
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic urushiols from biorenewable carbon resources: chemical conversion of enzymatic degradation products of wood lignin to an ancient yet future coating material†
Katsuhiro
Isozaki
 *ab,
Hiroshi
Matsuda
a,
Ryosuke
Agata
a,
Jaeyoung
Jeon
a,
Beiling
Wu
ab,
Francesca
Pincella
ab,
Makoto
Ikenaga
c,
Yoichi
Tachibana
c,
Yukari
Ohta
d and
Masaharu
Nakamura
*ab
*ab,
Hiroshi
Matsuda
a,
Ryosuke
Agata
a,
Jaeyoung
Jeon
a,
Beiling
Wu
ab,
Francesca
Pincella
ab,
Makoto
Ikenaga
c,
Yoichi
Tachibana
c,
Yukari
Ohta
d and
Masaharu
Nakamura
*ab
aInternational Research Center for Elements Science, Institute for Chemical Research, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan. E-mail: kisozaki@scl.kyoto-u.ac.jp; masaharu@scl.kyoto-u.ac.jp
bDepartment of Energy and Hydrocarbon Chemistry, Graduate School of Engineering, Kyoto University, Nishikyoku, Kyoto 615-8510, Japan
cKyoto Municipal Institute of Industrial Technology and Culture, 91 Awatacho, Chudo-ji, Shimogyoku, Kyoto 600-8815, Japan
dGunma University Center for Food Science and Wellness, 4-2 Aramaki, Maebashi, Gunma 371-8510, Japan
First published on 25th March 2024
Abstract
An artificial urushi material was developed by chemical transformations of phenylpropanoids available from enzymatic degradation of woody lignin, demonstrating the potential of renewable biomass in materials science and technology. The succeeding five-step organic synthesis brings about new urushiol analogs, which possess curing and coating properties, such as hardness, comparable to those of the precious natural urushi materials. The present study exemplifies the application of woody biomass to the synthesis of sustainable and high-performance coating materials.
Sustainability spotlightLignin is one of the main components of renewable woody biomass and the most abundant aromatic resource on the earth. Nonetheless, it is burned primarily as an energy source in the pulping industry and therefore finally converted to CO2. In light of the current green transformation efforts worldwide, the valorization of lignin can be the key step for the development of a sustainable chemical industry fully reliant on a renewable aromatic feedstock. In this work, we demonstrate that lignin could serve as the chemical resource for the synthesis of an urushi-like coating material by combinations of enzymatic and chemical transformations. We hope that the present research will contribute to UN SDGs No. 9 (Industry, Innovation, and Infrastructure) and 12 (Responsible Consumption and Production). |
The need for sustainable materials to replace petroleum-derived ones has renewed the interest in a traditional resin used in Asian countries for millennia, urushi or Asian lacquer, originating from the sap of Toxicodendron vernicifluum.1–3Urushi is a natural product, a water-in-oil emulsion composed of urushiol (Fig. 1, 60–80%), water (10–30%), gum (3–6%) and enzymes (2–3%).4–8Urushi has long been employed as a coating and adhesive material for both practical and aesthetic purposes, but more recently applications of urushi in fields such as biomedical and electronics have been proposed and successfully demonstrated.9–14 The durability, high stability and aesthetic beauty of urushi, combined with its renewable nature, have made urushi a good candidate to become a building block for a more sustainable society. Nevertheless, several limitations exist to using natural urushi due to its time-consuming and labor-intensive collection and processing steps that require highly skilled labor.
