
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnthraquinone-catalyzed H2O2 electrosynthesis coupled with an advanced oxidation process for water treatment†
Pengdong
Liu‡
ab,
Haixing
Zhang‡
ab,
Yu
Chen‡
ab,
Yajing
Di
ab,
Zhilin
Li
ab,
Baoning
Zhu
*c,
Zheng
Liu
*d,
Zhengping
Zhang
 *ab and
Feng
Wang
ab
*ab and
Feng
Wang
ab
aState Key Laboratory of Chemical Resource Engineering, Beijing Key Laboratory of Electrochemical Process and Technology for Materials, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: zhangzhengping@mail.buct.edu.cn
bBeijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China
cBeijing Engineering Center for Environmental Pollution Control and Resource Utilization, Beijing University of Chemical Technology, Beijing 100029, P. R. China
dState Key Laboratory of Environmental Criteria and Risk Assessment, Chinese Research Academy of Environmental Sciences, Beijing 100012, P. R. China
First published on 21st December 2023
Abstract
Advanced oxidation processes (AOPs) coupled with in situ H2O2 electrosynthesis are highly desirable and safe as they do not involve H2O2 storage and transportation, and therefore, developing highly robust cathodic electrocatalysts is especially important. In this work, we developed a series of anthraquinone molecules as metal-free electrocatalysts to accelerate H2O2 production. The electrocatalytic performance, mechanism, and enhancement of these anthraquinone molecules were studied and compared to those of traditional thermal catalysts. It was found that the electrochemical hydrogenation and chemical dehydrogenation processes on anthraquinones are important to accelerate H2O2 electrosynthesis. To increase the materials' applicability in chemical engineering, we assembled anthraquinone-derived gas diffusion electrodes (GDEs). Owing to the strong electron donation property of amino groups, amino-anthraquinone-derived GDEs exhibited high performance for H2O2 electrosynthesis in electrolytes of various pH values (H2O2 productivity of 0.25 mg cm−2 min−1 at 30 mA cm−2, and the cumulative amount of 1077 mg L−1 during 150 min in a static electrolyzer without a membrane separator) and long-term durability (stable operation for 120 h). The in situ H2O2 electrosynthesis coupled with electro-peroxone was further employed for the degradation of various phenolic compounds, where the chemical oxygen demand (COD) removal reached above 90% in 120 min with ultra-low energy consumption (below 15 W h gCOD−1). The treatment of practical petrochemical wastewater was also evaluated. This work provides valuable insights into using metal-free catalysts for high-performance electrochemical applications.
Sustainability spotlightWater is the foundation of ecology, whether the water quality standard is directly related to the degree of satisfaction for the ecological environment. This study is focused on the degradation of phenolics in wastewater via the advanced oxidation process coupled in situ hydrogen peroxide electrosynthesis. In this study, we synthesized a series of anthraquinone molecules as metal-free electrocatalysts to accelerate H2O2 production. Different from traditional thermal catalysis based on anthraquinones, the study is focused on the electrocatalytic performance of anthraquinone molecules, which is a new research field in terms of anthraquinones or electrocatalysis. This study emphasizes the importance of the following UN Sustainable Development Goals: clean water and sanitation (SDG 6); affordable and clean energy (SDG 7); and industry, innovation, and infrastructure (SDG 9). |
1. Introduction
Advanced oxidation process (AOP) technologies,1–5 highly efficient methods for water treatment,6 work by utilizing hydroxyl radicals (˙OH, the redox potential as high as 2.8 V)7 or sulphate radicals8 to oxidize hazardous organics, such as phenolic compounds, a type of stable molecules with a benzene ring structure, and refractory pollutants with high biological toxicity. Among most AOP technologies, hydrogen peroxide (H2O2) is a very important reactant.9–12 Although it is difficult for H2O2 to directly oxidize or degrade organic matter by itself, the HO–OH bonding facilitates the conversion of H2O2 into highly active ˙OH via various chemical processes (e.g. Fenton,13 UV/H2O2,14 O3/H2O2 (ref. 15)). However, the safe storage and transportation of H2O2 is a big challenge owing to its instability.16 Meanwhile, precise H2O2 addition is also important considering the redox reaction between the generated ˙OH and H2O2.17,18In this case, AOPs coupled with in situ H2O2 electrosynthesis,19,20 where cathodes can reduce oxygen (O2) into H2O2,21–23 is highly desirable to effectively solve the above problems. The AOP-coupled electrochemical operation is unique, involving high working voltages (above 2.0 V for most electrochemical AOPs) and uncontrollable solutes, which decreases the life of the electrode and increases the probability of side reactions, including the undesirable four-electron oxygen reduction reaction (directly reducing O2 into H2O) and the hydrogen evolution reaction.24 Compared to metal-containing materials (e.g., novel metal alloys,25,26 transition metal compounds,27–29 and M–N–C catalysts30,31), metal-free electrocatalysts present the natural advantages of long-term durability and low cost, which make these materials exhibit high applicability in economical water treatments.32,33 In addition, carbon-based metal-free electrocatalysts can also avoid secondary water pollution by toxic metal ions,34,35 and hence, they are valuable in purifying the water environment for aquaculture and agriculture.36,37 To further develop electrochemical AOP technologies, we propose to use anthraquinone molecular catalysts to accelerate H2O2 electrocatalysis and couple the anthraquinone-derived electrolyzer with electro-peroxone (H2O2/O3, EP) to degrade the phenol-containing wastewater. Different from other (electro)catalysis processes, anthraquinone molecules showed a good conversion selectivity due to their different H2O2 generation steps, where the first step is hydrogenation and the second is oxygen-participated dehydrogenation (Fig. 1a). It can effectively avoid the undesirable side reactions. Although the anthraquinone molecules have been used in traditional (thermal) catalysis, electrocatalysis research, and applications are hindered due to the limited electrical conductivity and poor processability caused by the lower melting point compared to that of polytetrafluoroethylene (the industrial electrode binders). In this case, we employed low-pressure rotary evaporation technology to support a series of anthraquinone molecules (including anthraquinone, AQ; methyl anthraquinone, MAQ; ethyl anthraquinone, EAQ; and amino anthraquinone, AAQ) on graphitic carbon black. To realize the electrode applications of these molecular catalysts, these anthraquinone-derived gas diffusion electrode (GDE) is also developed through the hot-pressing process using polyvinylidene fluoride (PVDF, melting point of 171 °C) as binders on porous nickel substrates to improve the low-cost processability and mechanical properties. The GDE technology, where massive oxygen can be transferred from the back of porous electrode to the reactive interface (i.e., catalyst layers), also endows these metal-free molecular electrocatalysts with a good performance for H2O2 electrosynthesis, and the corresponding EP-coupled electrolyzers further confirms the technical and economic feasibility for practical applications.
