
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ion-combination specific effects driving the enzymatic activity of halophilic alcohol dehydrogenase 2 from Haloferax volcanii in aqueous ionic liquid solvent mixtures†
Alexandra
Schindl
 *abcde,
M. Lawrence
Hagen
a,
Isabel
Cooley
h,
Christof M.
Jäger
af,
Andrew C.
Warden
gi,
Mischa
Zelzer
b,
Thorsten
Allers
c and
Anna K.
Croft
*h
*abcde,
M. Lawrence
Hagen
a,
Isabel
Cooley
h,
Christof M.
Jäger
af,
Andrew C.
Warden
gi,
Mischa
Zelzer
b,
Thorsten
Allers
c and
Anna K.
Croft
*h
aSustainable Process Technologies Group, Department of Chemical and Environmental Engineering, University of Nottingham, Nottingham NG7 2RD, UK
bSchool of Pharmacy, University of Nottingham, University Park Campus, Nottingham NG7 2RD, UK
cSchool of Life Sciences, University of Nottingham, Queen's Medical Centre, Nottingham NG7 2UH, UK
dSchool of Molecular and Cellular Biology, University of Leeds, Leeds LS2 9JT, UK
eAstbury Centre for Structural Molecular Biology, Faculty of Biological Sciences, University of Leeds, Leeds LS2 9JT, UK
fData Science and Modelling, Pharmaceutical Sciences, R&D, AstraZeneca Gothenburg, Pepparedsleden 1, SE-431 83 Mölndal, Sweden
gCSIRO Environment, Commonwealth Scientific and Industrial Research Organization (CSIRO), Research and Innovation Park, Acton, Canberra, ACT 2600, Australia
hDepartment of Chemical Engineering, Loughborough University, LE11 3TU, UK. E-mail: a.k.croft@lboro.ac.uk
iAdvanced Engineering Biology Future Science Platform, Commonwealth Scientific and Industrial Research Organisation (CSIRO), Research and Innovation Park, Acton, Canberra, ACT 2600, Australia
First published on 8th July 2024
Abstract
Biocatalysis in ionic liquids enables novel routes for bioprocessing. Enzymes derived from extremophiles promise greater stability and activity under ionic liquid (IL) influence. Here, we probe the enzyme alcohol dehydrogenase 2 from the halophilic archaeon Haloferax volcanii in thirteen different ion combinations for relative activity and analyse the results against molecular dynamics (MD) simulations of the same IL systems. We probe the ionic liquid property space based on ion polarizability and molecular electrostatic potential. Using the radial distribution functions, survival probabilities and spatial distribution functions of ions, we show that cooperative ion–ion interactions determine ion–protein interactions, and specifically, strong ion–ion interactions equate to higher enzymatic activity if neither of the ions interact strongly with the protein surface. We further demonstrate a tendency for cations interacting with the protein surface to be least detrimental to enzymatic activity if they show a low polarizability when combined with small hydrophilic anions. We also find that the IL ion influence is not mitigated by the surplus of negatively charged residues of the halophilic enzyme. This is shown by free energy landscape analysis in root mean square deviation and distance variation plots of active site gating residues (Trp43 and His273) demonstrating no protection of specific structural elements relevant to preserving enzymatic activity. On the other hand, we observe a general effect across all IL systems that a tight binding of water at acidic residues is preferentially interrupted at these residues through the increased presence of potassium ions. Overall, this study demonstrates a co-ion interaction dependent influence on allosteric surface residues controlling the active/inactive conformation of halophilic alcohol dehydrogenase 2 and the necessity to engineer ionic liquid systems for enzymes that rely on the integrity of functional surface residues regardless of their halophilicity or thermophilicity for use in bioprocessing.
Sustainability spotlightAchieving technoeconomically relevant space–time yields from biocatalysis can often be frustrated by issues in solubility of reagents, stability of the biocatalyst, and general compatibility with process conditions. As such, ionic liquids are being increasingly used to enhance biocatalytic function, although rules for selection of appropriate ionic liquids are not always available, especially for new enzymes. Examining the interactions of ionic liquid ions with halophilic enzyme ADH 2 from Haloferax volcanii, as an exemplar for a high-salt tolerant biocatalyst, we identify general trends and mechanisms governed by ion–ion association, with implications for future selection of ionic liquids suitable for biocatalysis. This outcome aligns with the sustainable development goals 9, 12 and 13 by facilitating improved biocatalytic outcomes and consequent utilization of bioprocesses. |
Introduction
Environmentally benign routes to replace existing chemical processes are a high priority to achieve a sustainable and circular economy. Enzymes can be deployed as biocatalysts for the synthesis of pharmaceuticals, biofuels, fine chemicals, and other industrially relevant molecules.1,2 Biocatalytic processes are often less energy demanding and polluting than traditional chemical synthetic processes since they function under temperatures and pressures below the boiling point of water, they are selective and have good specificity towards substrates and products, and the catalysts themselves are renewable.3 To ensure good utilization of biocatalytic solutions in industry, however, drawbacks such as catalyst instability and difficulties with dissolution of/access to substrates and product recovery need to be addressed.The use of ionic liquids (ILs) as co-solvents in aqueous media has shown great promise in enhancing biocatalytic outcomes.4–6 There are now many studies highlighting how enzyme stability can be improved through the application of ILs,7–9 the advantages conferred by improvements in solubility mediated by the tunability of solvation properties of the ILs,10,11 and how ILs allow for novel mechanisms for product recovery.12,13 Combined with the process advantages of ILs,14 these solvents are attractive to further expand the breadth of chemistries available to bioprocessing.
Usually, binary solutions of IL and water are applied for biocatalytic processes, since there are only a few pure ILs known to dissolve enzymes whilst not inactivating them. In addition, many enzymatic reactions in ILs are heterogeneous due to the low solubility.15 IL–water mixtures add a layer of complexity compared to the application of pure ILs, since solvent physicochemical properties are significantly altered,16,17 but this allows access to an even greater property space. In that sense, deep eutectic solvents, which contain charged and uncharged components, show similar properties to ILs and are similarly applied for biocatalysis.18 Being referred to as the 4th generation of ILs, deep eutectic solvents may be well suited for redox biocatalysis particularly to improve the solubility of substrates.19 Complexity in IL mixtures stems from specific ion effects, which were first described by Hofmeister for ionic salts,20 but ILs have so far defied a quantitative predictive theory. The most widely discussed theories are Collins' law of matching water affinity, where ion pair formation depends on the ions' matching enthalpies,21 and Pearson's very similar concept of hard (polarisable) and soft (non-polarisable) acids and bases.22 Moreover, specific ion effects have not only been observed in dilute aqueous solutions, but also in pure ILs,23 hence ion hydration alone as proposed by Hofmeister cannot explain this effect. Therefore, the exact mechanisms for the interaction between ionic liquid ions and proteins remain poorly understood and finding an adequate system wherein a given biocatalyst remains stable and soluble remains challenging. For this, biocatalysts derived from extremophilic organisms are of substantial interest as they remain functional under harsh conditions, such as high temperatures,24 extreme pH25 or salinity,26 without being specifically engineered. Hence, extremozymes (enzymes derived from extremophiles) act as promising initial candidates for enzyme engineering where challenging reaction conditions are necessary.
To maximise the scope of biosynthetic outcomes, a combination of approaches can be beneficial. Bioprocesses harnessing the inherent properties of halophilic proteins are so far scarce, with the most successful example combining ionic liquids and halophilic cellulases in the saccharification of pretreated lignocelluloses (for examples see ref. 27–33). Studies of a halophilic protease from Salinivibrio sp.,34 a halophilic phenylalanine dehydrogenase from Natranaerobius thermophilus35 and an engineered halophilic malate dehydrogenase36 found ionic liquid systems wherein the enzyme showed increased activity compared to the free enzyme. Halophilic organisms thrive in high salt environments and have adapted their proteins to intracellular molar concentrations of salt. The main structural adaptations are an excess of negatively charged residues located at the protein surface and a reduction of aromatic hydrophobic residues in the core.37,38 A comprehensive study looking at improved IL tolerance of a positively and negatively charge engineered B. subtilis lipase A found electrostatic repulsions of both IL ions.39 Halophilic proteins might thus be expected to be an existing match to avoid problems with ionic liquid compatibility that are seen in mesophilic enzymes.
This study focuses on the industrially significant enzyme alcohol dehydrogenase, here from the archaeal species Haloferax volcanii (HvADH2). HvADH2 forms a homo-tetramer under native conditions and is in its tetrameric form around two thirds more active than as a homo-dimer.40 The archaeal enzyme has been previously found to exert a preference for haloalkaliphilic conditions (4 M KCl, pH 10) when catalysing the oxidative conversion of alcohol substrates to ketones or aldehydes, and slightly acidic (pH 6) for the catalysis of the reductive reaction.40 It exhibits a remarkable thermoactivity with a maximum at 90 °C and its binding pocket can accommodate bulky substrates.41 Under bioprocess conditions, pH can be controlled to allow for the tuning of the reaction equilibrium in favour of the desired product. Glycine–KOH buffer has been routinely used to characterise HvADH2.40 The same buffer conditions have been used to characterise enzymatic activity in a range of organic solvents and the enzyme demonstrated remarkable resilience, specifically in dimethyl sulfoxide and methanol.42 Commercially, the use of co-solvents is a necessity for certain reactions to afford maximum yields and in this case, if sparingly water-soluble ketones are to be used as substrates, they are indispensable. Substitution of organic co-solvents with ionic liquids that can be more easily recycled and endure higher temperatures therefore has good potential for industrial adaptation.
A broad range of different IL ions acting as co-solvent additives were investigated and are depicted in Fig. 1. With regards to MD simulations, these ions represent a wider physicochemical space to study interactions between surface residues and biocatalyst structure and ionic liquid ions than has been reported to date in the literature.43–62
 | ||
| Fig. 1 Structures of ionic liquid ions used in this work. Cation classes comprise phosphonium, ammonium, sulfonium and imidazolium ions (blue). Anions comprise halides, bistriflimides, sulfonic acids, phosphatidic acids and carbonic acids (red). | ||
We report here both experimental results and extensive molecular dynamics simulations that together shed light on some of the key interactions and considerations needed when using ionic liquid co-solvents, including specific complexities of halophilic systems.