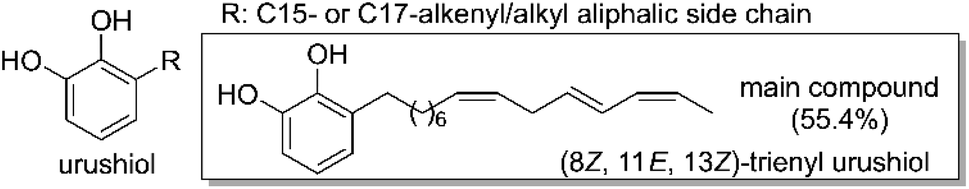 | ||
| Fig. 1 Molecular structure of natural urushiol. | ||
The limited access to this precious raw material has spawned considerable efforts to develop artificial urushi coating materials to date.12–21 Most of the recent reports focus on the synthesis of urushiol analogs (catechols bearing C15 or C17 unsaturated aliphatic side chains at the 3-position) since the enzymatic oxidative polymerization of this mixture is responsible for the hardness, solvent resistance and durability of this coating. Several urushiol analogs have been synthesized by combining catechol and unsaturated side chain moieties derived from vegetable fats.17–21
Nevertheless, to preserve the sustainability of artificial urushi, it is crucial to take advantage of abundant and renewable starting materials for its synthesis. A promising strategy would be to rely on lignocellulosic biomass, particularly lignin, its second principal component, as a renewable chemical feedstock.22–24 Lignin is currently underutilized and mostly burned as fuel in the paper industry or as waste, despite being the most abundant aromatic carbon resource on the earth. Lignin consists of a randomly polymerized phenylpropanoid backbone cross-linked by a variety of linkages, such as β-O-4, β-5, β–β, 5–5, 4-O-5, and β-1.25 The degradative conversion of lignin to several aromatic compounds has been studied extensively to date by biological and chemical approaches.26–29 We previously reported an enzymatic reaction degrading woody lignin efficiently to phenylpropanoid monomers, such as guaiacylhydroxypropanone (GHP) and syringylhydroxypropanone (SHP), by marine microorganisms.30,31
Herein, we report the first synthesis of lignin-derived urushiol analogs by installing vegetable oil-derived polyunsaturated side chains on G- and S-urushiol through a 5-step chemical transformation. The preparation and characterization of artificial urushi films confirmed the similar curing/polymerization properties of lignin-derived urushiol analogs to natural urushiol.
The synthesis of lignin-derived urushiol analogs started by preparing the trienyl side chain and the catechol/pyrogallol moiety and was completed in 5 steps. First, the lignin-derived aromatic monomer guaiacyl- and syringylhydroxypropanone GHP and SHP were dehydrated and protected by the TBS group according to a previously reported method,32 affording the corresponding TBS-protected enone products G-2 and S-2 (Scheme 1). In order to introduce the trienyl side chains, a natural trienyl fatty acid, linolenic acid, was converted to the corresponding Grignard reagent, (9Z,12Z,15Z)-octadeca-9,12,15-trienylmagnesium bromide (1) via linolenyl bromide (Scheme 1). The 1,4-addition of the Grignard reagent 1 was performed in the presence of chlorotrimethylsilane, copper bromide dimethyl sulfide complex, and lithium chloride, followed by acidic treatment giving the corresponding trienyl side chain-bearing products G-3 and S-3 in 56% and 52% yield, respectively. Both the reduction of the ketone group and the deprotection of the TBS group were performed at the same time by the reaction with triethylsilane in trifluoroacetic acid solution to afford the methyl-protected artificial urushiol analogs Me-G- and Me-S-urushiol in 65% and 72% yield, respectively. Finally, refluxing of a mesitylene solution of methylmagnesium iodide resulted in the removal of methyl groups, producing the guaiacyl (G)- and syringyl (S)-type artificial urushiol analogs G- and S-urushiol in 23% and 51% yield, respectively. It is noteworthy that 1H and 13C NMR revealed that no isomerization of the trienyl side chains had taken place under these reaction conditions.
