
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIon effects on minimally hydrated polymers: hydrogen bond populations and dynamics†
Eman
Alasadi
and
Carlos R.
Baiz
 *
*
Department of Chemistry, University of Texas at Austin, 105 E 24th St. A5300, Austin, TX 78712, USA. E-mail: cbaiz@cm.utexas.edu
First published on 7th October 2024
Abstract
Compared to bulk water, the effect of ions in confined environments or heterogeneous aqueous solutions is less understood. In this study, we characterize the influence of ions on hydrogen bond populations and dynamics within minimally hydrated polyethylene glycol diacrylate (PEGDA) solutions using Fourier-transform infrared (FTIR) and two-dimensional infrared (2D IR) spectroscopies. We demonstrate that hydrogen bond populations and lifetimes are directly related to ion size and hydration levels within the polymer matrix. Specifically, larger monovalent cation sizes (Li+, Na+, K+) as well as anion sizes (F−, Cl−, Br−) increase hydrogen bond populations and accelerate hydrogen bond dynamics, with anions having more pronounced effects compared to cations. These effects can be attributed to the complex interplay between ion hydration shells and the polymer matrix, where larger ions with diffuse charge distributions are less efficiently solvated, leading to a more pronounced disruption of the local hydrogen bonding network. Additionally, increased overall water content results in a significant slowdown of dynamics. Increased water content enhances the hydrogen bonding network, yet simultaneously provides greater ionic mobility, resulting in a delicate balance between stabilization and dynamic restructuring of hydrogen bonds. These results contribute to the understanding of ion-specific effects in complex partially-hydrated polymer systems, highlighting the complex interplay between ion concentration, water structuring, and polymer hydration state. The study provides a framework for designing polymer membrane compositions with ion-specific properties.
Introduction
Ions significantly impact the thermodynamics of aqueous systems by directly or indirectly altering the hydrogen-bond networks.1–4 One such class of multicomponent systems includes polymers in aqueous environments,5–7 a topic of increasing importance due to their relevance in both natural and engineered contexts.8–10 For instance, characterizing the effect of ions can inform the design and functionality of polymer membranes, which have wide-ranging applications, from advanced filtration technologies to energy-efficient fuel cells and targeted drug delivery systems.9–14 This relevance additionally extends to the design of ion-conducting polymers for battery materials, where understanding the conductivity changes in materials under different moisture levels can inspire new investigations and applications.9,15 Such complexity underscores the need for a deeper exploration of specific polymer–ion interactions, particularly in heterogeneous systems, to further our molecular understanding of these multicomponent systems.Polyethylene glycol (PEG)16–22 is a frequently studied polymer due to its chemical and physical properties. Its high solubility in water, attributable to the partial negative charges on the ether oxygens, facilitates PEG's incorporation within the hydrogen bond network of water. This high solubility makes PEG suitable for crosslinking processes, forming various membrane architectures.18,23 Given the high solubility of PEG in water, many studies have focused on well-hydrated environments, typically maintaining at least ∼3 water molecules per ether oxide unit,12,24 on the other hand, minimal hydration provides opportunities to further understand the complex interplay of interactions between polymers, ions, and water that may resemble more real-world applications. Specifically, the ‘salting-in’ and ‘salting-out’ effects, or the specific ion effects influencing these phenomena, are of particular interest given that the traditional roles of solute and solvent are challenged. Investigating polymers in dehydrated conditions not only allows for the identification of specific molecular interactions but also elucidates specific ions’ effects on polymer structures.
The Hofmeister series provides a conceptual framework for interpreting ion effects in aqueous solutions.17,21,24,25 Specifically, the Hofmeister series, as applied to protein solutions, orders salts by their ability to stabilize or destabilize proteins. Similarly the lyotropic series,26–28 originally pertaining to hydrophilic colloids, ranks ions based on their impact on the solubility and aggregation behavior of amphiphilic molecules. Historically these series are interpreted based on the influence of ions on the aqueous environment—from chaotropes, which disrupt the water's hydrogen-bonding networks, to kosmotropes, which have opposite effects (Fig. 1). We have revised the sentence to more accurately convey the intended meaning: however, these series, while insightful, do not fully capture the complexities of ion behavior, especially in systems where the distinctions between solute and solvent, as well as hydrophilic and hydrophobic interactions, are blurred.21,27 For these reasons it is key to specifically characterize, at the molecular level, the direct and indirect effects of ions on heterogeneous multicomponent systems, such as the minimally-hydrated polymer systems presented here.
 | ||
| Fig. 1 Schematic representation of the Hofmeister classification of the ions studied here. This figure presents a categorization based on their effect on aqueous solution structure. The yellow horizontal line demarcates the transition from kosmotropes, associated with structure stabilization, to chaotropes, which are linked to structure destabilization. The Hofmeister series for anions and cations is demonstrated in red and blue boxes, respectively. | ||
This study focuses on the subtleties of hydrogen bonding, by characterizing populations and dynamics, within polyethylene glycol diacrylate (PEGDA) in minimally hydrated solutions with only 0.53 to 2.62 water molecules per ether oxide unit.29,30 By emphasizing PEGDA's role as a quasi-solvent in these sparsely hydrated environments, our approach aligns with the framework devised to interpret ions in aqueous as well as non-aqueous systems. The choice of polymer (PEGDA) presents a unique model as PEGDA is similar to PEG, but with terminal ester carbonyl groups, which serve as local vibrational probes, as well as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds. The highly hydrophilic nature of PEGDA makes it highly responsive to changes in hydration level.
C double bonds. The highly hydrophilic nature of PEGDA makes it highly responsive to changes in hydration level.
To measure hydrogen bond populations and dynamics, we use Fourier Transform Infrared (FTIR) spectroscopy.31,32 which reports on the average number of water–carbonyl hydrogen bonds, revealing how these interactions are modulated by different ions as well as water content. Two-dimensional infrared (2D IR)33 spectroscopy provides probes of the dynamics of the hydrogen bond lifetimes at the ester carbonyl termini—a local probe of the surrounding solvation environment. Combined, these spectroscopies will thus serve as a foundation for understanding the intricate chemical interplay dictating the behavior of PEGDA in the presence of specific ionic species in minimally hydrated solutions.
Methods
Polymer sample preparation
Poly(ethylene glycol) diacrylate (average molecular mass: 700 g mol−1, corresponding to ∼13 ether oxide units) was purchased from Sigma Aldrich. First, salt-free PEGDA/D2O solutions were prepared at different weight fractions (e.g., 0, 0.20, 0.40, 0.60, 0.80, and 1.0 g D2O/g PEGDA).15 Corresponding to these weight fractions, the number of water molecules per ether oxide unit were calculated as 0, 0.53, 1.07, 1.56, 2.08, and 2.62, respectively. Samples were prepared volumetrically at room temperature and vortexed to ensure complete mixing. Next, salt-doped polymer/D2O solutions were prepared at concentrations of 0.1 mol salt per L with LiCl, NaCl, KCl, NaF, and NaBr. For each salt, samples were prepared at a constant salt concentration (0.1 mol L−1), with varying weight fractions of PEGDA/D2O solutions (0, 0.20, 0.40, 0.60, 0.80, and 1.0 g D2O/g PEGDA). D2O is used to avoid interference from the strong H–O–H bending vibration near 1640 cm−1. H2O presents combination bands that can extend into the critical carbonyl stretching region (1700–1750 cm−1). These spectral features could overlap with and obscure the carbonyl stretching vibrations of the polymer, complicating our analysis. By contrast, D2O shifts the O–D stretching vibration to a lower frequency (∼2500 cm−1) and its bending vibration to around 1210 cm−1, thereby providing a clear spectral window for observing the carbonyl region without overlap.Fourier transform infrared (FTIR) spectroscopy
The samples were placed between two flat CaF2 windows without a spacer, giving an approximate path length of 6 μm, which is required to maintain a maximum absorbance of <1 in the CO stretching region. The requirement for thin sample path lengths is the primary reason why polymer solutions were used instead of polymer films. FTIR spectra were measured at 1 cm−1 resolution using a Bruker InvenioS FTIR spectrometer. The spectrometer chamber was purged with dry air. Each spectrum is an average of 32 scans. All spectra were collected at room temperature.