Results
Selection of ionic liquids for aqueous mixtures
To effectively probe ion–protein interactions, we selected a range of ions and ion combinations according to the two ion descriptors, polarizability and the range of the molecular electrostatic potential (MEPrange) (Fig. 2). Both have been shown to be of great importance to IL ion behaviour in solution, at surfaces, and in directly influencing reaction kinetics.35,64,65 Anions were selected based on their increasing polarizability, starting with monatomic anions [Cl]− and [I]−, followed by [MeSO4]−, [(MeO)2OPO]−, [bitartrate]−, [MeOEtSO4]− and [NTf2]− in this order. Anions were then combined with different cations diverging in their polarizability and/or MEPrange. Cations included two imidazolium cations ([DiMIM]+ and [BMIM]+), hydroxyl-functionalised cations ([tetrakis]+ and [choline]+), sulfonium based small hydrocarbon cations ([Me3S]+ and [Me3SO]+), and phosphonium or nitrogen based cations with medium-to-long alkyl chains ([N1,1,1,4]+, [N1,1,8,8]+, [P4,4,4,4]+ and [P6,6,6,14]+). Through these combinations we have covered a majority of the descriptor space. A detailed description of these combinations is given in the ESI† section ‘Ionic liquid descriptor space’. | ||
| Fig. 2 Ion descriptor distribution for increasing polarizability (squares) and MEPrange (diamonds) calculated with EMPIRE.63 | ||
Activity of HvADH2 in aqueous ionic liquid solvent systems
Initially, the influence of aqueous ionic liquid mixtures on HvADH2 was investigated by measuring the effect of added ILs on enzyme activity using UV spectroscopy. The relative activity of HvADH2 was assayed by monitoring the formation of NADPH (Fig. 3). Ionic liquid concentrations of 25 mM, 150 mM and 750 mM were assayed for [Me3S]+[MeSO4]−, [tetrakis]+[Cl]−, [choline]+[Cl]−, [DiMIM]+[MeSO4]−, [Me3S]+[NTf2]−, [N4,1,1,1]+[(MeO)2OPO]−, [P6,6,6,14]+[NTf2]− and [Me3S]+[I]−. For a detailed composition of mixtures see the ESI† section ‘Preparation of ionic liquid mixtures’. The relative activities of HvADH2 in systems containing imidazolium ions are consistent with studies reporting inhibitory effects on ADH enzymes.66,67 In our experiments, sulfonate ions [MeSO4]− and [MeOEtSO4]−, when paired with imidazolium ions, are detrimental for HvADH2 activity, despite reports of improvements on conversion rates for ADHs in non-imidazolium and imidazolium-paired sulfonate ion ILs.68,69 In hydroxy-functionalised ILs [tetrakis]+[Cl]− and [choline]+[bitartrate]−, HvADH2 showed the lowest activities. Sulfonium based ions appear compatible with HvADH2, since the enzyme showed the highest tolerance in aqueous ionic liquid mixtures of [Me3S]+[MeSO4]−, [Me3S]+[I]− and [Me3SO]+[I]− with comparable activity to the buffer system. These activities were eclipsed only by the exceptional activity increase of ∼150% for aqueous mixtures of [P6,6,6,14]+[NTf2]−. | ||
| Fig. 3 Ionic liquid effect on the activity of HvADH2 in buffer (blue) and in aqueous ionic liquid mixtures (orange-hues) for all ionic liquid systems (duplicates, error bars indicate estimated standard deviations). Assays containing IL aqueous mixtures were compared to native conditions measured in glycine–KOH buffer at pH 10 containing 4 M KCl at 50 °C. Glycine–KOH buffer was used to make up IL aqueous mixtures for experimental assays, since it has been routinely used in previous studies.40,42 | ||
Additional and repeat concentrations were measured for systems [Me3S]+[MeSO4]−, [P6,6,6,14]+[NTf2]− and [Me3S]+[I]−, since the activity of these initial assays was highest for these aqueous IL mixtures (ESI Fig. S1†). These experiments showed that the enzymatic activity in aqueous mixtures of either [Me3S]+[MeSO4]− or [P6,6,6,14]+[NTf2]− does not follow a continuous decrease with increasing ionic liquid concentration. Activity in [Me3S]+[MeSO4]− plateaued between 150 mM and 300 mM, although the activity was decreased for both compared to the lower concentrations of 25 mM and 75 mM, and activity diminished to below 10% at 600 mM. Despite the formation of an emulsion for all concentrations, the enzymatic activity in [P6,6,6,14]+[NTf2]− also followed a non-continuous concentration dependence. Enzymatic activity decreased between 25 mM and 75 mM but increased at 150 mM compared to 75 mM and increased further at 300 mM, where enzymatic activity was highest. In contrast to [Me3S]+[MeSO4]− and [P6,6,6,14]+[NTf2]−, enzymatic activity decreased continuously with increasing concentrations of [Me3S]+[I]−. The non-linear changes in activity for the aqueous mixture of [P6,6,6,14]+[NTf2]− might be rationalised by the relative solubility of the cofactor (Table 1), which preferably remains in the aqueous phase in both its oxidised and reduced forms when compared to the other applied ILs, according to COSMO-RS calculations. The ADH-catalysed reaction shows improvement over buffer and miscible ILs, similar to previous studies with non-polar biphasic systems.71,72 An optimum volume-ratio of a water-immiscible IL is thus likely to enhance this effect, and accounts for the non-linearity.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) values using OpenCOSMO-RS70 for infinite dilution in pure ILs compared to pure water
P) values using OpenCOSMO-RS70 for infinite dilution in pure ILs compared to pure water
| IL | −logPNADPH |
−logPNADP+ |
|
|---|---|---|---|
| [P4,4,4,4]+[Cl]− | −44.956 | −62.975 | NADP+ more soluble than NADPH in IL |
| [N1,1,8,8]+[MeSO4]− | −19.314 | −27.867 | |
| [N4,1,1,1]+[(MeO)2OPO]− | −37.684 | −45.741 | |
| [Me3S]+[NTf2]− | −3.136 | −9.493 | |
| [P6,6,6,14]+[NTf2]− | +7.038 | +1.307 | NADPH/NADP+ both more soluble in water than IL |
| [BMIM]+[MeOEtSO4]− | −19.368 | −24.832 | |
| [Choline] + [bitartrate] − | −18.387 | −18.706 | NADP + and NADPH display similar solubility |
| [DiMIM]+[MeSO4]− | −22.901 | −21.577 | |
| [Me3S]+[MeSO4]− | −21.201 | −16.184 | |
| [Tetrakis]+[Cl]− | −13.708 | −7.397 | |
| [Choline]+[Cl]− | −49.939 | −42.073 | |
| [Me3S]+[I]− | −34.748 | −19.966 | |
| [Me3SO]+[I]− | −23.702 | −6.173 | NADPH more soluble than NADP+ in IL |
Multiple studies have found ADH activity enhanced at very low IL concentrations of different ions but dropping particularly rapidly at higher concentrations.69,73 A study on a zinc finger protein proposed a transition point in water structuring, affecting the electrostatic interactions and residence times of ions at the protein surface and consequentially, the secondary structure of the protein.53,74,75 The same mechanism could help explain the non-linear decrease in activity in [Me3S]+[MeSO4]− at 300 mM. As such, a molecular-level insight is likely to prove valuable in teasing out these possibilities but will require a comparison of different concentrations of IL [P6,6,6,14]+[NTf2]− and [Me3S]+[MeSO4]−.
MD simulations
Results from activity assays were compared with MD simulations of the enzyme in aqueous solutions containing 4 M KCl and 0.15 M ionic liquid in order to obtain structural insights of specific effects of different IL ions. Comparisons demonstrate good agreement between systems showing a high/low enzymatic activity in assays and IL ion interaction trends with regards to ion specific coordination to residues directly implicated in the substrate conversion mechanism.Fluctuations in distance between residues of the catalytic triad of HvADH2 are increased and become less static demonstrating the impact of IL ions on the active centre
The impact of the presence of ionic liquid ions on the active centre of HvADH2 was assessed from the distance of residues making up the catalytic triad (Ser40, His59 and Asp155) and the catalytic zinc coordinating residue (Cys38) of monomer B (NAD+ absent) and monomer D (NAD+ bound) (ESI Fig. S5†). While distances of these residues stay approximately within 2 Å for the native system containing 4 M KCl, distances in ionic liquid mixtures are increased for all residues with the exception of Asp155–His59 in monomer B of [Me3S]+[MeSO4]− and monomer D of [Me3SO]+[I]− as well as Cys38–Ser40 of monomer D of [Me3SO]+[I]−. Although Asp155–His59 of monomer B of [P4,4,4,4]+[Cl]− and [P6,6,6,14]+[NTf2]− stays within this range too, the distance range over time is less static for these systems in comparison to the native system. For all systems the range between Cys38 and His59 remains similarly static to the native system with the exception of [P4,4,4,4]+[Cl]− for monomer B. Overall, an increase in frequency of rearrangements of residues is observed leading to two regimes that by themselves assume similar distance ranges of ∼2 Å as comparable residues in the native system, although frequently switching between these regimes. This is especially visible for the plotted distance for Cys38–Ser40 in monomer B of [choline]+[bitartrate]−, [BMIM]+[MeOEtSO4]− and [P6,6,6,14]+[NTf2]− and is absent for Asp155–His59 in monomer B of [Me3S]+[MeSO4]−, [Me3SO]+[I]− and [P6,6,6,14]+[NTf2]− and Cl-containing IL systems, as well as Cys38–Ser40 of monomer D for [P6,6,6,14]+[NTf2]−. IL [Me3SO]+[I]− starts out switching between regimes, but over the course of the simulation becomes similarly static as comparable residues in the native system. Taken together, the active centre is impacted by the presence of IL ions in all systems, but the lowest impact is observed for systems containing [Me3S]+[MeSO4]−, [P6,6,6,14]+[NTf2]−, [Me3SO]+[I]− and [Me3S]+[I]−.Active-site gating residues are targeted for π-stacking interactions by IL ions
Work from Klinman et al.76 on a thermophilic ADH (htADH) from B. stearothermophilus identified residues Trp49 and Phe272 as involved in a π-stacking interaction that has a direct effect on the active site microenvironment. These residues are not directly located at the proteins' active site but on the surface, connecting homo-monomers. Closer inspection of the HvADH2 structure shows a similar π-stacking interaction between residues Trp43 and His273, albeit within the same monomer (Fig. 4 and S5†). This interaction correlates with the open, or inactive, state of HvADH2 in the native system and appears to stabilise the apo-enzyme. MD simulations were started with three of the four subunits having NAD+ bound (monomers A, C and D), and, over the course of the trajectory, NAD+ dissociated from two of the three subunits (monomers A and C), whereby only one subunit expelled the cofactor completely (monomer C). Both vanguard residues Trp43 and His273 are involved in guiding the cofactor through π-stacking out of the binding pocket. In a first step His273 pulls the ribose-ring of NAD+ out of the immediate vicinity of the catalytic zinc, followed by a takeover of NAD+ by Trp43 through the interaction with the pyridinium ring of NAD+. NAD+ is then passed back to His273via the ribose-ring and released into an outer cavity on the surface of the nicotinamide binding domain (NBD), where it stays for the rest of the trajectory (monomer A). After NAD+ is expelled, residues His273 and Trp43 assume the π-stacking interaction, which stays undisturbed until the end of the trajectory, and is also found in monomers B and C. In comparison, monomer D remains as a holo-enzyme during the whole simulation and residues Trp43 and His273 do not assume the π-stacking interaction (ESI Fig. S5B†). By comparison, in aqueous mixtures of IL systems [Me3S]+[MeSO4]−, [tetrakis]+[Cl]−, [P4,4,4,4]+[Cl]−, [P6,6,6,14]+[NTf2]−, [Me3SO]+[I]− and [Me3S]+[I]− the cofactor does not dissociate from any of the monomers, while in none of the systems do all cofactors dissociate from monomers. In the [choline]+[bitartrate]− system the cofactor gets pulled farther into the binding cleft separating the substrate and the nucleotide binding domain in monomer D, while it gets removed to the outer cavity in both other monomers. | ||
| Fig. 4 Residues Trp43 and His273 in monomer A of the native HvADH2 system jointly coordinating NAD+ out of the binding pocket. Coordination starts at around 10 ns, and expelling the co-factor is complete after around 20 ns. However, π-stacking between vanguard residues is only established at around 100 ns. (A) NAD+ is coordinated in the vicinity of the nicotine binding domain (NBD) (ice-blue) in close proximity to the catalytic zinc, which is located in the substrate binding domain (SBD) (mauve). (B) NAD+ has been expelled from the catalytic cleft and residues Trp43 and His273 have assumed the π-stacking interaction. | ||
The multimer set-up of the enzyme enables IL ion interactions with the gating residues to be observed for both the closed and open conformations of the enzyme. A close coordination of ions to residues Trp43 and His273 is seen, disrupting the π-stacking interaction in monomers A, B and C, while coordination to Trp43 is also frequent in monomer D (Fig. 5).
 | ||
| Fig. 5 Examples of ions coordinating to gating residues via π-stacking over a prolonged time in monomers B (black) and D (grey). | ||
Coordination takes place in all IL systems and for all ions; however, in some systems ([Me3SO]+[I]− and [Me3S]+[I]−) ions coordinate for short times (<5 ns) and infrequently (less than 10 times over the course of the trajectory) to vanguard residues Trp43 and His273. Other systems ([tetrakis]+[Cl]−, [N4,1,1,1]+[(MeO)2OPO]−, [DiMIM]+[MeSO4]−, [BMIM]+[MeOEtSO4]−, [Me3S]+[NTf2]− and [P4,4,4,4]+[Cl]−) instead exhibit ion coordination over a prolonged time (>50 ns). These interactions take place within the π-stacking distance of 3.7–6 Å.77 In turn, oppositely charged ions directly coordinate to the ions which interact with residues Trp43 or His273, thereby crowding the binding pocket of NAD+. These multi-ion arrangements can persist over a prolonged time (∼50 ns). Especially, systems containing [DiMIM]+[MeSO4]−, [BMIM]+[MeOEtSO4]− and [Me3S]+[NTf2]− show these multi-ion arrangements at gating residues. Furthermore, in the aqueous mixture of [BMIM]+[MeOEtSO4]− the cation is observed to coordinate preferentially to the catalytic residue His59, and in the aqueous mixture of [tetrakis]+[Cl] the cation is observed to coordinate to active Zn2+ ions. Systems wherein ions associate over a prolonged time to vanguard residues or catalytic residues correlate with lower relative activity from activity assays. In the aqueous mixture of [tetrakis]+[Cl]−, wherein HvADH2 showed no activity, [tetrakis]+ cations, for example, coordinate throughout the trajectory and stay associated for up to a third of the trajectory (∼60 ns) (Fig. 5). A synergistic effect of ion combinations is demonstrated by sulfonium ion containing IL mixtures (Fig. 6). Sulfonium ions showed high mobility at vanguard residues in systems paired with [I]−, while π-stacking was observed for the aqueous mixture of [Me3S]+[MeSO4]−. These ions were found in frequent proximity (>10 Å) to vanguard residues and the catalytic zinc, although direct coordination was less frequent and lasted below 5 ns. Contrary to this, in the system paired with the anion [NTf2]−, [Me3S]+ shows a >50 ns long interaction with Trp43.
 | ||
| Fig. 6 Distances between IL cations (blue) and anions (red) and gating residue Trp43 are depicted, and plotted over the course of the trajectory. Cartoon representation shows tetrameric HvADH2 in ILs [Me3S]+[MeSO4]− (top), [Me3S]+[I]− (middle) and [Me3S]+[I]− (bottom). Plots demonstrate the difference in interaction of cation [Me3S]+ within the different systems and its dependence on its counter-ion. | ||
To further assess the ion impact on vanguard residues and hence potential ion influence on activity, the change in distance between the two gating residues was analysed and plotted over the course of the trajectory, comparing IL containing systems to the native system (ESI Fig. S6†). The change in distance of the residues in the native system reflects the dissociation of NAD+ for monomer A, where rearrangements are greatest in the beginning of the trajectory until expulsion of NAD+ is established when the π-stacking interaction fixates the distance between Trp43 and His273 for the rest of the trajectory. For monomer D, change in distance between Trp43 and His273 remains roughly constant throughout the trajectory. All IL mixtures distort distances. A distance trajectory similar to that seen for native conditions is observed for monomer A for ILs [P6,6,6,14]+[NTf2]− and [Me3S]+[MeSO4]−. The most constant narrow distance is observed for system [Me3S]+[I]−, while the most static systems are [Me3S]+[NTf2]− and [choline]+[bitartrate]−.