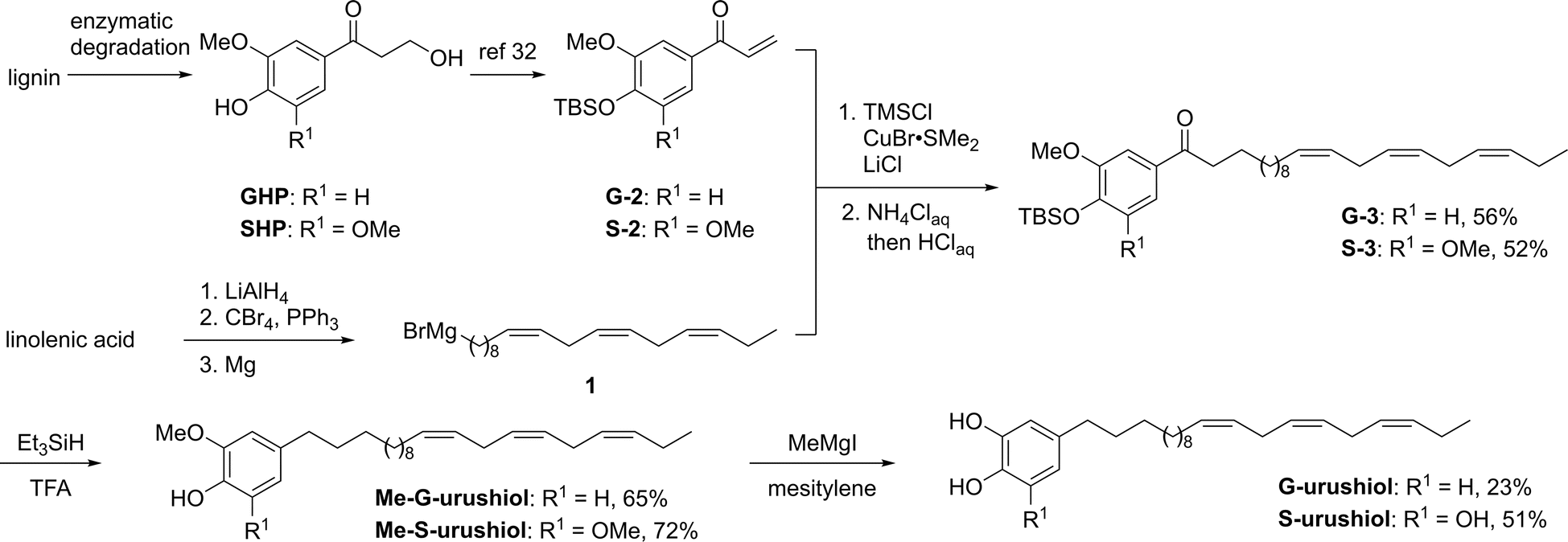 | ||
| Scheme 1 Synthesis of lignin-derived urushiol analogs. | ||
The synthesized urushiol analogs were applied for the preparation of artificial urushi films. The raw urushi-like liquids were prepared by mixing the urushiol analogs, protein hydrolysate, and water in the ratio of approximately 70, 7, and 23 wt% in the presence of laccase. The prepared artificial urushi liquids were coated on glass substrates and were kept at 30 °C in 80% relative humidity for several days. The lignin-derived S-type urushiol analog S-urushiol bearing the C21 trienyl side chain at the 5-position of pyrogallol was instantaneously cured after the addition of laccase, indicating the extremely high reactivity of the electron-rich pyrogallol core toward oxidation (Table 1, entry 1). This curing reaction was slowed down by the addition of terpene oil (Table 1, entry 2), suggesting a possible application of S-urushiol for the preparation of rapidly curing coating/adhesive materials. In the case of the lignin-derived G-type urushiol analog G-urushiol (Table 1, entry 3), the curing reaction successfully proceeded showing a hardening profile relatively similar to that of natural urushi. In the natural lacquer, the curing mechanism relies on the formation of micelles where the oil phase, or the continuous phase, contains catechols, while the water phase, or the dispersed phase, contains the enzymes. Radicals are first formed by the laccase-catalyzed oxidation of urushiol at the interface layer, and then transferred to the urushiol oil phase, enabling its polymerization. Molecular oxygen can continuously penetrate the aqueous phase to oxidize the reduced laccase, allowing the urushiol polymerization to continue smoothly, mostly leading to the formation of C–C and C–O–C nuclei crosslinks involving nuclei and side chains. Given the high number of polyunsaturations on the catechol side chains, auto-oxidative radical chain reactions proceed as well, enabling the formation of chemical crosslinks between the aliphatic chains.33 As can be seen from the time-course profile of Martens hardness (Fig. 2), the initial oxidation reaction proceeded very slowly, which is compatible with the slow enzymatic oxidation of the catechol core. After 10 days, the hardness started to increase remarkably, suggesting that the dominant process in the reaction had shifted to the faster auto-oxidation of unsaturated side chains (Fig. S3†).2–4 Finally, after 73 days, the Martens hardness reached a plateau value of 147.4 ± 0.9 N mm−2 (Table 1, entry 