
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-catalytic regioselective synthesis of trans bis-pyrrolo isoxazole cycloadducts in water†
Alhussein Arkan
Majhool
a,
Assad Abbas
Khalaf
b,
Iman Sabeeh
Hasan
c,
Ranvijay
Kumar
d,
Sandeep
Kaushal
 *ef and
Rahul
Badru
*e
*ef and
Rahul
Badru
*e
aDepartment of Environmental Health, College of Applied Medical Sciences, University of Karbala, Karbala, Iraq
bCollege of Nursing, University of Karbala, Karbala City, Iraq
cDepartment of Pharmacy, Al-Zahrawi University College, Karbala, Iraq
dUniversity Centre of Research and Development, Chandigarh University, Gharuan, Mohali, Punjab, India
eDepartment of Chemistry, Sri Guru Granth Sahib World University, Fatehgarh Sahib, Punjab, India-140406. E-mail: rahulbadru@gmail.com; kaushalsandeep33@gmail.com
fDESM, Regional Institute of Education, NCERT, Ajmer, Rajasthan, India
First published on 23rd January 2024
Abstract
Pyrrolo isoxazoles are key structural motifs of many drugs and pharmacologically active compounds and are often synthesized by one-pot cycloaddition reactions under thermal conditions. In the present study, a series of new symmetric bis-pyrrolo isoxazole cycloadducts have been synthesized by cycloaddition of bis-nitrones of glyoxal with N-substituted maleimides in water under non-catalytic conditions. The thermal cycloaddition reactions conducted in an aqueous medium at 70 °C afforded a single regioisomer of symmetric bis-pyrrolo isoxazoles in a shorter reaction period of 10–15 minutes. The formation of only the trans diastereoisomer, as confirmed by 1H-NMR spectral analysis, was attributed to the involvement of endo-transition mode, where a succinimide moiety and N-phenyl ring of the nitrone being on the same side stabilized the transition state by maximum π–π overlap. Several pyrrolo isoxazole derivatives have been synthesized using this methodology. The present approach for pyrrolo isoxazole synthesis, being one-pot, facile, non-catalytic, and conducted at low temperature and under aqueous conditions, can be counted as a step towards sustainable and green synthetic chemistry.
Sustainability spotlightIn our continuous efforts to develop green and sustainable methods for synthesis of heterocyclic compounds of pharmacological importance, in this paper, we report the novel diastereoselective synthesis of bis-pyrrolo isoxazoles via 1,3-dipolar cycloadditions of glyoxal bis-nitrones with N-substituted maleimides in water. This study is significant because of the formation of a single regio-isomer (trans isomer) exclusively, in very short reaction times (10–15 minutes). Moreover, the binitrone and cycloaddition reactions have been performed in water. Thus, this protocol of synthesis of bis cyclo-adducts of pyrrolo isoxazoles is facile, efficient, selective and eco-friendly. |
1. Introduction
The isoxazoles are crucial five-membered heterocycles with nitrogen and oxygen atoms at adjacent positions. They are an appealing target in organic, bioorganic, and pharmaceutical chemistry, and building blocks of a diverse array of pharmaceutically important compounds. They have been shown to exhibit a wide range of biological properties including antifungal,1 antimicrobial,2 antiviral,3 antimycobacterial,4 antioxidant,5 anticonvulsant,6 and antitumor.7 They have also been found to have an inhibition towards HDAC,8 protein-tyrosine phosphatase,9 and COX-2 isozyme.10 They are nematicidal11 and have been used to treat leishmaniasis12 and arthritis.13While there exist several additional techniques for synthesizing oxazoles and isoxazoles, a multicomponent reaction among β-ketoesters, hydroxylamine hydrochloride, and aromatic aldehydes produces the product in higher yields in a single step. Several acidic and basic catalysts including boric acid,14 potassium hydrogen phthalate,15 DABCO,16 metal-oxide nanoparticles,17 pyridine,18,19 and citrazinic acid20 have been reported to catalyze the process.
Pyrrolo oxazoles and isoxazoles are an important class of pharmaceutical compounds that have been established as antimitotic,21 antibacterial,22 anti-stress,23 anti-inflammatory,24 anti-tubercular,25 and anti-cancer agents.26 In pyrrolo isoxazoles, a pyrrole ring is joined to an isoxazole moiety. The pyrrolo isoxazoles are often synthesized by one-pot cycloadditions of nitrones with various maleimide substrates.27 The advantages of synthesizing them via the cycloaddition mode lies in that the regioselectivity of the reaction can be controlled to obtain either of the regioisomers as the product.27,28 But in few cases the regioselectivity is lost due to the isolation of a mixture of both cis and trans regioisomers.23