Secondary structure and free energy landscapes of HvADH2 are altered in all IL systems
To assess the overall structural impact of IL ions on HvADH2 we derive free energy landscapes (FELs) and assign the secondary structure of HvADH2 using the DSSP (Dictionary of Secondary Structure in Proteins) algorithm (Fig. 7 and S7†). FELs represent the conformational space the enzyme adopts during simulation. A higher number of separated minima indicates greater flexibility of the protein, but if the barrier between minima is low, it is an indication of a transition into non-native and possibly non-active states. The native system of ADH2 descends into multiple local higher energy minima before occupying the final lowest minimum. The second lowest minimum is separated via a higher energy bridge and local minima from the lowest minimum, indicating a stabilisation of the structure followed by structural rearrangements into higher energy states before its final descent (Fig. 7 top). All IL systems force HvADH2 into non-native states. The general trend shown by the native system is preserved for all IL systems except for [Me3SO]+[I]−, and is best preserved by [Me3S]+[MeSO4]−. ILs can be grouped into those preventing ADH2 from reaching stabilising minima before its final descent (Fig. 7 middle) ([choline]+[bitartrate]−, [DiMIM]+[MeSO4]−, [BMIM]+[MeOEtSO4]−, [P4,4,4,4]+[Cl]−, [P6,6,6,14]+[NTf2]−, and [Me3SO]+[I]−) and those that show a broadening and an increased number of troughs of local minima (Fig. 7 bottom) ([Me3S]+[MeSO4]−, [tetrakis]+[Cl]−, [choline]+[Cl]−, [Me3S]+[NTf2]−, [N1,1,8,8]+[MeSO4]−, [N4,1,1,1]+[(MeO)2OPO]−, and [Me3S]+[I]−). The former appear to trap HvADH2 in non-native states and the latter appear to destabilise the native state. Secondary structural plots confirm the maintenance of the overall integrity of HvADH2 throughout the trajectory in all ILs. Matching observations from FELs we find a stabilisation of the helix and bent content in systems such as [DiMIM]+[MeSO4]− (Fig. 7 middle) and a destabilisation of the helix and bent content in systems such as [Me3S]+[I]− (Fig. 7 bottom) when compared to the native system (Fig. 7 top). | ||
| Fig. 7 FELs (left, A, C and E) and corresponding secondary structure plots (right, B, D and F) of HvADH2 in buffer (A and B) and aqueous mixtures of ILs [DiMIM]+[MeSO4]− (C and D) and [Me3S]+[I]− (E and F). | ||
Cooperative ion–ion interactions modulate interaction with protein residues
We derive binding energies (Table 2) from radial distribution functions (RDFs) of IL ions around acidic residues (ESI Fig. S8†) and between IL ion pairs (ESI Fig. S10†) and combine them with survival probability values (SPs) between ions (ESI Fig. S10†) alongside a visual inspection of trajectories to investigate ion–ion interaction effects on ion–protein surface interaction in more detail. We find that binding energies of ions to the protein surface are influenced by the binding energies and survival probabilities to their respective counter-ions (Tables 2 and S4† and Fig. 8). Binding energies are in good agreement with the literature.78| Ionic liquid | Binding energies [kJ mol−1] | Inter ion survival probability (ranked) | ||
|---|---|---|---|---|
| Inter ion (one shell) | Ion–acidic residues (combined shells within 15 Å) | |||
| Cations | Anions | |||
| [P6,6,6,14]+[NTf2]− | 181.8 | 2.5 ± 3.9 | 2.9 ± 4.8 | 1 |
| [Me3S]+[I]− | 10.5 | 8.3 ± 0.8 | 21.4 ± 3.4 | 11 |
| [Me3SO]+[I]− | 11.3 | 8.7 ± 0.5 | 20.5 ± 2.2 | 12 |
| [Me3S]+[NTf2]− | 29.5 | 9.6 ± 1.1 | 23.7 ± 5.2 | 6 |
| [P4,4,4,4]+[Cl]− | 5.6 | 19.2 ± 5.1 | 4.4 ± 0.3 | 3 |
| [Choline]+[Cl]− | 11.5 | 12.4 ± 0.9 | 4.8 ± 0.2 | 13 |
| [Me3S]+[MeSO4]− | 16.2 | 7.2 ± 0.3 | 18.2 ± 4.1 | 10 |
| [Tetrakis]+[Cl]− | 7.9 | 21.6 ± 2.0 | 4.8 ± 0.1 | 9 |
| [N4,1,1,1]+[(MeO)2OPO]− | 23.1 | 14.9 ± 1.8 | 15.3 ± 0.7 | 5 |
| [DiMIM]+[MeSO4]− | 21.7 | 12.8 ± 1.9 | 20.7 ± 5.8 | 7 |
| [N1,1,8,8]+[MeSO4]− | 118.9 | 10.1 ± 7.6 | 16.1 ± 5.4 | 2 |
| [Choline]+[bitartrate]− | 14.2 | 11.8 ± 1.9 | 22.5 ± 7.2 | 8 |
| [BMIM]+[MeOEtSO4]− | 16.3 | 19.0 ± 3.4 | 25.1 ± 1.2 | 4 |
 | ||
| Fig. 8 Summary of the general trend of ion–protein and ion–ion interaction inferred from the binding energies of RDFs in combination with SPs and observations from trajectories. Negatively charged residues and anions (triangles) are shown in red, while positively charged residues and cations (squares) are in blue. Functional residues His59, Trp43 and His273 are shown in mint-green. Central surface representation of HvADH2 shows the proportion of negatively charged (red) and positively charged (blue) residues. Catalytic zinc is shown in orange (sphere). | ||
The largest divergence of binding energy between systems for the same ion is observed for anion [NTf2]−. Confinement of [NTf2]− within the hydrophobic chains of [P6,6,6,14]+ prevents frequent access of the anion to the protein surface (Fig. 8A). A similar confinement takes place within system [N1,1,8,8]+[MeSO4]− where the cation [N1,1,8,8]+ has a preference for interacting with the protein surface (Fig. 8K), potentially due to the shortened methyl chains when compared to [P6,6,6,14]+. Binding energies for anion [MeSO4]− diverge between ion combinations too and suggest that in the system [DiMIM]+[MeSO4]− it is the cation that draws the anion to the surface. A strong ion–ion interaction in systems [Me3S]+[NTf2]− and [Me3S]+[MeSO4]− leads to multi-ion associations with protein residues (Fig. 8D and G). Higher binding energies of anions [NTf2]− and [I]− around acidic residues draw the cation [Me3S]+ closer to acidic residues (Table 2). We further find that hydroxy groups on [tetrakis]+ lead to direct coordination of the anion to the Zn2+ ion in the active centre (Fig. 8H), while this is not observed for [P4,4,4,4]+. The stronger binding energy between ions [choline]+ and [Cl]− draws the anion to the surface (Table 2 and Fig. 8F). System [N4,1,1,1]+[(MeO)2OPO]− has the most similar binding energies between counter ions, apart from [P6,6,6,14]+[NTf2]−, based on a strong ion–ion interaction, suggesting a cooperative interaction with the protein surface (Fig. 8I). A similar cooperative strong interaction with the protein surface is observed for [BMIM]+[MeOEtSO4]−, as well as frequent π-stacking interactions between [BMIM]+ and the catalytic His59 (Fig. 8M) and gating residues.
Survival probability values reflect the length of interaction between ions and between ions and the protein surface (ESI Fig. S11†). In [P6,6,6,14]+[NTf2]−, ions show the longest (highest) SP, and in [choline]+[Cl]−, ions show the shortest (lowest) SP. In conjunction with binding energies, this reflects on how frequently the exchange of ions on the protein surface takes place and if ions are present as single molecules or in multi-ion associations. For instance, although the interaction between [P4,4,4,4]+ and [Cl]− is long-lived (third highest SP of all ions), it is a rare encounter (lowest RDF of all ions) (Table 2). Anions and cations of ionic liquid [P6,6,6,14]+[NTf2]− form one big hydrophobic patch that occasionally interacts with a part of the protein surface (Fig. 8A). [Me3S]+ and [Me3SO]+ cations and [I]− anions interact as loose ion pairs (Fig. 8B and C) and [NTf2]− anions and [Me3S]+ cations form ion patches (Fig. 8D). We further observe for hydroxyl-functionalised ions [choline]+, [tetrakis]+ and [bitartrate]− an intrusion below the solvation shell and an interaction with counter-charged protein residues. The association of ions with residues increases with an increased number of OH groups (ESI Fig. S8†). Ions of [choline]+[bitartrate]− interact as ion pairs, while [choline]+ and [tetrakis]+ interact as loose ion pairs with [Cl]− ions. [Me3S]+ is mostly paired with two [MeSO4]− (Fig. 8G). Finally, ions of the systems [N4,1,1,1]+[(MeO)2OPO]−, [DiMIM]+[MeSO4]− and [N1,1,8,8]+[MeSO4]− form ion patches (Fig. 8K and J) and cations of IL [P4,4,4,4]+[Cl]− form patches on the protein surface (Fig. 8E). Systems wherein cations show the lowest binding energy to acidic residues correlate with the highest activities of HvADH2 from assays in IL mixtures, while systems containing cations with the highest binding energies to acidic residues impair activity significantly.