2D IR spectroscopy
Ultrafast 2D IR spectra were measured using a custom-built spectrometer described previously.34 In brief, 100-fs mid-IR pulses, centered at 5.8 μm, generated using an OPA/DFG, are split into excitation (pump), and detection (probe) pulses. The coherence time (t1) is scanned using a mid-IR pulse shaper (Quick Shape, Phase Tech Inc) and is numerically Fourier transformed to generate the excitation frequency (ω1). The t1 time was scanned to 6 ps in 150 fs steps, and spectra were measured in a 1450 cm−1 rotating frame. The probe pulse is measured using a grating spectrometer in a 128–128-pixel MCT array, to generate the detection frequency (ω3). Spectra were measured at selected waiting times from 150 fs to 6 ps. All spectra were measured using a perpendicular pump–probe polarization condition to minimize the scattered light at the detector. The 1690–1750 cm−1 region was analyzed via center-line slope (CLS) analysis.35,36 The CLS relaxation as a function of waiting time is fit to an exponential function, and error bounds are computed using a statistical bootstrapping method.Results
Hydrogen-bond populations
Since the populations of different species are dictated by a balance of polymer–water, polymer–ion, and ion–water interactions, it is important to first characterize the effects of ions on the hydration of the polymer. To this end, we use FTIR spectroscopy to directly measure the average number of water–carbonyl hydrogen bonds in PEGDA as a function of salt identity and water content. Specifically, we probe the interactions between water and the terminal carbonyl groups of PEGDA. This measurement is reported as the number of water molecules per ether oxide unit, which serves as a metric for quantifying water content in these systems.8,37,38 While these measurements offer valuable insights into the local solvation environment around the ester carbonyl group, they do not directly probe the interactions involving the ethylene oxide (EO) units within the polymer chain. Therefore, the discussion of PEG–water interactions involving EO units is thus inferred from existing literature and the established behavior of PEG in aqueous environments, rather than directly observed in this work.The ester carbonyl terminus provides a local probe for the solvation environment around the polymer. Carbonyl spectra report directly on the hydrogen bond populations, as a CO···H–O–H hydrogen bond shifts the carbonyl vibrational frequency given that the polymer lacks polar hydrogens, only polar interactions with water are captured in the spectra. Thus, the lineshapes report specifically on the fraction of “free” carbonyls (that is, carbonyls without hydrogen bonds) versus “1-HB”, where a carbonyl accepts a single hydrogen bond from water.39,40 Additionally, hydrogen bonds can induce further shifts, but these are not observed in the present experiments.37
Initially, we compare the measured FTIR line shapes at varying weight fractions of the alkali chloride salts (Fig. 2) at a concentration of 0.1 mol L−1. In general, higher water content results in increased “1-HB” populations as there is more water available to interact with the polymer. Specifically, between the solutions with water-to-ether oxide ratios of 0.53 to 2.62, we observe a 56–63% increase in hydrogen bond population for the cations Li+, Na+, and K+, respectively. In addition, we observe a 58%, 58%, and 62% increase in hydrogen bond population for anions F−, Cl−, and Br−, respectively (Tables S1–S6, ESI†). This means the water molecules can surround and interact with the PEGDA chains and salt ions, reducing direct interactions between the polymers and salts. This increased hydration can diminish the individual impact of different salts, leading to similar spectra across different salt types at high water content. We observe the same trend for sodium halides as well (Fig. 2).
 | ||
| Fig. 2 FTIR spectra of polymer–salt solutions with water-to-ether oxide ratios of 0.53, 1.56, and 2.62. Left Panel. FTIR spectra alkali chloride salts. Right Panel. FTIR spectra of sodium halide salts. | ||
FTIR spectra reveal that all cations and anions increase the number of hydrogen bonds to the carbonyls compared to salt-free samples. Hydrogen-bond populations, extracted from spectra are shown in Fig. 3 (Gaussian fits in Section S1, ESI†). Additionally, at a fixed water content, larger ions result in more CO hydrogen bonds, as seen by the trends for cations (KCl > NaCl > LiCl, Fig. 3a) and anions (NaBr > NaCl> NaF, Fig. 3b). Moreover, the spectra show an increase in the number of CO hydrogen bonds as water content increases. Taken together, at a fixed water content (Fig. 2 and 3), PEGDA containing smaller ions exhibits fewer water–carbonyl hydrogen bonds than membranes containing less hydrated ions. As the water content of the solution increases at a fixed salt concentration (0.1 M), the average hydrogen bond populations all increase and eventually converge, indicating the influence of salt identity on hydrogen bond populations becomes diminished.
 | ||
| Fig. 3 Hydrogen bond populations for solutions with increasing water content. The figure compares the hydrogen bond populations for the cation (left panel) and anions (right panel) series at a constant 0.1 M salt concentration. | ||
This convergence can be attributed to the increasing dilution of the polymer, leading to a more uniform solvation structure. As the polymer becomes more dilute, its solvation structure changes less with additional water content, resulting in the observed convergence of hydrogen bond populations. This suggests that at higher water contents, the influence of salt identity on hydrogen bond populations diminishes. Further, we can consider the role of PEG's higher hydrogen bond acceptor ability compared to water, based on the Kamlet–Taft parameters for short-chain PEGs.41 This suggests that water molecules are more likely to form hydrogen bonds with PEG rather than with each other. This propensity likely explains why the introduction of salts, which disrupt water–water hydrogen bonding, enhances the availability of water to interact more with PEG rather than remaining self-associated. This effect is supported by the fact that ions in solution are predominantly solvated by water, as evidenced by the significantly negative ΔH of solvation for water compared to ethylene glycol. This differential solvation highlights the ionic influence in promoting water–PEG interactions over water–water interactions, aligning with the observed trends in hydrogen bonding in our experiments. These considerations help rationalize why, at higher water contents, we see an increase in CO hydrogen bonds and a convergence in hydrogen bond populations across different salts, indicating a diminishing influence of salt identity under these conditions.
