two phenolic hydroxyl groups with excess 4-pentenoyl chloride or 10-undecenoyl chloride in the presence of pyridine in chloroform (Fig. 1a). The reactions were performed under ambient conditions and allowed to react for 24–48 hours yielding the difunctional alkenes (UA-1 and UA-2, Fig. 1b). The disappearance of the phenolic hydroxyl hydrogens before and after the reaction is confirmed via1H NMR (Fig. 2). The enolic hydroxyl group does not seem to react under these experimental conditions which is supported by literature precedent when reacting UA with aliphatic acyl chlorides.50 One explanation of this phenomenon may be due to tautomerization which could make the enolic hydroxyl less reactive when compared to the phenolic hydroxyl groups. The chemical structures of UA-1 and UA-2 were further confirmed by 1H NMR, 13C NMR, and mass spectrometry. For detailed spectra and synthetic details, see ESI.†
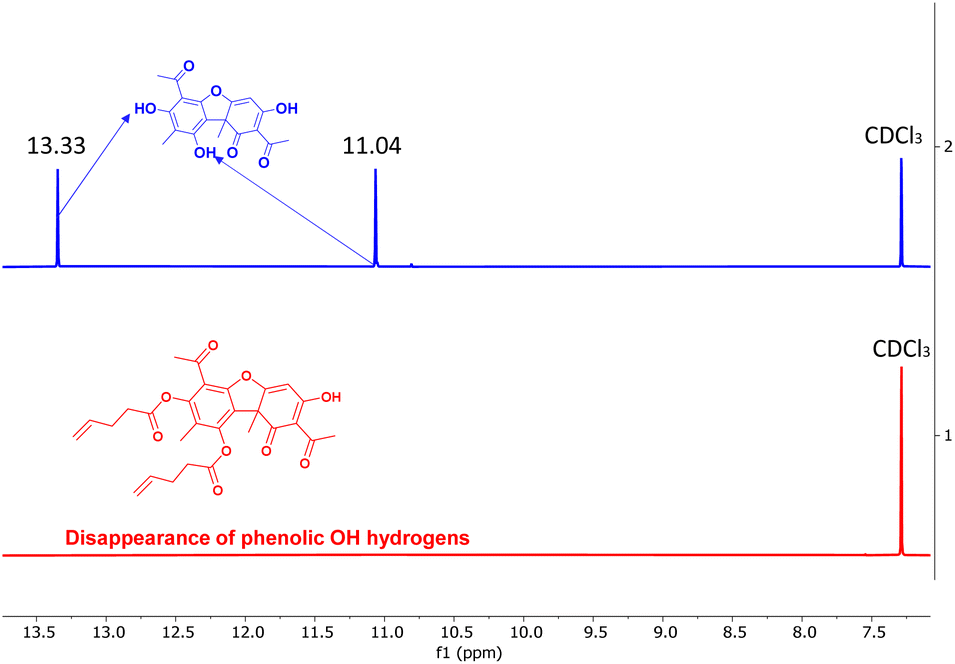 | ||
| Fig. 2 1H NMR spectrum of usnic acid showing the phenolic hydroxyl hydrogens at 11.04 and 13.33 ppm (top, blue) and their respective disappearance in UA-1 (bottom, red). | ||
Over the past several years, there has been a growing interest in utilizing ‘click’ chemistry as a technique to synthesize polymers from renewable sources.51–54 ‘Click’ reactions are a subset of organic chemical reactions that are selective, high yielding, rapid and able to proceed under simple conditions.55–57 These types of reactions have been used as a method to synthesize cross-linked and other polymer architectures.58,59 Thiol–ene ‘click’ chemistry entails the reaction between an alkene group and a thiol moiety which occurs through sulphur–hydrogen bond cleavage, promoted through heat or light produced nucleophilic radicals.56,60 To test the ability of UA-1 and UA-2 to polymerize, an equimolar amount of ene to thiol functional groups from either monomer was added to Pentaerythritol tetrakis(3-mercaptopropionate) (PETMP, Fig. 1b), a commercially available tetrafunctional thiol that is commonly used in thiol–ene polymerizations. The thiol–ene polymerization through an A4 + B2 step-growth approach is illustrated in Fig. 1c. We attempted to photo-induce the polymerization of our monomers to afford spatial and temporal control over this reaction. To realize this goal, we measured the UV-vis absorption of our synthesized monomers, UA-1 and UA-2, between 250 and 850 nm as shown in Fig. 3. These monomers showed strong absorbance in the UV range and violet visible range (up to ∼400 nm).