All ILs distort the native solvation shell of HvADH2
To specifically investigate the impact of IL ions on the solvation shell of the halophilic enzyme we firstly characterised the native system using RDFs and SPs and compared our results to literature reported solvent structure surrounding proteins and single acidic amino acids in high salt. Our results confirm that the presence of K+ ions around negatively charged residues ‘breaks’, or rather, mediates the solvation shell, and, through the increased number of acidic residues, allows a cumulative effect to offset salting-out conditions through localised ion association. In addition to literature reports we observe that the protein surface introduces a higher ordering of molecules for the association shell between OW and OW, when compared to free floating aspartic amino acid molecules.79 We further observe a difference for the RDF between K and OW resulting in peak splitting of the first peak into two solvation shells due to the presence of the protein surface,79 and furthermore, a higher ordering into better defined shells of K+ arrangements at the protein surface (15 Å) compared to the profile of K+–K+ within 100 Å. The RDF between potassium and carboxylate groups of Asp/Glu residues determined in this study (ESI Fig. S12†) matches the same positions for all three coordination shell peaks as found in a study by Warden et al., constituting sodium ions in various salts at the surface of an engineered halo-tolerant carbonic anhydrase.80 RDF distances between OW and C4/C5 of Asp/Glu, respectively, match solvation shells found at protein surfaces reported in the literature.81 However, in addition, we observe an increase of OW at acidic residues of the first coordination shell at ∼2 Å compared to non-halophilic CALB81 as well as at non-acidic surface residues of HvADH2 (ESI Fig. S13†).Based on our simulations, we calculated probability densities around charged (Fig. 9) and uncharged residues (ESI Fig. S14†). These indicate coordination of water structure by K+ at the protein surface regardless of surface charge. For instance, coordination of water around Thr1073 by K+ demonstrates these site-specific influences particularly well (ESI Fig. S14†). We can demonstrate a direct interaction (∼2.7 Å) between K+ ions and specific buried Glu residues (Fig. 9A), which is established over a prolonged time. The very same K+ ions stay associated with Glu1292 in monomer D, as well as its equivalent in monomer A, Glu245, over the whole trajectory (ESI Fig. S15†). This prolonged coordination of K+ ions to Glu245 and Glu1292 is unchanged or only occasionally exchanged in most systems ([Me3S]+[MeSO4]−, [choline]+[Cl]−, [choline]+[bitartrate]−, [N1,1,8,8]+[MeSO4]−, [P4,4,4,4]+[Cl]−, [N4,1,1,1]+[(MeO)2OPO]−, and [P6,6,6,14]+[NTf2]−), perturbed in some ([tetrakis]+[Cl]−, [DiMIM]+[MeSO4]−, and [Me3SO]+[I]−) or entirely prevented in others ([Me3S]+[NTf2]−, [BMIM]+[MeOEtSO4]−, and [Me3S]+[I]−). These Glu1292 associated K+ ions in turn coordinate water molecules, which stay associated for about half the trajectories before being exchanged. In systems [Me3S]+[I]− and [Me3SO]+[I]−, however, in the absence of K+, interstitial water molecules still coordinate over a prolonged period of time (∼25 ns). By comparison, acidic surface residues exchange their K+ frequently (every few fs). We find that in the native HvADH2 system the decay of K+ around Glu and Asp is slower by approximately a factor of 4 compared to its decay around positively charged residues, thus indicating a prolonged interaction between K+ ions and acidic residues (ESI Fig. S16†). Compared to the native system, SPs of K+ ions around acidic residues are decreased in all IL systems and increased for all ILs around basic residues, except for hydroxy-functionalised ILs, where they are slightly decreased (ESI Fig. S17†). This indicates faster dynamics of K+ ions around acidic residues and slower dynamics of K+ ions around basic residues in IL systems. RDFs of K+ ions in IL systems show the same profile as the native system, but are increased for all ILs around acidic (Fig. 9) as well as basic residues within 4 Å of the residues (ESI Fig. S18†), while Cl− ions are slightly removed from basic as well as acidic residues (ESI Fig. S19†). This indicates a tighter positively charged ion lattice at the protein surface regardless of charge for all IL systems. Taken together this suggests that all IL systems influence the dynamics of K+ around charged residues leading to faster dynamics and a greater total number of K+ surrounding negatively charged residues, while mitigating dynamics between K+ and positively charged residues.
 | ||
| Fig. 9 Plotted RDFs and visualised SDFs of K+ (blue mesh or solid) and H2O (orange mesh or solid) molecules surrounding carboxylic acid residues in the native HvADH2 system. (A) Direct association of K+ with COO− occurs. Directly associated K+ at 2.7 Å may account for the distance of the second hydration shell of H2O around COO− at 2.9–3.1 Å consistent with the distance between K+ and H2O in their first hydration shell at 2.9 Å. (B) Neither K+ nor H2O associate directly. Hydration of COO− is established by the fourth association distance of H2O at >4.1 Å, since H2O molecules get pulled towards the associated K+ ions. (C) Direct association of water with COO− at 1.5–2.1 Å may account for the second coordination shell of K+ around COO− at 3.7–4.3 Å, in concordance with the first hydration shell of H2O around K+ of 2.9 Å. (D) No direct association takes place. K+ ions either strip H2O molecules partially from COO− residues or are themselves removed behind a water barrier. (E) Direct association of water in its second hydration shell at 2.9 Å with COO− may associate K+ ions according to the fourth association distance between H2O and K+ at 4.5 Å. | ||
Water has a similar short permanence time (survival probability, SP) around negatively or positively charged residues, indicating that the dynamics (mobility) of the water network is not altered by the prolonged presence of K+ ions at carboxylate groups (ESI Fig. S16†). SP of H2O around acidic as well as basic residues is decreased in all IL systems (ESI Fig. S20†). RDFs of H2O are decreased for all systems around acidic residues, around K+ ions and between H2O molecules (Fig. 10). In this, the dynamics of H2O are altered in all IL systems similarly, becoming generally faster, and molecules become more dispersed.
 | ||
| Fig. 10 (A) Total number of salt bridges (grey) between protein residues of HvADH2 monomers calculated by VMD from MD simulations for the native system in high salt (4 M KCl) in comparison to IL systems. Within all IL systems novel salt bridges are formed (red), while some native salt bridges are lost (blue) for all systems, with the exception of IL [Me3S]+[MeSO4]−, where none are lost. (B–E) Plots show RDFs of K+ and water around acidic residues in IL mixtures (coloured lines) in comparison to the native system (black line). While K+ ions are increased around acidic residues in all IL systems, water coordination is overall decreased. | ||
ILs compromise the structural integrity of the native protein salt bridge networks
To investigate the impact of altered spatial correlations and residence times of K+ and Cl− ions and water molecules on the solvation shell of HvADH2, the total number of salt bridges of the tetramer (intermolecular and intramolecular) and of the monomers (intramolecular) with an O–N distance cut-off of 3.2 Å was calculated for the native system and compared to IL systems. All ILs lead to an increase in the total number of salt-bridges, but not all native salt-bridges are conserved, with the exception for system [Me3S]+[MeSO4]−. New salt-bridges are formed in all ILs (Fig. 10). This implies an interruption of the native salt-bridge network for all IL mixtures and supports the distortion observed in the secondary and tertiary structure of HvADH2 (Fig. 7).RDF profiles of H2O around acidic residues indicate that all aqueous IL mixtures lead to a decrease in water content around acidic acid residues compared to the native system (Fig. 10). A possible explanation is that water is either displaced on the surface by K+ or IL ions, since RDF profiles of K+ around acidic residues are increased. This stripping of water molecules and increase of K+ ion presence at the protein surface could be an explanation for the overall increase in salt bridges observed for IL systems.
While there are new salt bridges formed in the system [Me3S]+[MeSO4]−, none of the native salt bridges are lost. This suggests that the formation of new salt bridges and the loss of native salt bridges are founded in two different ion–water–surface interaction mechanisms. [Me3S]+[I]− interferes most with the hydration of K+ around acidic residues and [choline]+[bitartrate]− acts most severely on the water structure itself leading to the greatest difference in RDF of OW around acidic residues compared to the native system. Surprisingly, [Me3S]+[MeSO4]− is one of the systems that interferes most with both water and ion structure. This suggests that the specificity of interactions between IL ions and residues plays a role in compromising native salt bridges and thus potentially the enzyme activity.
Discussion
Individual ionic liquid ion combinations offer a vast combinatorial property space. However, an outstanding challenge remains to choose an adequate IL system for a given protein and application. Our work provides insight into multifaceted interactions taking place between ions, the solvation shell and protein residues. We are able to show that all IL systems have an influence on the solvation of cofactors, the solvation shell and the secondary structure of the protein. Interaction with residues directly implicated in maintaining activity of the enzyme is affected in all ILs; however, while some ion combinations synergistically prevent co-ions from specifically interacting with these residues, others synergistically coordinate to them. Ions of systems containing ionic liquids [Me3S]+[I]− and [Me3SO]+[I]− for instance interact infrequently with gating residues, and [Me3S]+[MeSO4]− does not disrupt native salt bridges; on the other hand, IL systems such as [Me3S]+[NTF2]− form multi-ion arrangements through π-stacking with activity conferring aromatic residues, despite consisting of the same cation. This can be explained by the inter-ion interaction strength through derived binding energies. In turn, binding energies between ions correlate to some degree with MEPrange ratios between anions and cations (Fig. 11).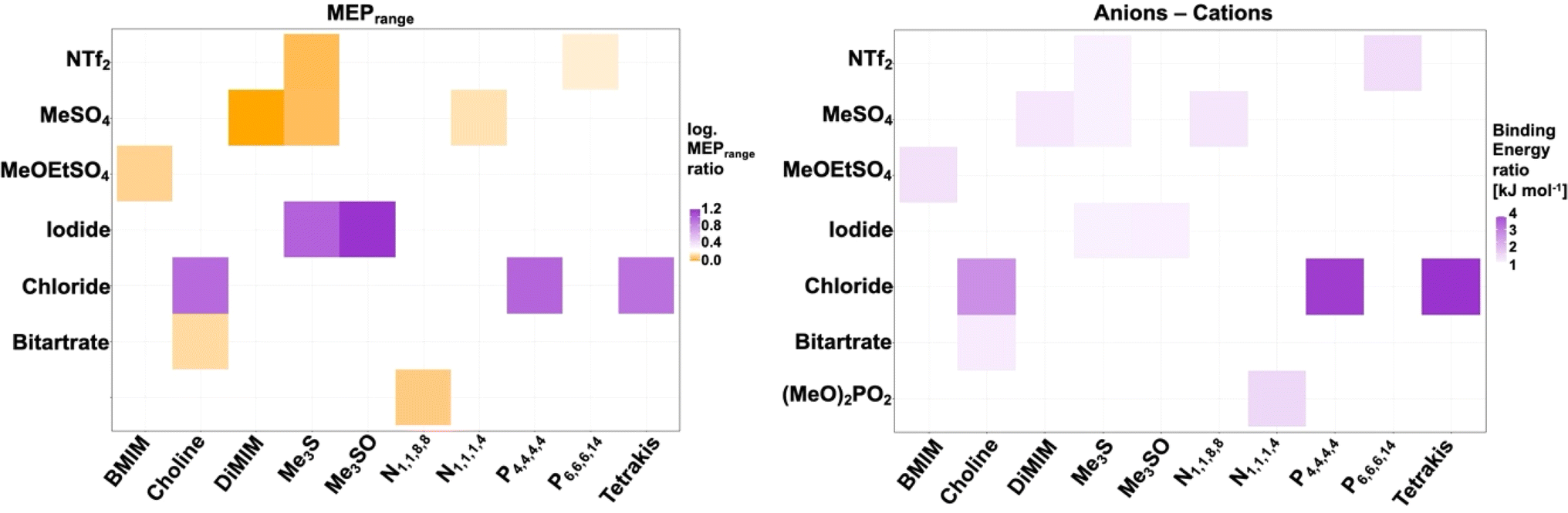 | ||
| Fig. 11 MEPrange ratio of anions and cations (left) and ratio of binding energies between anions and cations (right). A greater difference in MEPrange between the anion and the cation correlates loosely with a smaller binding energy between paired counter-ions. | ||
Although we are unable to show a clear correlation between polarizability and MEPrange associated with either an increased or decreased enzyme activity, our results demonstrate that cooperative ion–ion interactions strongly influence ion–protein interactions and can help explain enzymatic activity found in assays. We observe for ions that show a high inter-ion binding energy and a high inter-ion SP (close to 1) that these have a close interaction with the protein surface. This surface interaction modulates the overall interaction with protein residues, while the influence of individual ions on the protein surface is greater for ions that share a low SP (close to 0) and low binding energies with one another.
For example, while the strong hydrophobic interaction between [P6,6,6,14]+ and [NTf2]− shields the anion from the protein surface, the comparatively decreased interaction with the cation [Me3S]+ allows [NTf2]− to strongly interact with positive residues at the protein surface. This strong inter-ion interaction, as is the case for [P6,6,6,14]+[NTf2]−, can be harnessed to favour biocatalytic processes. Here, phase separation means the protein is mainly excluded from the hydrophobic patch of the IL, whilst providing an advantage for the partition coefficient of the cofactor in the aqueous phase, and hence the agglomeration of [P6,6,6,14]+ and [NTf2]− correlates with a much higher relative activity. This system consists of ions with the highest polarizability and a high MEPrange. However, as a general trend, a low polarizability and a low MEPrange of ions seems to be less detrimental to enzymatic activity of HvADH2. Small anions of hydrophilic ILs were shown to interact as mediators between water molecules and cations, which are to some extent expelled and form clusters similar to micelles.82 The [Me3S]+[MeSO4]− ions, which form ion patches, and [Me3S]+[iodide]−, [choline]+[Cl]− and [Me3SO]+[iodide]−, which form loose ion pairs, show low polarizability and rank best in terms of enzymatic activity behind the superior system [P6,6,6,14]+[NTf2]−. This suggests that small, highly dynamic cations with low polarizability paired with small charge-dense anions help preserve catalytic activity in HvADH2 best. By comparison, when paired with a highly polarisable cation ([N1,1,8,8]+) enzymatic activity is impaired for the anion [MeSO4]−, unlike in [Me3S]+[MeSO4]−. The atomic structure of the ions plays an equally important role. The cation in system [DiMIM]+[MeSO4]−, which forms ion patches similar to [Me3S]+[MeSO4]−, has a comparable polarizability to cation [Me3S]+. However, the presence of the planar structure of [DiMIM]+ led to a 5-fold decrease in enzymatic activity when compared to the tetrahedral structure possessing cation [Me3S]+. The formation of multi-ion structures is driven by entropic forces, since apolar domains minimise the disturbance of the H-bond network of the water molecules. This surfactant effect becomes stronger as the cationic alkyl chain becomes longer and this can affect the stability and activity of proteins positively through suppression of protein–protein interactions, preventing aggregation.83 However, if coulombic interactions between ions are strong and moreover if such ions are substituted with hydrophobic tails, strong dispersion forces are observed.83 While this does not have immediate implications for proteins if these ion clusters are located in the bulk solvent, it is likely that such ions are expelled from the hydrogen bond network at interfaces and may act as surfactants for solvated proteins. We find this to be the case for systems [N1,1,8,8]+[MeSO4]−, [P4,4,4,4]+[Cl]−, [DiMIM]+[MeSO4]− and [N4,1,1,1]+[(MeO)2OPO]−, which form small patches all over the protein surface, and these systems show a decreased enzymatic activity. By comparison, [MeOEtSO4]− and [BMIM]+ form ion pairs, but because both ions interact strongly with the protein surface, their synergistic effect on the protein is detrimental, since a greatly diminished relative activity (<5 U mg−1 at 150 mM) is observed for this system.