The use of nitrone as a 1,3-dipole for cycloaddition processes dominates nitrone chemistry. Nitrone cycloaddition has been utilized to develop a number of natural products, versatile synthetic intermediates and biologically interesting compounds.29,30 Aside from a vast range of five membered heterocycles created using nitrone cycloaddition, there are fewer reports on the synthesis of polymeric compounds utilizing nitrones via this approach.31–33 Furthermore, only a few studies report the synthesis of bis-nitrones and their usage as a dipole substrate to synthesize macro sized molecules. The use of nitrones in polymer synthesis dates back to 1971, when Manecke et al.34 for the first time reported the synthesis of α-(p-maleinimidophenyl)-N-(phenylnitrone) that was subjected to dipolar cycloaddition with the maleinimido group in the same molecule and this upon repeated addition led to the formation of macromolecules. Later, Heaney et al. in 2001 (ref. 35) and Vretik et al. in 2003,36 reported the synthesis of bisisoxazolidines from bisnitrones and bis-maleimides. However, an insight into the surveyed literature showed that the usage of glyoxal bis-nitrones in the construction of macromolecules has not been described so far. Only a single paper by Chakraborty et al.37 in 2014 reported the preliminary results for the synthesis of bisisoxazolidines from glyoxal derived nitrones. Although the authors reported the synthesis in water, the reaction time is too long i.e. 3–5 hours, whereas with the current protocol, bis pyrrolo isoxazoles were obtained within 10–15 minutes of addition of glyoxal solution to an aqueous solution of in situ generated phenylhydroxyl amines. The crude bis pyrrolo isoxazole product is insoluble, and precipitated out of the aqueous medium in 10–15 min. Moreover, the authors reported the synthesis of glyoxal-based bis-nitrones in organic solvents, whereas in the present study the same has been obtained by the condensation of glyoxal with in situ generated phenylhydroxyl amines. In the present case, a thermodynamically more stable regioisomer i.e. the trans cycloadduct has been formed.
Thus, with the aim of and in continuation to our interest to develop hetero-structures incorporating isoxazolidine rings,23,27,38 the current study reports the synthesis of some novel symmetrical bis-pyrrolo isoxazoles via thermal 1,3-dipolar cycloaddition of glyoxal bis-nitrones with N-substituted maleimides. The glyoxal-based bis nitrones were synthesized by condensation of glyoxal with N-phenyl hydroxylamines in an aqueous medium only. And the cycloadditions too were performed in aqueous media. The formation of all the products is supported by spectral studies such as FTIR, NMR, elemental analysis, and mass spectrometry.
2. Experimental
2.1. General
The melting points reported here are uncorrected. All the chemicals used in the present study were of analytical grade and used without any further purification. FTIR spectra were recorded on a Bruker Alpha-T FTIR spectrophotometer. NMR analysis of the synthesized compounds was performed using a Bruker Avance-II 500 MHz FTNMR spectrometer, with tetramethylsilane as the internal standard. The mass spectral analysis was performed using an LCMS model LTQ-XL, from Thermo Scientific. Elemental analysis of the compounds was performed on an Elementar Vario MICRO cube CHN analyzer. Monitoring of the reaction was performed using TLC plates coated with silica gel (G254 grade). As the bis nitrones and bis isoxazolidine compounds synthesized are symmetrical, NMR spectral data for half of the structures are reported. To get the exact number of protons and carbon atoms, one needs to multiply by a factor of 2 in each case.2.2. General procedure for the synthesis of nitrones (azomethine N-oxides)
For the synthesis of azomethine N-oxide, the partial reduction of nitrobenzene has been carried out at 60–70 °C under aqueous conditions. For this, 4 mL (42 mmol) of nitrobenzene was added to a beaker containing 100 mL water, followed by addition of 2.5 g of NH4Cl (45 mmol). Thereafter, a pinch of Zn dust (out of the total 6 g, 84 mmol) was added to it and the contents were stirred mechanically. Zn dust (6 g) was added in portions to avoid an increase in temperature above 70 °C. The reduction is completed when no more nitrobenzene seems to be floating on the water surface. The filtrate is separated from solid ZnO and it was cooled to room temperature. A stoichiometric amount of glyoxal solution (3 mL, 40%) was added to the filtrate and stirred. Nitrone of glyoxal precipitated out as a yellow solid in just 15–20 min. The solid was then filtered, dried and recrystallized from chloroform. The formation of nitrone products was confirmed by their spectral and elemental analyses.2.2.1.1 Bis(N-phenyliminium)ethane-N,N′-dioxide. Yellow solid, mp 165–66 °C; FTIR (ν cm−1): 1706, 1783 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR (500 MHz, CDCl3), δ: 7.27–7.41 (m, 5H); 7.88 (s, 1H, –CHN); 13C NMR (125 MHz, CDCl3), δ: 123.8, 129.1, 134.7, 143.8; MS: m/z: 240 [M]+; anal. calcd for C14H12N2O2: C, 70.00; H, 5.00; N, 11.67, found: C, 69.79; H, 4.91; N, 11.56.