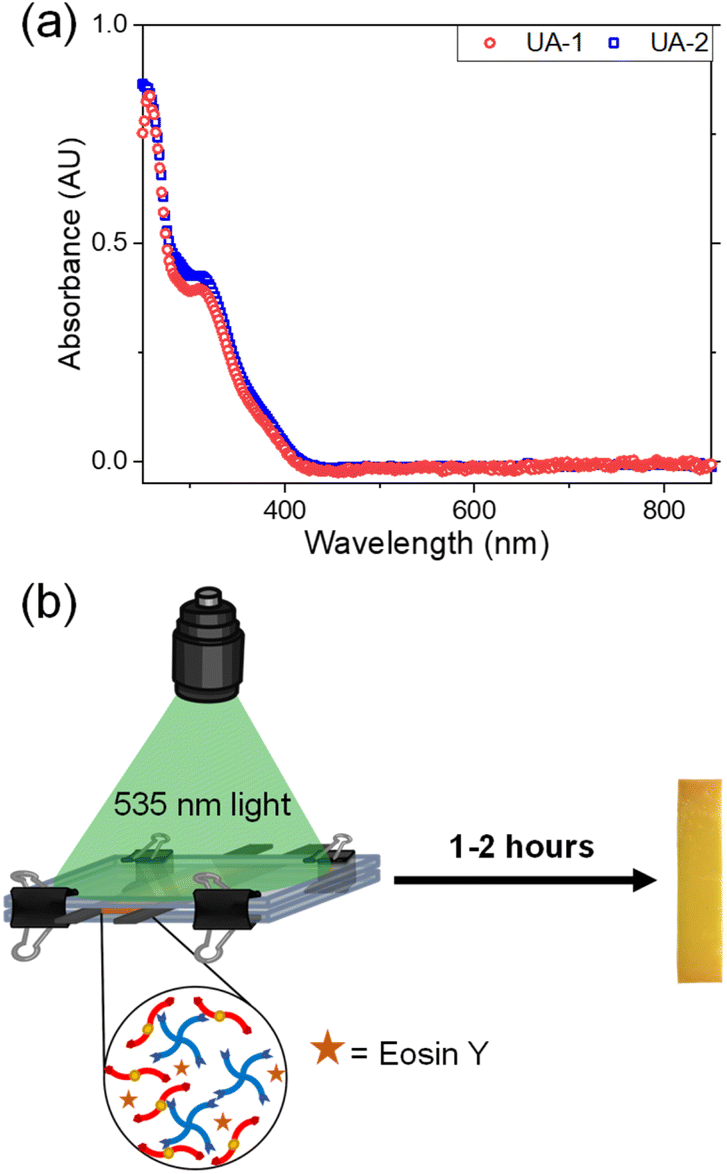 | ||
| Fig. 3 (a) UV-vis absorbance of a 50 mg mL−1 of UA-1 and UA-2 in chloroform showing absorbance in the UV and near UV regions (between 250 and 410 nm). (b) PETMP and UA-1 or UA-2 are sandwiched between glass slides in the presence of Eosin Y. Green light is irradiated and a yellow polymer network is formed after one to two hours. | ||
Accordingly, we hypothesized that common thiol–ene photoinitiators such as 2,2-dimethoxy-2-phenylacetophenone (DMPA) or bis(2,4,6-trimethylbenzoyl)-phenylphosphineoxide (BPO), norrish type I photoinitiators, would be unable to fully initiate the polymerization due to overlap in absorbance wavelength between the monomers and the photoinitiators. Indeed, when these photointiators were added to the reaction mixture in amounts ranging from 1 to 10 weight percent (wt%) with UV-light (365 nm) for DMPA or 405 nm visible light for BPO irradiated onto the sample for over two hours, the polymerization was either unsuccessful or incomplete when tested by the inverted vial method. In other words, when the vial which contained the monomers and photoinitiator was inverted after two hours of irradiation, flow was still observed which indicates incomplete polymerization. Eosin Y (Fig. 1b) was then tested as a suitable photoinitiator due to its absorbance at higher wavelengths between 500 and 600 nm which is not in the range of absorbance of the UA-1 and UA-2 monomers and has demonstrated success in initiating thiol–ene reactions.61,62 When 3 wt% of Eosin Y was dissolved in either UA-1 or UA-2 and PETMP in the presence of 10 wt% of methanol, polymer networks were formed after 535 nm light irradiation for one to two hours as depicted in Fig. 3b.