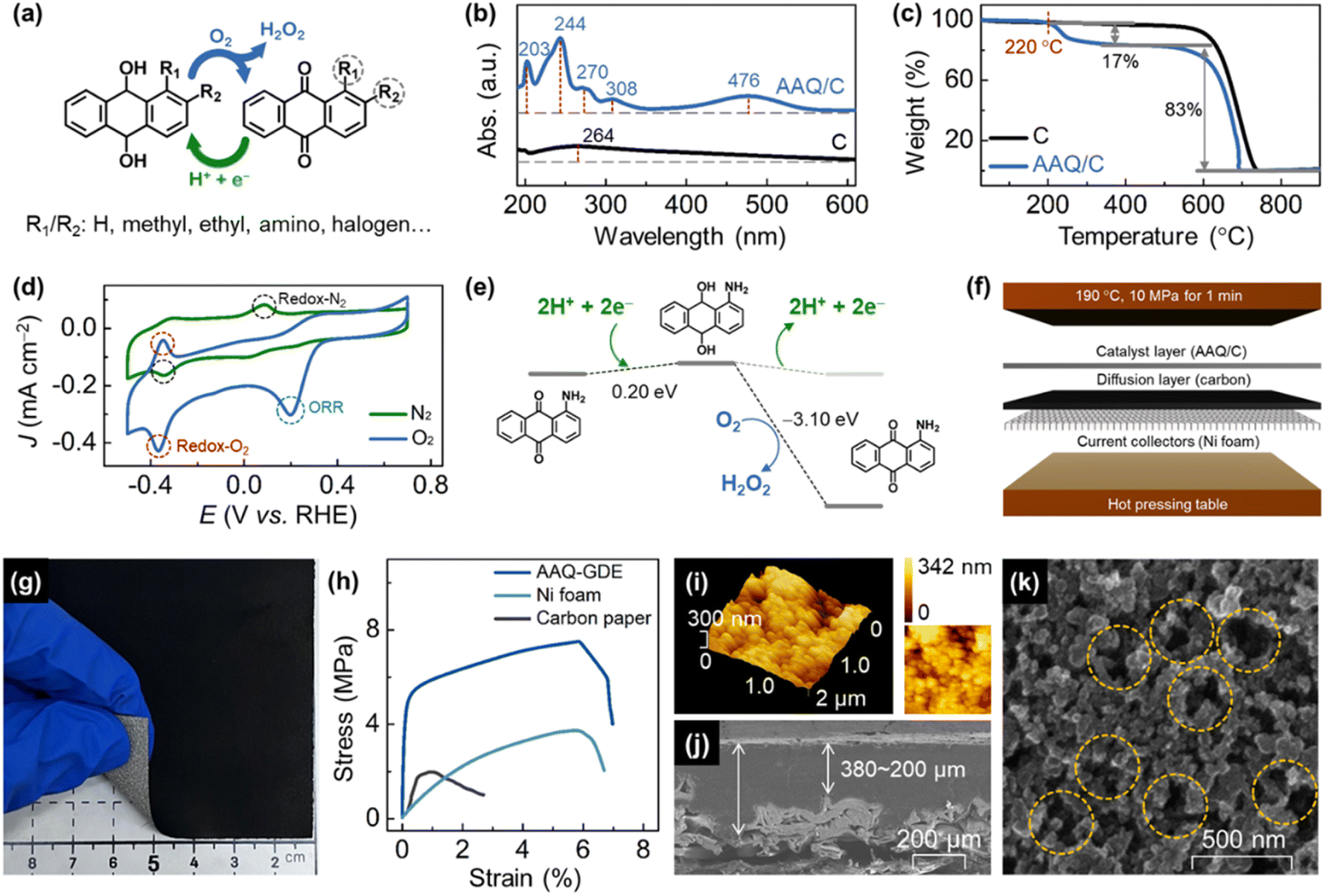 | ||
| Fig. 1 (a) Illustrations of the hydrogenation and dehydrogenation of anthraquinone compounds. (b) Thermal gravimetric analysis curves and (c) UV-vis spectra of AAQ/C and carbon black. (d) CV curves of AAQ/C in O2-saturated or N2-saturated 0.1 M Na2SO4 solution at a sweep rate of 50 mV s−1. (e) The calculated free energy diagrams of H2O2 production by AAQ. (f) Schematic preparation and (g) a digital photo of the as-prepared AAQ-GDE. (h) Stress–strain curves of AAQ GDE, Ni foam, and carbon paper. (i) AFM image and (j) cross-sectional and (k) in-plane SEM images of AAQ-GDE. | ||
2. Results and discussion
A series of graphitic carbon black supported anthraquinone molecules (i.e., AQ/C, MAQ/C, EAQ/C, and AAQ/C) and anthraquinone molecules-derived GDEs (AQ-GDE, MAQ-GDE, EAQ-GDE, and AAQ-GDE) were prepared (the detailed preparation is provided in ESI†). AAQ-GDE exhibited the best performance for H2O2 electrosynthesis (detailed discussion infra), and hence, only the AAQ/C electrocatalyst and AAQ-GDE electrode were used for the subsequent analysis. Multiple characterizations were performed on AAQ/C to confirm that the AAQ molecules were successfully dispersed on carbon blacks by rotary evaporation technology. Scanning electron microscope (SEM) images showed that there was no obvious other aggregated matter except carbon blacks in AAQ/C (Fig. S1†). As shown in ultraviolet-visible (UV-vis) spectra (Fig. 1b), the absorption peaks at ca. 270 nm were attributed to the quinone structure, and the other two absorption peaks at ca. 244 nm and 308 nm were ascribed to benzene structure. The additional absorption peak at 476 nm is attributed to the influence of the α-position amino groups.38 Infrared absorption spectra (Fig. S2†) revealed that the peaks at 1668 cm−1, 1590 cm−1, and 3415 cm−1, which are attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, aromatic ring, and N–H stretching in AAQ, respectively.39,40 Due to the π–π interaction between the anthracene structure and graphitization layers, the introduced AAQ molecules reduced the disorder and structural defects evidenced from the decreased peak-intensity ratio of the D band (ca. 1330 cm−1) to the G band (ca. 1580 cm−1) from 1.16 (C) to 1.01 (AAQ/C) in Raman spectra (Fig. S3†).41 The mass content of the molecular compound was detected by a thermogravimetric analyzer (Fig. 1c). The AAQ/C composite showed the first weight-loss step at 220 °C due to the thermal decomposition of AAQ, and the molecular loading was measured as around 20 wt%.