O); 1H NMR (500 MHz, CDCl3), δ: 7.27–7.41 (m, 5H); 7.88 (s, 1H, –CHN); 13C NMR (125 MHz, CDCl3), δ: 123.8, 129.1, 134.7, 143.8; MS: m/z: 240 [M]+; anal. calcd for C14H12N2O2: C, 70.00; H, 5.00; N, 11.67, found: C, 69.79; H, 4.91; N, 11.56.
2.2.1.2 Bis(N-tolyliminium)ethane-N,N′-dioxide. Light-yellow solid, mp 140–42 °C; FTIR (ν cm−1): 1706, 1783 (C
O); 1H NMR (500 MHz, CDCl3), δ: 2.23 (s, 3H); 7.27 (m, 2H, Ar–H); 7.64 (m, 2H, Ar–H); 7.86 (s, 1H, –CHN); 13C NMR (125 MHz, CDCl3), δ: 17.6, 123.7, 128.4, 143.6, 144.4; MS: m/z: 268 [M]+; anal. calcd for C16H16N2O2: C, 71.64; H, 5.97; N, 10.44, found: C, 71.15; H, 5.99; N, 10.32.
2.2.1.3 Bis(N-4-chlorophenyliminium)ethane-N,N′-dioxide. Light-yellow solid, mp 207–08 °C; FTIR (ν cm−1): 1706, 1783 (C
O); 1H NMR (500 MHz, CDCl3), δ: 7.33 (m, 2H, Ar–H); 7.82 (m, 2H, Ar–H); 7.89 (s, 1H, –CHN); 13C NMR (125 MHz, CDCl3), δ: 123.7, 129.8, 145.9, 146.3; MS: m/z: 309 [M]+; anal. calcd for C14H10Cl2N2O2: C, 54.37; H, 3.24; N, 9.06, found: C, 54.04; H, 3.18; N, 9.26.
2.3. General procedure for synthesis of N-arylmaleimide
50 mmol each of maleic anhydride and amine were dissolved separately in 50 mL toluene and then mixed at room temperature to obtain the corresponding maleamic acid. In the next step, the obtained maleamic acid was filtered, dried and converted to maleimide in the presence of 10 mmol of anhydrous sodium acetate in acetic anhydride (20 mL). The contents were refluxed on a boiling water bath for 1.5 hour and then poured onto ice-cold water taken in a beaker. The white precipitates of maleimide were filtered and dried.2.4. General procedure for the cycloaddition reaction
To the aqueous solution of bis-nitrone of glyoxal (∼5 mmol) (1), 10 mmol of N-arylmaleimide (2) were added and the contents of the flask were refluxed at 70 °C after being equipped with a reflux water condenser. Just after refluxing for 10–15 min, a white solid separated out of the mixture. The white precipitates of the cycloadduct were filtered and further purified through recrystallization from an ethanol–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture.
:1) mixture.
2.5. The spectral data for the various cycloadducts
As the bis pyrroloisoxazole compounds synthesized are symmetrical, NMR spectral data for half of the structures are reported. To get the exact number of protons and carbon atoms, one needs to multiply by a factor of 2 in each case.O); 1H NMR (500 MHz, CDCl3), δ: 3.87 (d, 1H); 4.59 (s, 1H); 5.21 (d, 1H); 6.97–7.24 (m, 10H); 13C NMR (125 MHz, d6-DMSO), δ: 51.5, 69.0, 72.8, 112.9, 121.9, 126.9, 128.1, 128.5, 134.3, 149.0, 173.6, 174.4; MS: m/z: 586 [M]+; anal. calcd for C34H26N4O6: C, 69.62; H, 4.47; N, 9.55, found: C, 70.19; H, 4.59; N, 9.51.
O); 1H NMR (500 MHz, CDCl3), δ: 2.12 (s, 3H); 3.83 (d, 1H); 4.66 (s, 1H); 5.14 (d, 1H); 6.93–7.31 (m, 9H); 13C NMR (125 MHz, d6-DMSO), δ: 17.4, 54.8, 65.2, 72.3, 123.3, 124.9, 125.7, 126.1, 129.5, 133.4, 136.0, 172.8, 173.9; MS: m/z: 614 [M]+; anal. calcd for C36H30N4O6: C, 70.35; H, 4.92; N, 9.12, found: C, 71.09; H, 4.90; N, 9.14.
O); 1H NMR (500 MHz, CDCl3), δ: 3.9 (d, 1H); 4.1 (s, 1H); 4.79 (d, 1H); 7.08–7.38 (m, 9H); 13C NMR (125 MHz, d6-DMSO), δ: 51.6, 67.5, 77.8, 113.6, 126.0, 128.7, 128.9, 129.0, 131.4, 138.1, 146.9, 173.2, 174.4; MS: m/z: 655 [M]+; anal. calcd for C36H30N4O6Cl2: C, 63.07; H, 4.41; N, 8.17, found: C, 62.89; H, 4.33; N, 8.15.
O); 1H NMR (500 MHz, CDCl3), δ: 3.83 (d, 1H, J = 7.76 Hz); 4.66 (s, 1H); 5.14 (d, 1H, J = 7.72 Hz); 6.84–7.23 (m, 9H); 13C NMR (125 MHz, d6-DMSO), δ: 19.1, 56.5, 65.3, 72.9, 123.3, 124.9, 125.7, 126.2, 129.4, 133.5, 134.7, 172.9, 174.0; MS: m/z: 614 [M]+; anal. calcd for C36H30N4O6: C, 70.35; H, 4.92; N, 9.12, found: C, 70.71; H, 4.93; N, 9.04.
O); 1H NMR (500 MHz, CDCl3), δ: 2.12 (s, 3H); 3.86 (d, 1H); 4.51 (s, 1H); 5.03 (d, 1H); 6.90–7.28 (m, 8H); 13C NMR (125 MHz, d6-DMSO), δ: 17.8, 19.5, 56.7, 65.8, 73.0, 123.3, 124.5, 125.1, 129.9, 133.5, 134.9, 173.4, 174.1; MS: m/z: 642 [M]+; anal. calcd for C38H34N4O6: C, 71.01; H, 5.33; N, 8.72, found: C, 70.86; H, 5.29; N, 8.75.
O); 1H NMR (500 MHz, CDCl3), δ: 3.81 (d, 1H, J = 7.28 Hz); 4.52 (s, 1H); 5.22 (d, 1H, J = 7.36 Hz); 6.93–7.28 (m, 8H); 13C NMR (125 MHz, d6-DMSO), δ: 17.6, 54.9, 63.7, 71.5, 124.3, 125.6, 126.2, 127.4, 128.9, 133.5, 134.9, 171.7, 173.9; MS: m/z: 683 [M]+; anal. calcd for C36H28N4O6Cl2: C, 63.26; H, 4.13; N, 8.20, found: C, 63.51; H, 4.11; N, 8.17.
O); 1H NMR (500 MHz, CDCl3), δ: 3.83 (d, 1H); 4.22 (s, 2H); 4.66 (s, 1H); 5.14 (d, 1H); 6.90–7.28 (m, 10H); 13C NMR (125 MHz, d6-DMSO), δ: 14.2, 23.2, 46.1, 60.4, 113.7, 126.6, 127.7, 128.9, 129.1, 171.6; MS: m/z: 614 [M]+; anal. calcd for C36H30N4O6: C, 70.35; H, 4.92; N, 9.12, found: C, 70.55; H, 4.92; N, 9.07.
O); 1H NMR (500 MHz, CDCl3), δ: 2.99 (s, 3H); 3.84 (d, 1H); 4.10 (s, 2H); 4.61 (s, 1H); 5.18 (d, 1H); 7.00–7.37 (m, 9H); 13C NMR (125 MHz, d6-DMSO), δ: 20.9, 26.9, 48.3, 56.2, 114.0, 127.3, 127.5, 128.3, 128.6, 129.0, 131.8, 148.1, 173.2, 174.9; MS: m/z: 642 [M]+; anal. calcd for C38H34N4O6: C, 71.01; H, 5.33; N, 8.72, found: C, 70.83; H, 5.35; N, 8.74.
O); 1H NMR (500 MHz, CDCl3), δ: 3.86 (d, 1H); 4.22 (s, 2H); 4.66 (s, 1H); 5.14 (d, 1H); 6.84–7.32 (m, 9H); 13C NMR (125 MHz, d6-DMSO), δ: 27.4, 31.9, 54.9, 63.6, 124.4, 125.1, 126.3, 127.0, 128.7, 129.0, 133.5, 171.9, 174.1; MS: m/z: 683 [M]+; anal. calcd for C36H28N4O6Cl2: C, 63.26; H, 4.13; N, 8.20, found: C, 63.46; H, 4.09; N, 8.16.
O); 1H NMR (500 MHz, CDCl3), δ: 1.04–1.76 (m, 7H), 1.7 (m, 2H), 2.7 (m, 2H), 3.83 (d, 1H); 4.28 (s, 1H); 4.81 (d, 1H); 6.95–7.67 (m, 5H); 13C NMR (125 MHz, d6-DMSO), δ: 18.4, 19.8, 20.5, 51.6, 52.0, 55.9, 67.5, 68.2, 77.8, 78.4, 118.7, 129.0, 129.3, 129.4, 147.4, 171.9, 173.8; MS: m/z: 598 [M]+; anal. calcd for C34H38N4O6: C, 68.21; H, 6.40; N, 9.36, found: C, 68.37; H, 6.44; N, 9.38.
O); 1H NMR (500 MHz, CDCl3), δ: 1.04–1.38 (m, 5H), 1.50–1.68 (m, 2H), 1.73 (m, 2H); 2.52 (s, 2H), 2.89 (s, 3H); 3.83 (d, 1H); 4.66 (s, 1H); 5.21 (d, 1H); 6.93–7.21 (m, 4H); 13C NMR (125 MHz, d6-DMSO), δ: 17.9, 24.8, 26.1, 28.5, 28.8, 52.3, 53.4, 65.0, 123.5, 125.2, 126.4, 132.6, 172.7, 173.8; MS: m/z: 626 [M]+; anal. calcd for C36H42N4O6: C, 68.99; H, 6.75; N, 8.94, found: C, 69.17; H, 6.71; N, 9.00.
O); 1H NMR (500 MHz, CDCl3), δ: 1.04–1.22 (m, 5H), 1.37–1.58 (m, 2H), 1.73–1.80 (m, 2H); 2.66 (m, 2H); 3.83 (d, 1H); 4.59 (s, 1H); 5.14 (d, 1H); 7.06–7.52 (m, 4H); 13C NMR (125 MHz, d6-DMSO), δ: 26.1, 30.6, 34.9, 48.4, 60.0, 65.4, 109.2, 129.0, 129.5, 131.3, 141.2, 178.4; MS: m/z: 667 [M]+; anal. calcd for C34H36N4O6Cl2: C, 61.17; H, 5.44; N, 8.39, found: C, 60.99; H, 5.39; N, 8.43.
3. Results and discussion
3.1. Synthesis
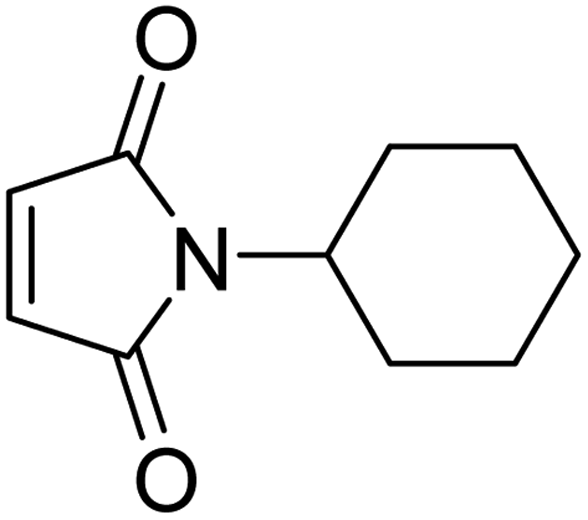
Initially, the dipolarophile maleimides to be used in this cycloaddition were synthesized using a process similar to that described in the literature.39 The condensation reaction between amines and maleic anhydride, followed by dehydration using sodium acetate and anhydrous acetic anhydride afforded cyclised maleimides (Scheme 1). The formation of the maleimide products has been confirmed on the basis of their melting points (compared with that of authentic samples) and by comparing their spectral data.39 | ||
| Scheme 1 Synthesis of N-substituted maleimide. | ||
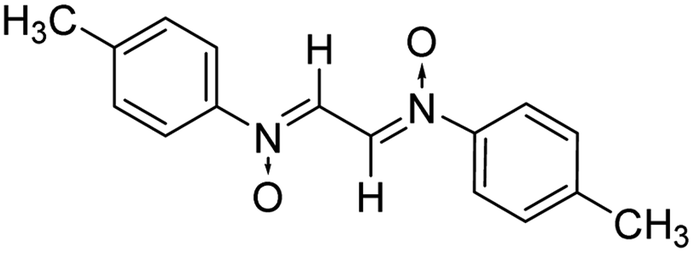
Another reactant i.e. bis-nitrones, to be used in the 1,3-dipolar cycloadditions, were synthesized by condensing glyoxal solution with the mono substituted phenyl hydroxylamines in a 1:2 ratio in an aqueous medium (Scheme 2). Phenyl hydroxylamines were prepared by the partial reduction of substituted nitrobenzene with zinc dust and ammonium chloride in an aqueous medium as described in the literature.40 The subsequent condensation reaction of the above synthesized N-phenyl hydroxylamines with glyoxal solution in an aqueous medium afforded bis-nitrones (Table 1) as ascertained by their FTIR and 1H-NMR analyses. All the bis-nitrones obtained were yellow in colour and were crystallized out of ethanol prior to their use in cycloadditions.
 | ||
| Scheme 2 Synthesis of bis azomethine N-oxides. | ||
| Entry | R2 | % Yield | MP | Time (in min) | m/z (M+) |
|---|---|---|---|---|---|
| 1 | H | 92 | 165–66 | 15 | 240 |
| 2 | CH3 | 96 | 140–42 | 15 | 268 |
| 3 | Cl | 89 | 207–08 | 20 | 309 |
After synthesizing the reactants, their dipolar cycloaddition was performed in aqueous media at 70 °C. For this, a 2 equiv. of N-aryl maleimide were added to an aqueous solution of bis-nitrones of glyoxal and heated to 70 °C (Scheme 3). Upon refluxing for just 15–20 min, a solid appeared in the flask. The white solid thus obtained was filtered, purified and dried in vacuo and subjected to spectral analysis.
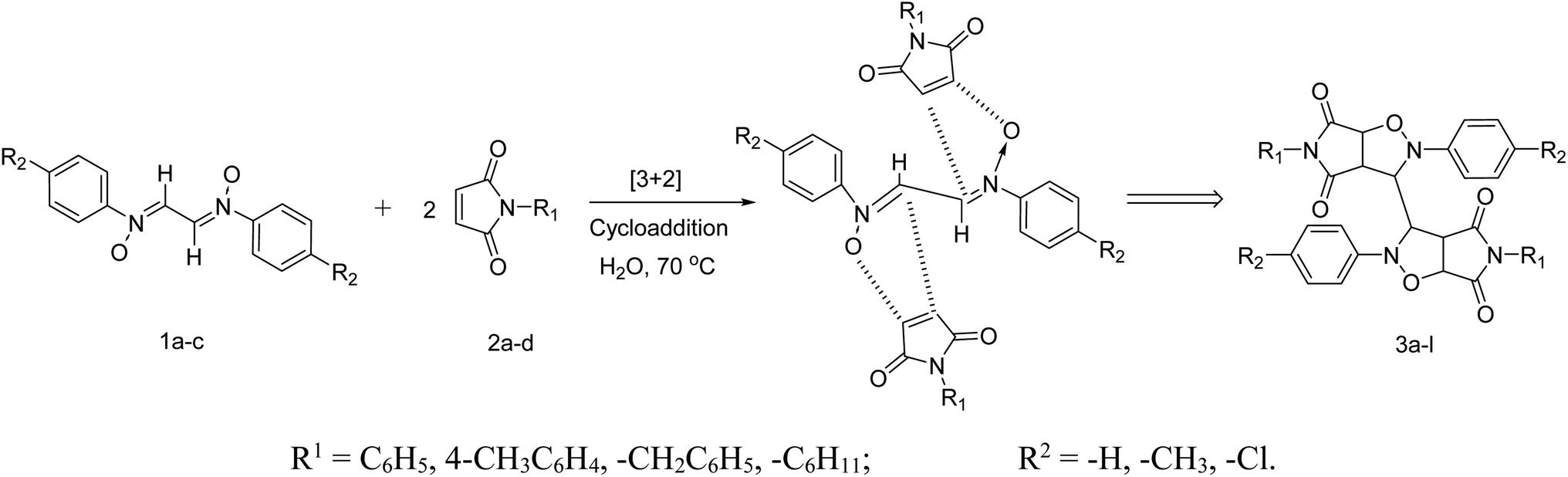 | ||
| Scheme 3 Synthesis of symmetrical bis isoxazolidines. | ||
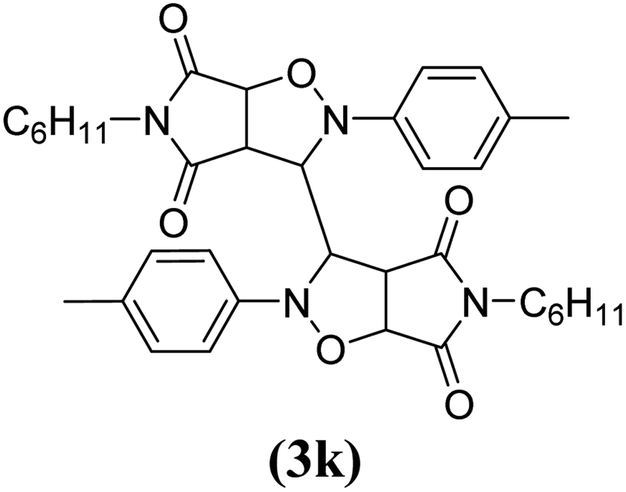
The products have been characterized as bis trans-2,5-diaryl-pyrroloisoxazole-4,6-diones (Table 2) through their melting point, and FTIR, NMR, mass spectral and elemental analyses. The position of the C3–H proton in relation to the C3a–H and C6a–H protons in the 1H-NMR spectrum was used to ascribe the trans configuration to these products. Furthermore, the C3–H and C3′–H protons were also found to be trans to each other, which may be due to steric reasons. However, no product with a cis configuration was isolated.
| Entry | Nitrone | Maleimide | Product | Time (min) | % Yield | MP |
|---|---|---|---|---|---|---|
| 1 |
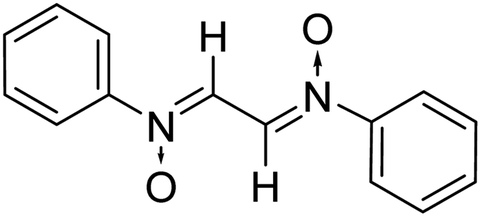
|