We sought to characterize the thermomechanical properties of the synthesized networks. Rectangular samples were prepared by sandwiching the monomer mixture in between glass slides and subsequent 535 nm irradiation as illustrated in Fig. 3b. Once formed, the polymer networks (UA-1-Net and UA-2-Net) were further dried in the oven at 120 °C for post-curing for an additional two hours to remove the methanol and react any residual functional groups. The resulting polymer samples were rigid and glassy to the touch.
Differential Scanning Calorimetry (DSC) confirmed that both samples had a glass transition temperature (Tg) above room temperature (Fig. 4, and Table 1). This temperature signifies a transition of the cross-linked polymer from a glassy state to a rubbery state. The Tg of UA-1-Net was within the standard error to that of UA-2-Net (34 ± 1 vs. 31 ± 4 °C), indicating that the main contributor to the glass transition temperature was the rigid dibenzofuran structure rather than the length of the alkenyl chain. Indeed, when compared with 1,2-diallyl phthalate (DAP), a difunctional ene monomer with a phenyl ring in its core, and under identical polymerization conditions, the Tg decreased to 5 °C (Fig. 4). These results taken together indicate that the rigid structure of dibenzofuran in UA can be utilized to increase the Tg of thiol–ene cross-linked polymer networks.
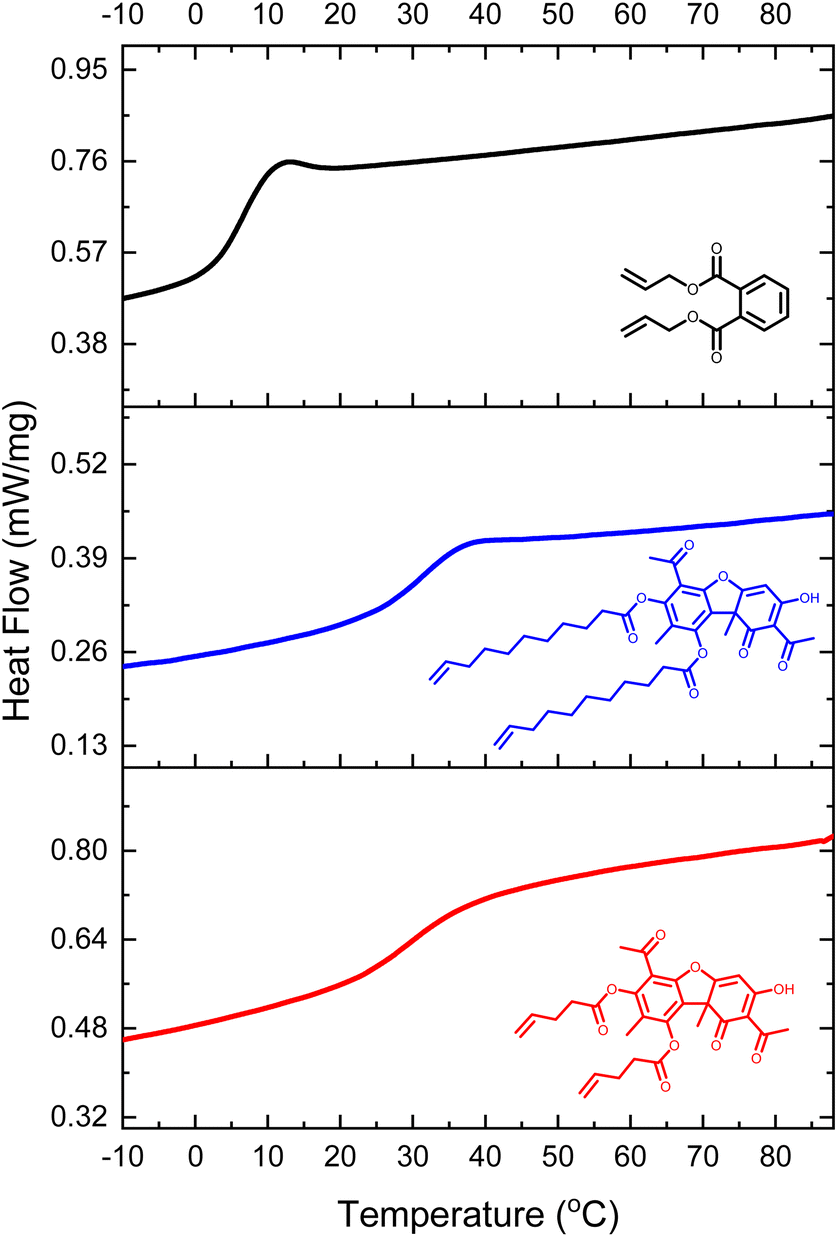 | ||
| Fig. 4 Representative differential scattering calorimetry (DSC) plots with exotherm down depicting the heat flow of the polymer networks with the control DAP-Net (top, black line), UA-2-Net (middle, blue line) and UA-1-Net (bottom, red line). | ||
Monomer conversion was investigated through gel fraction studies. The gel fraction of the polymer samples was calculated through swelling experiments where 30 to 40 mg of the network was swollen in toluene over 24 hours, followed by drying the sample in an oven at 140 °C for an additional 24 hours and reweighing the sample (see ESI† for more details). UA-1-Net and UA-2-Net had gel fractions that ranged between 83 and 93% indicating high monomer conversion of these networks (Table 1). These gel fractions were similar to networks formed from DAP as a cross-linker under identical conditions where the gel fraction was ∼85%. These results indicate that the UA synthesized monomers reach high conversions when polymerized with PETMP when compared to a similar difunctional alkene. The disappearance of the PETMP thiol peak at 2570 cm−1 before and after irradiation is confirmed via FTIR spectroscopy, supporting the conversion of the thiol groups when reacted with the UA-1 and UA-2 alkene groups (see ESI†).