O, aromatic ring, and N–H stretching in AAQ, respectively.39,40 Due to the π–π interaction between the anthracene structure and graphitization layers, the introduced AAQ molecules reduced the disorder and structural defects evidenced from the decreased peak-intensity ratio of the D band (ca. 1330 cm−1) to the G band (ca. 1580 cm−1) from 1.16 (C) to 1.01 (AAQ/C) in Raman spectra (Fig. S3†).41 The mass content of the molecular compound was detected by a thermogravimetric analyzer (Fig. 1c). The AAQ/C composite showed the first weight-loss step at 220 °C due to the thermal decomposition of AAQ, and the molecular loading was measured as around 20 wt%.
In order to investigate whether the AAQ/C composite could show hydrogenation/oxygen-participated dehydrogenation in the electrochemical process, cyclic voltammetry (CV) measurements in either N2-saturated or O2-saturated electrolyte were performed (Fig. 1d). The redox peaks in the N2-saturated electrolyte were located at −0.343 V (reduction, vs. RHE, the same as below) and 0.089 V (oxidation) attributed the hydrogenation/dehydrogenation process without the participation of O2. However, the redox behavior of AAQ/C in O2-saturated electrolyte is different, where the redox peaks were located at −0.367 V/−0.351 V (reduction/oxidation). The much negatively shifted oxidation peaks indicated that the O2 molecules participated in the dehydrogenation process. Meanwhile, the enlarged area of the redox peaks in the O2-saturated electrolyte also demonstrated that the enhanced hydrogenation/dehydrogenation process was due to the participation of O2. In addition, the oxygen electroreduction peak was also observed for the AAQ/C composites in the O2-saturated electrolyte, but not in the N2-saturated electrolyte. To analyze the electrochemical results in detail, density functional theory (DFT) calculations were employed to investigate the hydrogenation/dehydrogenation process. The DFT calculated free energies (ΔG) are shown in Fig. 1e, S4 and Table S1.† After participating with protons and electrons, AAQ could be reduced into hydro-AAQ, and then the H2O2 molecule was released during the dehydrogenation process when hydro-AAQ interacted with O2 molecules. Compared with the O2-free condition (ΔG = −0.201 eV), the oxygen-participated H2O2 production was a more favorable thermodynamic process (ΔG = −3.097 eV).
After the confirmation of the electrochemical property of the AAQ/C composite, the molecular catalyst-derived GDEs were further developed in this work to realize the electrode applications of these molecular catalysts. Considering the relatively low thermal stability of AAQ (below 220 °C), we applied PVDF as the binder, whose processing temperature was around 170 °C. After the spray and rapid hot pressing (T = 190 °C, p = 10 MPa, t = 1 min, Fig. 1f) on nickel foam, the AAQ-GDE was successfully prepared (7 × 7 cm2), and the comparative samples (pure carbon black loaded on GDE, C-GDE) were also prepared. The tightly-gripped coating layer with the porous substrate ensured a good mechanical strength, which allowed AAQ-GDE could partially bend as shown in the digital photo in Fig. 1g. Besides, the mechanical properties of the hot-pressed AAQ-GDE were also measured in comparison with the Ni foam substrates and the traditional gas diffusion materials (i.e., carbon paper, Fig. 1h). Both metal-substrate electrodes exhibited the larger elongation than carbon paper, and the enhanced tensile strength of AAQ-GDE to Ni foam could be attributed to the cold processing and the possible composite interaction. The good mechanical properties indicated that the as-prepared GDE possessed good self-supporting behaviour, which could provide an advantage in further large-size electrode applications.
Then, the morphologies were studied from the optical microscope photograph (Fig. S5†) and the SEM image (Fig. S6†), both of which showed that the AAQ-GDE surface was smooth at macroscale or microscale. To investigate the profilometry and quantitative roughness in details, topographic measurements were further performed using atomic force microscope (AFM, Fig. 1i). The arithmetic mean deviation of roughness (ra) and the root-mean-square deviation of the roughness (rq) were also measured to quantitatively evaluate the roughness of surface, and it was found that AAQ-GDE presented a similar value of ra (ca. 46.81 nm) and rq (ca. 153.4 nm) compared with ra (ca. 36.53 nm) and rq (ca. 120.5 nm) of C-GDE (Fig. S7 and Table. S2†). The SEM cross-sectional image (Fig. 1j) exhibited the total thickness of the coating layer, including that of the catalyst layer and the gas diffusion layer, was about 200–380 μm, which was tightly combined with Ni foam. There was no obvious stratified structure remaining between the catalyst layer and the gas diffusion layer or between the gas diffusion layer and the metal collector. The SEM in-plane image of AAQ-GDE in high resolution (Fig. 1k) demonstrated the high porosity and well contacted electrocatalysts on the electrode surface, which could both enhance mass transport and electrical conductivity.
To further gain insights into the porous structure, the mercury intrusion porosimeter measurements were performed to analyze the porous structure of AAQ-GDE in comparison to C-GDE (Fig. 2a). A full pore-size range showed pore diameter peaks at around 40 nm, 7 μm, and 30 μm. The 40 nm pores accounted for 17–19% of the total pore volume, and were caused by primary pores of AAQ/C; the secondary pores of 50–1000 nm (22–24%) were mainly formed by the aggregations of the catalyst particles themselves with each other and/or binders. The larger pores (above 1 μm) were mainly owing to the porous metal substrates, which accounted for 57–59% of the total pore volume. In addition, we also applied the water contact angle measurements to estimate the hydrophobicity of the as-prepared GDEs. As shown in Fig. 2b, AAQ-GDE and C-GDE were above 130°, ensuring their hydrophobicities. Meanwhile, the electrical conductivity of AAQ-GDE (1.45 S cm−1) and C-GDE (8.76 S cm−1) were further measured (Fig. S8†). The chemical structure of AAQ-GDE was detected by X-ray photoelectron spectroscopy (XPS, Fig. 2c), proving the existence of the C, F, and O elements in both electrodes (i.e., AAQ-GDE and C-GDE). The element F originated from the PVDF resin, and an additional N 1s peak in AAQ-GDE (the inset of Fig. 2c) was attributed to the –NH2 species in AAQ.42 The X-ray diffraction (XRD) patterns (Fig. S9†) showed a similar structure of AAQ-GDE and C-GDE without the additional AAQ crystallizations. The peaks at 44.5°, 51.8°, and 76.4° were indexed to (111), (200), and (220) planes of metallic nickel (JCPDS no. 04-0850), respectively. The peak at 18.2° was the characteristic peak of PTFE (JCPDS no. 54-1595), while the rest of the broadened diffraction peak at 22.1° was attributed to carbon (JCPDS no. 50-0926). The poor crystallinity of PVDF meant that no characteristic peaks appeared in the XRD pattern. The obvious N XPS signal and non-AAQ diffraction peaks demonstrated that the hot pressing would not break the molecular structure or make the AAQ recrystallize during the electrode processing.