Hydrogen-bond dynamics
Two-dimensional infrared (2D IR) spectra measure dynamic aspects of polymer–water–ion interactions.33,42,43 Specifically, the center line slope (CLS) method extracts a frequency–fluctuation correlation function (FFCF) from measurements, which is subsequently fit to a monoexponential decay. Example 2D IR spectra are shown in Fig. 4, and all the measured spectra are shown in Section S3 (ESI†). The FFCF can then be related to the local dynamics of water molecules, as discussed previously in the context of surfactants, polymers, and lipids.38,44–47 Shorter decay constants indicate faster dynamics. We analyze both the zero and one hydrogen bond peaks for samples of PEGDA within the cation (Li+, Na+, K+) and anion (F−, Cl−, Br−) series at water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ether oxide ratios of 1.29 and 2.62 (50% and 100% solutions respectively). For these solutions, we focus on the one hydrogen bond feature since the zero hydrogen bond feature shows no appreciable dynamics in the timescale of the measurements. The remainder of the results, alongside the fitting range and parameters are included in Section S3 (ESI†).
:ether oxide ratios of 1.29 and 2.62 (50% and 100% solutions respectively). For these solutions, we focus on the one hydrogen bond feature since the zero hydrogen bond feature shows no appreciable dynamics in the timescale of the measurements. The remainder of the results, alongside the fitting range and parameters are included in Section S3 (ESI†).
 | ||
| Fig. 4 Snapshots of 2D IR spectra. 2D IR spectra at 1 ps for anion-series (left panel) and cation-series (right panel) polymer solution at water:EO ratio of 1.29 are shown. In 2D IR spectroscopy, the spectra are plotted with excitation frequency on the x-axis and detection frequency on the y-axis, capturing the vibrational dynamics and coupling within the sample. The red peaks represent the 0 → 1 transition, indicating the population of vibrational modes excited from the ground state to the first excited state, while the blue peaks correspond to the 1 → 2 transition, associated with excited state absorption. The red line illustrates the center line slope (CLS) fitting of the one hydrogen bond feature. All other 2D IR spectra are shown in Section S3 (ESI†). | ||
The one hydrogen bond feature observed in the 1700–1750 cm−1 range signifies a carbonyl group involved in a single hydrogen bond, either with another molecule of water or with functional groups on the polymer. This state is more typical in hydrated polymer systems. In this context, the dynamics are shaped by water–water and water–polymer/ion interactions. The strength, number, and nature of these interactions dictate the timescales of hydrogen–bond fluctuations and molecular reorientation, as reflected in the decay constants of the one hydrogen bond feature. Notably, the presence of ions, particularly larger ions or those with specific charge distributions, can disrupt the hydrogen bonding network, altering the mobility and arrangement of water molecules. Therefore, by analyzing these two hydrogen bond features, we obtain a molecular picture of the water dynamics. Our results for the one hydrogen bond feature will be discussed in this section.
We start our investigation with the anion-series solutions. For the 1.29 and 2.62 water:EO ratio 2D IR spectra, a notable faster hydrogen bond dynamics is observed with larger ions (Fig. 5). These observations imply that larger ions disrupt the local hydrogen bonding network, leading to bifurcated, or otherwise weaker hydrogen bonds with shorter lifetimes. The Hofmeister conceptual framework can further support these results.17,21 Fluoride (F−) is a kosmotropic ion, which tends to stabilize water structure and strengthen hydrogen bonds. Chloride (Cl−) is less kosmotropic than fluoride, and bromide (Br−) is considered chaotropic. Therefore from F− to Cl− to Br−, the water structure becomes increasingly disrupted, leading to a decrease in the strength and lifetime of hydrogen bonds.10,48–50 This results in faster hydrogen bond dynamics, as sensed by the carbonyl probes. It is important to note that the anions are primarily solvated by water, and not by the polymer backbone. Thus, ions accelerate hydrogen bond dynamics as they disturb the hydrogen bonding network, leading to shorter-lived hydrogen bonds to the carbonyls. The ions, primarily solvated by water, also affect local dynamics as water molecules reorient around the ions and carbonyl groups of PEGDA. In addition, these results align with molecular dynamics simulations which suggest that larger halide ions can alter the arrangement and hydration-shell dynamics more so than smaller halide ions.49
 | ||
| Fig. 5 Frequency fluctuation correlation function time constants for anion-series (left panel) and cation-series (right panel) at 1.29 and 2.62 water:EO ratios as indicated in the plot. For 1.29 solutions, the time constant decreases for larger anions (F− > Cl− > Br−). The same observation is made for the 2.62 solutions. For the same ion with 1.29 and 2.62 water:ether oxide ratio, the decay constant increases, demonstrating a slowdown in dynamics with increased water content. For cations, the correlation time decreases for larger cations (Li+ > Na+ > K+−). The same observation is made for 2.62 solutions. For the same ion, the decay constant increases, demonstrating a slowdown in dynamics with increased water content. | ||
Next, we examine the cation-series solutions. Similar to the anion results, larger ions lead to faster dynamics. Therefore, by increasing the radius from Li+ to Na+ to K+, the decay constant is shorter, implying that hydrogen bond dynamics become faster (Fig. 5). This observation can be interpreted within the solvation sphere surrounding the cations. This observation can be interpreted within the solvation sphere surrounding the cations, where the difference in structuring ability is attributed to their hydration characteristics. Li+, with its smaller size and higher charge density, forms a more structured and tightly bound hydration shell compared to the larger K+ ion. In contrast, the larger K+ ion results in a less structured water environment, leading to the observed faster dynamics of hydrogen bonding. The notable differences in ΔH° of hydration for Li+ (−5.8 kJ mol−1) compared to K+ (−3.2 kJ mol−1),3,51,52 and the corresponding Gibbs free energies of solvation (ΔG°) for Li+ (2.5 kJ mol−1) and K+ (6.3 kJ mol−1)3,51 underscore this behavior. The less negative ΔG° for K+ indicates weaker interaction with water, resulting in a more dynamic and less structured solvation shell. Consequently, Li+'s more structured hydration shell leads to stronger and slower hydrogen bond dynamics, whereas K+'s looser shell correlates with faster dynamics. Though modest in absolute terms, these differences in hydration enthalpy and Gibbs free energy play a crucial role in shaping the solvation and hydrogen bond dynamics observed in our study.21,53,54
Discussion
We analyzed the dynamic behavior of ions in minimally hydrated poly(ethylene glycol) diacrylate (PEGDA) systems. Our studies position PEGDA as a minimally hydrated medium where ion concentration is kept constant, ensuring sufficient ion solvation. This unique dual characteristic enables us to isolate specific effects of ion hydration, independent of extensive bulk water interactions, thus providing more direct insight into ion-polymer dynamics. The focus was on three specific cations (Li+, Na+, K+) and three anions (F−, Cl−, Br−), examining their behavior within the solvent matrix. The results demonstrate that the dynamics of these ions conform to established ionic series: anions adhere to the Hofmeister series, where larger anions exhibit faster dynamics, while cations follow the reverse lyotropic series—also interpretable under the Hofmeister framework—where larger cations similarly show increased dynamic behavior. Additionally, we observed that as hydration increases, there is a marked slowdown in its dynamics. This underscores the significant impact of hydration on the PEGDA matrix, illustrating the interplay between hydrogen bond structure and dynamics.Hydration effects