3), which is comparable to the hardness of the commercially available polyvinyl chloride film (144.6 ± 0.4 N mm−2, Table S1†), demonstrating a good potential for practical applications. In contrast, the methoxy-protected derivative Me-G-urushiol was not cured under the same conditions (Table 1, entry 4), indicating that the catechol moiety is crucial to the initial polymerization step, in line with the current knowledge regarding natural urushiol.10–12 The urushiol analog 4 bearing the C18 trienyl side chain at the 3-position of catechol34 (for the synthetic protocol see ESI†) was also cured and resulted in a Martens hardness of 140.4 ± 1.5 N mm−2 after 120 days (Table 1, entry 5). The comparable value of hardness for compounds 4 and G-urushiol reveals the negligible contribution of the alkyl chain length and substitution position towards the curing properties of the artificial urushi film.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Compound | R1 | R2 | Days after sample preparation | Martens hardness (N mm−2) |
| a Terpene oil was added. | |||||
| 1 | S-urushiol | OH | OH | — | Hardened too quick |
| 2 | S-urushiol | OH | OH | 73 | Gradually hardened |
| 3 | G-urushiol | H | OH | 73 | 147.4 ± 0.9 |
| 4 | Me-G-urushiol | H | OMe | 73 | Not hardened |
| 5 | 4 | — | — | 120 | 140.4 ± 1.5 |
| 6 | Chinese urushiol | — | — | 73 | 176.7 ± 2.3 |
| 7 | Cambodian thitsiol | — | — | 73 | 58.5 ± 0.6 |
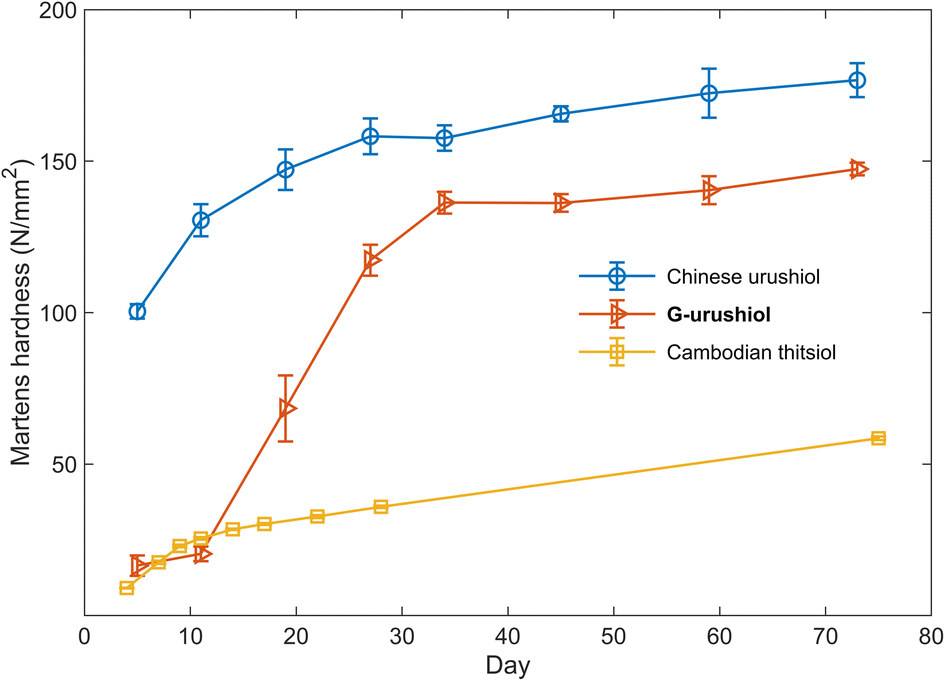 | ||
| Fig. 2 Time-course curing properties of the film obtained from G-urushiol (red line), natural Chinese urushiol (blue line) and natural Cambodian thitsiol (yellow line). | ||
The crosslinking reaction was also investigated using natural urushiol and thitsiol extracted from Chinese raw urushi and Cambodian raw thitsi, in which the major constituents are catechols bearing the C17 trienyl side chain at the 3-position and catechols bearing the C17 dienyl side chain at the 4-position, respectively.5,35,36 The structural difference is clearly represented by their different curing properties, in agreement with previous reports,1,35,37–39 resulting in hardness of 176.7 ± 2.3 for Chinese urushiol and 58.5 ± 0.6 N mm−2 for Cambodian thitsiol (Table 1, entries 6 and 7).35,39 The comparable hardness of the artificial urushi film from G-urushiol to that from Chinese urushiol suggests similar curing properties of lignin-derived urushiol analogs. Despite the comparable hardness of the artificial urushi film from G-urushiol to that from Chinese urushiol, the curing reaction proceeds slowly at the initial stage. The