|
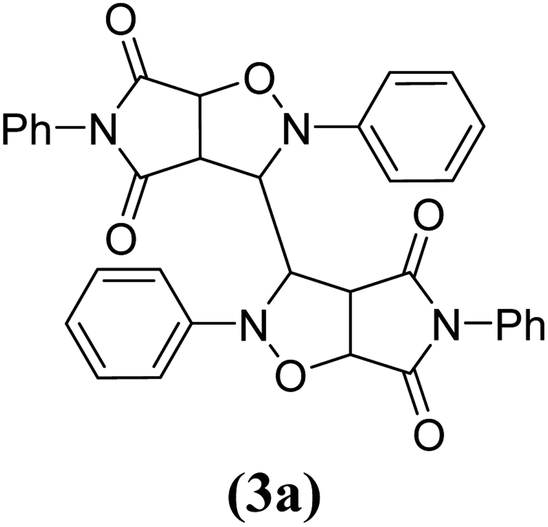
|
10 | 84 | 285–286 |
| 2 |

|

|

|
10 | 88 | 280–281 |
| 3 |

|
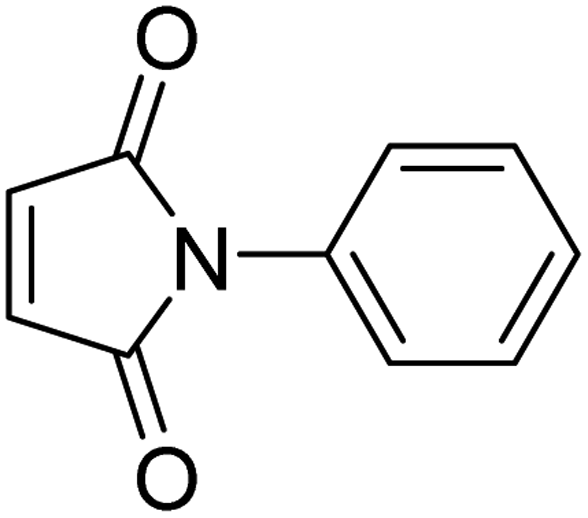
|
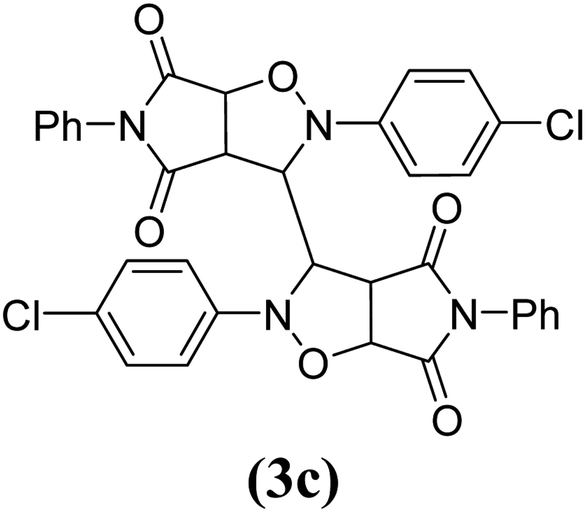
|
12 | 84 | 291–292 |
| 4 |

|

|

|
10 | 87 | 272–273 |
| 5 |
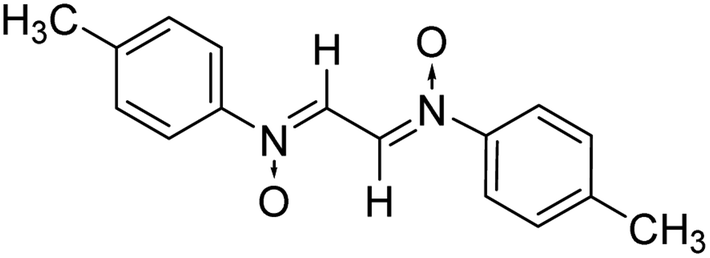
|
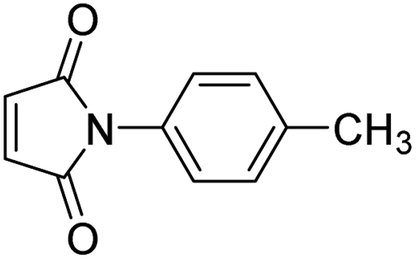
|
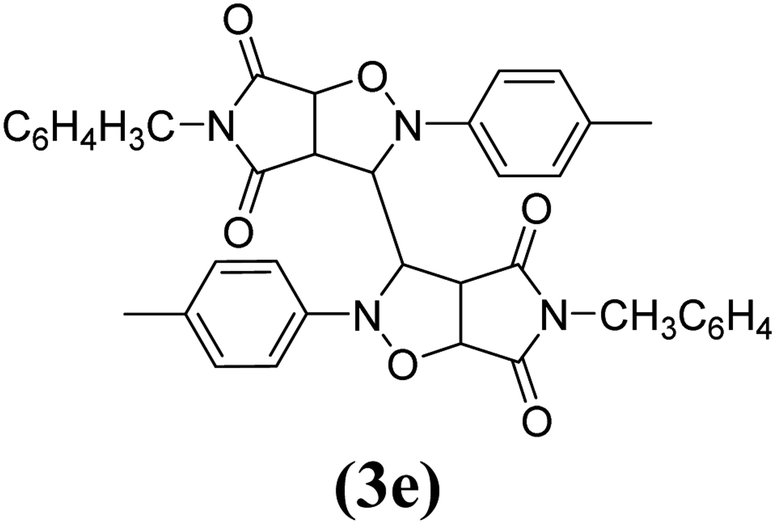
|
10 | 90 | 267–268 |
| 6 |
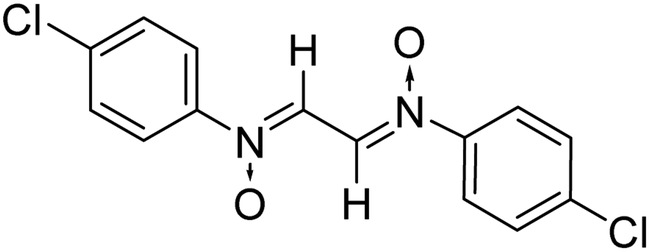
|
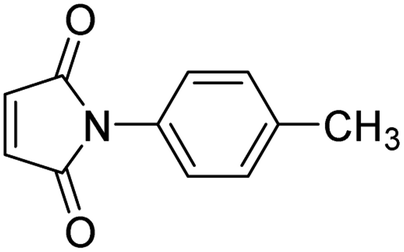
|

|
14 | 82 | 274–275 |
| 7 |
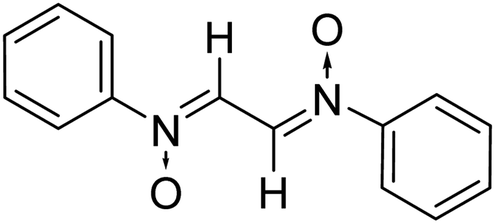
|

|
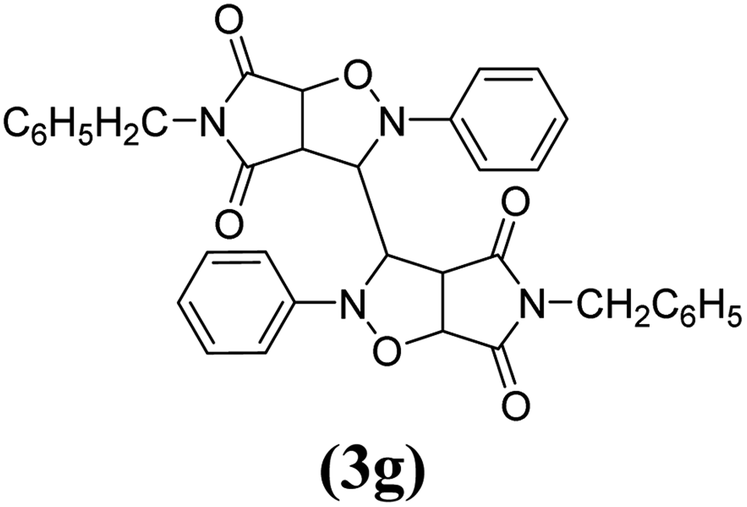
|
10 | 88 | 264–265 |
| 8 |
|

|

|
10 | 90 | 269–270 |
| 9 |

|

|

|
12 | 86 | 295–296 |
| 10 |

|
|

|
12 | 88 | 290–291 |
| 11 |

|

|
|
12 | 84 | 291–292 |
| 12 |

|

|

|
15 | 80 | 297–298 |
3.2. Spectral studies
The spectral data of one of the synthesized compounds, pyrrolo isoxazole 3g are described here. In the infrared absorption spectrum of compound 3g (Fig. 1), an absorption band in the region of 3063–2977 cm−1 has been linked to the C–H stretching vibration. Two absorption bands at 1784 and 1714 cm−1 correspond to the two carbonyls of the succinimide moiety. The absorption bands due to the CC bond of the aromatic ring and C–N bond appeared at 1598 cm−1 and 1398 cm−1, respectively. The C–O bond stretching appeared in the form of medium intensity bands at 1261 and 1181 cm−1.
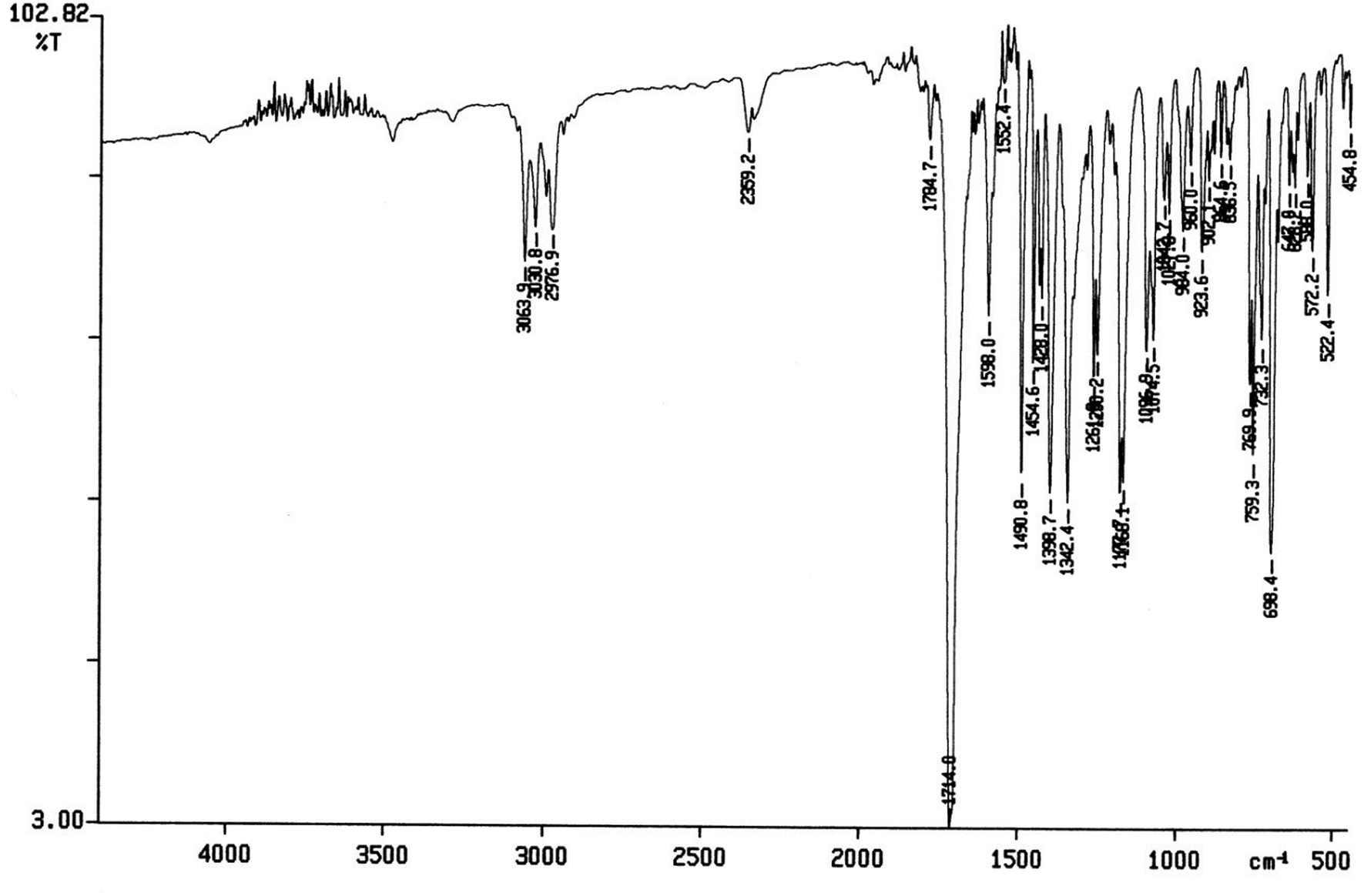 | ||
| Fig. 1 FTIR spectrum of the synthesized compound 3g. | ||
In the proton NMR spectrum of compound 3g (Fig. 2), a doublet at δ 3.83 Hz was ascribed to the C3a–H proton. The C3–H proton appeared in the form of a singlet at δ 4.66 as it does not couple with the C3–H of the other half of the molecule due to dihedral angle between the two. Another doublet at δ 5.14 (J = 7.64 Hz) has been assigned to the C6a–H proton. The benzylic protons appeared in the form of a singlet at δ 4.22, while the multiplet of aromatic protons appeared between δ 6.9 to δ 7.3, in the NMR spectrum.
 | ||
| Fig. 2 1H NMR spectrum of the synthesized compound 3g. | ||
The mass spectra of the compound 3g (Fig. 3) show a molecular ion peak at m/z 614, thereby confirming that the bis 1,3-dipolar cycloaddition has taken place. And since the molecule is symmetrical around one of the aliphatic carbons, a peak corresponding to half of the molecular mass i.e. at m/z 307, was also observed (Fig. 3).
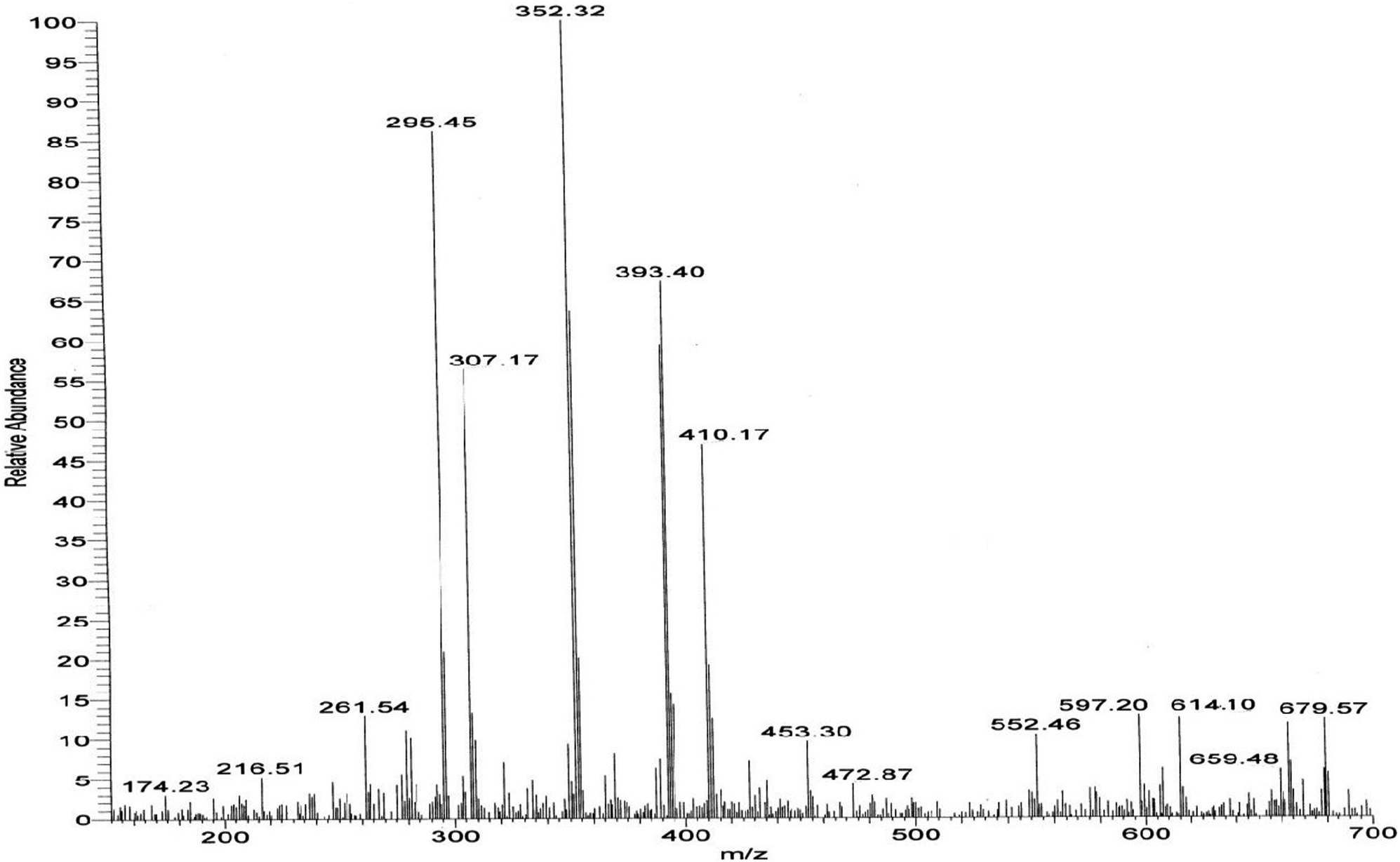 | ||
| Fig. 3 Mass spectrum of the synthesized compound 3g. | ||
Mechanistically, the formation of only a trans diastereoisomer (Fig. 4) can be explained on the basis of the endo-transition mode, in which the succinimide moiety and N-phenyl ring of the nitrone lie on the same side, and thus stabilizes the transition state due to maximum π–π overlap. These 1,3-dipolar cycloadditions provide trans diastereoisomer exclusively as the single product as steric interactions seem to be maximum in the transition state leading to the cis isomer. Moreover, the large sized bicyclic pyrrolo isoxazole moiety could not be on the same side due to steric hindrance as they are located on adjacent carbon atoms of the glyoxal precursor, which ruled out the formation of a cis isomer.
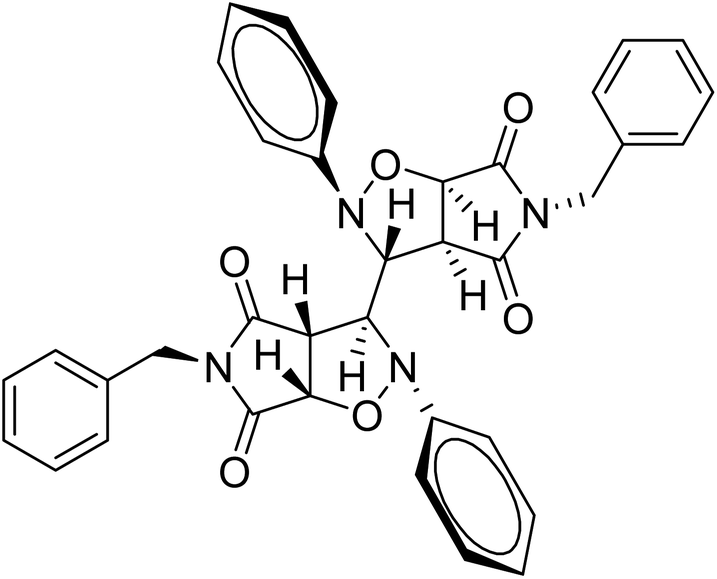 | ||
| Fig. 4 Dreiding model of compound 3g (trans). | ||
4. Conclusion
To sum up, the synthesis of bis-pyrrolo isoxazole cycloadducts reported here is facile, efficient, regioselective and eco-friendly. An attractive feature of this protocol is that it generates only the trans stereoisomer without the involvement of any chiral catalyst or specialised reaction conditions. The formation of only one trans-diastereoisomer was confirmed through IR and NMR spectroscopy. Moreover, the cycloadducts were formed in a very short interval of 15–20 minutes and that too under aqueous conditions. Further work to synthesize bis-pyrrolo isoxazoles replacing glyoxal nitrones with other bis dipole substrates is in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge SGGSW University, Fatehgarh Sahib, Punjab (India), for lab facilities and SAIF Lab, Punjab University, Chd. for NMR spectral analysis.References
- M. M. M. Santos, N. Faria, J. Iley, S. J. Coles, M. B. Hursthouse, M. L. Martins and R. Moreira, Reaction of naphthoquinones with substituted nitromethanes. Facile synthesis and antifungal activity of naphtho[2,3-d]isoxazole-4,9-diones, Bioorg. Med. Chem. Lett., 2010, 20, 193–195 CrossRef CAS PubMed.
- Y. Prashanthi, K. Kiranmai and N. J. P. Subhashini, Synthesis, potentiometric and antimicrobial studies on metal complexes of isoxazole Schiff bases, Spectrochim. Acta, Part A, 2008, 70, 30–35 CrossRef CAS PubMed.