To contextualize the hydration effects within the broader scope of ion-specific effects in polymer systems, it is essential to discuss size-specific characteristics and how these relate to hydrogen bond dynamics in minimally hydrated systems. In cations, water coordinates through the oxygen atom, resulting in tightly bound hydration shells that significantly immobilize solvation-shell waters, promoting a more structured solvation shell for smaller ions. In contrast, larger cations exhibit weaker coordination allowing water molecules to remain “free” to interact with the polymer matrix or other water molecules, essentially exhibiting “bulk-like” behavior. Hydrogen bond populations showed that there was increased hydrogen bonding to the carbonyl group for larger cations and less for smaller cations. This is consistent with the interpretation that having a more structured solvation shell prevents water from forming favorable H-bond configurations with the polymer. Further, unfavorable overlaps in the hydration zones of ions and PEGDA, as well as direct polymer–ion interactions could lead to partial dehydration. This effect is more pronounced with smaller cations due to their denser hydration shells which limits their overlap with the carbonyl solvation shell. On the other hand, larger cations interact more weakly with water, leaving more mobile water molecules to induce hydrogen bonding with the PEGDA carbonyls.55 In anions, though the water–ion interactions are different, the argument is similar, larger anions contain more tightly bound shells, that constrain water molecules, leading to fewer hydrogen bonds with the polymer.Hydrogen bond dynamics
2D IR revealed that larger anions lead to faster dynamics. For cations, larger ions result in faster dynamics. This trend can also be explained using size-specific effects on the hydration shell. In PEGDA, smaller cations such as Li+ create a more structured environment around the polymer, while larger, less hydrated cations like K+ have a less structuring effect. The tightly bound water around Li+ results in constrained geometries that both lower hydration and increase hydrogen bond lifetime as water lacks both proper tetrahedral orientations and flexibility that is required for hydrogen bond switching. With anions, this trend can be similarly interpreted through the effect on local solvation shells. Stronger hydrogen solvation shells and lower hydration lead to longer-lived hydrogen bonds due to poor geometric alignments as the molecular configurations lack proper tetrahedral alignment for hydrogen bond switching.Recent studies on non-aqueous solvents27 offer a useful point of comparison, particularly in illustrating specific-ion effects like electrostriction and variations in standard molar volumes within non-aqueous solvents. Interestingly, monovalent ion behavior consistently aligns with the Hofmeister series across various solvents, highlighting the universal nature of ion-specific effects and the strong relationship between ion charge density and observed effects. In contrast, our system involves water solvation. In particular, enthalpy changes associated with hydrating ions3,52,56–60—compared to those from solvating ions with ether oxide—suggest that water interactions drive the observed dynamics (i.e. the ions remain mostly hydrated). This distinction underscores that while Hofmeister effects can be useful for bulk water and for non-aqueous solvents, the dynamics in our study are primarily driven by local interactions.
Ion effects on PEGDA
In mixed-solvent environments, ions tend to be preferentially solvated by one solvent species. Recent simulations61 on single salts in PEGDA show that the binding of ions by ether oxygens in equilibrated membranes and the distribution of water–water and water–ether oxygen contacts demonstrate significant interactions between ions and the polymer matrix. Therefore, while ions are preferentially solvated by water, as water content decreases, there is a shift towards more direct ion–polymer interactions. Consequently, these interactions can affect the dynamics within the system. Under highly dehydrated conditions, ions may exclude part of the solvation shell, leading to selective solvation by the polymer. The extent to which cations and anions are preferentially solvated differs given the electrostatics of the PEGDA backbone, meaning that the overall effect is influenced by the nature of both ions. This preferential solvation leads to deviations from their behavior in bulk solvents.In addition, we can compare the relative effects of cations versus anions. The standard hydration free energy and partial molar volumes reveal that anions can more effectively influence the solubility of non-electrolytes due to their relatively weaker solvation.59,60,62 Larger ions generally have less favorable solvation energies.56–58 Comparing our 2D IR measurements of hydrogen bond dynamics (Fig. 5), this effect becomes clear: anions like Br− have a greater impact on disrupting the water structure compared to cations like Na+. This observation is consistent with the known asymmetry in water's hydrogen bonding, where water can donate hydrogen bonds to anions but not to cations, leading to a more significant disruption of the hydrogen bond network by anions. These findings align with the concept of preferential solvation and the importance of understanding these interactions to accurately interpret ion behavior in mixed-solvent systems. From our work, we observe that both ion identity and their size and hydration play critical roles in interpreting ion behavior in mixed-solvent systems.
Guided by the above observations, we postulate a set of mechanisms by which anions and cations interact with the polymer matrix in minimally-hydrated polymer systems. Our direct measurements using FTIR spectroscopy focus on the interactions between water and the ester carbonyl groups of PEGDA, which provide insights into the local solvation environment and ion interactions with these terminal groups. However, it is important to note that the interactions involving the ether oxide units of the polymer chain, such as the proposed interactions shown in Fig. 6, are not directly observed in this study. These inferred interactions are based on established concepts and literature from related systems where such behaviors have been documented.22,48,63
 | ||
| Fig. 6 Schematic representation of anion and cation interactions. (A) Anion-induced polarization of water molecules within the shared solvation shells in the polymer. (B) Anions altering hydrophobic hydration by modifying hydrogen bond networks around the hydrophobic segments of the polymer, (C) direct binding of anions to the polymer's functional groups. (D) Cations maintaining hydration shells that stabilize the polymer structure and interact with ether oxygens, (E) direct interactions of relatively dehydrated cations with the polymer chains via binding to ether oxygens. (F) Cation–π interaction between cations and the negatively charged π-electrons the PEGDA termini. | ||
1. Polarization of water molecules: anions can alter the hydrogen bonding network by polarizing adjacent water molecules (Fig. 6A). This polarization primarily refers to electronic polarization, where the electron cloud around the water molecules is distorted by the electric field of the anion. This effect can lead to smaller anions producing longer-lived hydrogen bonds, as seen in our 2D IR data, which shows slower hydrogen bond dynamics for solutions with smaller anions.
2. Modification of hydrophobic hydration: anions influence the hydrophobic hydration of the polymer by altering the tetrahedral hydrogen bond networks around the hydrophobic segments (Fig. 6B).24,26,48,64 This is deduced from the general behavior of ions in hydrophobic environments and is supported by trends observed in our hydrogen bond population data.
3. Direct binding to the polymer: anions can interact with the conjugated ends of the polymer through van der Waals forces. These interactions could impact the polymer matrix, manifesting as salting-in or salting-out effects which could create heterogeneous domains enriched in polymer (Fig. 6C). This is suggested by the variations in hydrogen bond populations across different anions, as well as by the shifts in FTIR spectra.
Cations also demonstrate diverse interaction patterns in PEGDA solutions, influenced strongly by their hydration states:
4. Hydration shell dynamics: cations such as Li+ and Na+ maintain their tightly bound hydration shells. The water molecules within this hydration shell are constrained, resulting in slower dynamics for smaller cations (Fig. 6D). This is directly observed in the 2D IR spectra where smaller cations exhibit slower hydrogen bond dynamics compared to larger cations like K+.
5. Direct interactions with ether oxygens: under low hydration, direct interaction with ether oxide oxygens is likely (Fig. 6E). Smaller ions have increased preferential interactions with the negatively charged ether oxides in PEGDA. Direct interactions constrain the polymer resulting in slower dynamics and this aligns with our data showing slower dynamics for smaller cations in minimally hydrated environments.