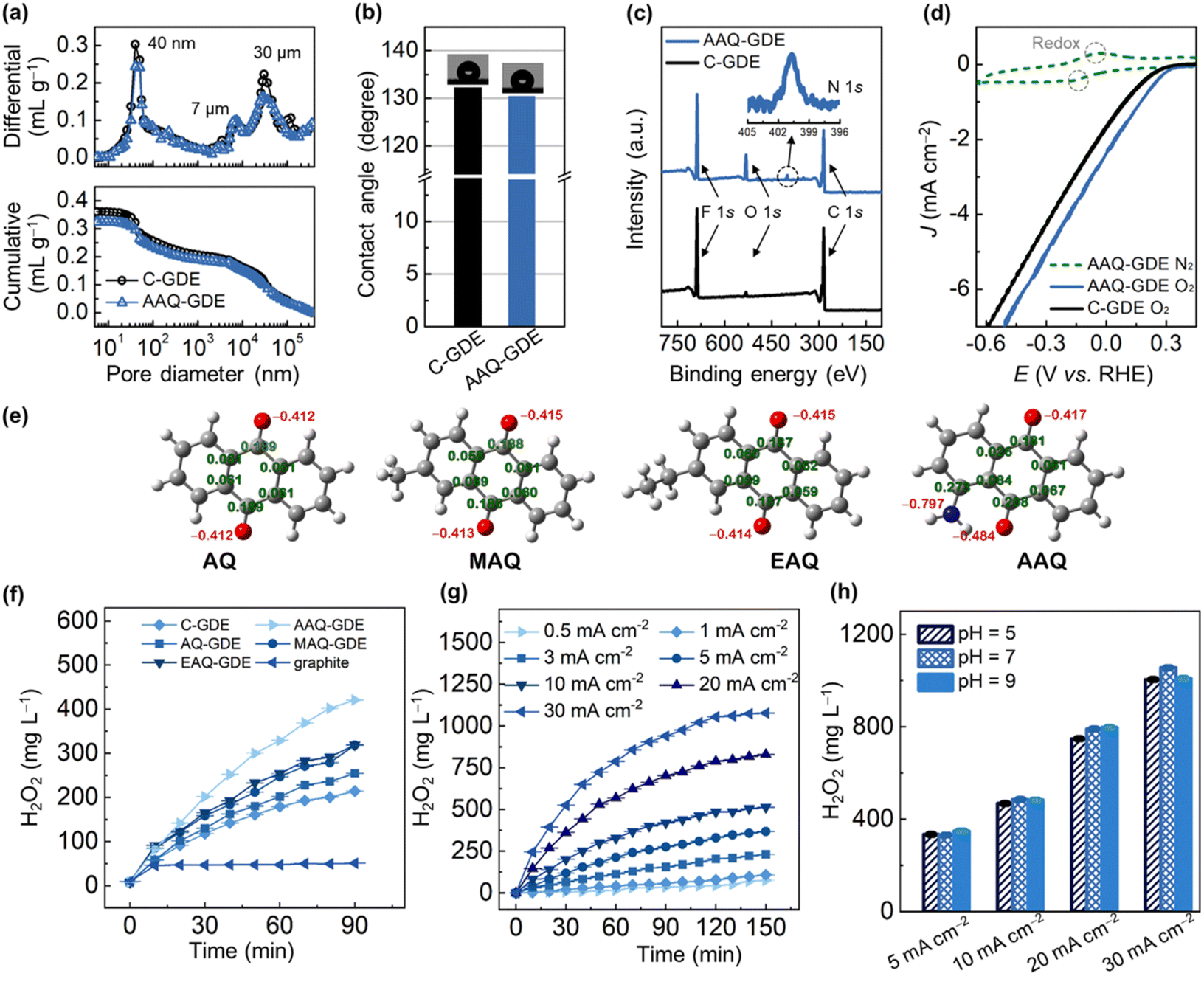 | ||
| Fig. 2 (a) Pore size distribution (top) and cumulative pore volume (bottom) measured by mercury injection, (b) diagram of water contact angles, and (c) XPS survey spectra of AAQ-GDE and C-GDE. The inset is the high-resolution XPS N 1s spectrum of AAQ-GDE. (d) CV curves of AAQ-GDE and C-GDE in 0.1 M Na2SO4 at a scan rate of 50 mV s−1, where O2 or N2 was supplied at the electrode back at a flow rate of 10 mL min−1. (e) DFT-optimized anthraquinone molecules, where the gray balls represent carbon, red oxygen, blue nitrogen, and light gray hydrogen. Accumulated H2O2 production of (f) different cathodes at 10 mA cm−2 and (g) AAQ-GDE at different current densities. (h) H2O2 production of AAQ-GDE in different operations for 120 min. The above electrochemical measurement was carried out in a 200 mL static and membrane-free electrolyzer with BDD as the anode. The working electrode area was 4 × 4 cm2. | ||
To evaluate the electrochemical performance, the CV curves of AAQ-GDE were tested in N2 and O2-supplied conditions, which were also compared with C-GDE. Due to the conservation of AAQ molecules, the CV curve of AAQ-GDE also showed the redox peaks when the N2 was supplied (Fig. 2d). While the O2 was supplied at the back of the electrodes, AAQ-GDE exhibited a greater current response than that of C-GDE, implying that AAQ-GDE showed a better device performance than C-GDE. To detect the ability for H2O2 production, we prepared a series of anthraquinone molecules-derived GDEs (i.e., AQ-GDE, MAQ-GDE, and EAQ-GDE) as the comparative samples, and these anthraquinone molecules were analysed as shown in Fig. 2e. Compared with the graphite electrode (oxygen was supplied in the front of the electrode surface) and C-GDE, all the anthraquinone-derived GDEs showed the enhanced performance on H2O2 production (Fig. 2f), which was attributed to their intrinsic activities. Among these anthraquinone-derived GDEs, the AAQ-GDE showed the highest H2O2 accumulations of 420 mg L−1 in the static and membrane-free electrolyzer after 90 min. The enhanced performance on H2O2 production should be due to the more negatively charged quinone group (Mulliken charge of −0.484, Fig. 2e and S10†) induced by the α-position amino group. It implied that these quinone groups on AAQ molecules were more beneficial to adsorb protons, and hence accelerate the hydrogenation process, which should be the rate-determination step for the total reaction.43,44
Furthermore, H2O2 accumulations over time for AAQ-GDE were studied at various current densities to evaluate its production efficiency (Fig. 2g). After the electrochemical operation for 150 min, the AAQ-GDE showed the largest H2O2 accumulations of 75, 108, 232, 369, 520, 830, and 1077 mg L−1 at 0.5, 1, 3, 5, 10, 20, and 30 mA cm−2, respectively. The H2O2 productivity was also increased with the increased current densities (Table S3†). When the current density increased at 30 mA cm−2, the initial H2O2 productivity could be as high as 0.25 mg cm−2 min−1. However, along with the prolonged time, the H2O2 productivity in the static and membrane-free electrolyzer was slowed down, implying that the anode consumed the generated