In order to determine potential processability and applicability of these polymers, thermal degradation studies were performed by using thermogravimetric analysis (TGA). Thermogravimetric studies of samples ranging between 10 and 20 mg of UA-1-Net and UA-2-Net in an N2 environment were conducted. Both thermosets presented 50% weight loss temperatures (Td50%) at high temperatures above 405 °C. The thermal stability of these materials was furthermore assessed by Td10% which represented 10% weight loss. Both samples showed high thermal stability with Td values above 280 °C. Specific TGA values are found in Table 1 and ESI.† The thermal results are comparable to other biobased thiol–ene polymer networks.63,64
Finally, the rubbery plateau modulus (E′) of the networks was measured using dynamic mechanical analysis at a temperature of 100 °C. Both UA-1 and UA-2 had a rubbery modulus between 6 and 8 MPa (Table 1). These values are consistent with other thiol–ene networks synthesized from PETMP and a difunctional alkene.63
In conclusion, we have shown that lichen secondary metabolites can be utilized as a naturally occurring source of polymer synthesis. Specifically, we have demonstrated that usnic acid, a common and commercially available secondary metabolite of many lichen species, can be chemically modified and utilized as a monomer in a thiol–ene polymerization reaction. Moreover, due to usnic acid's broad antimicrobial capabilities and its UV-absorption, we see this work as a stepping stone for utilizing usnic acid in targeted applications in biobased synthesis of antimicrobial and UV-protective polymers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from NC Biotech Flash Grant (2022-FLG-3836). This work was performed in part at the Joint School of Nanoscience and Nanotechnology, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant ECCS-2025462). We acknowledge the support of internal Davidson College programs, specifically the Research In Science Experience (RISE) and the Davidson Research Initiative (DRI) both of which provided student funding. Finally, we thank Prof. David Blauch for his help in measuring MS-MALDI data, and Prof. Nicole Snyder for assistance with automated flash column chromatography.Notes and references
- H. Uyama, Polym. J., 2018, 50, 1003–1011 CrossRef CAS.