6. Cation–π interactions: cations can directly interact with the negatively charged π-electrons the PEGDA termini (Fig. 6F). Cation–π interactions are well documented in literature, particularly involving larger aromatic systems, and are inferred here as a potential interaction based on the observed trends in the spectra.
Ion-specific interactions
Recent studies have illuminated the complexity of specific ion effects, revealing that ion behavior is influenced by a multitude of factors beyond just ion size and hydration. Gregory et al.65 demonstrated that radial charge density and site-specific electrostatic interactions are pivotal in understanding specific ion effects, particularly in how ions interact with surfaces and solvents. This nuanced understanding challenges the traditional view that ion size is the primary determinant of solvation dynamics. Mazzini and Craig27 extended this perspective by exploring the role of electrostriction and the variations in standard molar volumes, emphasizing that ion-specific effects are deeply rooted in the balance between electrostatic forces and the intrinsic properties of ions. Additionally, Parsons et al.66 highlighted the significance of nonelectrostatic potentials, such as ionic polarizability and van der Waals forces, in influencing Hofmeister effects, particularly in colloidal systems. These factors, combined with surface hydration dynamics, suggest that the manifestation of specific ion effects is a result of a complex interplay between multiple physicochemical properties. Our findings, while primarily focused on ion size and hydration, align with this broader context, acknowledging the multifaceted nature of ion behavior as it relates to the stability and dynamics of hydrogen bonds in confined environments.Ion-independent hydration effects
Another important observation in our systems is the dynamics of the same ion at different hydration levels. A pronounced slowdown in hydrogen bond dynamics was observed with an increase in water content for the same ion. Increased water–water hydrogen bonding typically exhibits slower dynamics.8,40,44 This observation is consistent with the understanding that sufficient water is necessary to ensure proper hydrogen bonding geometries of bound water or within hydration shells. As the water content increases, the hydration shells around the polymer become more structured, leading to slower dynamics as observed in Fig. 5.5,10,49,67 In brief, with an abundance of water, each oxygen atom is more likely to engage in a robust hydrogen bond network compared to dehydrated environments where water is crowded by the polymer, not able to engage in proper hydrogen bonding. This enhanced network at higher water contents forms geometrically favorable hydrogen bonds, which inherently exhibit slower dynamics. This effect can be attributed to the increased likelihood of optimal orbital overlap between the polymer lone pairs and the hydrogen atoms of water.68,69 The resulting matrix is one where water molecules are more tightly bound, which is reflected in the extended hydrogen-bond lifetimes, characteristic of more ordered systems (Fig. 7).22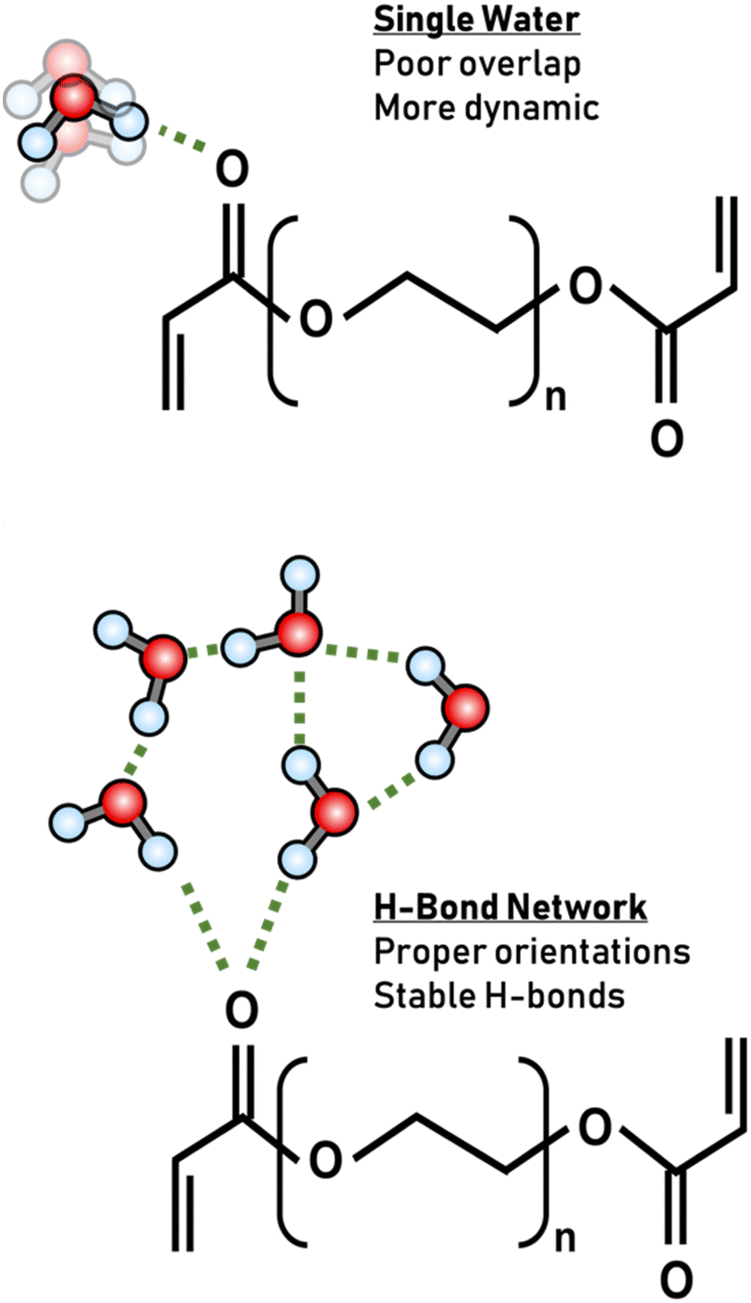 | ||
| Fig. 7 Schematic representation of hydrogen bonding interactions with ester carbonyl groups in different water environments. The top panel illustrates a scenario with lower hydration levels, where the ester carbonyl group forms fewer hydrogen bonds with water molecules, leading to more dynamic behavior as water molecules lack proper geometries due to crowding by the polymer. The bottom panel depicts a scenario with higher hydration levels, where the ester carbonyl group is involved in extensive and stable hydrogen bonding with multiple water molecules, resulting in slower dynamics. | ||
In addition to the enthalpic contributions discussed, the dynamics of hydrogen bonding in our system are also influenced by entropic factors, particularly under confined conditions. As water content increases, the hydration shell around the polymer becomes more structured, leading to a reduction in entropy (ΔS) due to decreased molecular freedom. This reduction in entropy, in conjunction with favorable enthalpic interactions, results in a lower free energy (ΔG = ΔH − TΔS), thereby stabilizing the hydrogen bond network. The interplay of these thermodynamic factors—where the enthalpic gains from optimized hydrogen bonds are counterbalanced by entropic penalties—provides a comprehensive explanation for the slower hydrogen bond dynamics observed at higher water content.