Ion–water interactions negatively affected protein–water interactions in all IL systems, and hydroxy-functionalised ILs did not stabilise the surrounding water network. Indeed, hydroxy-functionalised systems performed the worst of all ILs when multiple functionalised groups were present, with only [choline]+[Cl]− performing well. This may be due to increased protonation/deprotonation events at the protein surface84 or may be a more general outcome for all ILs due to ion type dependent alignment of water at interfaces.85 The tight binding of hydration water around carboxylic acid residues in the native system, consistent with previous reports,79,86–88 is disturbed in all ILs, as is the selective breaking of water structure induced by the coordination of potassium around carboxylic acid residues.88 An increased presence of potassium ions at the protein surface is observed and displaces water molecules, thereby altering the native salt bridge network. Overall, this increases salt bridges in all IL systems. This finding is consistent with an MD study on lactalbumin in different concentrations of [BMIM]−[BF4]+, which found an increase in the number of salt bridges with increasing concentration of IL, and an increased strength of the salt bridge bond.58 Inter-subunit salt bridges have been shown to be increased for halophilic proteins when compared to their mesophilic counterparts89–91 and it was suggested that oligomerisation acts as a stabilisation mechanism in extremophiles.92 This perhaps helps explain the well preserved activity of HvADH2 in aqueous mixtures of [Me3S]+[MeSO4]−, which is the only system wherein none of the native salt bridges are broken.
The negatively charged residues of halophilic protein surfaces cannot protect enzymatic activity from the influence of IL ions. Unlike an engineered B. subtilis lipase, where 20 amino acids were specifically selected and substituted, the elevated charge is present throughout the protein surface in HvADH2 and makes up 27% of the total amino acid content, whereby all charged residues are located on the surface, except one amino acid per monomer.39,93 Perhaps this explains the difference in electrostatic repulsion and attraction between B. subtilis lipase and HvADH2, since the same group derived a ranking of amino acid substitutions, which do not nearly cover the surface as extensively as negative charges cover the surface of HvADH2.94 Unlike potassium interacting specifically with carboxylate residues, IL ions interact with the protein surface depending on both their physico-chemical properties and their inter-ion interactions. In contrast to a study where the local structural stability of B. subtilis lipase is modified by IL ions inducing long-range perturbations of noncovalent interactions, which eventually reach the active site, we observe a direct interaction with active site residues and apo enzyme structure-determining residues.95 However, this does not mean that there are no such long-range perturbations in our system. For enzymes such as dehydrogenases that have a less robust catalytic mechanism, which depends very much on the integrity of their quaternary structure,40 and for which the active and inactive conformations rely on the functionality of specific (aromatic) residues located on the surface of the protein, a fine-tuning of IL ions is required in the same way for halophilic as well as non-halophilic proteins.
Conclusion
Taken together, our data highlights the tuneability of the influence of ions on protein activity depending on co-ion interaction. The same anion or cation exhibits a different influence on the protein when paired with a different co-ion, mainly depending on the strength of ion–ion interaction. The quality of ion–ion interactions depends on polarizability and the resulting molecular electrostatic potential, and binding energies between ions correlate loosely with MEPrange ratios between anions and cations. We find that, for halophilic proteins, cations with low polarizability are better suited to maintaining activity; however, ion structure needs to be taken into account, since specific ions and ion-combinations target specific residues, such as aromatics, and thereby elicit a decrease in activity. Future experiments on structure/stability relationships should be undertaken, which will provide a fully conclusive and complementary support for the extensive computational and activity data presented, and provide further illumination of the important roles of ion combinations in protein–solvent interactions.Materials and methods
Reagents and culture conditions
All chemical reagents, unless stated otherwise, were purchased as analytical grade from Sigma-Aldrich. All restriction enzymes were purchased from New England Biolabs. Standard molecular cloning techniques were used. PCR amplification used Q5® Hot Start High-Fidelity DNA polymerase. H. volcanii strains were grown at 45 °C on complete (Hv-YPC) or Cas-amino acid (Hv-Ca) agar or broth as described previously.96 Isolation of genomic and plasmid DNA as well as transformation of H. volcanii strains were carried out as described previously.97,98Plasmid construction
All primers were designed using MacVector Version 14.5.2 (MacVector, Inc.) and synthesized by Eurofins MWG, Germany. All plasmids were confirmed by sequencing. Construction of expression-plasmid pTA1205 for 6xHis-ADH2 expression and deletion-plasmids pTA1229 and pTA1230 for the deletion of adh1 and adh2 genes, respectively, were described previously.40Strain construction
H. volcanii Δadh1, Δadh2, ΔtnaA and ΔgabT1 mutant strains were generated using previously described gene knock-out systems.97,99H. volcanii strain H1895 (ΔpyrE2, Nph-pitA, Δmrr, ΔhdrB, Cdc48d-Ct, ΔpilB3C3)100 was the source strain for generating the expression strain H2974 (ΔpyrE2, Nph-pitA, Δmrr, ΔhdrB, Cdc48d-Ct, ΔpilB3C3, Δadh1, Δadh2, ΔtnaA, ΔgabT1). Deletions were confirmed using colony hybridisation and Southern blotting. The H. volcanii strain H2974 was transformed with pTA1205 to obtain strain H3094 (ΔpyrE2, Nph-pitA, Δmrr, ΔhdrB, Cdc48d-Ct, ΔpilB3C3, Δadh1, Δadh2, ΔtnaA, ΔgabT1) for overexpression of 6xHis-ADH2.Protein expression and purification
An overnight starter culture (5 ml) was diluted (1:100) at OD600 ∼0.1 into 5 ml and again grown until OD600 ∼0.1. Cultures were diluted 1:100 in 50 ml and incubated for 24 h at 150 rpm until an OD600 of ∼0.5 was reached. Cultures were then diluted (1:50) into 333 ml YPC broth and induced with 0.1 g tryptophan and incubated at 150 rpm until an OD600 of ∼1.2–1.5 was reached (24–36 h). Cells were harvested by centrifugation and the resultant pellet was either frozen for later use or resuspended in 5 ml wash buffer (20 mM HEPES, pH 7.5, 2 M NaCl). Cells were disrupted by sonication on ice (∼3 × 30 s at 6 W) until the lysate appeared clear. After centrifugation (48000g for 30 min, 4 °C) the supernatant was filtered and loaded onto a HisTrap HP (GE Healthcare) immobilised metal-chelate affinity chromatography (IMAC) column pre-charged with NiSO4 (0.2 M) at a flow rate of 0.5 ml min−1 using loading buffer (20 mM HEPES, pH 7.5, 2 M NaCl, 20 mM imidazole). Elution buffer (20 mM HEPES, pH 7.5, 2 M NaCl, 50 mM EDTA) was applied to the IMAC column to obtain 2 ml fractions. Fractions were assayed spectrophotometrically for alcohol dehydrogenase activity. Selected fractions were pooled and then dialysed and concentrated using Viva Spin columns (Sartorius) using 3 M glycine–KOH buffer (pH 8). Purified protein samples were analysed by SDS-PAGE and protein concentration was determined using the Bradford protein assay dye reagent (Bio-Rad Laboratories GmbH, Germany).
Activity assays
Relative activity was assayed spectrophotometrically by monitoring the increase in absorbance of the cofactor NADPH at 340 nm using an Epoch2 Microplate spectrophotometer (Biotek) with UV-transparent 96-well plates. The reaction mixture routinely contained ethanol (100 mM), NADP+ (1 mM) and enzyme sample (∼450 nM) and 50 mM glycine–KOH (pH 10.0) buffer containing 4 M KCl. Experiments were carried out at 50 °C for 20 minutes.Ionic liquids
Aqueous ionic liquid mixtures used for experimental activity assays as well as MD simulations are summarised in ESI Table S1.† For activity assays [Me3S]+[MeSO4]−, [tetrakis]+[Cl]−, [choline]+[Cl]− and [choline]+[bitartrate]− were purchased from Acros Organics. [DiMIM]+[MeSO4]− and [P6,6,6,14]+[NTf2]− were purchased from Fluka. [Me3S]+[NTf2]− was purchased from Solvent Innovation. [BMIM]+[MeOEtSO4]−, [N1,1,8,8]+[MeSO4]− and [N4,1,1,1]+[(MeO)2OPO]− were donated by the Sustainable Process Technologies (SPT) group at the University of Nottingham. [P4,4,4,4]+[Cl]− was purchased from QUILL. [Me3SO]+[I]− and [Me3S]+[I]− were purchased from Sigma Aldrich. All ionic liquids were used as received without further purification.Characterisation of HvADH2 activity in ILs
All ionic liquid mixtures were made up in 50 mM glycine–KOH (pH 10.0) buffer containing 4 M KCl, unless stated otherwise. Oxidative reactions of HvADH2 were assayed using ethanol (100 mM) and NADP+ (1 mM) in aqueous ionic liquid mixtures. For a detailed composition of mixtures see the ESI.†Protein sequences and structures
The protein sequence of HvADH2 was retrieved from the National Centre for Biotechnology (NCBI) database with deposit number ELY36761.1. Homology models were built using Swiss-Model,101–105 Phyre2,106 and I-TASSER.107,108 Models were assessed via structural alignment (BLAST) and visualisation (PyMOL). The best model was selected based on the preservation of conserved residues of the catalytic triad coordinating the catalytic zinc (CYS-89, CYS-92, CYS-95 and CYS-103). Following this, the homology model from I-TASSER with the highest C-score (1.62) was chosen to build the tetrameric structure using BIOVIA Discovery Studios Visualizer.109 The tetrameric structure of TADH from Thermus sp. Atn1 was retrieved from the protein data bank (RCSB) with the deposit reference 4cpd, and was used as a template to superimpose the monomeric model built by I-TASSER to obtain coordinate positions of the four homo-tetramer units. The geometrically cleaned-up structure was then validated by PROCHECK.110 The obtained Ramachandran plot reported the dihedral angles at 74.7% in the most favoured region, 20.1% in the allowed region, 2.8% in the generously allowed region and 2.4% in the disallowed region (see ESI Fig. S21†). The model was then used to create necessary topology files and was processed with AMBER18 (Assisted Model Building with Energy Refinement),111 as described below, to run MD simulations. From 200 ns MD time the root mean square deviation (RMSD) was calculated for backbone atoms of the four monomers (ESI Fig. S2†) as well as the root mean square fluctuations (RMSF) (ESI Fig. S3†) to validate the model stability during simulation. RMSF was plotted as standard deviation between the four monomers.Molecular dynamics simulation and preparation
The protonation states of amino acid side chains of the I-TASSER HvADH2 model were adjusted to pH 10 conditions using the H++ server. The software suite used for running all simulations was AMBER18,111 whereby xLEaP, antechamber and parmchk were used as preparatory programs, pmemd.cuda 9.2 was used to run simulations and cpptraj was used to transform obtained trajectories for visualisation. To obtain MD simulation input files (.inpcrd and .mdcrd) the routinely applied gaff force field was used.8,112,113 The forcefield leaprc.ff14SB was used for non-coordinating protein residues114 and the zinc Amber forcefield (ZAFF)115 was used for Zn2+ coordinating residues, whereby CY4 and HD2 parameters were applied for catalytic residues and CY1 parameters were chosen for structural residues.A recent study by Daronkola et al. found that unoptimized anion–cation parameters for the interaction of potassium with acetate applied in the Amber-GAFF force fields can misrepresent activity derivatives varying with concentration.116 They found that contact-shared ion pairs (CIPs) are overestimated, while solvent-shared ion pairs (SIPs) are slightly underestimated. RDF values produced in their study with their newly chosen scaling factor showed the same peak position, but coordination numbers were overall lower for potassium around acetate compared to the amber scaling factor. They still found an increased proximal number density for K+ ions and H2O molecules around halophilic proteins when compared to their mesophilic counterparts. Use of the amber GAFF force field for the present study thus presents a limitation to quantitation that we acknowledge in the context of this finding. We have thus presented comparative data rather than quantitative, to ensure that our findings remain representative.