H2O2, especially for the high concentration of H2O2 (i.e., the high-current-density operation). The environment adaptability of AAQ-GDE was also evaluated from operations in different pH electrolytes (Fig. 2h and S11†). It was found that AAQ-GDE presented steady H2O2 production in either weakly acidic or alkaline environments at low or high current densities, which was different from other electrocatalysts for H2O2 production.45,46 The low pH sensitivity indicated that AAQ-GDE could be applicable in treating the majority of wastewater.
Before the application was coupled with the electro-peroxone (H2O2/O3, EP) techniques, the AAQ-GDE-derived electrolyzer was ameliorated. Considering that the degradation electrolyzer is difficult to equip with membrane separators due to the complexity of wastewater, we evaluated the serviceability of different anodes for long-term operations. As shown in Fig. 3a, S12 and S13,† the AAQ-GDE-derived electrolyzer with the boron-doped diamond (BDD) anode showed a higher H2O2 production than that with the dimensionally stable anode (DSA and RuTi-containing), which was attributed to the poor H2O2 decomposition on the inert surface (i.e., BDD). However, the inert surface of BDD also greatly increased the cell voltage of the electrolyzer, leading to much higher energy consumption on H2O2 production than that was applied to DSA. When the energy consumption were close (e.g., DSA at 20 mA cm−2 and BDD at 10 mA cm−2, the energy consumption was ca. 10 W h gH2O2−1), DSA (550 mg L−1) presented a much higher H2O2 production than that of BDD (330 mg L−1). Hence, the DSA electrode was employed as the anode of the AAQ-GDE-derived electrolyzer for further evaluation. The electrochemical stability of AAQ-GDE was verified. The flow electrolyzer testing (pumping 0.1 M Na2SO4 electrolyte at the speed of 0.4 L h−1, stirring it evenly, and pumping it out at a fixed level to ensure that 200 mL solution was maintained in the electrolyzer) was employed (Fig. 3b). The H2O2 concentration was stabilized at 300–340 mg L−1, and the current efficiency could reach 60–70%. During the 120 hour operation, the cell voltage was approximately 3.8 V at 20 mA cm−2, indicating that the AAQ-GDE and AAQ-GDE//DSA electrolyzer had good long-term durability. After the long-term operation, FT-IR, UV-vis, and XPS spectra were collected to investigate the operated AAQ-GDE, and it was found that anthraquinone molecules were still preserved even after the 120 hour operation (Fig. S14†).
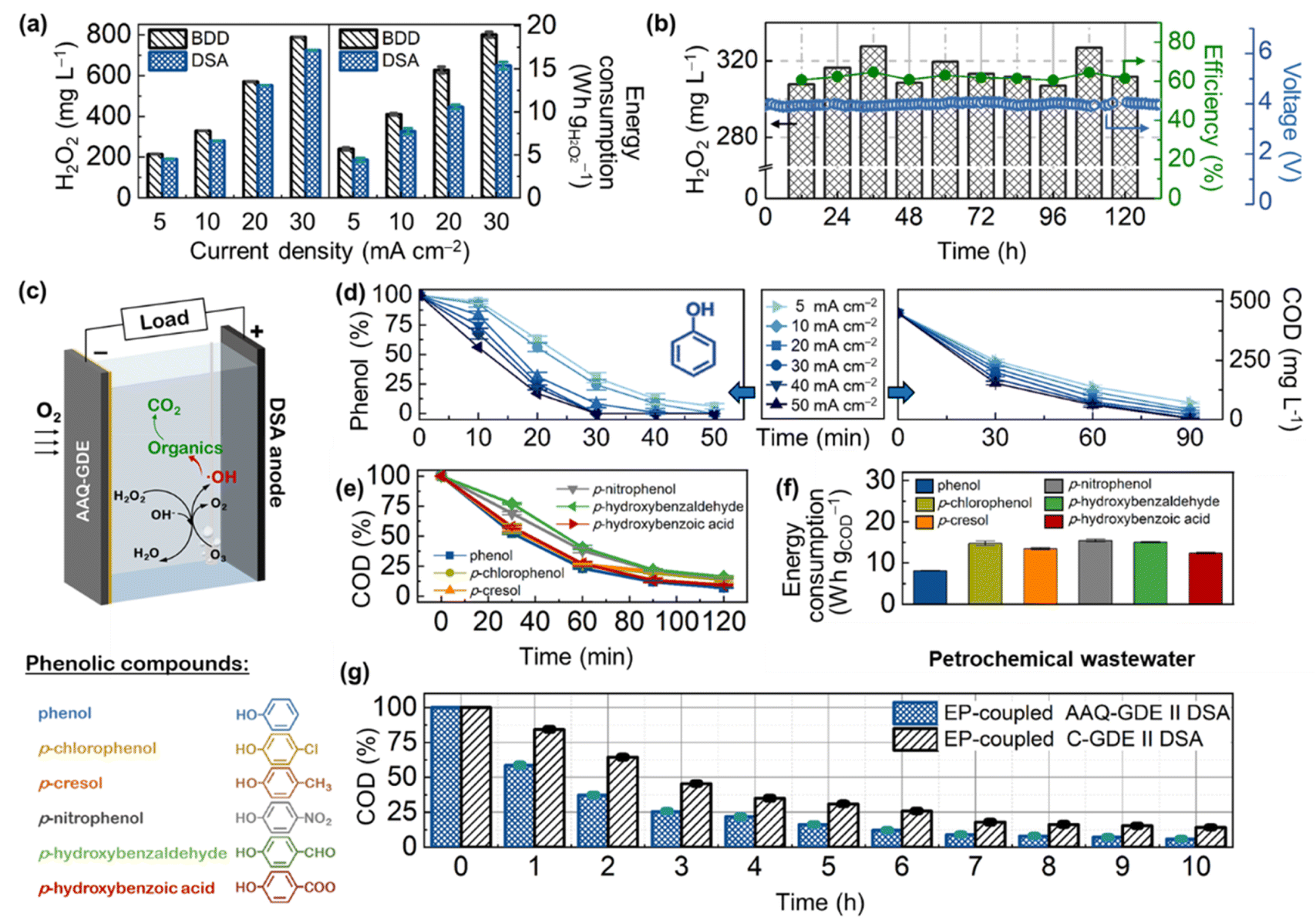 | ||
| Fig. 3 (a) H2O2 production (for 60 min) and the corresponding energy consumption of AAQ-GDE as a cathode with BDD or DSA as anodes in a 200 mL static electrolyzer. (b) H2O2 production, faradaic efficiency, and cell voltage of the long-term operation at 20 mA cm−2 on the flow cell with DSA as the anode in flowing 0.1 M Na2SO4 solution. (c) Illustration of EP-coupled AAQ-GDE//DSA for phenolic compounds degradation. (d) Phenol and COD degradation at different current densities (pH = 7). (e) COD degradation and (f) energy consumption (for 120 min) on different phenolic compounds (pH = 13). (g) COD degradation of petrochemical wastewater using the EP-coupled AAQ-GDE//DSA device at the operation of in comparison of EP-coupled C-GDE//DSA. Other testing conditions: current density = 10 mA cm−2, electrode area = 4 × 4 cm2; [Na2SO4] = 0.1 M; [phenolic compounds]0 = 150 mg L−1; [O3] = 30 g m−3. | ||