- S. Iravani and R. S. Varma, Green Chem., 2019, 21, 4839–4867 RSC.
- B. P. Mooney, Biochem. J., 2009, 418, 219–232 CrossRef CAS PubMed.
- M. R. Islam, M. D. H. Beg and S. S. Jamari, J. Appl. Polym. Sci., 2014, 131, 9016–9028 CrossRef.
- A. Gandini, T. M. Lacerda, A. J. F. Carvalho and E. Trovatti, Chem. Rev., 2016, 116, 1637–1669 CrossRef CAS PubMed.
- D. Araújo, I. C. Ferreira, C. A. V. Torres, L. Neves and F. Freitas, J. Chem. Technol. Biotechnol., 2020, 95, 1277–1289 CrossRef.
- Z. Li, J. Yang and X. J. Loh, NPG Asia Mater., 2016, 8, e265 CrossRef CAS.
- M. Rose and R. Palkovits, Macromol. Rapid Commun., 2011, 32, 1299–1311 CrossRef CAS PubMed.
- A. L. Holmberg, K. H. Reno, N. A. Nguyen, R. P. Wool and T. H. Epps, ACS Macro Lett., 2016, 5, 574–578 CrossRef CAS PubMed.
- A. L. Holmberg, N. A. Nguyen, M. G. Karavolias, K. H. Reno, R. P. Wool and T. H. Epps, Macromolecules, 2016, 49, 1286–1295 CrossRef CAS.
- K. H. Nicastro, C. J. Kloxin and T. H. Epps, ACS Sustain. Chem. Eng., 2018, 6, 14812–14819 CrossRef CAS.
- N. Lee, Y. T. Kim and J. Lee, Polymers, 2021, 13, 364 CrossRef CAS PubMed.
- A. Grossman and V. Wilfred, Curr. Opin. Biotechnol., 2019, 56, 112–120 CrossRef CAS PubMed.
- F. Della Monica and A. W. Kleij, Polym. Chem., 2020, 11, 5109–5127 RSC.
- A. C. Weems, K. R. Delle Chiaie, J. C. Worch, C. J. Stubbs and A. P. Dove, Polym. Chem., 2019, 10, 5959–5966 RSC.
- M. E. G. Mosquera, G. Jiménez, V. Tabernero, J. Vinueza-Vaca, C. García-Estrada, K. Kosalková, A. Sola-Landa, B. Monje, C. Acosta, R. Alonso and M. Á. Valera, Sustainable Chem., 2021, 2, 467–492 CrossRef CAS.
- S. Curia, S. Dautle, B. Satterfield, K. Yorke, C. E. Cranley, B. E. Dobson, J. J. La Scala, L. Soh, M. B. Gordon and J. F. Stanzione, ACS Sustain. Chem. Eng., 2019, 7, 16371–16381 CrossRef CAS.
- C. Di Mauro, S. Malburet, A. Genua, A. Graillot and A. Mija, Biomacromolecules, 2020, 21, 3923–3935 CrossRef CAS PubMed.
- A. Abbasi, M. M. Nasef and W. Z. N. Yahya, Sustainable Chem. Pharm., 2019, 13, 100158 CrossRef.
- E. Nekhavhambe, H. E. Mukaya, D. B. Nkazi and C. E. Hembe Mukaya, J. Adv. Manuf. Process., 2019, 1, e10030 CrossRef CAS.
- H. Liang, Y. Li, S. Huang, K. Huang, X. Zeng, Q. Dong, C. Liu, P. Feng and C. Zhang, ACS Sustain. Chem. Eng., 2020, 8, 914–925 CrossRef CAS.
- T. Spribille, V. Tuovinen, P. Resl, D. Vanderpool, H. Wolinski, M. C. Aime, K. Schneider, E. Stabentheiner, M. Toome-Heller, G. Thor, H. Mayrhofer, H. Johannesson and J. P. McCutcheon, Science, 2016, 353, 488–492 CrossRef CAS PubMed.
- V. Tuovinen, S. Ekman, G. Thor, D. Vanderpool, T. Spribille and H. Johannesson, Curr. Biol., 2019, 29, 476–483 CrossRef CAS PubMed.
- S. T. Bates, G. W. G. Cropsey, J. G. Caporaso, R. Knight and N. Fierer, Appl. Environ. Microbiol., 2011, 77, 1309–1314 CrossRef CAS PubMed.
- G. Tagirdzhanova, P. Saary, J. P. Tingley, D. Díaz-Escandón, D. W. Abbott, R. D. Finn and T. Spribille, Genome Biol. Evol., 2021, 13, evab047 CrossRef PubMed.
- I. M. Brodo, S. D. Sharnoff and S. Sharnoff, Lichens of North America, Yale University Press, 2001 Search PubMed.
- J. D. Lawrey, Bryologist, 1986, 89, 111–122 CrossRef CAS.
- J. Rikkinen, What's behind the pretty colours? A study on the photobiology of lichens, Bryobrothera, 1995, 4, 1–239 Search PubMed.
- A. L. Demain and A. Fang, Adv. Biochem. Eng./Biotechnol., 2000, 69, 1–39 CrossRef CAS PubMed.
- S. Huneck and I. Yoshimura, Identification of Lichen Substances, Springer Berlin, 1996, 11–123 Search PubMed.
- M. Cocchietto, N. Skert, P. Nimis and G. Sava, Naturwissenschaften, 2002, 89, 137–146 CrossRef CAS PubMed.
- K. Ingólfsdóttr, Phytochemistry, 2002, 61, 729–736 CrossRef PubMed.
- C. Kohlhardt-Floehr, F. Boehm, S. Troppens, J. Lademann and T. G. Truscott, J. Photochem. Photobiol., B, 2010, 101, 97–102 CrossRef CAS PubMed.
- A. Galanty, J. Popiół, M. Paczkowska-Walendowska, E. Studzińska-Sroka, P. Paśko, J. Cielecka-Piontek, E. Pękala and I. Podolak, Molecules, 2021, 26, 5224 CrossRef CAS PubMed.
- M. Varol, T. Tay, M. Candan, A. Türk and A. T. Koparal, Phytother. Res., 2016, 30, 9–15 CrossRef CAS PubMed.
- R. B. Johnson, G. Feldott and H. A. Lardy, Arch. Biochem., 1950, 28, 317–323 CAS.
- M. Ghione, D. Parrello and L. Grasso, Chemioterapia, 1988, 7, 302–305 CAS.
- M. Lauterwein, M. Oethinger, K. Belsner, T. Peters and R. Marre, Antimicrob. Agents Chemother., 1995, 39, 2541–2543 CrossRef CAS PubMed.
- H. Elo, J. Matikainen and E. Pelttari, Naturwissenschaften, 2007, 94, 465–468 CrossRef CAS PubMed.
- M. Mayer, M. A. O'Neill, K. E. Murray, N. S. Santos-Magalhães, A. M. A. Carneiro-Leão, A. M. Thompson and V. C. L. Appleyard, Anticancer Drugs, 2005, 16, 805–809 CrossRef CAS PubMed.