Ionic liquid structures were built in Avogadro117 and geometry optimised using Gaussian B3LYP/6-31G(d).118 The atomic point charges for the ILs were obtained by following the standard restrained electrostatic potential (RESP) fitting procedure,119 followed by scaling the charges by applying a factor of 0.8 in accordance with established literature.112 Leaprc.gaff was used to produce .mol2 and .lib files and antechamber was used to convert the .mol2 file into a .pdb file. Packmol was used to assemble molecules for simulations in variable numbers reflecting different ion concentrations.120 Molecule numbers were calculated according to Nion = NA × cion (mol l−1) × V (l), where Nions is the number of ions, NA is the Avogadro number (6.022 × 1023), cion the concentration of the ion and V the volume. The protein structure was placed in a quadratic box (100 × 100 × 100 Å) and neutralised by adding K+ and Cl− ions using xLEaP. Following neutralisation, ionic liquid molecules were inserted to a concentration of 150 mM (a total number of 80 ion pairs) using packmol. In a final preparation step, the box was solvated with a minimum distance of 2 Å between water molecules using the TIP3P121 water model in xLEaP and K+ and Cl− ions were added to a concentration of 4 M (total number was around 1800) using the addionsrand command. The forcefield leaprc.gaff with amber parameters sourced from frcmod.ionsjc_tip4pew was used in the production of .mdcrd and .inpcrd files for input in simulations.
Long-range electrostatic interactions and non-bonded interactions were modelled using the particle-mesh Ewald method for periodic boundaries with a cut-off of 10 Å. The first energy minimisation run (50000000 steps, steepest descent algorithm for the first 20000000 steps, then switched to conjugate gradient algorithm) was performed only on water with a restraining force of 10 kcal mol−1 on all other molecules. This was followed by a first heating step of 10 ps to 323.15 K applied to water molecules and a restraining force of 10 kcal mol−1 on all other molecules. The overall system was integrated to a temperature isotherm using the Berendsen thermostat with a close coupling of 0.5 ps. A second minimisation step was performed and applied to all atoms using no restraints, followed by a second heating step of the whole system for 0.25 ns under NPT conditions (constant volume in periodic boundaries, Berendsen thermostat) and a subsequent density equilibration step applying Langevin dynamics for 2.5 ns under NTP conditions (constant pressure in periodic boundaries) with restraining forces of 10 kcal mol−1 on the protein. Molecular dynamics production run simulations were subsequently run with unrestrained systems for between 150 and 200 ns.
Analysis of MD simulations
Trajectories were visualised using VMD.122 The physical parameters RMSD,123,124 SP125 and RDFs were calculated using python package MDAnalysis.126,127 Principal component analyses on Cα atoms to obtain eigenvalues and eigenvectors were calculated using GROMACS128,129 and the first two eigenvectors, PC1 and PC2, which describe >55% of the structural transitions of the overall protein for every system were used to construct free energy landscapes. Post analysis of the molecular dynamics data was graphically represented with Matlab and python packages (MDAnalysis, matplotlib, numpy). TRAVIS was used to calculate SDFs,130 which were visualised in VMD with isovalues of 10 for water and 3 for potassium.Calculations of local properties and descriptors
The molecular electrostatic potential range (MEPrange) and the polarizability for IL cations and anions were calculated using the Cepos Insilico software packages EMPIRE.63 Cube files were obtained through an eh5cube.sh off the EMPIRE wavefunction output file and visualised in VMD with isovalues of ∼0.02 e Å−3.Calculation of relative solubilities using OpenCOSMO-RS
The relative solubilities of NADPH and NADP+ between water and the ILs were calculated using the COnductor-like Screening MOdel for Real Solvents (COSMO-RS) approach,131–134 as implemented in the open source openCOSMO-RS software.70 The COSMO-RS method and openCOSMO-RS implementation have been described in detail previously.135 In brief, a selection of conformers was generated using the RDKit python package,136 followed by a workflow of quantum chemical calculations using the Orca software package,137,138 to generate a conformer ensemble for COSMO-RS calculations. To refine the ensemble, conformers were initially eliminated according to energy and similarity. For those conformers that remained, screening charge density was obtained with a COSMO single point calculation using the BP86 method and def2-TZVPD basis set. COSMO-RS calculations were then applied. Populations of conformers in the ensemble were determined according to the Boltzmann distribution by an iterative procedure based on the COSMO-RS chemical potential.135 The iterative system was considered converged if, among conformers, the mean chemical potential difference was below 1 × 10−6 kJ mol−1 and the highest chemical potential difference was below 3 × 10−6 kJ mol−1. For each IL, the weighted average by population of the converged conformer COSMO-RS chemical potentials, was used to obtain γinf(NADPH) and γinf(NADP+), the infinite dilution activity coefficients of NADPH and NADP+, respectively. Calculations were performed with respect to a pure water reference state. Therefore, infinite dilution IL/water partition coefficients are reported as −ln(γinf(NADPH)) and −ln(γinf(NADP+)).Data availability
Data are available both within the ESI† and within separate files and repositories as detailed. Data points obtained from measuring spectrophotometrically the increase in NADPH are provided in the Excel sheet named “Experimental_data_ADH2_IL_ActivityAssays.xlsx”. Amber input files for simulations (xxx.prmtop and xxx.inpcrd) alongside files derived from packmol (xxx_box.pdb) and xLEaP (xxx.pdb, HvADH2net.pdb) are deposited at the Zenodo repository under https://doi.org/10.5281/zenodo.10066549. Since xxx.mdcrd files were too big to be deposited (some >50 GB), files were converted into gromacs trajectory files xxx.xtc and xxx.gro using the trajectory converter from the python MDAnalysis suite in Jupyter Notebook. These are deposited at the Zenodo repository under https://doi.org/10.5281/zenodo.4706937. VMD output .dat files of salt-bridges occurring for each step of the trajectory are zipped in the file “saltbridges.zip” and deposited at the Zenodo repository under https://doi.org/10.5281/zenodo.11916740. Scripts derived from the python library ‘MD Analysis’ to calculate RDFs, SPs and RMSDs and the respective data points stored in .csv sheets used to plot RDFs, SPs and RMSDs are zipped in the file “MDAnalysis_Calculated_Plotted_Data.zip”. Calculated MEPrange, polarisability and other ion descriptors in form of .csv files as well as .cube files and others necessary for visualisation of descriptors are contained in “IonDescriptors.zip”. The .lib, .frcmod and .pdb files required to parametrise IL ions for simulations are contained in “ILsource.zip”.Author contributions
Alexandra Schindl: conceptualisation (equal), formal analysis (lead), investigation (lead), visualisation (lead), methodology (lead), writing – original draft preparation (lead), writing – review and editing (equal); M. Lawrence Hagen: formal analysis (support), visualisation (support); Isabel Cooley: solubility analysis (lead), solubility methodology (writing); Christof M. Jäger: methodology (support), writing – review and editing (equal); Andrew C. Warden: methodology (support); Mischa Zelzer: conceptualisation (equal), supervision (equal), writing – review and editing (equal); Thorsten Allers: conceptualisation (equal), supervision (equal), methodology (support), writing – review and editing (equal); Anna K. Croft: conceptualisation (equal), supervision (equal), writing – original draft preparation (support), writing – review and editing (equal).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to gratefully acknowledge the support of the BBSRC/EPSRC Synthetic Biology Research Centre – Nottingham (BB/L013940/1) and the University of Nottingham for the financial support provided to AS, and the BBSRC for the award of a Doctoral Training Grant (BB/M008770/1, Nottingham-Rothamsted Doctoral Training Partnership) for MLH. AKC would like to gratefully acknowledge the support of a Leverhulme Trust Research Fellowship RF/2022-625. AKC and IC acknowledge support from the EPSRC (EP/W017032/1) and Physical Sciences Data Infrastructure Phase 1b (EP/X032701/1). AS gratefully thanks Ricardo Parra-Cruz for assistance with Free Energy Landscape calculations and discussions, and Jennifer Cassidy for assistance and advice on protein purification, as well as Karin S. Schaffarczyk McHale for supplying a manual on IL ion preparation for MD simulation. The work was further supported by access to the University of Nottingham's High Performance Computing Facility.References
- S. Simić, et al., Shortening Synthetic Routes to Small Molecule Active Pharmaceutical Ingredients Employing Biocatalytic Methods, Chem. Rev., 2021, 122(1), 1052–1126 CrossRef PubMed.
- J. Chapman, A. Ismail and C. Dinu, Industrial Applications of Enzymes: Recent Advances, Techniques, and Outlooks, Catalysts, 2018, 8(6), 238 CrossRef.
- P. Domínguez de María, Green solvents and biocatalysis: A bigger picture, EFB Bioeconomy J., 2023, 3, 100056 CrossRef.
- A. Schindl, et al., Proteins in Ionic Liquids: Reactions, Applications, and Futures, Front. Chem., 2019, 7, 347 CrossRef CAS PubMed.
- M. L. Hagen, J. B. Harper and A. K. Croft, Recent advances in the use of ionic liquids as solvents for protein-based materials and chemistry, Curr. Opin. Green Sustainable Chem., 2022, 36, 100637 CrossRef CAS.
- R. A. Sheldon, Biocatalysis in ionic liquids: state-of-the-union, Green Chem., 2021, 23(21), 8406–8427 RSC.
- N. Byrne, et al., Reversible folding--unfolding, aggregation protection, and multi-year stabilization, in high concentration protein solutions, using ionic liquids, Chem. Commun., 2007, 2714–2716 RSC.
- S. Summers, et al., Enhanced Activity and Stability of Acidothermus cellulolyticus Endoglucanase 1 in Ionic Liquids via Engineering Active Site Residues and Non-Native Disulfide Bridges, ACS Sustain. Chem. Eng., 2020, 8(30), 11299–11307 CrossRef CAS.
- J. Ramos-Martín, et al., Biocatalysis at Extreme Temperatures: Enantioselective Synthesis of both Enantiomers of Mandelic Acid by Transesterification Catalyzed by a Thermophilic Lipase in Ionic Liquids at 120 \$$\backslash$,\^$\backslash$circ\$C, Catalysts, 2020, 10(9), 1055 CrossRef.
- D. W. Shin, et al., Enhanced lipase-catalyzed synthesis of sugar fatty acid esters using supersaturated sugar solution in ionic liquids, Enzyme Microb. Technol., 2019, 126, 18–23 CrossRef CAS PubMed.
- Z. Jarin and J. Pfaendtner, Ionic Liquids Can Selectively Change the Conformational Free-Energy Landscape of Sugar Rings, J. Chem. Theory Comput., 2014, 10(2), 507–510 CrossRef CAS PubMed.
- E. Alvarez, et al., Clean Enzymatic Production of Flavor Esters in Spongelike Ionic Liquids, ACS Sustain. Chem. Eng., 2019, 7(15), 13307–13314 CrossRef CAS.
- R. Villa, et al., Ionic liquids as an enabling tool to integrate reaction and separation processes, Green Chem., 2019, 21, 6527–6544 RSC.
- R. Villa, et al., Chapter 14 – Biocatalytic processes in ionic liquids and supercritical carbon dioxide biphasic systems, in Biocatalysis in Green Solvents, ed. P. Lozano, Academic Press, 2022, pp. 403–433 Search PubMed.
- Z. Hua, Methods for stabilizing and activating enzymes in ionic liquids—a review, J. Chem. Technol. Biotechnol., 2010, 85(7), 891–907 CrossRef.
- L. Cammarata, et al., Molecular states of water in room temperature ionic liquids, Phys. Chem. Chem. Phys., 2001, 3(23), 5192–5200 RSC.
- N. Yaghini, L. Nordstierna and A. Martinelli, Effect of water on the transport properties of protic and aprotic imidazolium ionic liquids – an analysis of self-diffusivity, conductivity, and proton exchange mechanism, Phys. Chem. Chem. Phys., 2014, 16(20), 9266–9275 RSC.
- L. Cicco, et al., Advances in deep eutectic solvents and water: applications in metal- and biocatalyzed processes, in the synthesis of APIs, and other biologically active compounds, Org. Biomol. Chem., 2021, 19(12), 2558–2577 RSC.
- V. Gotor-Fernández and C. E. Paul, Deep eutectic solvents for redox biocatalysis, J. Biotechnol., 2019, 293, 24–35 CrossRef PubMed.