We employed the EP-coupled AAQ-GDE//DSA electrolyzer to degrade phenol and a series of phenolic compounds (Fig. 3c). The efficiency of the operation current on the phenol degradation was first conducted for phenol degradation. As illustrated in the left panel in Fig. 3d, the phenol removal was increased from 69.7% to 100% after 30 min, when the current densities were increased from 5 to 50 mA cm−2. Analogously, the chemical oxygen demand (COD) removal was increased from 44.8% to 65.9% after 30 min (from 84.3% to 99.9% after 90 min) with the increased current densities (the right panel in Fig. 3d). Combined with the electrochemical testing for H2O2 production, the increased current densities could produce more H2O2, which could satisfy the H2O2 demand for the EP process to degrade the phenolic organics. On the other hand, the pH effect on phenol removal was also explored (Fig. S15†). The enhanced phenol removal (from 55.7% to 100% in 20 min) was followed by the increased pH value. The increased pH value also enhanced the COD removal (pH = 5.0, 85.2%; pH = 7.0, 89.5%; pH = 9.0, 92.4%; pH = 11.0, 95.5%; and pH = 13.0, 97.8%) at the constant current density of 20 mA cm−2 after 90 min. The significantly enhanced degradation rate in the highly alkaline electrolyte was owing to the large amount of production of HO2− after the deprotonation of H2O2. The deprotonated HO2− was more efficient in cooperating with O3 to produce the strong oxidizing ˙OH.47
Therefore, the degradation of various phenolic compounds, including (phenol, p-chlorophenol, p-cresol, p-nitrophenol, p-hydroxybenzaldehyde, and p-hydroxybenzoic acid), was operated in the electrolyte of pH = 13.0 at 10 mA cm−2 (Fig. 3e). After 120 min of the degradation reaction, the COD removal of these phenolic compounds was about 90%. Meanwhile, energy consumption on COD degradation in our in situ devices. As shown in Fig. 3f, the cost per energy per mass of these phenolic compounds was below 15 W h gCOD−1. The excellent degradation performance was attributed to the high efficiency of the AAQ-GDE for H2O2 production. To better elucidate the advantages of AAQ-GDE on phenolic degradation, we also conducted the EP-coupled AAQ-GDE//DSA devices to treat high-concentration petrochemical wastewater. Various organic pollutants, including phenol, m-cresol, o-cresol, 2,5-methylphenol, and furan 2,3-dihydro-4-(1-methylethyl), were analyzed in the high-concentration petrochemical wastewater by total ion chromatography (Fig. S16†) and mass spectrometry (Fig. S17†). As the reaction time prolonged, the contents of these phenolic pollutants gradually decreased after 2 h and transformed into some organic acids (valeric acid, heptanoic acid). It was attributed to the oxidation attack of hydroxyl radicals. Meanwhile, the COD testing was conducted. Compared with the EP-coupled C-GDE//DSA, the AAQ-GDE-derived devices showed a rapid degradation rate, and the COD removal could reach beyond 90% after 7 hours (Fig. 3g).