Anomalous ion behavior
In addition to the general trends observed with varying ion size and hydration, it is noteworthy that fluoride (F−) and lithium (Li+) ions, which typically exhibit anomalous behavior in many systems, conform more closely to expected trends within our study. This behavior is often attributed to the high charge density of F− and the small, hydrated ion size of Li+, leading to strong interactions with water molecules that deviate from those of other ions. However, in our study, the reduced solvation environment may be mitigating these effects, allowing these ions to behave in a manner more consistent with the other ions studied. This observation suggests that the solvation state plays a role in modulating the ion-specific effects, potentially masking the unique properties of these ions.Conclusion
This study on hydration dynamics within PEGDA solutions explored the influence of ions on polymer systems. FTIR and 2D IR spectroscopy revealed the impact of anions and cations on populations and dynamics across varying hydration levels. Larger ions, regardless of charge, accelerate hydrogen bond dynamics within PEGDA as evidenced by the shorter frequency fluctuation correlation decays observed between smaller, structure-stabilizing ions like Li+ and F−, and larger, structure-disrupting ions such as K+ and Br−. Notably, these observations persist irrespective of the hydration levels. Further, independent of ion identity increased hydration leads to slower dynamics compared to dehydrated conditions. These findings underscore the importance of ion identity in dictating the organization of water molecules. These studies further our understanding of ion behavior in heterogeneous multi-component systems where water molecules are highly confined, revealing the interplay between ion concentration, hydration level, and polymer interactions. The insights pave the way for future investigations aimed at exploiting these interactions in the design of new polymer-based materials and technologies.Author contributions
CRB conceptualized the research and designed the experiments. EA carried out the measurements and analyzed the data. Both authors contributed to making figures and writing the manuscript. Both authors approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the National Science Foundation (CHE-1847199) the Welch Foundation (F-1891), and by the Sloan Foundation.References
- V. Balos, N. K. Kaliannan, H. Elgabarty, M. Wolf, T. D. Kühne and M. Sajadi, Time-Resolved Terahertz–Raman Spectroscopy Reveals That Cations and Anions Distinctly Modify Intermolecular Interactions of Water, Nat. Chem., 2022, 14(9), 1031–1037, DOI:10.1038/s41557-022-00977-2.
- K. D. Collins, G. W. Neilson and J. E. Enderby, Ions in Water: Characterizing the Forces That Control Chemical Processes and Biological Structure, Biophys. Chem., 2007, 128(2), 95–104, DOI:10.1016/j.bpc.2007.03.009.
- Y. Marcus, Effect of ions on the structure of water, Pure Appl. Chem., 2010, 82(10), 1889–1899, DOI:10.1351/PAC-CON-09-07-02.
- Z. Yang, Hofmeister Effects: An Explanation for the Impact of Ionic Liquids on Biocatalysis, J. Biotechnol., 2009, 144(1), 12–22, DOI:10.1016/j.jbiotec.2009.04.011.
- S.-H. Chong and S. Ham, Dynamics of Hydration Water Plays a Key Role in Determining the Binding Thermodynamics of Protein Complexes, Sci. Rep., 2017, 7(1), 8744, DOI:10.1038/s41598-017-09466-w.
- A. Ghosh, J. S. Ostrander and M. T. Zanni, Watching Proteins Wiggle: Mapping Structures with Two-Dimensional Infrared Spectroscopy, Chem. Rev., 2017, 117(16), 10726–10759, DOI:10.1021/acs.chemrev.6b00582.
- P. K. Nandi, N. J. English, Z. Futera and A. Benedetto, Hydrogen-Bond Dynamics at the Bio–Water Interface in Hydrated Proteins: A Molecular-Dynamics Study, Phys. Chem. Chem. Phys., 2016, 19(1), 318–329, 10.1039/C6CP05601F.
- P. Garrett and C. R. Baiz, Dynamic Effect of Polymers at the Surfactant–Water Interface: An Ultrafast Study, Soft Matter, 2022, 18(9), 1793–1800, 10.1039/D1SM01651B.
- X. Ge, F. Zhang, L. Wu, Z. Yang and T. Xu, Current Challenges and Perspectives of Polymer Electrolyte Membranes, Macromolecules, 2022, 55(10), 3773–3787, DOI:10.1021/acs.macromol.1c02053.
- F. Foglia, Q. Berrod, A. J. Clancy, K. Smith, G. Gebel, V. G. Sakai, M. Appel, J.-M. Zanotti, M. Tyagi, N. Mahmoudi, T. S. Miller, J. R. Varcoe, A. P. Periasamy, D. J. L. Brett, P. R. Shearing, S. Lyonnard and P. F. McMillan, Disentangling Water, Ion and Polymer Dynamics in an Anion Exchange Membrane, Nat. Mater., 2022, 21(5), 555–563, DOI:10.1038/s41563-022-01197-2.
- X. Xu, Y. Yang, T. Liu and B. Chu, Cost-Effective Polymer-Based Membranes for Drinking Water Purification, Giant, 2022, 10, 100099, DOI:10.1016/j.giant.2022.100099.
- B. A. Rogers, H. I. Okur, C. Yan, T. Yang, J. Heyda and P. S. Cremer, Weakly Hydrated Anions Bind to Polymers but Not Monomers in Aqueous Solutions, Nat. Chem., 2022, 14(1), 40–45, DOI:10.1038/s41557-021-00805-z.
- W. B. Liechty, D. R. Kryscio, B. V. Slaughter and N. A. Peppas, Polymers for Drug Delivery Systems, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 149–173, DOI:10.1146/annurev-chembioeng-073009-100847.
- G. Geise, H. B. Park, A. Sagle, B. Freeman and J. Mcgrath, Water Permeability and Water/Salt Selectivity Tradeoff in Polymers for Desalination, Fuel Energy Abstr., 2011, 369, 130–138, DOI:10.1016/j.memsci.2010.11.054.
- E.-S. Jang, J. Kamcev, K. Kobayashi, N. Yan, R. Sujanani, T. J. Dilenschneider, H. B. Park, D. R. Paul and B. D. Freeman, Influence of Water Content on Alkali Metal Chloride Transport in Cross-Linked Poly(Ethylene Glycol) Diacrylate.2. Ion Diffusion, Polymer, 2020, 192, 122316, DOI:10.1016/j.polymer.2020.122316.
- C. Kojima, Y. Suzuki, Y. Ikemoto, M. Tanaka and A. Matsumoto, Comparative Study of PEG and PEGylated Dendrimers in Their Eutectic Mixtures with Water Analyzed Using X-Ray Diffraction and Infrared Spectroscopy, Polym. J., 2023, 55(1), 63–73, DOI:10.1038/s41428-022-00700-5.
- A. K. Borkowski and W. H. Thompson, Shining (Infrared) Light on the Hofmeister Series: Driving Forces for Changes in the Water Vibrational Spectra in Alkali–Halide Salt Solutions, J. Phys. Chem. B, 2022, 126(35), 6700–6712, DOI:10.1021/acs.jpcb.2c03957.
- S. Di Fonzo, B. Bellich, A. Gamini, N. Quadri and A. Cesàro, PEG Hydration and Conformation in Aqueous Solution: Hints to Macromolecular Crowding, Polymer, 2019, 175, 57–64, DOI:10.1016/j.polymer.2019.05.004.
- A. Patra, A. Bandyopadhyay, S. Roy and J. A. Mondal, Origin of Strong Hydrogen Bonding and Preferred Orientation of Water at Uncharged Polyethylene Glycol Polymer/Water Interface, J. Phys. Chem. Lett., 2023, 14(50), 11359–11366, DOI:10.1021/acs.jpclett.3c03098.
- A. A. Steuter, A. Mozafar and J. R. Goodin, Water Potential of Aqueous Polyethylene Glycol, Plant Physiol., 1981, 67(1), 64–67 CrossRef CAS PubMed.
- K. P. Gregory, G. R. Elliott, H. Robertson, A. Kumar, E. J. Wanless, G. B. Webber, V. S. J. Craig, G. G. Andersson and A. J. Page, Understanding Specific Ion Effects and the Hofmeister Series, Phys. Chem. Chem. Phys., 2022, 24(21), 12682–12718, 10.1039/D2CP00847E.
- S. Z. Moghaddam and E. Thormann, The Hofmeister Series: Specific Ion Effects in Aqueous Polymer Solutions, J. Colloid Interface Sci., 2019, 555, 615–635, DOI:10.1016/j.jcis.2019.07.067.
- S. Lee, X. Tong and F. Yang, Effects of the Poly(Ethylene Glycol) Hydrogel Crosslinking Mechanism on Protein Release, Biomater. Sci., 2016, 4(3), 405–411, 10.1039/c5bm00256g.
- H. I. Okur, J. Hladílková, K. B. Rembert, Y. Cho, J. Heyda, J. Dzubiella, P. S. Cremer and P. Jungwirth, Beyond the Hofmeister Series: Ion-Specific Effects on Proteins and Their Biological Functions, J. Phys. Chem. B, 2017, 121(9), 1997–2014, DOI:10.1021/acs.jpcb.6b10797.
- H. J. Bakker, Structural Dynamics of Aqueous Salt Solutions, Chem. Rev., 2008, 108(4), 1456–1473, DOI:10.1021/cr0206622.
- B. Kang, H. Tang, Z. Zhao and S. Song, Hofmeister Series: Insights of Ion Specificity from Amphiphilic Assembly and Interface Property, ACS Omega, 2020, 5(12), 6229–6239, DOI:10.1021/acsomega.0c00237.
- V. Mazzini and V. Craig, What Is the Fundamental Ion-Specific Series for Anions and Cations? Ion Specificity in Standard Partial Molar Volumes of Electrolytes and Electrostriction in Water and Non-Aqueous Solvents, Chem. Sci., 2017, 8, 10.1039/C7SC02691A.
- N. A. Avramenko, E. J. Hopkins, P. Hucl, M. G. Scanlon and M. T. Nickerson, Effect of Salts from the Lyotropic Series on the Handling Properties of Dough Prepared from Two Hard Red Spring Wheat Cultivars of Differing Quality, Food Chem., 2020, 320, 126615, DOI:10.1016/j.foodchem.2020.126615.
- M. Hakim Khalili, R. Zhang, S. Wilson, S. Goel, S. A. Impey and A. I. Aria, Additive Manufacturing and Physicomechanical Characteristics of PEGDA Hydrogels: Recent Advances and Perspective for Tissue Engineering, Polymers, 2023, 15(10), 2341, DOI:10.3390/polym15102341.
- T. Şener Raman, M. Kuehnert, O. Daikos, T. Scherzer, C. Krömmelbein, S. G. Mayr, B. Abel and A. Schulze, A Study on the Material Properties of Novel PEGDA/Gelatin Hybrid Hydrogels Polymerized by Electron Beam Irradiation, Front. Chem., 2023, 10, 1094981, DOI:10.3389/fchem.2022.1094981.
- S. Errico, M. Moggio, N. Diano, M. Portaccio and M. Lepore, Different Experimental Approaches for Fourier-Transform Infrared Spectroscopy Applications in Biology and Biotechnology: A Selected Choice of Representative Results, Biotechnol. Appl. Biochem., 2023, 70(3), 937–961, DOI:10.1002/bab.2411.
- K. Fahmy, Fourier Transform Infrared Spectroscopy for Biophysical Applications: Technical Aspects, in Encyclopedia of Biophysics, Roberts, G. C. K., ed., Springer, Berlin, Heidelberg, 2013, pp. 844–852 DOI:10.1007/978-3-642-16712-6_113.
- Y. S. Kim and R. M. Hochstrasser, Applications of 2D IR Spectroscopy to Peptides, Proteins, and Hydrogen-Bond Dynamics, J. Phys. Chem. B, 2009, 113(24), 8231–8251, DOI:10.1021/jp8113978.
- S. C. Edington, A. Gonzalez, T. R. Middendorf, D. B. Halling, R. W. Aldrich and C. R. Baiz, Coordination to Lanthanide Ions Distorts Binding Site Conformation in Calmodulin, Proc. Natl. Acad. Sci., 2018, 115(14), E3126–E3134, DOI:10.1073/pnas.1722042115.
- N. T. Hunt, Using 2D-IR Spectroscopy to Measure the Structure, Dynamics, and Intermolecular Interactions of Proteins in H2O, Acc. Chem. Res., 2024, 57(5), 685–692, DOI:10.1021/acs.accounts.3c00682.
- F. Šanda, V. Perlík, C. N. Lincoln and J. Hauer, Center Line Slope Analysis in Two-Dimensional Electronic Spectroscopy, J. Phys. Chem. A, 2015, 119(44), 10893–10909, DOI:10.1021/acs.jpca.5b08909.
- S. C. Edington, J. C. Flanagan and C. R. Baiz, An Empirical IR Frequency Map for Ester CO Stretching Vibrations, J. Phys. Chem. A, 2016, 120(22), 3888–3896, DOI:10.1021/acs.jpca.6b02887.
- P. Garrett, J. C. Shirley and C. R. Baiz, Forced Interactions: Ionic Polymers at Charged Surfactant Interfaces, J. Phys. Chem. B, 2023, 127(12), 2829–2836, DOI:10.1021/acs.jpcb.2c08636.
- G. Li, Y.-Y. Zhang, Q. Li, C. Wang, Y. Yu, B. Zhang, H.-S. Hu, W. Zhang, D. Dai, G. Wu, D. H. Zhang, J. Li, X. Yang and L. Jiang, Infrared Spectroscopic Study of Hydrogen Bonding Topologies in the Smallest Ice Cube, Nat. Commun., 2020, 11(1), 5449, DOI:10.1038/s41467-020-19226-6.
- X. You and C. R. Baiz, Importance of Hydrogen Bonding in Crowded Environments: A Physical Chemistry Perspective, J. Phys. Chem. A, 2022, 126(35), 5881–5889, DOI:10.1021/acs.jpca.2c03803.
- J. J. Black, A. Dolan, J. B. Harper and L. Aldous, Kamlet–Taft Solvent Parameters, NMR Spectroscopic Analysis and Thermoelectrochemistry of Lithium–Glyme Solvate Ionic Liquids and Their Dilute Solutions, Phys. Chem. Chem. Phys., 2018, 20(24), 16558–16567, 10.1039/C8CP02527D.
- I. Noda, Two-Dimensional Infrared (2D IR) Spectroscopy: Theory and Applications, Appl. Spectrosc., 1990, 44(4), 550–561, DOI:10.1366/0003702904087398.
- R. Fritzsch, S. Hume, L. Minnes, M. J. Baker, G. A. Burley and N. T. Hunt, Two-Dimensional Infrared Spectroscopy: An Emerging Analytical Tool, The Analyst, 2020, 145(6), 2014–2024, 10.1039/C9AN02035G.
- C. P. Baryiames, E. Ma and C. R. Baiz, Ions Slow Water Dynamics at Nonionic Surfactant Interfaces, J. Phys. Chem. B, 2020, 124(52), 11895–11900, DOI:10.1021/acs.jpcb.0c09086.
- C. P. Baryiames and C. R. Baiz, Slow Oil, Slow Water: Long-Range Dynamic Coupling across a Liquid–Liquid Interface, J. Am. Chem. Soc., 2020, 142(18), 8063–8067, DOI:10.1021/jacs.0c00817.
- M. L. Valentine, M. K. Waterland, A. Fathizadeh, R. Elber and C. R. Baiz, Interfacial Dynamics in Lipid Membranes: The Effects of Headgroup Structures, J. Phys. Chem. B, 2021, 125(5), 1343–1350, DOI:10.1021/acs.jpcb.0c08755.
- C. P. Baryiames, M. Teel and C. R. Baiz, Interfacial H-Bond Dynamics in Reverse Micelles: The Role of Surfactant Heterogeneity, Langmuir, 2019, 35(35), 11463–11470, DOI:10.1021/acs.langmuir.9b01693.
- Y. Zhang and P. S. Cremer, Interactions between Macromolecules and Ions: The Hofmeister Series, Curr. Opin. Chem. Biol., 2006, 10(6), 658–663, DOI:10.1016/j.cbpa.2006.09.020.
- S. Chowdhuri and A. Chandra, Dynamics of Halide Ion−Water Hydrogen Bonds in Aqueous Solutions: Dependence on Ion Size and Temperature, J. Phys. Chem. B, 2006, 110(19), 9674–9680, DOI:10.1021/jp057544d.
- D. J. Tobias and J. C. Hemminger, Getting Specific About Specific Ion Effects, Science, 2008, 319(5867), 1197–1198, DOI:10.1126/science.1152799.
- Y. Marcus, Ions in Solution and Their Solvation, 2015, p. 293 DOI:10.1002/9781118892336.
- Y. Marcus, Water Structure Enhancement in Water-Rich Binary Solvent Mixtures, J. Mol. Liq., 2011, 158(1), 23–26, DOI:10.1016/j.molliq.2010.10.002.
- M. L. Valentine, A. E. Cardenas, R. Elber and C. R. Baiz, Calcium-Lipid Interactions Observed with Isotope-Edited Infrared Spectroscopy, Biophys. J., 2020, 118(11), 2694–2702, DOI:10.1016/j.bpj.2020.04.013.
- B. C. Gibb, Hofmeister's Curse, Nat. Chem., 2019, 11(11), 963–965, DOI:10.1038/s41557-019-0355-1.
- S. Z. Moghaddam and E. Thormann, Hofmeister Effect of Salt Mixtures on Thermo-Responsive Poly(Propylene Oxide), Phys. Chem. Chem. Phys., 2015, 17(9), 6359–6366, 10.1039/C4CP05677A.
- H. Piekarski, Thermochemistry of Electrolyte Solutions, J. Therm. Anal. Calorim., 2012, 108(2), 537–545, DOI:10.1007/s10973-011-2019-2.
- H. Piekarski and K. Kubalczyk, Thermochemical Properties of Ions in Acetonitrile–Organic Cosolvent Mixtures at 298.15 K, J. Mol. Liq., 2005, 121(1), 35–40, DOI:10.1016/j.molliq.2004.08.028.
- H. Piekarski and A. Pietrzak, Enthalpic Interaction Coefficients of NaI–Alkanediol Pairs in Methanol and in Water, J. Therm. Anal. Calorim., 2012, 110(2), 917–922, DOI:10.1007/s10973-011-1603-9.
- G. T. Hefter, J.-P. E. Grolier, A. H. Roux and G. Roux-Desgranges, Apparent Molar Heat Capacities and Volumes of Electrolytes and Ions in Acetonitrile-Water Mixtures, J. Solution Chem., 1990, 19(3), 207–223, DOI:10.1007/BF00650455.
- G. T. Hefter, J.-P. E. Grolier and A. H. Roux, Apparent Molar Heat Capacities and Volumes of Electrolytes and Ions Int-Butanol-Water Mixtures, J. Solution Chem., 1989, 18(3), 229–248, DOI:10.1007/BF00652986.
- E. S. Zofchak, A. E. Quigley, J. G. Yoh, H. S. Sachar, K. K. Reimund, S. T. Milner, B. D. Freeman and V. Ganesan, Molecular and Electrostatic Origins of Mixed Salt Partitioning Phenomena in Uncharged Poly(Ethylene Oxide)-Based Membranes, J. Membr. Sci., 2024, 702, 122800, DOI:10.1016/j.memsci.2024.122800.
- P. Wang, J. J. Kosinski, A. Anderko, R. D. Springer, M. M. Lencka and J. Liu, Ethylene Glycol and Its Mixtures with Water and Electrolytes: Thermodynamic and Transport Properties, Ind. Eng. Chem. Res., 2013, 52(45), 15968–15987, DOI:10.1021/ie4019353.
- M. González-Jiménez, Z. Liao, E. L. Williams and K. Wynne, Lifting Hofmeister's Curse: Impact of Cations on Diffusion, Hydrogen Bonding, and Clustering of Water, J. Am. Chem. Soc., 2024, 146(1), 368–376, DOI:10.1021/jacs.3c09421.
- X. He and A. G. Ewing, Hofmeister Series: From Aqueous Solution of Biomolecules to Single Cells and Nanovesicles, ChemBioChem, 2023, 24(9), e202200694, DOI:10.1002/cbic.202200694.
- K. P. Gregory, E. J. Wanless, G. B. Webber, V. S. J. Craig and A. J. Page, The Electrostatic Origins of Specific Ion Effects: Quantifying the Hofmeister Series for Anions, Chem. Sci., 2021, 12(45), 15007–15015, 10.1039/D1SC03568A.
- D. F. Parsons, M. Boström, P. L. Nostro and B. W. Ninham, Hofmeister Effects: Interplay of Hydration, Nonelectrostatic Potentials, and Ion Size, Phys. Chem. Chem. Phys., 2011, 13(27), 12352–12367, 10.1039/C1CP20538B.
- J. T. Titantah and M. Karttunen, Water Dynamics: Relation between Hydrogen Bond Bifurcations, Molecular Jumps, Local Density & Hydrophobicity, Sci. Rep., 2013, 3(1), 2991, DOI:10.1038/srep02991.
- E. Brini, C. J. Fennell, M. Fernandez-Serra, B. Hribar-Lee, M. Lukšič and K. A. Dill, How Water's Properties Are Encoded in Its Molecular Structure and Energies, Chem. Rev., 2017, 117(19), 12385–12414, DOI:10.1021/acs.chemrev.7b00259.
- K. D. Collins, Charge Density-Dependent Strength of Hydration and Biological Structure, Biophys. J., 1997, 72(1), 65–76 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00830h |
| This journal is © The Royal Society of Chemistry 2024 |