- W. Kunz, J. Henle and B. W. Ninham, Zur Lehre von der Wirkung der Salze’ (about the science of the effect of salts): Franz Hofmeister's historical papers, Curr. Opin. Colloid Interface Sci., 2004, 9(1), 19–37 CrossRef CAS.
- K. D. Collins, Charge density-dependent strength of hydration and biological structure, Biophys. J., 1997, 72(1), 65–76 CrossRef CAS PubMed.
- R. G. Pearson, Hard and Soft Acids and Bases, J. Am. Chem. Soc., 1963, 85(22), 3533–3539 CrossRef CAS.
- V. Mazzini and V. S. J. Craig, Specific-ion effects in non-aqueous systems, Curr. Opin. Colloid Interface Sci., 2016, 23, 82–93 CrossRef CAS.
- J. W. Schuliger, Purification and characterization of a novel amylolytic enzyme from ES 4, a marine hyperthermophilic archaeum, Molecular Marine Biology and Biotechnology, ed. S. H. Brown, 1993. p. 76–87 Search PubMed.
- M. Falb, et al., Living with two extremes: Conclusions from the genome sequence of Natronomonas pharaonis, Genome Res., 2005, 15(10), 1336–1343 CrossRef CAS PubMed.
- M. Mevarech, F. Frolow and L. M. Gloss, Halophilic enzymes: proteins with a grain of salt, Biophys. Chem., 2000, 86(2–3), 155–164 CrossRef CAS PubMed.
- T. Zhang, et al., Identification of a haloalkaliphilic and thermostable cellulase with improved ionic liquid tolerance, Green Chem., 2011, 13(8), 2083 RSC.
- A. A. N. Gunny, et al., Potential halophilic cellulases for in situ enzymatic saccharification of ionic liquids pretreated lignocelluloses, Bioresour. Technol., 2014, 155, 177–181 CrossRef CAS PubMed.
- B. Zhao, et al., Efficient enzymatic saccharification of alkaline and ionic liquid-pretreated bamboo by highly active extremozymes produced by the co-culture of two halophilic fungi, Bioresour. Technol., 2021, 319, 124115 CrossRef CAS PubMed.
- N. Raddadi, et al., Halo-alkalitolerant and thermostable cellulases with improved tolerance to ionic liquids and organic solvents from Paenibacillus tarimensis isolated from the Chott El Fejej, Sahara desert, Tunisia, Bioresour. Technol., 2013, 150, 121–128 CrossRef CAS PubMed.
- I. Ben Hmad and A. Gargouri, Ionic liquid-tolerant cellulase system of Stachybotrys microspora exploited in the in situ saccharification of lignocellulosic biomass, J. Mol. Liq., 2020, 310, 113167 CrossRef CAS.
- N. M. Mesbah and J. Wiegel, A Halophilic, Alkalithermostable, Ionic Liquid-Tolerant Cellulase and Its Application in In Situ Saccharification of Rice Straw, BioEnergy Res., 2017, 10(2), 583–591 CrossRef CAS.
- S. Rezaei, A. R. Shahverdi and M. A. Faramarzi, Isolation, one-step affinity purification, and characterization of a polyextremotolerant laccase from the halophilic bacterium Aquisalibacillus elongatus and its application in the delignification of sugar beet pulp, Bioresour. Technol., 2017, 230, 67–75 CrossRef CAS PubMed.
- H. R. Karbalaei-Heidari, M. Shahbazi and G. Absalan, Characterization of a Novel Organic Solvent Tolerant Protease from a Moderately Halophilic Bacterium and Its Behavior in Ionic Liquids, Appl. Biochem. Biotechnol., 2013, 170(3), 573–586 CrossRef CAS PubMed.
- K. Liu, et al., Effect of ionic liquids on catalytic characteristics of hyperthermophilic and halophilic phenylalanine dehydrogenase and mechanism study, Biochem. Eng. J., 2021, 176, 108175 CrossRef CAS.
- Z. Ji, et al., Design of robust malate dehydrogenases by assembly of motifs of halophilic and thermophilic enzyme based on interaction network, Biochem. Eng. J., 2023, 190, 108758 CrossRef CAS.
- O. Dym, M. Mevarech and J. L. Sussman, Structural Features That Stabilize Halophilic Malate Dehydrogenase from an Archaebacterium, Science, 1995, 267(5202), 1344–1346 CrossRef CAS PubMed.
- W. V. Ng, et al., Genome sequence of Halobacterium species NRC-1, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(22), 12176–12181 CrossRef CAS PubMed.
- S. Pramanik, et al., How Does Surface Charge Engineering of Bacillus subtilis Lipase A Improve Ionic Liquid Resistance? Lessons Learned from Molecular Dynamics Simulations, ACS Sustain. Chem. Eng., 2022, 10(8), 2689–2698 CrossRef CAS.
- L. M. Timpson, et al., A comparison of two novel alcohol dehydrogenase enzymes (ADH1 and ADH2) from the extreme halophile Haloferax volcanii, Appl. Microbiol. Biotechnol., 2012, 97(1), 195–203 CrossRef PubMed.
- D. Alsafadi, S. Alsalman and F. Paradisi, Extreme halophilic alcohol dehydrogenase mediated highly efficient syntheses of enantiopure aromatic alcohols, Org. Biomol. Chem., 2017, 15(43), 9169–9175 RSC.
- D. Alsafadi and F. Paradisi, Effect of organic solvents on the activity and stability of halophilic alcohol dehydrogenase (ADH2) from Haloferax volcanii, Extremophiles, 2012, 17(1), 115–122 CrossRef PubMed.
- E. M. Nordwald and J. L. Kaar, Stabilization of enzymes in ionic liquids via modification of enzyme charge, Biotechnol. Bioeng., 2013, 110(9), 2352–2360 CrossRef CAS PubMed.
- V. Jaeger, P. Burney and J. Pfaendtner, Comparison of Three Ionic Liquid-Tolerant Cellulases by Molecular Dynamics, Biophys. J., 2015, 108(4), 880–892 CrossRef CAS PubMed.
- N. Mohammadyazdani, M. R. Bozorgmehr and M. Momen-Heravi, Conformation changes and diffusion of α-amylase in 1-hexyle-3-methylimidazolium chloride ionic liquid: A molecular dynamics simulation perspective, J. Mol. Liq., 2016, 221, 463–468 CrossRef CAS.
- H.-J. Tung and J. Pfaendtner, Kinetics and mechanism of ionic-liquid induced protein unfolding: application to the model protein HP35, Mol. Syst. Des. Eng., 2016, 1(4), 382–390 RSC.
- J. Zhao, et al., Ionic liquid activatedBacillus subtilislipase A variants through cooperative surface substitutions, Biotechnol. Bioeng., 2015, 112(10), 1997–2004 CrossRef CAS PubMed.
- J. Zhao, et al., Unraveling the effects of amino acid substitutions enhancing lipase resistance to an ionic liquid: a molecular dynamics study, Phys. Chem. Chem. Phys., 2018, 20(14), 9600–9609 RSC.
- M. Jafari, et al., Molecular level insight into stability, activity, and structure of Laccase in aqueous ionic liquid and organic solvents: An experimental and computational research, J. Mol. Liq., 2020, 317, 113925 CrossRef CAS.
- N. M. Micaêlo and C. M. Soares, Protein Structure and Dynamics in Ionic Liquids. Insights from Molecular Dynamics Simulation Studies, J. Phys. Chem. B, 2008, 112(9), 2566–2572 CrossRef PubMed.
- Q. Shao, On the influence of hydrated imidazolium-based ionic liquid on protein structure stability: A molecular dynamics simulation study, J. Chem. Phys., 2013, 139(11), 115102 CrossRef PubMed.
- H. S. Kim, et al., The relationship between enhanced enzyme activity and structural dynamics in ionic liquids: a combined computational and experimental study, Phys. Chem. Chem. Phys., 2014, 16(7), 2944 RSC.
- M. Haberler, C. Schröder and O. Steinhauser, Solvation studies of a zinc finger protein in hydrated ionic liquids, Phys. Chem. Chem. Phys., 2011, 13(15), 6955 RSC.
- A. M. Figueiredo, et al., Protein destabilisation in ionic liquids: the role of preferential interactions in denaturation, Phys. Chem. Chem. Phys., 2013, 15(45), 19632 RSC.
- S. Ghosh, et al., Ionic liquid induced dehydration and domain closure in lysozyme: FCS and MD simulation, J. Chem. Phys., 2015, 143(12), 125103 CrossRef PubMed.
- S. M. Ghorbani, M. R. Housaindokht and M. R. Bozorgmehr, Investigating the effect of 1-Butyl-3-methylimidazolium bromide and 1-Butyl-3-methylimidazolium methyl sulfate ionic liquids on structure and function of Chloroproxidase by molecular dynamics simulation, J. Mol. Liq., 2021, 332, 115850 CrossRef CAS.
- S. Ghanbari-Ardestani, et al., The effect of different percentages of triethanolammonium butyrate ionic liquid on the structure and activity of urate oxidase: Molecular docking, molecular dynamics simulation, and experimental study, J. Mol. Liq., 2019, 292, 111318 CrossRef CAS.
- K. P. Ghanta, et al., Microscopic Understanding of the Effect of Ionic Liquid on Protein from Molecular Simulation Studies, J. Phys. Chem. B, 2020, 124(19), 3909–3921 CrossRef CAS PubMed.
- F. Janati-Fard, et al., The influence of two imidazolium-based ionic liquids on the structure and activity of glucose oxidase: Experimental and theoretical studies, Int. J. Biol. Macromol., 2018, 114, 656–665 CrossRef CAS PubMed.
- S. Ghaedizadeh, et al., Understanding the molecular behaviour of Renilla luciferase in imidazolium-based ionic liquids, a new model for the α/β fold collapse, Biochem. Eng. J., 2016, 105, 505–513 CrossRef CAS.
- A. Tarannum, et al., Role of Preferential Ions of Ammonium Ionic Liquid in Destabilization of Collagen, J. Phys. Chem. B, 2016, 120(27), 6515–6524 CrossRef CAS PubMed.
- O. Singh, et al., Dual mechanism of ionic liquid-induced protein unfolding, Phys. Chem. Chem. Phys., 2020, 22(35), 19779–19786 RSC.
- M. Hennemann and T. Clark, EMPIRE: a highly parallel semiempirical molecular orbital program: 1: self-consistent field calculations, J. Mol. Model., 2014, 20, 2331 CrossRef PubMed.
- J. Gäding, et al., Impact of confinement and polarizability on dynamics of ionic liquids, J. Chem. Phys., 2022, 156(6), 064703 CrossRef PubMed.
- A. Schindl, et al., Controlling the outcome of SN2 reactions in ionic liquids: from rational data set design to predictive linear regression models, Phys. Chem. Chem. Phys., 2020, 22, 23009–23018 RSC.
- B. Dabirmanesh, et al., Inhibition mediated stabilization effect of imidazolium based ionic liquids on alcohol dehydrogenase, J. Mol. Liq., 2012, 170, 66–71 CrossRef CAS.
- Y. Zhang, X. Huang and Y. Li, Negative effect of [bmim][PF6] on the catalytic activity of alcohol dehydrogenase: mechanism and prevention, J. Chem. Technol. Biotechnol., 2008, 83(9), 1230–1235 CrossRef CAS.
- G. de Gonzalo, et al., Asymmetric biocatalytic reduction of ketones using hydroxy-functionalised water-miscible ionic liquids as solvents, Tetrahedron: Asymmetry, 2007, 18(21), 2541–2546 CrossRef CAS.
- C. Kohlmann, et al., Ionic liquid facilitates biocatalytic conversion of hardly water soluble ketones, J. Mol. Catal. B: Enzym., 2011, 68(2), 147–153 CrossRef CAS.
- T. Gerlach, et al., An open source COSMO-RS implementation and parameterization supporting the efficient implementation of multiple segment descriptors, Fluid Phase Equilib., 2022, 560, 113472 CrossRef CAS.
- M. M. Musa, K. I. Ziegelmann-Fjeld, C. Vieille and R. S. Phillips, Activity and selectivity of w110a secondary alcohol dehydrogenase from Thermoanaerobacter ethanolicus in organic solvents and ionic liquids: Mono- and biphasic media, Org. Biomol. Chem., 2008, 6, 887–892 RSC.
- M. Eckstein, et al., Use of an ionic liquid in a two-phase system to improve an alcohol dehydrogenase catalysed reduction, Chem. Commun., 2004,(9), 1084–1085 RSC.
- X.-A. Shi, M.-H. Zong and W.-Y. Lou, Effect of Ionic Liquids on Catalytic Characteristics of Horse Liver Alcohol Dehydrogenase, Chin. J. Chem., 2006, 24(11), 1643–1647 CrossRef CAS.