- Y. S. Lee, S. M. Park and B. H. Kim, Synthesis of 5-isoxazol-5-yl-2′-deoxyurid-ines exhibiting antiviral activity against HSV and several RNA viruses, Bioorg. Med. Chem. Lett., 2009, 19, 1126–1128 CrossRef CAS PubMed.
- C. Changtam, P. Hongmanee and A. Suksamrarn, Isoxazole analogs of curcuminoids with highly potent multidrug-resistant antimycobacterial activity, Eur. J. Med. Chem., 2010, 45, 4446–4457 CrossRef CAS PubMed.
- A. Padmaja, T. Payani, G. D. Reddy and V. Padmavathi, Synthesis, antimicrobial and antioxidant activities of substituted pyrazoles, isoxazoles, pyrimidine and thioxopyrimidine derivatives, Eur. J. Med. Chem., 2009, 44, 4557–4566 CrossRef CAS PubMed.
- S. Balalaie, A. Sharifi and B. Ahangarian, Solid phase synthesis of isoxazole and pyrazole derivatives under microwave irradiation, Indian J. Heterocycl. Chem., 2000, 10, 149–150 CAS.
- D. Patrizia, A. Carbone, P. Barraja, G. Kelter, H. H. Fiebig and G. Cirrincione, Synthesis and antitumor activity of 2,5-bis(3′-indolyl)-furans and 3,5-bis(3′-indolyl)-isoxazoles, nortopsentin analogues, Bioorg. Med. Chem., 2010, 18, 4524–4529 CrossRef PubMed.
- P. Conti, L. Tamborini, A. Pinto, L. Sola, R. Ettari, C. Mercurio and C. De Micheli, Design and synthesis of novel isoxazole-based HDAC inhibitors, Eur. J. Med. Chem., 2010, 45, 4331–4338 CrossRef CAS PubMed.
- B. Kafle and H. Cho, Isoxazolone derivatives as potent inhibitors of PTP1B, Bull. Korean Chem. Soc., 2012, 33, 275–277 CrossRef CAS.
- J. J. Talley, D. L. Brown, J. S. Carter, M. J. Graneto, C. M. Koboldt, J. L. Masferrer, W. E. Perkins, R. S. Rogers, A. F. Shaffer, Y. Y. Zhang, B. S. Zweifel and K. Seibert, 4-[5-Methyl-3-phenylisoxazol-4-yl]-benzene-sulfonamide, valdecoxib: a potent and selective inhibitor of COX-2, J. Med. Chem., 2000, 43, 775–777 CrossRef CAS PubMed.
- A. Srinivas, A. Nagaraj and C. S. Reddy, Synthesis and in vitro study of methylene-bistetrahydro[1,3]thiazolo[4,5-c]isoxazoles as potential nematicidal agents, Eur. J. Med. Chem., 2010, 45, 2353–2358 CrossRef CAS PubMed.
- S. N. Suryawanshi, A. Tiwari, N. Chandra and S. Ramesh, Chemotherapy of leishmaniasis. Part XI: synthesis and bioevaluation of novel isoxazole containing heteroretinoid and its amide derivatives, Bioorg. Med. Chem. Lett., 2012, 22, 6559–6562 CrossRef CAS PubMed.
- W. Knecht and M. Loffler, Species-related inhibition of human and rat dihydroorotate dehydrogenase by immunosuppressive isoxazol and cinchoninic acid derivatives, Biochem. Pharmacol., 1998, 56, 1259–1264 CrossRef CAS PubMed.
- H. Kiyani and F. Ghorbani, Boric acid-catalyzed multi-component reaction for efficient synthesis of 4H-isoxazol-5-ones in aqueous medium, Res. Chem. Intermed., 2015, 41, 2653–2664 CrossRef CAS.
- H. Kiyani and F. Ghorbani, Efficient tandem synthesis of a variety of pyran-annulated heterocycles, 3,4-disubstituted isoxazol-5(4H)-ones, and α,β-unsaturated nitriles catalyzed by potassium hydrogen phthalate in water, Res. Chem. Intermed., 2015, 41, 7847–7882 CrossRef CAS.
- X. Li, P. Zhai, Y. Fang, W. Li, H. Chang and W. Gao, Synthesis of isoxazolidines via catalyst-free one-pot three-component cycloaddition of sulfoxonium ylides, nitrosoarenes and alkenes, Org. Chem. Front., 2021, 8, 988–995 RSC.
- H. Kiyani and F. Ghorbani, Expeditious green synthesis of 3,4-disubstituted isoxazole-5(4H)-ones catalyzed by nano-MgO, Res. Chem. Intermed., 2016, 42, 6831–6844 CrossRef CAS.
- Z. Faramarzi and H. Kiyani, Organocatalyzed three-component synthesis of isoxazol-5(4H)-ones under aqueous conditions, Heterocycles, 2021, 102, 1779–1790 CrossRef CAS.
- Z. Faramarzi and H. Kiyani, Steglich’s base catalyzed three-component synthesis of isoxazol-5-ones, Polycyclic Aromat. Compd., 2022, 43, 3099–3121 CrossRef.
- H. Ostadzadeh and H. Kiyani, Synthesis of isoxazole-5(4H)-ones using citrazinic acid as an organocatalyst in aqueous conditions, Org. Prep. Proced. Int., 2023, 55, 538–548 CrossRef CAS.
- V. Spano, R. Rocca and M. Berreca, et al., Pyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazoles, a new class of antimitotic agents active against multiple malignant cell types, J. Med. Chem., 2020, 63, 12023 CrossRef CAS PubMed.