3. Conclusion
In this work, we successfully developed a series of anthraquinone molecules supported on carbon blacks as metal-free electrocatalysts to accelerate H2O2 production for the AOP-coupled electrochemical devices. Combined with the electrochemical measurements and DFT calculations, the enhancement of H2O2 electrosynthesis was owing to the electrochemical hydrogenation and oxygen-participating dehydrogenation process on anthraquinone molecules. For the applicability in industrial engineering, we also assembled the anthraquinone-derived GDEs through the hot-pressing process. The resulting anthraquinone-derived GDEs not only exhibited good physical properties, including mechanical strength, hydrophobicities, electrical conductivities, and porosities but also the structure of anthraquinone was maintained. Owing to the strong electron donation of α-position amino groups, AAQ-GDE exhibited a higher performance on H2O2 electrosynthesis compared to other anthraquinone-derived GDEs, where the H2O2 productivity was as high as 0.25 mg cm−2 min−1 in various pH electrolytes; the large cumulative H2O2 amount was 1077 mg L−1 in 150 min, and the long-term durability for 120 h with the high and stable current efficiency was more than 60%. Moreover, the EP-coupled in situ AAQ-GDE//DSA was further employed for degrading phenol and phenolic compounds, where the COD removal could be above 90% in 120 min with an ultra-low energy consumption. The in situ treatment of petrochemical wastewater also confirmed the superior treatment performance of AAQ-GDE to the comparative C-GDE sample. The low-cost materials and devices for the high-performance and energy-saving devices in this work provide a new route to develop more efficient electrochemical devices for wide applications in environmental protection and other fields.4. Experimental section
4.1 Preparation of carbon black-supported aminoanthraquinone (AAQ/C)
All chemicals were purchased from commercial sources and used without further purification in all experiments. Detailed information on all chemicals is provided in the ESI.† For the synthesis of AAQ/C, 0.80 g graphitized carbon black and 0.22 g AAQ were mixed in 200 mL of isopropyl alcohol with ultrasonication for 1 h and stirring for 3 h. Then, the solid product was separated by rotary evaporation in a vacuum at 50 °C, and further dried at 70 °C for 12 h. The obtained product was designated as AAQ/C. Carbon-supported anthraquinone (AQ/C), methyl anthraquinone (MAQ/C), and ethyl anthraquinone (EAQ/C) were prepared in the same process using 0.20 g AQ, MAQ, and EAQ instead of AAQ.4.2 Preparation of AAQ-derived GDE (AAQ-GDE)
The composite substrate with a gas diffusion layer was constructed using nickel foam collectors, carbon black, and polytetrafluoroethylene (see details in the ESI†). The catalyst ink, including 80 mL of isopropyl alcohol, 0.75 g PVDF, and 0.75 g the catalyst (i.e., AAQ/C, AQ/C, MAQ/C, EAQ/C, and pure carbons), was prepared by ultrasonic cell crushing for 20 min and high-speed stirring for 30 min. Then, the catalyst ink was evenly sprayed on the composite substrate with a solid content (including PVDF and the catalyst) of 10 mg cm−2. After completely drying at 50 °C, the precursors were hot pressed at 190 °C and 10 MPa for 1 min, and AAQ-GDE, AQ-GDE, MAQ-GDE, EAQ-GDE, and C-GDE were generated.Author contributions
Pengdong Liu: methodology, formal analysis, investigation, data curation, writing – original draft. Haixing Zhang: methodology, formal analysis, investigation, data curation, writing – review & editing. Yu Chen: data curation, writing – review and editing. Yajing Di: resources and DFT calculation. Zhilin Li: conceptualization, review. Baoning Zhu: conceptualization, review. Zheng Liu: conceptualization, review. Zhengping Zhang: conceptualization, supervision, writing – review and editing. Feng Wang: conceptualization, review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFE0110400); the National Natural Science Foundation of China (52122207, 52173245, U20A20337, 52130206, and 52221006); and the Fundamental Research Funds for the Central Universities (CLYY2022).References
- B. C. Hodges, E. L. Cates and J. H. Kim, Nat. Nanotechnol., 2018, 13(8), 642–650 CrossRef CAS PubMed.
- V. I. Parvulescu, F. Epron, H. Garcia and P. Granger, Chem. Rev., 2022, 122(3), 2981–3121 CrossRef CAS PubMed.
- Y. Shang, X. Xu, B. Gao, S. Wang and X. Duan, Chem. Soc. Rev., 2021, 50(8), 5281–5322 RSC.
- Y. Zeng, E. Almatrafi, W. Xia, B. Song, W. Xiong, M. Cheng, Z. Wang, Y. Liang, G. Zeng and C. Zhou, Coord. Chem. Rev., 2023, 475, 214874 CrossRef CAS.
- K. Wang, C. Han, Z. Shao, J. Qiu, S. Wang and S. Liu, Adv. Funct. Mater., 2021, 31(30), 2102089 CrossRef CAS.
- K. Zuo, S. Garcia-Segura, G. A. Cerrón-Calle, F.-Y. Chen, X. Tian, X. Wang, X. Huang, H. Wang, P. J. J. Alvarez and J. Lou, et al. , Nat. Rev. Mater., 2023, 8(7), 472–490 CrossRef.
- Q. Zhang, M. Zhou, G. Ren, Y. Li, Y. Li and X. Du, Nat. Commun., 2020, 11(1), 1731 CrossRef CAS PubMed.
- C. Wang, J. Kim, V. Malgras, J. Na, J. Lin, J. You, M. Zhang, J. Li and Y. Yamauchi, Small, 2019, 15, 1900744 CrossRef PubMed.
- X. Shi, S. Back, T. M. Gill, S. Siahrostami and X. Zheng, Chem, 2021, 7(1), 38–63 CAS.
- K.-H. Wu, D. Wang, X. Lu, X. Zhang, Z. Xie, Y. Liu, B.-J. Su, J.-M. Chen, D.-S. Su and W. Qi, et al. , Chem, 2020, 6(6), 1443–1458 CAS.
- A. T. Murray, S. Voskian, M. Schreier, T. A. Hatton and Y. Surendranath, Joule, 2019, 3(12), 2942–2954 CrossRef CAS.
- Y. Bu, Y. Wang, G. F. Han, Y. Zhao, X. Ge, F. Li, Z. Zhang, Q. Zhong and J. B. Baek, Adv. Mater., 2021, 33(49), e2103266 CrossRef PubMed.
- J. Xu, Q. Zhang, X. Gao, P. Wang, H. Che, C. Tang and Y. Ao, Angew Chem. Int. Ed. Engl., 2023, 62(32), e202307018 CrossRef CAS PubMed.
- Z. Teng, Q. Zhang, H. Yang, K. Kato, W. Yang, Y.-R. Lu, S. Liu, C. Wang, A. Yamakata and C. Su, et al. , Nat. Catal., 2021, 4(5), 374–384 CrossRef CAS.
- S. Li, J. Huang, Y. Wang and G. Yu, Appl. Catal., B, 2022, 304, 120930 CrossRef CAS.
- S. Qu, H. Wu and Y. H. Ng, Adv. Energy Mater., 2023, 2301047 CrossRef CAS.