similar curing profile was observed with an urushiol analog synthesized from vanillyl alcohol,20 suggesting a slightly different initial reaction step for an artificial urushi prepared from synthetically pure urushiol analogs, compared to the one prepared from natural urushiols, which are a mixture of catechols bearing different side chains. The initial slow curing reaction resembles the auto-oxidation profile of vegetable oils,40 suggesting that a similar auto-oxidation of trienyl side chains takes place from the initial stage. In addition, the lower value of hardness obtained from Cambodian thitsiol bearing mainly dienyl side chains suggests that the number of unsaturated bonds in the side chain has a much stronger influence on the hardening process compared to the substituent position to the catechol core (Fig. 2).
In order to analyze the curing reaction of lignin-derived urushiol analog G-urushiol and natural urushiol extracted from Chinese urushi the prepared urushi films (after 73 days of curing) were characterized by FT-IR spectra (Fig. 3). Both the artificial and the natural urushi films appear translucent (Fig. 3, inset), but the artificial film presents a paler color (yellow) compared to the natural film (red/brown). From the IR spectra, we can notice that artificial urushi and natural urushi films differ mainly in the regions corresponding to the two vibrational bands at 1139 and 1504 cm−1; these two peaks are clearly observed in the artificial urushi film but not in the cured film from Chinese urushiol. These vibrations are assigned to the out-of-plane bending and aromatic stretching vibrations of 4-substituted catechol21 based on the DFT calculation (Fig. S1†). When comparing the liquid and cured films, we can notice that the aromatic vibrational band at 1608 cm−1 of G-urushiol was broadened after the polymerization, in a similar way to the 1593 and 1628 cm−1 bands in natural urushiol, in agreement with a previous report.41 An additional significant spectral change from liquid to cured film was observed for both natural and artificial urushi with the appearance of two peaks centered at 1635 and 1714 cm−1. These two strong and broad peaks originate from the formation of quinone and the oxidation of side chains.21 Furthermore, the disappearance of the oleofinic C–H vibration at 3011 cm−1 suggests the complete oxidation of the unsaturated side chains by autooxidation. These IR spectra demonstrate that the lignin-derived urushiol analog G-urushiol and natural urushiol undergo similar enzymatic polymerization reactions.
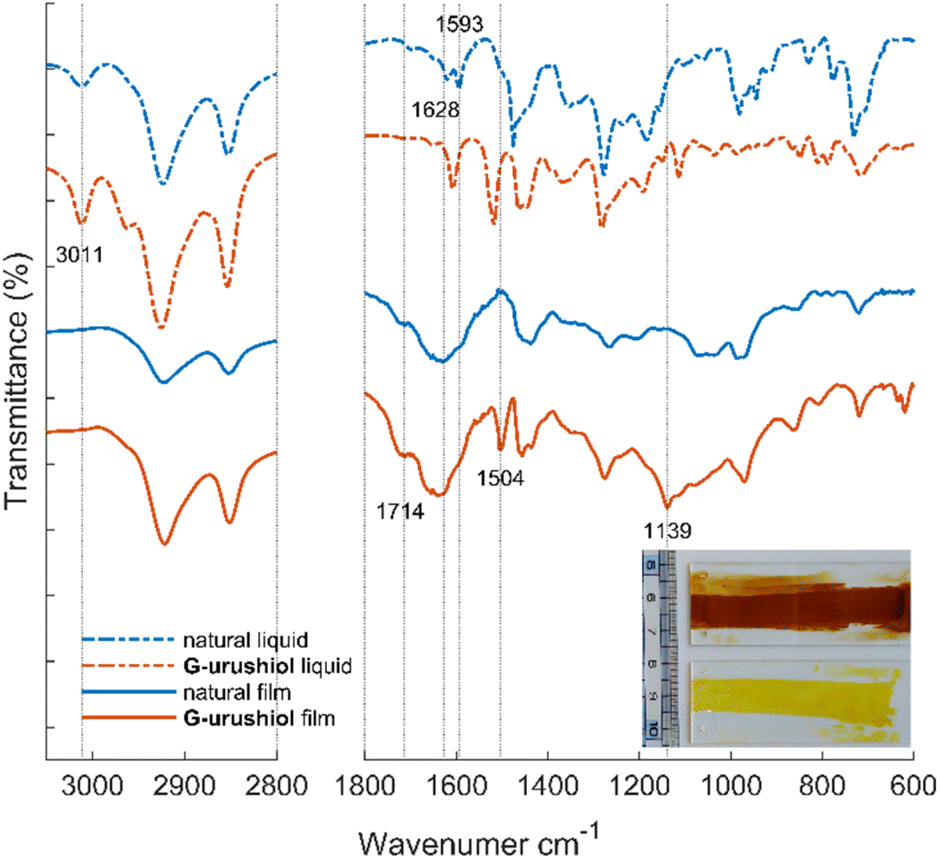 | ||
| Fig. 3 IR spectra of the liquid state (dotted line) and cured film (solid line) obtained from natural Chinese urushiol (blue line) and G-urushiol (orange line). All spectra have been shifted and normalized for clarity. Inset: photo of natural Chinese urushi film (top, red film) and artificial (G-urushiol) urushi film (bottom, yellow film) deposited on glass. | ||
In summary, artificial urushi materials were developed by utilizing lignin-derived phenylpropanoids as aromatic nuclei. Vegetable oil-derived polyunsaturated side chains were installed on lignin-derived aromatic compounds GHP and SHP to afford the lignin-derived urushiol analogs. Artificial urushi films were successfully prepared by mixing the lignin-derived urushiol analogs with protein hydrolysate and water in the presence of laccase enzyme. Time-course analysis of Martens hardness shows a stepwise oxidation reaction in the curing process, similar to natural urushi. The natural urushi-like properties were also confirmed by FT-IR analysis, suggesting remarkably similar molecular transformations during the curing process. The sufficiently high hardness of the lignin-derived artificial urushi film paves the way for future applications as an environmentally benign and sustainable coating material.