- N. Agarwal and P. Mishra, The synthetic and therapeutic expedition of isoxazole and its analogs, Med. Chem. Res., 2018, 27, 1309 CrossRef PubMed.
- R. Badru, P. Anand and B. Singh, Synthesis and evaluation of hexahydropyrrolo[3,4-d]isoxazole-4,6-diones as anti-stress agents, Eur. J. Med. Chem., 2012, 48, 81 CrossRef CAS PubMed.
- S. R. Pedada, N. S. Yarla and P. J. Tambade, et al., Synthesis of new secretory phospholipase A2-inhibitory indole containing isoxazole derivatives as anti-inflammatory and anticancer agents, Eur. J. Med. Chem., 2016, 112, 289 CrossRef CAS PubMed.
- K. M. Naidu, S. Srinivasarao and N. Agnieszka, et al., Seeking potent anti-tubercular agents: design, synthesis, anti-tubercular activity and docking study of various ((triazoles/indole)-piperazin-1-yl/1,4-diazepan-1-yl) benzo[d] isoxazole derivatives, Bioorg. Med. Chem. Lett., 2016, 26, 2245 CrossRef CAS PubMed.
- W. Shi, Z. Hu, N. Bao, D. Li, L. Chen and J. Sun, Design, synthesis and cytotoxic activities of scopoletin-isoxazole and scopoletin-pyrazole hybrids, Bioorg. Med. Chem. Lett., 2017, 27, 147 CrossRef CAS PubMed.
- R. Badru, S. Shah and B. Singh, 1,3-Dipolar cycloaddition reactions of 2-substituted azomethine N-oxides with N-benzyl maleimides leading to the synthesis of stereoisomers, J. Heterocycl. Chem., 2012, 49, 336 CrossRef CAS.
- H. Wang, R. Cheng, G. Wang, Y. Shi, J. Wang, H. Guo, L. Trigoura, Y. Xing and S. Sun, Synthesis of pyrrolo[3,4-d]isoxazolines via a one-pot radical functionalization/cycloaddition of methyl ketone, tert-butyl nitrite, and maleimide, Tetrahedron Lett., 2020, 61, 151652 CrossRef CAS.
- S. Thakur, A. Das and T. Das, 1,3-Dipolar cycloaddition of nitrones: synthesis of multisubstituted, diverse range of heterocyclic compounds, New J. Chem., 2021, 45, 11420 RSC.
- C. J. Gerry, Z. Yang, M. Stasi and S. L. Schreiber, DNA-compatible [3+2] nitrone-olefin cycloaddition suitable for DEL syntheses, Org. Lett., 2019, 21, 1325 CrossRef CAS PubMed.
- S. A. Ali, H. A. Al-Muallem, S. U. Rahman and M. T. Saeed, Bis-isoxazolidines: a new class of corrosion inhibitors of mild steel in acidic media, Corros. Sci., 2008, 50, 3070–3077 CrossRef CAS.
- Y. Luo, C. H. Chen, J. Q. Zhang, C. Liang and D. L. Mo, Synthesis of spirofluorenyl-1,2,4-oxadiazinan-5-ones through metal-free [3+3] cycloaddition of N-vinyl fluorenone nitrones with aza-oxyallyl cations, Synthesis, 2020, 52, 424–432 CrossRef CAS.
- M. Dickmeis, H. Cinar and H. Ritter, Bisnitrone: new starting material for heterocyclic poly(1,2,4-oxadiazolidin-5-one) via polycycloaddition with diisocyanate and urethane prepolymer, Macromolecules, 2012, 45, 3285 CrossRef CAS.
- V. G. Manecke and J. Klawitter, Zur ynthese von makromolekulen aus nitronen durch 1,3-dipolare cycloaddition. II, Makromol. Chem., 1971, 142, 253–257 CrossRef.
- F. Heaney, O. Rooney, D. Cunningham and P. McArdle, Simultaneous double 1,3-dipolar cycloaddition reactions involving bisnitrones or bisdipolarophiles. 1H NMR investigation of the conformational preferences of N-methyl- and N-phenyl-isoxazolidines, J. Chem. Soc., Perkin Trans. 2, 2001, 373 RSC.
- L. Vretik and H. Ritter, 1,3-Dipolar cycloaddition in polymer synthesis. 1. Polyadducts with flexible spacers derived from bis(N-methylnitrone)s and bis(N-phenylmaleimide)s, Macromolecules, 2003, 36, 6340 CrossRef CAS.
- B. Chakraborty and G. P. Luitel, Synthesis of some novel bisisoxazolidine derivatives from glyoxal-derived bisnitrones via simultaneous double cycloaddition reactions in water, J. Heterocycl. Chem., 2014, 52, 726 CrossRef.
- S. Shah, R. Badru and B. Singh, Diastereoselective synthesis of novel spiro-isoxazolidines via [3 + 2] cycloaddition, Synth. Commun., 2012, 43, 1073 CrossRef.
- N. E. Searle, Synthesis of N-aryl-maleimides, US Pat. 2444536, 1948Chem. Abstr. 1948, 42, 7340 Search PubMed.
- B. S. Furniss, A. J. Hannaford, V. Rogers, P. W. G. Smith and A. R. Tatchell, Vogel's Textbook of Practical Organic Chemistry, Longman Scientific & Technical, Essex: England, 5th edn, 1991; p. p. 955 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00396e |
| This journal is © The Royal Society of Chemistry 2024 |