- M. Haberler and O. Steinhauser, On the influence of hydrated ionic liquids on the dynamical structure of model proteins: a computational study, Phys. Chem. Chem. Phys., 2011, 13(40), 17994 RSC.
- M. Haberler, C. Schröder and O. Steinhauser, Hydrated Ionic Liquids with and without Solute: The Influence of Water Content and Protein Solutes, J. Chem. Theory Comput., 2012, 8(10), 3911–3928 CrossRef CAS PubMed.
- Z. D. Nagel, S. Cun and J. P. Klinman, Identification of a Long-range Protein Network That Modulates Active Site Dynamics in Extremophilic Alcohol Dehydrogenases, J. Biol. Chem., 2013, 288(20), 14087–14097 CrossRef CAS PubMed.
- D. T. Infield, et al., Cation-π Interactions and their Functional Roles in Membrane Proteins, J. Mol. Biol., 2021, 433(17), 167035 CrossRef CAS PubMed.
- E. I. Izgorodina, U. L. Bernard and D. R. MacFarlane, Ion-Pair Binding Energies of Ionic Liquids: Can DFT Compete with Ab Initio-Based Methods?, J. Phys. Chem. A, 2009, 113(25), 7064–7072 CrossRef CAS PubMed.
- S. Lenton, et al., Structural evidence for solvent-stabilisation by aspartic acid as a mechanism for halophilic protein stability in high salt concentrations, Phys. Chem. Chem. Phys., 2016, 18(27), 18054–18062 RSC.
- A. C. Warden, et al., Rational engineering of a mesohalophilic carbonic anhydrase to an extreme halotolerant biocatalyst, Nat. Commun., 2015, 6(1), 10278 CrossRef CAS PubMed.
- J. N. Dahanayake and K. R. Mitchell-Koch, Entropy connects water structure and dynamics in protein hydration layer, Phys. Chem. Chem. Phys., 2018, 20(21), 14765–14777 RSC.
- C. Schröder, et al., Collective rotational dynamics in ionic liquids: A computational and experimental study of 1-butyl-3-methyl-imidazolium tetrafluoroborate, J. Chem. Phys., 2007, 126(8), 084511 CrossRef PubMed.
- M. Tariq, et al., Surface tension of ionic liquids and ionic liquid solutions, Chem. Soc. Rev., 2012, 41(2), 829–868 RSC.
- A. P. A. Sthoer and E. C. Tyrode, Anion Specific Effects at Negatively Charged Interfaces: Influence of Cl$^--$, Br$^--$, I$^--$, and SCN$^--$ on the Interactions of Na$^+$ with the Carboxylic Acid Moiety, J. Phys. Chem. B, 2021, 125(44), 12384–12391 CrossRef CAS PubMed.
- J. Hunger, et al., Nature of Cations Critically Affects Water at the Negatively Charged Silica Interface, J. Am. Chem. Soc., 2022, 144(43), 19726–19738 CrossRef CAS PubMed.
- F. Frolow, et al., Insights into protein adaptation to a saturated salt environment from the crystal structure of a halophilic 2Fe-2S ferredoxin, Nat. Struct. Biol., 1996, 3(5), 452–458 CrossRef CAS PubMed.
- S. Arai, et al., Structure of a highly acidic β-lactamase from the moderate halophileChromohalobactersp. 560 and the discovery of a Cs$^+$-selective binding site, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71(3), 541–554 CrossRef CAS PubMed.
- R. Talon, et al., An experimental point of view on hydration/solvation in halophilic proteins, Front. Microbiol., 2014, 5, 66 Search PubMed.
- C. F. Aguilar, et al., Crystal structure of the β-glycosidase from the hyperthermophilic archeon Sulfolobus solfataricus: resilience as a key factor in thermostability, J. Mol. Biol., 1997, 271(5), 789–802 CrossRef CAS PubMed.
- M. Hennig, et al., 2.0 åstructure of indole-3-glycerol phosphate synthase from the hyperthermophile Sulfolobus solfataricus: possible determinants of protein stability, Structure, 1995, 3(12), 1295–1306 CrossRef CAS PubMed.
- V. Villeret, et al., The crystal structure of Pyrococcus furiosus ornithine carbamoyltransferase reveals a key role for oligomerization in enzyme stability at extremely high temperatures, Proc. Natl. Acad. Sci. U. S. A., 1998, 95(6), 2801–2806 CrossRef CAS PubMed.
- D. Madern, C. Ebel and G. Zaccai, Halophilic adaptation of enzymes, Extremophiles, 2000, 4(2), 91–98 CrossRef CAS PubMed.
- S. Pramanik, et al., How To Engineer Ionic Liquids Resistant Enzymes: Insights from Combined Molecular Dynamics and Directed Evolution Study, ACS Sustain. Chem. Eng., 2019, 7(13), 11293–11302 CrossRef CAS.
- H. Cui, et al., Enzyme Hydration: How to Retain Resistance in Ionic Liquids, ACS Sustain. Chem. Eng., 2022, 10(46), 15104–15114 CrossRef CAS.
- T. El Harrar, et al., Aqueous ionic liquids redistribute local enzyme stability via long-range perturbation pathways, Comput. Struct. Biotechnol. J., 2021, 19, 4248–4264 CrossRef CAS PubMed.
- C. P. Guy, et al., Interactions of RadB, a DNA Repair Protein in Archaea, with DNA and ATP, J. Mol. Biol., 2006, 358(1), 46–56 CrossRef CAS PubMed.
- T. Allers, et al., Development of Additional Selectable Markers for the Halophilic Archaeon Haloferax volcanii Based on the leuB and trpA Genes, Appl. Environ. Microbiol., 2004, 70(2), 943–953 CrossRef CAS PubMed.
- C. Norais, et al., Genetic and Physical Mapping of DNA Replication Origins in Haloferax volcanii, PLoS Genet., 2007, 3(5), e77 CrossRef PubMed.
- G. Bitan-Banin, R. Ortenberg and M. Mevarech, Development of a Gene Knockout System for the Halophilic Archaeon Haloferax volcanii by Use of the pyrE Gene , J. Bacteriol., 2003, 185(3), 772–778 CrossRef CAS PubMed.
- E. Strillinger, et al., Production of halophilic proteins using Haloferax volcanii H1895 in a stirred-tank bioreactor, Appl. Microbiol. Biotechnol., 2015, 100(3), 1183–1195 CrossRef PubMed.
- A. Waterhouse, et al., SWISS-MODEL: homology modelling of protein structures and complexes, Nucleic Acids Res., 2018, 46(W1), W296–W303 CrossRef CAS PubMed.
- S. Bienert, et al., The SWISS-MODEL Repository---new features and functionality, Nucleic Acids Res., 2017, 45(D1), D313–D319 CrossRef CAS PubMed.
- N. Guex, M. C. Peitsch and T. Schwede, Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective, Electrophoresis, 2009, 30(S1), S162–S173 CrossRef PubMed.
- G. Studer, et al., ProMod3---A versatile homology modelling toolbox, PLoS Comput. Biol., 2021, 17(1), e1008667 CrossRef CAS PubMed.
- P. Benkert, M. Biasini and T. Schwede, Toward the estimation of the absolute quality of individual protein structure models, Bioinformatics, 2011, 27(3), 343–350 CrossRef CAS PubMed.
- L. A. Kelley, et al., The Phyre2 web portal for protein modeling, prediction and analysis, Nat. Protoc., 2015, 10(6), 845–858 CrossRef CAS PubMed.
- Y. Zhang, I-TASSER server for protein 3D structure prediction, BMC Bioinf., 2008, 9(1), 40 CrossRef PubMed.
- J. Yang, et al., The I-TASSER Suite: protein structure and function prediction, Nat. Methods, 2015, 12(1), 7–8 CrossRef CAS PubMed.
- Materials Studio, v16.1.0.21, 2018, Dassault Systèmes, https://www.3ds.com/products/biovia/materials-studio Search PubMed.
- C. Colovos and T. O. Yeates, Verification of protein structures: Patterns of nonbonded atomic interactions, Protein Sci., 1993, 2(9), 1511–1519 CrossRef CAS PubMed.
- T.-S. Lee, et al., GPU-Accelerated Molecular Dynamics and Free Energy Methods in Amber18: Performance Enhancements and New Features, J. Chem. Inf. Model., 2018, 58(10), 2043–2050 CrossRef CAS PubMed.
- K. G. Sprenger, V. W. Jaeger and J. Pfaendtner, The General AMBER Force Field (GAFF) Can Accurately Predict Thermodynamic and Transport Properties of Many Ionic Liquids, J. Phys. Chem. B, 2015, 119(18), 5882–5895 CrossRef CAS PubMed.
- O. Guvench and A. D. MacKerell, Comparison of protein force fields for molecular dynamics simulations, Methods Mol. Biol., 2008, 443, 63–88 CrossRef CAS PubMed.
- J. A. Maier, et al., ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J. Chem. Theory Comput., 2015, 11(8), 3696–3713 CrossRef CAS PubMed.
- M. B. Peters, et al., Structural Survey of Zinc-Containing Proteins and Development of the Zinc AMBER Force Field (ZAFF), J. Chem. Theory Comput., 2010, 6(9), 2935–2947 CrossRef CAS PubMed.
- H. Geraili Daronkola and A. Vila Verde, Proteins maintain hydration at high [KCl] concentration regardless of content in acidic amino acids, Biophys. J., 2021, 120(13), 2746–2762 CrossRef CAS PubMed.
- M. D. Hanwell, et al., Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4, 1–17 Search PubMed.
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, Gaussian-4 theory, J. Chem. Phys., 2007, 126, 084108 CrossRef PubMed.
- C. I. Bayly, et al., A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model, J. Phys. Chem., 1993, 97(40), 10269–10280 CrossRef CAS.
- L. Martínez, et al., PACKMOL: A package for building initial configurations for molecular dynamics simulations, J. Comput. Chem., 2009, 30(13), 2157–2164 CrossRef PubMed.
- W. L. Jorgensen, et al., Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79(2), 926–935 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual molecular dynamics, J. Mol. Graphics, 1996, 14(1), 33–38 CrossRef CAS PubMed.
- D. L. Theobald, Rapid calculation of RMSDs using a quaternion-based characteristic polynomial, Acta Crystallogr., Sect. A: Found. Crystallogr., 2005, 61(4), 478–480 CrossRef PubMed.
- P. Liu, D. K. Agrafiotis and D. L. Theobald, Fast determination of the optimal rotational matrix for macromolecular superpositions, J. Comput. Chem., 2009, 31(7), 1561–1563 CrossRef PubMed.
- R. Araya-Secchi, et al., Characterization of a Novel Water Pocket Inside the Human Cx26 Hemichannel Structure, Biophys. J., 2014, 107(3), 599–612 CrossRef CAS PubMed.
- N. Michaud-Agrawal, et al., MDAnalysis: A toolkit for the analysis of molecular dynamics simulations, J. Comput. Chem., 2011, 32(10), 2319–2327 CrossRef CAS PubMed.
- R. Gowers, et al., MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations, in Proceedings of the Python in Science Conference, SciPy, 2016 Search PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, GROMACS: fast, flexible, and free, J. Comput. Chem., 2005, 1701–1718 CrossRef CAS PubMed.
- E. Lindahl, B. Hess and D. Van Der Spoel, GROMACS 3.0: a package for molecular simulation and trajectory analysis, Molecular Modeling Annual, 2001, pp. 306–317 Search PubMed.
- M. Brehm and B. Kirchner, TRAVIS - A Free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories, J. Chem. Inf. Model., 2011, 51(8), 2007–2023 CrossRef CAS PubMed.
- A. Klamt, Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitative Calculation of Solvation Phenomena, J. Phys. Chem., 1995, 99(7), 2224–2235 CrossRef CAS.
- A. Klamt, et al., Refinement and Parametrization of COSMO-RS, J. Phys. Chem. A, 1998, 102(26), 5074–5085 CrossRef CAS.
- F. Eckert and A. Klamt, Fast solvent screening via quantum chemistry: COSMO-RS approach, AIChE J., 2002, 48(2), 369–385 CrossRef CAS.
- A. Klamt, F. Eckert and W. Arlt, COSMO-RS: An Alternative to Simulation for Calculating Thermodynamic Properties of Liquid Mixtures, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 101–122 CrossRef CAS PubMed.
- A. Klamt, COSMO-RS : from Quantum Chemistry to Fluid Phase Thermodynamics and Drug Design, Elsevier, 2005 Search PubMed.
- RDKit: Open-source cheminformatics, v2023.09.4, https://www.rdkit.org, DOI:10.5281/zenodo.591637.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2012, 2(1), 73–78 CrossRef CAS.
- F. Neese, Software update: The ORCA program system—Version 5.0, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2022, 12(5), e1606 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00412k |
| This journal is © The Royal Society of Chemistry 2024 |