Author contributions
This work was carried out in collaboration between all authors. Authors H. M. and J. J. performed the synthetic experiments. Authors M. I. and Y. T. analyzed the curing properties and FT-IR of urushiol films. Authors K. I., Y. O., and M. N. designed and supervised the study. K. I., Y. O., Y. T., F. P., and M. N. prepared the manuscript. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported in part by the Advanced Low Carbon Technology Research and Development Program (ALCA JPMJAL1504) from the Japan Science and Technology Agency (JST), and New Energy and Industrial Technology Development Organization (NEDO). This work was supported by JST Grant Number JPMJPF2114. R. A. thanks ‘Research Fellowships of Japan Society for the Promotion of Science for Young Scientists’ (JSPS 11488). We are grateful to Integrated Research Consortium on Chemical Sciences (IRCCS). We are grateful to TSK Corporation, Tosoh Finechem Corporation, and Nissan Chemical Corporation for their financial support. This work was supported in part by the International Collaborative Research Program of Institute for Chemical Research (ICR-iJURC), Kyoto University (grant # 2019-24, 2020-22 and 2021-24). FT-ICR-MS analyses were also supported by ICR-iJURC. We are grateful to I. Bonaduce (Univ. of Pisa) for the fruitful discussion.Notes and references
- J. Kumanotani, Prog. Org. Coat., 1995, 26, 163–195 CrossRef CAS.
- R. Lu, T. Yoshida and T. Miyakoshi, Polym. Rev., 2013, 53, 153–191 CrossRef CAS.
- H. Uyama and S. Kobayashi, Enzyme-Catalyzed Synthesis of Polymers, 2006, pp. 51–67 Search PubMed.
- J. Kumanotani, Prog. Org. Coat., 1998, 34, 135–146 CrossRef CAS.
- Y. Kamiya and T. Miyakoshi, J. Oleo Sci., 2001, 50, 865–874 CrossRef CAS.
- Y. Kamiya, W. Saito and T. Miyakoshi, J. Oleo Sci., 2002, 51, 473–483 CrossRef CAS.
- T. Miyakoshi, H. Kobuchi, N. Niimura and Y. Yoshihiro, Bull. Chem. Soc. Jpn., 1991, 64, 2560–2562 CrossRef CAS.
- H. K. T. Miyakoshi, Nippon Kagaku Kaishi, 1990, 1271–1276 CrossRef.
- H. Jeong, J. Heo, B. Son, D. Choi, T. H. Park, M. Chang and J. Hong, ACS Appl. Mater. Interfaces, 2015, 7, 26117–26123 CrossRef CAS PubMed.
- Y. Zhao, X. He, H. Wang, J. Zhu, H. Wang, Y. Zheng, S. Zhu and Z. Cui, RSC Adv., 2021, 11, 18448–18457 RSC.
- Z. Qi, C. Wang and J. Jiang, Molecules, 2018, 23, 1074 CrossRef PubMed.
- X. Zheng, X. Xiong, J. Yang, D. Chen, R. Jian and L. Lin, Chem. Eng. J., 2018, 333, 153–161 CrossRef CAS.
- X. Zheng, J. Weng, S. Li, H. Liu, B. Hu, Y. Li, X. Meng and H. Ruan, Chem. Eng. J., 2014, 245, 265–275 CrossRef CAS.
- J. Yang, F. Shen, J. Deng, J. Cai, Q. Zhang and W. Liu, J. Appl. Polym. Sci., 2018, 135, 45865 CrossRef.
- T. Otsuka, S.-i. Fujikawa, H. Yamane and S. Kobayashi, Polym. J., 2017, 49, 335–343 CrossRef CAS.
- T. Tsujimoto, R. Ikeda, H. Uyama and S. Kobayashi, Chem. Lett., 2000, 29, 1122–1123 CrossRef.
- J. Zhou, Y. Liu, Y. Hu, C. Zhou, M. Chen, T. Yuan, C. Chen and Z. Yang, Ind. Crops Prod., 2016, 94, 424–430 CrossRef CAS.
- J. Y. Cho, K. Y. Park, S. J. Kim, S. Oh and J. H. Moon, Biosci., Biotechnol., Biochem., 2015, 79, 1915–1918 CrossRef CAS PubMed.
- J. Y. Kim, J. Y. Cho, Y. K. Ma, Y. G. Lee and J. H. Moon, Free Radical Biol. Med., 2014, 71, 379–389 CrossRef CAS PubMed.
- S. Kobayashi, R. Ikeda, H. Oyabu, H. Tanaka and H. Uyama, Chem. Lett., 2000, 29, 1214–1215 CrossRef.
- H. O. Y. A. M. Terada, J. Jpn. Soc. Colour Mater., 1994, 66, 681–687 CrossRef.
- W. Yang, H. Ding, D. Puglia, J. M. Kenny, T. Liu, J. Guo, Q. Wang, R. Ou, P. Xu, P. Ma and P. Jan Lamestra, SusMat, 2022, 2, 535–568 CrossRef CAS.
- R. R. Singhania, A. K. Patel, T. Raj, C.-W. Chen, V. K. Ponnusamy, N. Tahir, S.-H. Kim and C.-D. Dong, Fuel, 2022, 311, 122608 CrossRef CAS.
- Z.-H. Liu, N. Hao, Y.-Y. Wang, C. Dou, F. Lin, R. Shen, R. Bura, D. B. Hodge, B. E. Dale, A. J. Ragauskas, B. Yang and J. S. Yuan, Nat. Commun., 2021, 12, 3912 CrossRef CAS PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed. Engl., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- J. C. Chan, M. Paice and X. Zhang, ChemCatChem, 2019, 12, 401–425 CrossRef.
- X. Liu, X. Duan, W. Wei, S. Wang and B.-J. Ni, Green Chem., 2019, 21, 4266–4289 RSC.
- S. Gazi, Appl. Catal., B, 2019, 257, 117936 CrossRef CAS.
- M. V. Galkin and J. S. Samec, ChemSusChem, 2016, 9, 1544–1558 CrossRef CAS PubMed.
- Y. Ohta, S. Nishi, R. Hasegawa and Y. Hatada, Sci. Rep., 2015, 5, 15105 CrossRef CAS PubMed.
- Y. Ohta, R. Hasegawa, K. Kurosawa, A. H. Maeda, T. Koizumi, H. Nishimura, H. Okada, C. Qu, K. Saito, T. Watanabe and Y. Hatada, ChemSusChem, 2017, 10, 425–433 CrossRef CAS PubMed.
- O. S. Ojo, B. Nardone, S. F. Musolino, A. R. Neal, L. Wilson, T. Lebl, A. M. Z. Slawin, D. B. Cordes, J. E. Taylor, J. H. Naismith, A. D. Smith and N. J. Westwood, Org. Biomol. Chem., 2018, 16, 266–273 RSC.
- J. Yang, N. Chen, J. Zhu, J. Cai, J. Deng, F. Pan, L. Gao, Z. Jiang and F. Shen, Sci. Rep., 2020, 10, 12867 CrossRef CAS PubMed.
- T. Miyakoshi, K. Nagase and T. Yoshida, Urushi kagaku no shinpo, IPC, Japan, 2000, p. 43 Search PubMed.
- R. Lu, D. Kanamori and T. Miyakoshi, Int. J. Polym. Anal. Charact., 2011, 16, 86–94 CrossRef CAS.
- D. Tamburini, J. Anal. Appl. Pyrolysis, 2021, 157, 105202 CrossRef CAS.
- N. Niimura and T. Miyakoshi, J. Mass Spectrom. Soc. Jpn., 2003, 51, 439–457 CrossRef CAS.
- A. Heginbotham and M. Schilling, East Asian Lacquer: Material Culture, Science and Conservation, Archetype, London, 2011, pp. 92–106 Search PubMed.
- D. Tamburini, G. Pescitelli, M. P. Colombini and I. Bonaduce, J. Anal. Appl. Pyrolysis, 2017, 124, 51–62 CrossRef CAS.
- S. Pizzimenti, L. Bernazzani, C. Duce, M. R. Tinè and I. Bonaduce, Sci. Rep., 2023, 13, 8094 CrossRef CAS PubMed.
- J. Yang, J. Deng, Q. Zhang, Q. Shen, D. Li and Z. Xiao, Prog. Org. Coat., 2015, 78, 176–182 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00446e |
| This journal is © The Royal Society of Chemistry 2024 |