- Y. Song, F. Dai, D. Zhai, Y. Dong, J. Zhang, B. Lu, J. Luo, M. Liu and Z. Yi, Angiogenesis, 2012, 15, 421–432 CrossRef CAS PubMed.
- O. A. Luzina and N. F. Salakhutdinov, Expert Opin. Ther. Pat., 2018, 28, 477–491 CrossRef CAS PubMed.
- B. Guzow-Krzemińska, K. Guzow and A. Herman-Antosiewicz, Curr. Pharmacol. Rep., 2019, 5, 429–439 CrossRef.
- A. Pyrczak-Felczykowska, R. Narlawar, A. Pawlik, B. Guzow-Krzemińska, D. Artymiuk, A. Hać, K. Ryś, L. M. Rendina, T. A. Reekie, A. Herman-Antosiewicz and M. Kassiou, J. Nat. Prod., 2019, 82, 1768–1778 CrossRef CAS PubMed.
- K. Victor, L. Boris, G. Athina, P. Anthi, S. Marija, K. Marina, R. Oliver and S. Marina, Medchemcomm, 2018, 9, 870–882 RSC.
- I. Francolini, P. Norris, A. Piozzi, G. Donelli and P. Stoodley, Antimicrob. Agents Chemother., 2004, 48, 4360–4365 CrossRef CAS PubMed.
- R. B. Karabacak, T. Tay and M. Kivanç, React. Funct. Polym., 2014, 83, 7–13 CrossRef CAS.
- M. Rauschenbach, S. B. Lawrenson, V. Taresco, A. K. Pearce and R. K. O'Reilly, Macromol. Rapid Commun., 2020, 41, 2000190 CrossRef CAS PubMed.
- V. Taresco, I. Tulini, I. Francolini and A. Piozzi, Int. J. Mol. Sci., 2022, 23, 14339 CrossRef CAS PubMed.
- E. Erba, D. Pocar and L. M. Rossi, Farmaco, 1998, 53, 718–720 CrossRef CAS.
- S. Li, G. Li, B. Gao, S. P. Pujari, X. Chen, H. Kim, F. Zhou, L. M. Klivansky, Y. Liu, H. Driss, D. D. Liang, J. Lu, P. Wu, H. Zuilhof, J. Moses and K. B. Sharpless, Nat. Chem., 2021, 13, 858–867 CrossRef CAS PubMed.
- M. Röhrl, J. F. Ködel, R. L. Timmins, C. Callsen, M. Aksit, M. F. Fink, S. Seibt, A. Weidinger, G. Battagliarin, H. Ruckdäschel, R. Schobert, J. Breu and B. Biersack, ACS Omega, 2023, 8, 9889–9895 CrossRef PubMed.
- K. K. Tremblay-Parrado, C. García-Astrain and L. Avérous, Green Chem., 2021, 23, 4296–4327 RSC.
- Y. Han, L. Yuan, G. Li, L. Huang, T. Qin, F. Chu and C. Tang, Polymer, 2016, 83, 92–100 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- J. Dong, L. Krasnova, M. G. Finn and K. Barry Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- W. Liu and C. M. Dong, Macromolecules, 2010, 43, 8447–8455 CrossRef CAS.
- X. Chang and C. M. Dong, Biomacromolecules, 2013, 14, 3329–3337 CrossRef CAS PubMed.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- H. Shih and C. C. Lin, Macromol. Rapid Commun., 2013, 34, 269–273 CrossRef CAS PubMed.
- J. Xu and C. Boyer, Macromolecules, 2015, 48, 520–529 CrossRef CAS.
- M. Shibata, K. Sugane and Y. Yanagisawa, Polym. J., 2019, 51, 461–470 CrossRef CAS.
- C. Li, M. Johansson, R. J. Sablong and C. E. Koning, Eur. Polym. J., 2017, 96, 337–349 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Small molecule and polymer synthetic methods, H and C NMR data, DSC, TGA, Gel fraction, UV-vis, FTIR, E′ modulus, photographs of polymer networks. See DOI: https://doi.org/10.1039/d3su00453h |
| This journal is © The Royal Society of Chemistry 2024 |