- P. Mazierski, M. Nischk, M. Gołkowska, W. Lisowski, M. Gazda, M. J. Winiarski, T. Klimczuk and A. Zaleska-Medynska, Appl. Catal., B, 2016, 196, 77–88 CrossRef CAS.
- N. Zheng, X. He, Q. Zhou, R. Wang, X. Zhang, R. Hu and Z. Hu, Appl. Catal., B, 2022, 319, 121918 CrossRef CAS.
- W. Peng, J. Liu, X. Liu, L. Wang, L. Yin, H. Tan, F. Hou and J. Liang, Nat. Commun., 2023, 14(1), 4430 CrossRef CAS.
- Y. Tian, D. Deng, L. Xu, M. Li, H. Chen, Z. Wu and S. Zhang, Nano-Micro Lett., 2023, 15(1), 122 CrossRef CAS.
- Q. Zhao, Y. Wang, W.-H. Lai, F. Xiao, Y. Lyu, C. Liao and M. Shao, Energy Environ. Sci., 2021, 14(10), 5444–5456 RSC.
- S. Mavrikis, M. Göltz, S. C. Perry, F. Bogdan, P. K. Leung, S. Rosiwal, L. Wang and C. Ponce de León, ACS Energy Lett., 2021, 6(7), 2369–2377 CrossRef CAS.
- P. Cao, X. Quan, K. Zhao, X. Zhao, S. Chen and H. Yu, ACS Catal., 2021, 11(22), 13797–13808 CrossRef CAS.
- X. Zhang, X. Zhao, P. Zhu, Z. Adler, Z. Y. Wu, Y. Liu and H. Wang, Nat. Commun., 2022, 13(1), 2880 CrossRef CAS PubMed.
- J. Chen, Q. Ma, Z. Yu, M. Li and S. Dong, Angew. Chem., Int. Ed., 2022, 61(48), 10375–10383 Search PubMed.
- F. Xiao, Z. Wang, J. Fan, T. Majima, H. Zhao and G. Zhao, Angew. Chem., Int. Ed., 2021, 60(18), 10375–10383 CrossRef CAS PubMed.
- J. Du, G. Han, W. Zhang, L. Li, Y. Yan, Y. Shi, X. Zhang, L. Geng, Z. Wang and Y. Xiong, et al. , Nat. Commun., 2023, 14(1), 4766 CrossRef CAS PubMed.
- H. Sheng, A. N. Janes, R. D. Ross, D. Kaiman, J. Huang, B. Song, J. R. Schmidt and S. Jin, Energy Environ. Sci., 2020, 13(11), 4189–4203 RSC.
- L. Li, Z. Hu, Y. Kang, S. Cao, L. Xu, L. Yu, L. Zhang and J. C. Yu, Nat. Commun., 2023, 14(1), 1890 CrossRef CAS PubMed.
- P. Cao, X. Quan, X. Nie, K. Zhao, Y. Liu, S. Chen, H. Yu and J. G. Chen, Nat. Commun., 2023, 14(1), 172 CrossRef CAS PubMed.
- B.-H. Lee, H. Shin, A. S. Rasouli, H. Choubisa, P. Ou, R. Dorakhan, I. Grigioni, G. Lee, E. Shirzadi and R. K. Miao, et al. , Nat. Catal., 2023, 6(3), 234–243 CrossRef CAS.
- X. Shen, Z. Wang, H. Guo, Z. Lei, Z. Liu and L. Wang, Small, 2023, 2303156 CrossRef CAS PubMed.
- H. Yang, N. Lu, J. Zhang, R. Wang, S. Tian, M. Wang, Z. Wang, K. Tao, F. Ma and S. Peng, Carbon Energy, 2023, e337 CrossRef CAS.
- X. Duan, H. Sun and S. Wang, Acc. Chem. Res., 2018, 51(3), 678–687 CrossRef CAS PubMed.
- S. Huang, S. Lu, Y. Hu, Y. Cao, Y. Li, F. Duan, H. Zhu, Y. Jin, M. Du and W. Zhang, Small Struct., 2023, 2200387 CrossRef CAS.
- F. Liu, P. Zhou, Y. Hou, H. Tan, Y. Liang, J. Liang, Q. Zhang, S. Guo, M. Tong and J. Ni, Nat. Commun., 2023, 14(1), 4344 CrossRef CAS PubMed.
- D. Zhang, E. Mitchell, X. Lu, D. Chu, L. Shang, T. Zhang, R. Amal and Z. Han, Mater. Today, 2023, 63, 339–359 CrossRef CAS.
- Q. Lei, H. Yuan, J. Du, M. Ming, S. Yang, Y. Chen, J. Lei and Z. Han, Nat. Commun., 2023, 14(1), 1087 CrossRef CAS PubMed.
- P. Chen, W. Tao, M. Hu, Y. Xu, Z. Zhang, H. Yu, X. Fu and C. Zhang, Carbon, 2021, 171, 104–110 CrossRef CAS.
- H. Wang, M. Chen, Y. Zhu, Y. Li, H. Zhang and T. Shi, Org. Chem. Front., 2022, 9(5), 1254–1260 RSC.
- W. Zhang, H. Liu, H. Kang, S. Zhang, B. Yang and Z. Li, Electrochim. Acta, 2023, 448, 142194 CrossRef CAS.
- J. Dong, H. Peng, J. Wang, C. Wang, D. Wang, N. Wang, W. Fan, X. Jiang, J. Yang and Y. Qian, Energy Storage Mater., 2023, 54, 875–884 CrossRef.
- J. G. M. de Carvalho, R. A. Fischer and A. Pothig, Inorg. Chem., 2021, 60(7), 4676–4682 CrossRef CAS PubMed.
- X. Li, S. Tang, S. Dou, H. J. Fan, T. S. Choksi and X. Wang, Adv. Mater., 2022, 34(25), e2104891 CrossRef PubMed.
- Y. Guo, R. Zhang, S. Zhang, H. Hong, Y. Zhao, Z. Huang, C. Han, H. Li and C. Zhi, Energy Environ. Sci., 2022, 15(10), 4167–4174 RSC.
- C. Ponce de León, Nat. Catal., 2020, 3(2), 96–97 CrossRef.
- J. Xiao, Y. Xie, J. Rabeah, A. Bruckner and H. Cao, Acc. Chem. Res., 2020, 53(5), 1024–1033 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00386h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |