
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fragment-based approach to study fungicide-biomimetic membrane interactions†
Shishir
Jaikishan
a,
Marine
Lavainne
 a,
Henri K.
Ravald
a,
Kieran
Scobbie
a,
Filip
Dusa
b,
Rekha
Maheswari
a,
Jenni
Turpeinen
a,
Ian
Eikemans
a,
Rui
Chen
a,
Julia
Rantala
a,
Vladimir
Aseyev
a,
Norbert N.
Maier
a and
Susanne K.
Wiedmer
*a
a,
Henri K.
Ravald
a,
Kieran
Scobbie
a,
Filip
Dusa
b,
Rekha
Maheswari
a,
Jenni
Turpeinen
a,
Ian
Eikemans
a,
Rui
Chen
a,
Julia
Rantala
a,
Vladimir
Aseyev
a,
Norbert N.
Maier
a and
Susanne K.
Wiedmer
*a
aDepartment of Chemistry, A.I. Virtasen aukio 1, POB 55, 00014 University of Helsinki, Finland. E-mail: susanne.wiedmer@helsinki.fi
bInstitute of Analytical Chemistry, Czech Academy of Sciences, Veveří 97, Brno 60200, Czech Republic
First published on 10th July 2024
Abstract
In this study, the molecular interactions of the allylamine-type fungicide butenafine and a set of substructures (“fragments”) with liposomes mimicking biological membranes were studied to gain a better understanding of the structural factors governing membrane affinity and perturbation. Specifically, drug/fragment-membrane interactions were investigated using an interdisciplinary approach involving micro differential scanning calorimetry, open-tubular capillary electrochromatography, nanoplasmonic sensing, and quartz crystal microbalance. By incubating the drug and the fragment compounds with liposomes with varying lipid composition or by externally adding the compounds to preformed liposomes, a detailed mechanistic picture on the underlying drug/fragment-membrane interactions was obtained. The nature and the degree of ionisation of polar head groups of the lipids had a major influence on the nature of drug-membrane interactions, and so had the presence and relative concentration of cholesterol within the membranes. The in-depth understanding of drug/fragment-membranes interactions established by the presented interdisciplinary fragment-based approach may be useful in guiding the design and early-stage evaluation of prospective antifungal drug candidates, and the discovery of agents with improved membrane penetrating characteristics in general.
1 Introduction
The impacts of fungal infections on human health are of increasing concern, as they can cause diverse diseases ranging from allergic syndromes to life-threatening invasive fungal diseases. These diseases together affect more than a billion people worldwide, and resistance of pathogenic fungi to all antifungals presently in clinical use has been documented.1 Therefore, the development of new, alternative antifungal strategies is urgently needed. This study will generate information on interactions between the antifungal agent butenafine and other structurally similar molecules with bio-imitating membranes. The information can possibly be used for developing specific antifungal agents with desired membrane penetrating characteristics.Several classes of antifungal agents including imidazoles, triazoles and allylamines are currently available. Butenafine is an antifungal agent with a chemical structure and a mode of action similar to those of other allylamine-type drugs, which act by inhibiting squalene epoxidase, a key enzyme in sterol biosynthesis in fungi. Unlike imidazoles and triazoles, allylamine-type antifungals do not interfere with human cytochrome P450 system, thus not causing any undesired toxicity.2
Butenafine is a derivative of benzylamine with potent and broad antimycotic activity. Butenafine have been shown to be extremely effective in the treatment of superficial mycotic infections like tinea pedis, tinea cruris and tinea corporis. However, mycotic infections, especially invasive types (by Cryptococcus neoformans, Pneumocystis or Jirovecii, Mucormycetes) or resistance to antifungal treatments (by, e.g., Candida auris), have become a serious healthcare problem as the immunocompromised population increases. The limited clinical arsenal of antifungal agents contributes to the increasing morbidity and mortality of invasive fungal infections. The main challenge in the development of effective antifungal drugs lies in the similarity of mammalian and fungal cells in terms of cellular structure and metabolic targets, which calls for agents of high levels of target selectivity for fungi to avoid adverse affecting to mammalian host cells.
The importance of membrane interaction in fungicidal activity is not well established. In a study carried on butenafine and tolnaftate with Candida albicans strains, a dual mechanism of the antifungal activity of butenafine was reported, involving both specific enzyme inhibitory and nonspecific direct membrane-damaging effects.3 Some bactericidal agents act by causing membrane disruption and/or permeabilization. For example, some antimicrobial peptides function via rapid physical disruption of the microbial cell membranes causing cell leakage and death. Additional complex mechanisms of action of antimicrobial agents includes generation of reactive oxygen species, interaction with specific lipids, or programmed cell death.4
Piscidins, a histidine-enriched antimicrobial peptide, execute their fungicidal activity by disrupting fungal membrane through pore formation.5 Similarly, an antifungal polyene, amphotericin B, has also shown fungicidal activity by forming pores through bilayer insertion and increasing permeability of the membrane.6 However, to the best of our knowledge, direct interaction of butenafine and the respective fragment molecules with lipid bilayers has not been investigated through a fragment-based approach.
An in-depth understanding of how the individual structure elements of highly effective antifungal agents contribute synergistically to favourable membrane interactions would be invaluable for guiding the development of new and highly specific agents. This knowledge may be established by adopting a “fragment-based” approach for the assessment of the membrane interactions by a systematic study of molecules representing substructures of clinically successful drugs. Dahal et al. reported a fragment library screening approach to identify selective inhibitors against a fungal enzyme.7 Fragment-based drug discovery has recently gained attraction for developing low molar weight antimicrobial compounds by ‘optimization and streamline identification’ of fragments with higher ligand efficiency.8–11 The structure-interaction relationships of incremental contributions from this effort can be used to optimise both target affinity and specificity of promising candidates through informed structure-based fine-tuning of drug-membrane interaction characteristics.
Key to the success of such a fragment-based drug development strategy is a toolbox of robust experimental methods for a comprehensive characterisation of the underlying interactions, such as drug-membrane distribution, localised drug binding to membrane constituents, drug transport across and retention within membranes, and drug-induced membrane perturbation etc.
To enable in-depth investigations of structure–activity relationships of membrane-antifungal agent interactions, butenafine and a set of substructural fragments were synthesised. In terms of molecular structure, these substructures were designed to preserve the crucial amino function of the drug, but to differ from the parent compound in molecular size, basicity and lipophilicity by systematic deletion of given constituting substituents. For the investigation of the interaction mechanisms of butenafine and corresponding fragments with biomimicking membranes, primarily their interactions with phospholipid liposomes. Were studied. Phospholipids constitute about 55–75% of fungal membrane lipids with phosphatidylcholine (PC) phosphatidylinositol (PI), and phosphatidylethanolamine (PE) as the main components, in addition to other lipids like phosphatidic acid (PA), phosphatidylserine (PS), cardiolipin, and phosphatidylglycerol (PG).12–14
PC and PE, being important fungal (also mammalian) lipid components, were selected for the current research. The acyl chains of phospholipids of fungal membranes are mostly unsaturated.15 Hence, POPC and DOPE as unsaturated phospholipids were considered. In some cases, cholesterol (Chol) was also included in the liposomes to validate the condensing effects of Chol on membrane and drug interactions.
The repertoire of instrumental techniques and methods employed in this work for studying drug-biomimetic membrane interactions is summarised in Table 1. These were micro differential scanning calorimetry (MicroCal DSC), nanoplasmonic (NPS) sensing, quartz crystal microbalance (QCM) and open-tubular capillary electrochromatography (OT-CEC). The obtained data shows the complimentary properties of the used methodologies.
| Method | Liposome composition | Internala or externalb addition of drug | B | F1 | F2 | F3 | C |
|---|---|---|---|---|---|---|---|
| a The compound was mixed with the lipids before liposome preparation. b The compound was allowed to interact with preformed liposomes. | |||||||
| MicroCal DSC | DPPC | Internal | x | x | x | x | x |
| DPPC:Chol | Internal | x | x | x | x | x | |
| DPPC:DOPE | Internal | x | x | x | x | x | |
| DPPC:DOPE:Chol | Internal | x | x | x | x | x | |
| NPS | POPC | External | — | — | x | x | x |
| POPC:DOPE | External | — | — | x | x | x | |
| POPC:DOPE:Chol | External | — | — | x | x | x | |
| POPC | Internal | x | x | — | — | — | |
| QCM | POPC | Internal | x | x | — | — | — |
| OT-CEC | POPC | External | — | — | x | x | x |
| POPC:DOPE | External | — | — | x | x | x | |
| POPC:DOPE:Chol | External | — | — | x | x | x | |
2. Experimental
2.1. Materials and methods
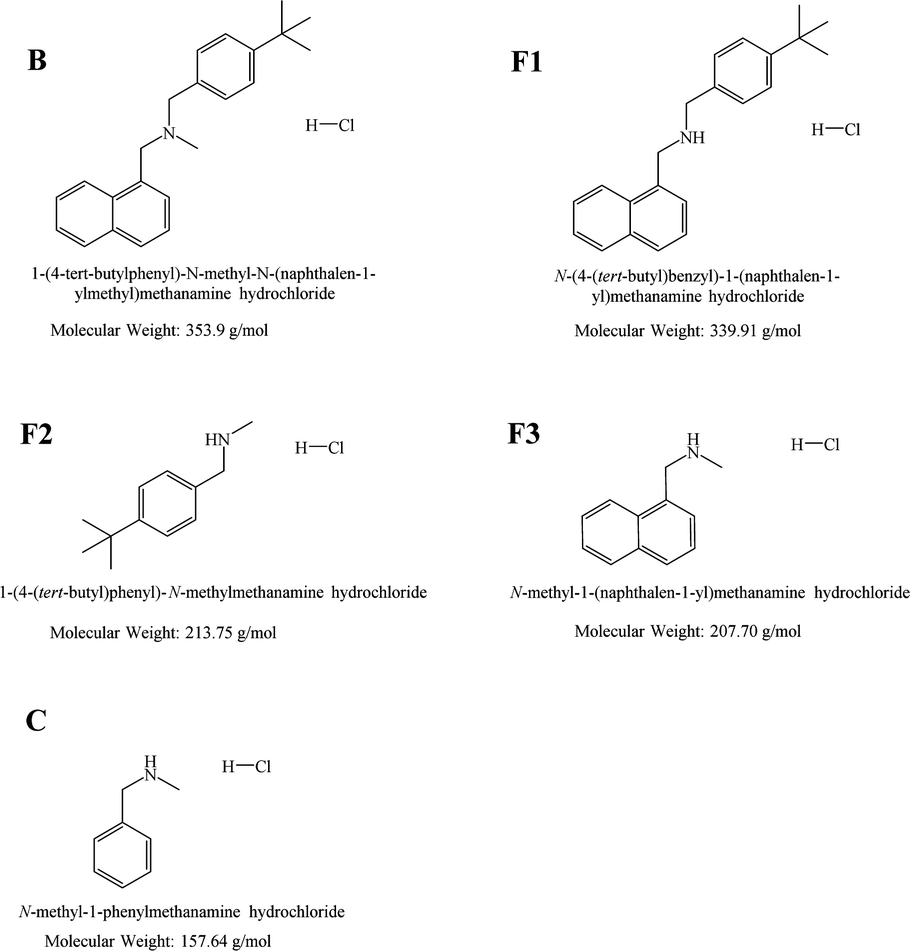 | ||
| Fig. 1 Chemical structures of butenafine (B), the corresponding substructures (fragments F1–F3), and the control molecule (C) including their abbreviations. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KD values by OT-CEC using different liposome compositions and experimentally determined apparent logDpH7.4 values. [parent drug, butenafine = B; fragment 1 = F1; fragment 2 = F2; fragment 3 = F3; control compound = C]
KD values by OT-CEC using different liposome compositions and experimentally determined apparent logDpH7.4 values. [parent drug, butenafine = B; fragment 1 = F1; fragment 2 = F2; fragment 3 = F3; control compound = C]
| Lipid/organic phase | B | F1 | F2 | F3 | C | |
|---|---|---|---|---|---|---|
| a Too low solubility in the aqueous phase – no visible peak in the electropherogram. b Too high solubility in the aqueous phase – did not interact with the immobilized liposome coating. | ||||||
| Experimental: OT-CEC | POPC (100 mol%) | 1.26 | 2.50 | |||
| POPC/DOPE (90:10 mol%) |
2.30 | 2.64 | ||||
| POPC/DOPE/Chol (80:10:10) |
2.22 | 2.67 | ||||
| Experimental calculated | logDpH7.4 octanol/PHB |
1.54 | 1.56 | 1.01 | 0.59 | −0.53 |
| logPo/w PubChem |
6.74 | 5.67 | 3.34 | 2.68 | 1.51 | |
| Calculated | logDpH7.4 Chemdraw |
4.72 | 3.98 | 1.05 | 0.50 | −0.76 |
All the solutions were filtered through a 0.45 μm membrane filter before CE analysis.
The phosphate buffer (c = 8.5 mM, I = 20 mM, pH 7.4) (PBS) was prepared by mixing appropriate amounts of Na2HPO4·12 H2O and NaH2PO4·H2O. The pH of the buffer solutions was checked with a WTW inoLab pH 7110 pH meter. The standard pH solutions (4.0, 7.0, and 10.0) used for pH meter calibration were purchased from Merck. An analytical balance Sartorius BP301S was used for weighing of the analytes. Purified water was procured from a water purification system Milli-Q Direct-Q 3 UV. 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (pH 7.4, I = 20 mM) was prepared by dissolving appropriate amount of HEPES in water and adjusting the pH with 1.0 M NaOH. The HEPES buffer containing 3 mM or 5 mM CaCl2 (calcium-HEPES) was prepared for OT-CEC experiments or NPS experiments, respectively, by adding an appropriate amount of CaCl2 to the buffer.
Vesicles for the DSC studies were prepared by bath sonication by hydrating in the phosphate buffer for at least 40 min at Tm + 20 °C to yield multilamellar vesicles. For NPS studies, the vesicles were extruded 11 times through Millipore (Bedford, MA, USA) 100-nm pore size polycarbonate filters using an Avanti® MiniExtruder (Avanti Polar Lipids, Alabaster, AL, USA) to yield unilamellar vesicles. The size distribution was checked using dynamic light scattering with a Zetasizer Nano ZS instrument (Malvern Instruments, Malvern, Worcestershire, UK). The average size (hydrodynamic diameter) was in the range of 100–130 nm. A back scattering angle of 173° was used.
:DOPE (90:10, mol%) and DPPC:compound/fragment (95:05, 90:10, 85:15, mol%); and the three-component systems DPPC:DOPE:Chol (80:10:10, mol%) or DPPC:Chol:compound (different compositions). Details on the experimental conditions are given in the ESI.†
3. Results and discussion
3.1. Distribution constants by liquid–liquid extraction and open-tubular capillary electrochromatography
In pharmaceutical research it is highly important to understand the drug partitioning between an aqueous phase and the lipid bilayers of cell membranes. The most common method to determine the lipophilicity of drugs is to calculate the octanol–water partition coefficient (logPo/w). However, this gives only a rough estimation of the affinity of the drugs for cell membranes since it does not account for the effect of electrostatic interactions. A slightly better distribution constant is obtained by calculating the logDpH7.4 value from the acid constant (pKa value) of the compound and its logPo/w value, however, these theoretical approaches are only rough estimates. Other methods, that are more appropriate for mimicking biological systems, often utilise liposomes, which are small artificial vesicles created from natural phospholipids or cholesterol. The structural similarities of liposomes to natural cell membranes have led them to be extensively applied in studies of analyte-membrane interactions using various analytical techniques.
Open-tubular capillary electrochromatography (OT-CEC), which belong to the class of capillary electromigration techniques (CE), is a highly suitable technique for studying drug-lipid membrane interactions. In OT-CEC, the capillary wall is dynamically or statically coated with a stationary phase, and the interactions can be evaluated by monitoring the changes in the analytes electrophoretic mobilities. This technique was developed by Yang et al. in 1998,22 who utilised the strong binding between avidin and biotin, and used biotinylated egg PC liposomes in the presence of low avidin concentration as a capillary coating material. Since then a number of studies have been utilizing this technique, with the coating procedure being based either on electrostatic interactions,23 physical adsorption,24 or covalent binding.25
Here, the apparent distribution constants logDpH7.4 were first determined by liquid–liquid extraction using 1-octanol as the organic phase and 20 mM phosphate buffer at pH 7.4 as the aqueous phase. The concentration and pH value of the buffer were selected to be similar to the conditions used in the capillary electromigration experiments. The logKD values by OT-CEC and the experimentally determined apparent logDpH7.4 values are shown in Table 2. The parent compound (butenafine) B and its methylated fragment F1 could not be dissolved in the buffer and therefore did not give any response in the electropherograms. The liquid–liquid extraction was done in a reversed mode, starting from the compound dissolved in the organic phase (1-octanol), and by this approach experimental apparent logDpH7.4 values could be estimated for all studied compounds. Comparing the calculated distribution constants (logPo/w by PubChem and logDpH7.4 by Chemdraw), one can see that the experimentally determined distribution constants were much lower than the calculated ones for all compounds. The difference in the values for the most hydrophobic compounds is remarkable. This difference is due to the improved solubility of the solid compounds as they were in the form of their hydrochloride salts.
All the distribution coefficients determined by OT-CEC using a lipid stationary phase were slightly greater for compound F3 than for compound F2, despite the experimentally determined apparent logDpH7.4 value being greater for F2. The 100 mol% POPC coating resulted in a much lower KD value for F2 than for F3. Substituting part of the POPC lipids with DOPE resulting in a much higher distribution constant for F2, whereas the effect was less pronounced for F3. Further making the liposome phase slightly more rigid by adding cholesterol had only a small effect on the distribution constants. No change in the electrophoretic mobility of control compound C was observed in the presence of the liposome-coatings, which was rather expected considering it hydrophilic character (apparent logDpH7.4 value of −0.52).
From the big variations in the presented values, it is obvious that (1) for such charged compounds the use of calculated logPo/w (PubChem) values gives poor predictions of the hydrophobicity; (2) the calculated logDpH7.4 (Chemdraw) values correspond rather well with the experimentally determined apparent logDpH7.4 values for the most polar compounds (F2, F3 and C), however, for the most hydrophobic (nonpolar) compounds (B and F1), the correlation is poor; (3) the pure phosphatidyl choline phase (POPC) is clearly less lipophilic than the phase with phosphatidyl ethanolamine and/or cholesterol, as seen in the distribution constants for the studied compounds F2 and F3 (much lower values using POPC than the DOPE or DOPE/Chol phases). This data clearly shows the precautions that should be taken when predicting the lipophilicity/hydrophobicity of compounds based on theoretical (calculated) values.
3.2. Micro differential scanning calorimetry measurements
Differential scanning calorimetry (DSC) is a thermal analysis technique for measuring physical transformations, such as phase transitions, of substances. It provides quantitative information about exothermic, endothermic heat capacity changes as a function of temperature and time. DSC measures the difference in the amount of heat required to increase the temperature of a sample and a reference material. High sensitivity microcalorimetric (MicroCal) DSC is specifically developed for dilute solutions and is frequently used in pharmaceutical thermal analysis because of its ability to provide detailed information about both the physical and energetic properties of a substance.26 Among other substances, this technique has been used to study interactions between analytes and liposomes.27–29 MicroCal DSC can be seen as a standard method for physicochemical characterisation of biomolecules like proteins and lipids or polymers.Important thermodynamic parameters like gel to liquid-crystalline phase transition (Tm), enthalpy values (ΔH), and cooperativity of pure as well as mixed components can be obtained by means of MicroCal DSC. Liposomes are excellent candidates for MicroCal DSC studies in interaction studies between lipid membranes and drug molecules. Ligands like drug molecules, induces various level of disruptions in the membrane due to van der Waals, hydrophobic and electrostatic interactions, formation of hydrogen bonds, and changes in the degree of the hydration. In this study, MicroCal DSC was used to study the thermotropic behaviour of liposomes, composed of DPPC, DPPC/DOPE and DPPC/DOPE/Chol and the effects of B, some of its fragments (F1–F3), and a control sample (C) (cf.Fig. 1) on the change in the Tm of liposomes, composed of DPPC, and DPPC/Chol.
Chol is an essential lipid component of mammalian membranes, which regulates the physical properties and lateral organization of all classes of lipids in the membrane bilayer. One of the major effects of Chol is the broadening and elimination of the cooperative gel-to-liquid crystalline phase transition with a substantial decrease in enthalpy values.30–32 Chol is responsible for increasing the orientational order (also termed as ‘ordering effect’) in the hydrophobic core of the bilayer with mostly unsaturated acyl chains33–35 and shows opposite effect by forming liquid-ordered domains when present with saturated phospholipids (like DPPC) and saturated sphingomyelins.36 Chol is also responsible for a decrease in the cross-sectional area of phospholipids (also termed as ‘condensing effect’) and restrict the mobility of acyl-chains of phospholipids.35,37 All such effects of Chol directly impact the permeability properties of the membranes and therefore it is highly relevant to study interactions of various drugs in the presence of Chol. The effect of phospholipid acyl-chain length as well as unsaturation and the presence of sterols in the membrane bilayer has previously been reported to affect the membrane properties.38,39
| Composition | Lipid or lipid:compound |
Composition (mol%) | Thermotropic behaviour | Area ΔH (kcal mol−1) | ΔT1/2 (°C) | Peak value (°C) | Entropy (J mol−1 K−1) |
|---|---|---|---|---|---|---|---|
| FWHM = fixed width at half maximum. | |||||||
| 1 | DPPC | 100 | Pre-transition | 0.67 | 2.00 | 35.5 | 9.1 |
| Main transition | 5.29 | 0.40 | 41.6 | 70.3 | |||
| 2 | DPPC:DOPE |
90:10 |
Pre-transition | 0.41 | 1.29 | 33.2 | 5.6 |
| Main transition | 4.10 | 2.75 | 38.9 | 55.0 | |||
| 3 | DPPC:DOPE:Chol |
80:10:10 |
Pre-transition | Not observed | Not observed | Not observed | 0.0 |
| Main transition | 2.61 | 8.02 | 37.6 | 35.2 | |||
| 4 | DPPC:B |
95:05 |
Pre-transition | 0.12 | 3.77 | 32.5 | 1.6 |
| Main transition | 4.94 | 0.97 | 41.3 | 65.8 | |||
| 5 | DPPC:B |
90:10 |
Pre-transition | 0.16 | 2.38 | 29.9 | 2.2 |
| Main transition | 4.38 | 2.55 | 40.1 | 58.5 | |||
| 6 | DPPC:B |
85:15 |
Pre-transition | 0.23 | 2.21 | 29.9 | 3.2 |
| Main transition | 3.55 | 4.42 | 37.4 | 47.8 | |||
| 7 | DPPC:F1 |
95:05 |
Pre-transition | 0.22 | 4.70 | 30.8 | 3.1 |
| Main transition | 4.66 | 1.63 | 41.0 | 62.0 | |||
| 8 | DPPC:F1 |
90:10 |
Pre-transition | Not observed | Not observed | Not observed | 0.0 |
| Main transition | 4.42 | 2.74 | 39.9 | 59.0 | |||
| 9 | DPPC:F1 |
85:15 |
Pre-transition | Not observed | Not observed | Not observed | 0.0 |
| Main transition | 5.34 | 5.46 | 39.2 | 71.5 | |||
| 10 | DPPC:F2 |
95:05 |
Pre-transition | 0.22 | 2.43 | 34.9 | 3.0 |
| Main transition | 4.72 | 0.97 | 41.6 | 62.8 | |||
| 11 | DPPC:F2 |
90:10 |
Pre-transition | 0.07 | 2.51 | 34.4 | 1.0 |
| Main transition | 4.67 | 1.72 | 41.5 | 62.0 | |||
| 12 | DPPC:F2 |
85:15 |
Pre-transition | 0.02 | 1.74 | 33.7 | 0.3 |
| Main transition | 4.33 | 2.04 | 41.2 | 57.6 | |||
| 13 | DPPC:F3 |
95:05 |
Pre-transition | 0.27 | 2.83 | 35.0 | 3.7 |
| Main transition | 4.93 | 0.79 | 41.6 | 65.5 | |||
| 14 | DPPC:F3 |
90:10 |
Pre-transition | 0.15 | 2.82 | 34.7 | 2.1 |
| Main transition | 4.58 | 1.25 | 41.5 | 60.9 | |||
| 15 | DPPC:F3 |
85:15 |
Pre-transition | Not observed | Not observed | Not observed | 0.0 |
| Main transition | 4.36 | 1.96 | 41.5 | 58.0 | |||
| 16 | DPPC:C |
95:05 |
Pre-transition | 0.58 | 2.56 | 35.5 | 7.9 |
| Main transition | 4.75 | 0.50 | 41.7 | 63.1 | |||
| 17 | DPPC:C |
90:10 |
Pre-transition | 0.17 | 2.74 | 35.1 | 2.3 |
| Main transition | 4.55 | 1.29 | 41.7 | 60.5 | |||
| 18 | DPPC:C |
85:15 |
Pre-transition | 0.08 | 2.57 | 35.1 | 1.1 |
| Main transition | 4.17 | 1.54 | 41.7 | 55.4 | |||
| 19 | DPPC:Chol:B |
85:10:05 |
Pre-transition | Not observed | Not observed | Not observed | 0.0 |
| Main transition | 3.35 | 2.94 | 39.6 | 44.8 | |||
| 20 | DPPC:Chol:B |
80:10:10 |
Pre-transition | Not observed | Not observed | Not observed | 0.0 |
| Main transition | 3.06 | 7.13 | 37.8 | 41.2 | |||
| 21 | DPPC:Chol:B |
75:10:15 |
Main transition | 1.18 | 3.50 | 34.1 | 16.1 |
| Main transition | 1.97 | 6.23 | 37.5 | 26.6 | |||
| 22 | DPPC:Chol:F1 |
85:10:05 |
Main transition | 0.87 | 8.54 | 41.9 | 11.5 |
| 23 | DPPC:Chol:F1 |
80:10:10 |
Main transition | 1.10 | 9.35 | 40.2 | 14.7 |
| 24 | DPPC:Chol:F1 |
75:10:15 |
Main transition | 3.60 | 9.64 | 36.7 | 48.6 |
| 25 | DPPC:Chol:F2 |
85:10:05 |
Main transition | 2.95 | 0.72 | 40.6 | 39.3 |
| 26 | DPPC:Chol:F2 |
80:10:10 |
Main transition | 2.96 | 0.74 | 40.5 | 39.5 |
| 27 | DPPC:Chol:F2 |
75:10:15 |
Main transition | 2.57 | 0.86 | 40.5 | 34.3 |
| 28 | DPPC:Chol:F3 |
85:10:05 |
Main transition | 3.14 | 0.47 | 40.7 | 41.9 |
| 29 | DPPC:Chol:F3 |
80:10:10 |
Main transition | 2.85 | 0.74 | 40.5 | 38.0 |
| 30 | DPPC:Chol:F3 |
75:10:15 |
Main transition | 2.65 | 1.00 | 40.5 | 35.4 |
| 31 | DPPC:Chol:C |
85:10:05 |
Main transition | 1.14 | 5.63 | 41.2 | 15.2 |
| 32 | DPPC:Chol:C |
80:10:10 |
Main transition | 1.14 | 5.32 | 41.6 | 15.2 |
| 33 | DPPC:Chol:C |
75:10:15 |
Main transition | 1.09 | 4.24 | 40.6 | 14.5 |
The nature of lipid melting or in general, the miscibility profile of lipid mixtures (or with analytes) can be inferred by the cooperativity value. Cooperativity gives an estimate of the number of molecules simultaneously involved in the gel to liquid-crystalline phase transition Tm. Cooperativity values can be semi-quantitatively evaluated from the full peak width at the half maximum of Tm (ΔT1/2). The cooperativity value of DPPC vesicles was 0.40, confirming the purity of DPPC. When DOPE was mixed with DPPC in a 90:10 mol% ratio, the Tm reduced to 38.9 °C (composition 2 in Table 3). The lowering of the gel-liquid transition temperature is due to the presence of two mono-unsaturated acyl chains in DOPE. Unsaturated acyl chains as well as the position of the double bonds interfere with the molecular packing and locally induce a disorder.46,47
The thermogram for the DPPC/DOPE mixture can be deconvoluted to give two components, suggesting some extent of phase separation and less than ideal miscibility. It is unclear whether the DOPE induced some domains in the DPPC bilayer or affected the pre-transition of the DPPC bilayer. For DPPC:DOPE:Chol (80:10:10 mol%) liposomes, the sterol induced a decrease in the values of the Tm (38.8 °C) and the enthalpy (2.61 kcal mol−1), as compared to DPPC (41.6 °C; 5.28 kcal mol−1) or DPPC/DOPE liposomes (38.9 °C; 4.10 kcal mol−1). This agrees with previous studies showing that Chol diminishes the Tm and enthalpy of phospholipids.48 The DSC data also indicated a wider and less cooperative melting behaviour of the complex liposome in the presence of Chol.
 | ||
| Fig. 2 Representative DSC heating thermograms of DPPC:compounds (B, F1, F2, F3 and C) in 95:5 mol% ratios (1st column), 90:10 mol% ratios (2nd column) and 85:15 mol% ratios (3rd column). The samples were heated at the rate of 1 °C min−1. The thermograms shown are the first heating scans. | ||
At 5 mol% of B in DPPC liposomes, the Tpre was reduced from 35.4 °C to 32.5 °C, while the Tm remained almost same, and the enthalpy value reduced from 5.28 to 4.94 kcal mol−1. An additional reduction in the thermotropic parameters was seen at 10 mol% B in DPPC liposomes; the Tpre was reduced to 29.9 °C and the enthalpy value reduced to 3.55 kcal mol−1. At higher concentrations (15 mol%), the effect on Tpre remained the same but the Tm reduced from 41.6 °C to 37.4 °C, probably due to the insertion of B into the acyl chain region of the bilayer. At 15 mol% B concentration, the sharp main transition was also broadened, as represented by ΔT1/2, which increased from 0.40 for pure DPPC vesicles to 4.42 for DPPC with 15% B.
This observation indicates that B was localised in the hydrophobic region of the bilayer, especially in the region of the C1–C8 carbon atoms of the acyl chain. Such position is expected because of the high hydrophobicity of B3, due to the presence of two aromatic rings. Our observations are in line with previous studies.50,51 Based on polarisation studies, fungicide B has shown interactions with both the hydrophilic head/interfacial region as well as the hydrophobic core of the bilayer.51 Molecular simulations or some additional studies are required to confirm the exact location of the drug in the hydrophobic region of the bilayer.52–54
 | ||
| Fig. 3 Representative DSC heating thermograms of DPPC:Chol with incorporated compounds (B, F1, F2, F3 and C (control)). The cholesterol fraction is 10% in each of the compositions. Apart from 10% cholesterol, the respective molar ratios of DPPC and compounds are (a) 85:05 molar ratios, (b) 80:10 molar ratios and (c) 75:15 molar ratios. The samples were heated at the rate of 1 °C min−1. The thermograms shown are the first heating scans. | ||
The addition of 10 mol% of Chol eliminated the pre-transition and substantially reduced the main phase transition of the DPPC bilayers containing 5% of B (Table 3; composition 19). Two main peaks were observed at 15 mol% of B in DPPC:Chol membrane, 34.0 °C and 37.5 °C. The cooperativity values for DPPC:Chol steadily increased upon inclusion of B from 2.94 (5 mol%) to 6.23 (15 mol%). This clearly indicates that fungicide B can permeate the membrane and to some extent destabilise the membrane bilayer.
In the presence of 10 mol% of Chol in the liposome, F1 was able to destabilise the membrane at the highest tested concentration (15 mol%), as indicated by a decrease in the Tm. However, F2 and F3 showed sharp transition peaks at Tm at all concentrations (5–15 mol%). The Tm peak continuously decreased with increasing concentration of the compounds. However, the enthalpy values reduced noticeably. The control sample C did not noticeable penetrate the membrane in the presence of Chol. The Tm peak was almost lost for C at all tested concentrations.
3.3. Nanoplasmonic sensing
Nanoplasmonic sensing (NPS) is an analytical technique based on the optical phenomenon of localised surface plasmon resonance of metallic nanostructures. In NPS experiments, the plasmonic particles are inert and adjacent to the material of interest to probe a process occurring either in the material investigated or on the surface of the material. Changes in the nanoparticle properties cause a shift in the maximum-extinction wavelength, which is monitored and recorded in real-time. Nanoplasmonic sensors have become widely used for the label-free detection of biomolecules across medical, biotechnology, and environmental science applications, with detection capabilities down to single-molecule level.55 Phospholipid characterisation using NPS56–58 has been summarised in some recent review papers.55,59,60 | ||
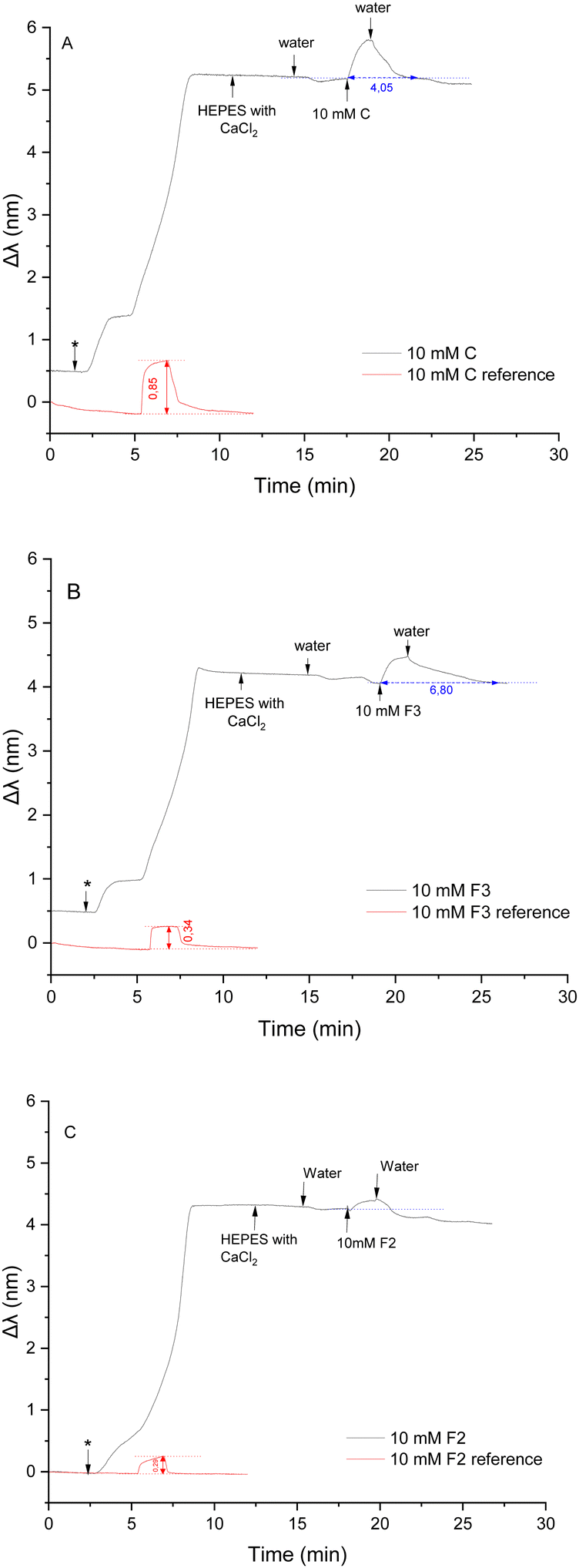
| Fig. 4 NPS interactions between POPC membrane and 10 mM of compounds (A) C, (B) F3 and (C) F2. The ‘*’ denotes the introduction of liposomes composed of POPC onto the sensors. The arrow mark indicates the introduction of solvent systems or fragment molecule on the sensor. | ||
The compounds F2, F3 and C are hydrophilic compounds with good water solubility, and therefore, a concentration of 10 mM was chosen for the study of their interactions. The measurement cell was flushed with Milli-Q water at the end of the injection until a stable Δλ response was reached. The injection of 150 μL of 10 mM of the control compound C induced a red shift of 0.45 nm (Fig. 4A), indicating a change in the density on the sensor's surface. The fragment penetrated the membrane through hydrophobic interactions, which increased the density inside the membrane and increased the mass on the sensor. This effect was expected considering the structure of compound C.
With its secondary amino group, the molecule can interact with the hydrophilic head group of the POPC lipids through hydrogen bonds, although this interaction is quite weak.51 The hydrophobic interactions between the benzyl group and the acyl chain regions of the lipid bilayer dominate over the electrostatic interactions with the hydrophilic head groups, allowing compound C to penetrate the bilayer. Once the injection was finished, the sensor was flushed with Milli-Q water until a stable baseline was reached. The water washed away C molecules from the membrane, inducing a slow decrease of the NPS signal. The Δλ signal came back to a stable plateau 3 min after the end of the injection of the compound on average, which indicates that all the analyte molecules were washed away from the membrane. The obtained plateau was slightly lower than the baseline obtained before the injection. When the membrane was flushed with water after the liposome immobilisation, the signal slightly decreased due to the high flow rate (100 μL min−1).
Interactions between compound F3 and the POPC liposomes was next investigated (Fig. 4B). The introduction of F3 induced a significant red shift of 0.50 nm. This change was even more significant than the shift observed with C, implying that compound F3 distributes even more easily and in higher quantity into the membrane. This difference is due to the greater hydrophobic nature of compound F3 as compared to C, hence involving stronger hydrophobic interactions. The naphthalene ring present in F3 possibly distributes over the wide domain of the membrane from the hydrophobic core to the hydrophilic region, with the amine group exhibiting a weak preferential orientation toward the hydrophilic headgroup.30 The sensor was subsequently rinsed with water and compound F3 was slowly flushed from the membrane for about 5 minutes until a stable baseline was reached. The stabilisation time was longer than for compound C, probably due to a higher volume of compounds partitioning into the membrane but also due to favourable interactions between compound F3 and the POPC membrane.
Finally, interactions between compound F2 and the POPC liposomes were investigated (Fig. 4C). The introduction of F2 induced a small red shift (0.36 nm) compared to the significant change in the signal observed with compounds F3 and C. A limited quantity of molecules of F2 could partition into the POPC membrane. This behaviour was not expected from compound F2 as this molecule is more hydrophobic than compound F3. It seems that the hydrophobic interactions between the tert-butyl benzyl group in F2 and the acyl chains of the phospholipids is weaker, allowing lesser partitioning of F2 into the POPC bilayer, as compared to F3. After the injection, the sensor was washed with water for 4 min on average until the NPS signal reached a stable baseline, which was similar to the measurements with control compound C. Since hydrophobic interactions between F2 and POPC are greater than those observed with C, stabilisation of the signal (i.e., washing out the compound) took a longer time.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :DOPE (90:10 mol%) membrane.
A binary lipid mix containing POPC and DOPE in a 90:10 molar ratio was studied to evaluate the effect of DOPE on the interactions between the studied compounds and the bilayer. The evaluation of PE interactions is important as PE is the second most common phospholipid in the plasma membrane.
:DOPE (90:10 mol%) membrane.
A binary lipid mix containing POPC and DOPE in a 90:10 molar ratio was studied to evaluate the effect of DOPE on the interactions between the studied compounds and the bilayer. The evaluation of PE interactions is important as PE is the second most common phospholipid in the plasma membrane.
First, 150 μL of a 10 mM solution of compound C was injected into the measurement cell after immobilisation of the liposomes (Fig. 5A). A significant red shift appeared after the introduction of the fragment, indicating that a large quantity of the molecules penetrated the membrane. The DOPE lipids clearly seem to help the compounds from entering the bilayer. This result was expected regarding DOPE's function in the membrane. DOPE increases the permeability of the membrane by inducing hexagonal structures within the membrane which relax the tight packing of the bilayer formed by PC phospholipids.40,63,64 By doing so, DOPE promotes membrane fusion and fission events and facilitates the integration of molecules. Once the injection was finished, the sensor was flushed with water. It took around 5 minutes for the NPS signal to go back to its original baseline, which was longer than that observed with the POPC membrane. It appears that DOPE lipids might also prevent the fragments from exiting the membrane.
 | ||
| Fig. 5 NPS interactions between POPC:DOPE membrane and 10 mM of compounds (A) C (control), (B) fragment F3 and (C) fragment F2. The asterisk (*) denotes the introduction of liposomes composed of POPC:DOPE (9:1 molar ratio) onto the sensors. The arrow mark indicates the introduction of solvent systems or compounds on the sensor. | ||
Compound F3 was then tested (Fig. 5B). A significant red shift, higher than with compound C, was obtained. This observation confirmed the role of DOPE to enhance the permeability of the membrane. The sensor was then flushed with water until a stable Δλ was obtained. The length of time necessary to wash away the fragments was 7 min on average, which is similar to the flushing time needed with the POPC membrane. With fragment F3, the DOPE lipids did not seem to influence the escape of the analyte out of the membrane.
Next, compound F2 was injected into the measurement cell after immobilisation with liposomes (Fig. 5C). The introduction of F2 also induced an important red shift, indicating that F2 was able to enter the membrane in the presence of DOPE. The new membrane structure created by DOPE facilitated the interactions between F2 and the hydrophobic region, allowing the penetration of F2 into the membrane. After the injection, the sensor was flushed with water and the signal slowly became stable after 8 min. Since F2 is hydrophobic, the flushing time needed to wash away the molecules was the longest.
:DOPE:Chol (80:10:10 mol%) membrane.
Chol has a vital function in the permeability of the membrane. Therefore, a tertiary mix containing POPC, DOPE and Chol in 80:10:10 mol% was tested to study the influence of Chol on the interactions between the compounds and the lipid membrane. Control compound C was first tested. 150 μL of a 10 mM solution was injected after immobilisation with liposomes (Fig. 6A). A red shift (0.61 nm) was induced, indicating an increase in the density on the sensor's surface. This shift was smaller than the previous shift observed with the membrane containing DOPE but bigger than the shift obtained with a membrane constituted of POPC only. As previously reported,65 Chol thickens the membrane, which has a stiffening effect and decreases its permeability, making it more difficult for external molecules to penetrate. Indeed, one of Chol's role is to hinder the penetration of foreign molecules that could be harmful to the cell to enter.66 DOPE favourited the integration of a higher quantity of compounds in the bilayer than with the POPC membrane, but Chol limited this effect by thickening the membrane. At the end of the injection, C molecules were washed away from the membrane after flushing for 3 minutes. The presence of both DOPE and Chol did not seem to influence the removal of the compounds from the membrane.
 | ||
| Fig. 6 NPS interactions between POPC:DOPE:CHOL membrane and 10 mM of compounds (A) C (control), (B) F3 and (C) F2. The ‘*’ denotes the introduction of liposomes composed of POPC:DOPE:CHOL (80:10:10 molar ratios) onto the sensors. The arrow mark indicates the introduction of solvent systems or fragment molecules on the sensor. | ||
Next, the more hydrophobic compound F3 was injected into the measurement cell after immobilisation with liposomes (Fig. 6B). A significant red shift (0.41 nm) was observed, which was bigger than the shift induced by interaction with POPC but smaller than the shift induced by interactions with a membrane enriched with DOPE.
The addition of Chol had the same effect on interactions with F3 than with C. After the injection, the sensor was flushed for an average time of 8 min 30 s before reaching a stable Δλ. Additional time was necessary to completely wash away F3 compounds from the membrane in the presence of Chol. It appears that Chol retain compounds C and F3 in the bilayer.
Finally, interactions between the lipid membrane and compound F2 were studied (Fig. 6C). The introduction of F2 on the sensor induced a very small red shift (0.18 nm). This shift is even smaller than the shift observed using the immobilised POPC bilayer. In some repetition measures, no shift was observed. It appears that in the presence of Chol, compound F2 is unable to penetrate the membrane despite the presence of DOPE.
3.4. Nanoplasmonic and quartz crystal microbalance study of compound filled POPC liposomes
Due to the insolubility of B and F1 molecules in aqueous buffers, an alternative approach was considered to investigate their interactions with POPC liposomes. Using NPS technique, such an approach has never been reported earlier as per literature survey during the writing of the article. For this method, liposomes filled with B and F1 were prepared. 5 and 10% B and F1 (from 10 mM stock solutions in MeOH) were mixed with POPC prior to hydration. The remaining liposome preparation steps remained the same, as stated in the experimental section.Both B and F1 at 5 mol% were able to penetrate the POPC bilayers (Fig. 7a and b), as observed by the increase in the Δλ in comparison to unfilled POPC vesicles. When present at 5 mol%, F1 and B appeared to interact with the POPC membranes.
 | ||
| Fig. 7 NPS study of drug prefilled vesicles composed of POPC, where the compound was mixed with the lipid before liposome preparation. (a) NPS curve for POPC vesicles prefilled with 5 mol% of compounds B (red) and F1 (blue), compared to POPC (black) vesicles. (b) NPS curve for POPC vesicles prefilled with 10 mol% of compounds B (red) and F1 (blue), compared to POPC (black) vesicles. | ||
Comparatively, the Δλ value was higher for F1 filled vesicles than for B filled vesicles. At lower concentrations, both drug molecules are expected to interact with POPC membranes electrostatically, where the amine group of both the fungicides are interacting with the carbonyl group of the phospholipids. Yet, F1 may slip into POPC bilayers due to lack of a methyl group in F1, allowing its easy access into POPC membranes. Lack of a methyl group in F1 may reduce the steric hindrance as compared to B. Previous studies have also shown that the presence of a methyl group in the acyl chain region creates steric hindrance and affect membrane properties.36
However, at 10 mol%, B was able to penetrate more into the POPC bilayers (Fig. 7b), as observed by higher Δλ value for B filled liposomes than F1 filled liposomes. At higher concentrations, the electrostatic interactions are superseded by van der Waals interactions between the naphthalene ring (here of B and F1 molecules) and the hydrophobic acyl chain region of phospholipids. B is expected to be more hydrophobic than F1 (from logDpH7.4 Chemdraw and logPo/w PubChem theoretical values). However, the octanol–water partition experiments in the present work showed that the B and F1 have comparable hydrophobicity. Hence, B filled POPC bilayers showed comparatively higher shift in Δλ value from 5% to 10% than in case of F1 filled POPC bilayers. The same observation was also supported by DSC in the present work.
Quartz crystal microbalance (QCM) is an acoustic sensing technique which can be used for label-free probing of subtle changes in mass on the surface of a sensor chip. It has been used for the analysis of lipid interactions with proteins, nanoparticles, peptides, and small compounds such as ionic liquids.67–70 Changes in mass adsorbed on the surface and changes in the viscosity in the proximity to the surface can be monitored by this technique, which renders it suitable for exploration of effects of small molecules on the adsorbed lipid layers or whole vesicles.70–73
The QCM sensor was surface coated with SiO2 instead of with Si3N4 as in NPS. This resulted in a different adsorption dynamic of POPC-based liposomes. When the liposome dispersion was applied, a slow adsorption of vesicles was observed which took about 30 min before the negative frequency change leveled out, suggesting the attainment of a full coverage of the sensor surface with vesicles and the formation of a supported vesicle layer (SVL) (Fig. 8). However, an abrupt increase in frequency was detected upon rinsing the SVL with H2O starting from the 46th minute. This could be explained by breakage of the adsorbed vesicles, probably induced by osmotic stress, and a subsequent formation of a SLB. This is also supported by the frequency leveling off at the lower frequency value of minus 40 to 50 Hz after 80 minutes (Fig. 8). In the case of 5% of B and F1 in the liposomes, more mass was observed on the surface after SLB induction compared to the parent POPC SLB. In contrary, 10% of B and F1 in the liposomes showed almost no change compared to parent POPC SLB. A possible explanation for this behavior can be found in the DSC results, which indicate that an increase of the content of B and F1 from 5% to 10% resulted in a deeper insertion of the compounds into the membrane (see discussions in the previous sections). It is justified to assume that this would also force the charge-carrying parts of the molecules deeper into the lipid bilayer, and the drug-loaded liposome would then display interactions with the sensor surface more similar to those of unloaded POPC vesicles. With the 5% B, and especially with F1, the positive charge of the compounds influenced the SLB formation by causing an uneven layer or remnants of vesicles to be present on the sensor surface. This was not observed in NPS, probably due to method-inherent limits in effective sensing depth, which is with NPS in the range of 10 nm and with QCM approximately 200 nm.74,75
 | ||
| Fig. 8 QCM change in resonance frequency (delta frequency) of liposomes filled with compounds B and F1 in the molar ratios of 5% (A) and 10% (B). | ||
Fragment-based drug discovery is a well-established concept for the discovery of active-site binders in current pharmacological research and has delivered a considerable number of promising candidates for various targets. However, in current fragment-based drug discovery emphasis is primarily placed on identifying structural motifs favouring interactions with target binding sites, while largely neglecting the equally important biophysical characteristics of the studied fragment structures in terms of biophysical properties responsible for distribution to and retention within target tissues. Clearly, and especially for drugs acting on membrane-located targets, such as allyl or benzyl amine-type antifungals, not only functional and spatial active-site complementarity but also their inherent membrane affinity contributes to efficacious and sustained drug action. In this context, the current study is a preliminary effort to expand the fragment-based drug discovery concept by a comprehensive assessment of fragment-membrane interactions to provide complementary guiding information concerning early-stage fragment selection and/or prioritisation of promising fragment structure repertories. Indeed, the combined finding of the present interdisciplinary fragment-based membrane-drug interaction study confirms the concept of a dual activity mechanism for butenafine.
Thus, the most lipophilic compounds B and F1 behaved similarly and interacted strongly with all studied liposomes primarily by hydrophobic interactions due to the naphthalene group in B and F1. These hydrophobic interactions are responsible for the slow release of butenafine and its fragments from the lipid membranes, explaining the favourable pharmacological profile of the parent drug. In the presence of Chol in the liposome, both B and F1 were able to slightly destabilise the lipid membrane. However, the interactions between fragments F2 and F3 and the lipid membranes strongly depended on the composition of the liposomes. Compounds F3 and C, containing a naphthalene and a benzyl group, respectively, could easily be incorporated in the POPC liposome membranes, while compound F2 carrying a tert-butyl benzyl group had difficulties in interacting with the membrane. Compounds F2, F3 and C interacted with the lipid bilayer without disintegrating the membrane.
The introduction of DOPE in the liposome composition enhanced the fluidity of the membrane, which facilitated the partition of all tested compounds into the membrane. The presence of DOPE also assisted in retaining the fragments inside the membrane. The introduction of Chol had a condensing effect on the membrane, which levelled the interaction-promoting effect of DOPE and thus prevented the fragments from penetrating the membrane. The fact that the current study could identify membrane-perturbation for butenafine and the structurally closely related F1 provides evidence that the fragment-based approach may have considerable merits in the development of new antifungal agents. It is also worth mentioning that our findings are consistent with those reported for a comparative pharmacological study carried for butenafine and tolnaftate with Candida albicans strains, in which a dual mechanism of the antifungal activity of butenafine was reported, involving both specific enzyme inhibitory and nonspecific membrane-damaging effects.3
4. Conclusions
In this study, the molecular interactions between the antifungal agent butenafine and its substructure fragments and biomimetic membranes were investigated using an interdisciplinary approach. Specifically, this study characterised the interactions of the parent compound, three substructure fragments (F1, F2 and F3) and a control compound (C) with compositional distinct biomimicking membranes using MicroCal DSC, OT-CEC, NPS and QCM. The employed liposomes were composed of three natural lipids; PCs, PEs and Chol. The vesicles were either in free solution (MicroCal) or immobilised on fused silica capillaries (OT-CEC) or sensors (NPS, QCM) in the form of a supported lipid bilayer.In summary, the interdisciplinary approach presented here using MicroCal DSC, OT-CEC, NPS and QCM for the study of the molecular interactions of an antifungal drug and its substructure fragments with liposomes provides detailed insights into the structural requirements for favourable membrane integration. As an added benefit, the combined use of these complementary methods provides a better understanding of individual merits and limitations of these methods when employed to the study of drug-membrane interactions.
While the current study was carried out on biomimicking membranes containing Chol, i.e. a model relevant for mammalian cells, future research will focus on liposomes incorporating ergosterol for generating insights relevant to fungal membranes. In addition, in future research an expanded repertoire of antifungal drugs/fragments will be probed to test the general utility of the fragment-based drug discovery concept.
Author contributions
Shishir Jaikishan: conceptualisation, investigation, supervision, visualisation, writing – original draft, writing – review & editing; Marine Lavainne: investigation, visualisation, writing – original draft; Henri K. Ravald: investigation, visualisation, writing – original draft; Kieran Scobbie: investigation; Filip Dusa: conceptualisation, investigation, visualisation, writing – original draft; Rekha Maheswari: investigation; Jenni Turpeinen: investigation; Ian Eikemans: investigation; Rui Chen: investigation, visualisation; Julia Rantala: investigation; Vladimir Aseyev: conceptualisation, writing – original draft, writing – review & editing; Norbert N. Maier: conceptualisation, supervision, writing – original draft, writing – review & editing; Susanne K. Wiedmer: conceptualisation, funding acquisition, project administration, supervision, writing – original draft, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
MSc Sanni Raskala is acknowledged for assistance with the synthesis of the compounds. MSc Arina Sukhova and PhilLic Matti Jussila are acknowledged for assisting with the mass spectrometric analysis of the compounds. Financial support from the Finnish Society of Sciences and Letters under grant number 4709946 (SKW) and from the Waldemar von Frenckell's foundation under grant number 4706562 (SKW) are acknowledged.References
- M. C. Fisher, A. Alastruey-Izquierdo, J. Berman, T. Bicanic, E. M. Bignell, P. Bowyer, M. Bromley, R. Bruggemann, G. Garber, O. A. Cornely, S. J. Gurr, T. S. Harrison, E. Kuijper, J. Rhodes, D. C. Sheppard, A. Warris, P. L. White, J. Xu, B. Zwaan and P. E. Verweij, Nat. Rev. Microbiol., 2022, 20, 557–571 CrossRef CAS.
- W. McNeely and C. M. Spencer, Drugs, 1998, 55, 405–412 CrossRef CAS ; discussion 413.
- W. Iwatani, T. Arika and H. Yamaguchi, Antimicrob. Agents Chemother., 1993, 37, 785–788 CrossRef CAS.
- C. Struyfs, B. P. A. Cammue and K. Thevissen, Front. Cell Dev. Biol., 2021, 9, 649875 CrossRef.
- W. S. Sung, J. Lee and D. G. Lee, Biol. Pharm. Bull., 2008, 31, 1906–1910 CrossRef CAS PubMed.
- E. Grela, M. Wieczór, R. Luchowski, J. Zielinska, A. Barzycka, W. Grudzinski, K. Nowak, P. Tarkowski, J. Czub and W. I. Gruszecki, Mol. Pharmaceutics, 2018, 15, 4202–4213 CrossRef CAS.
- G. P. Dahal and R. E. Viola, SLAS Discovery, 2018, 23, 520–531 CrossRef CAS PubMed.
- M. I. Konaklieva and B. J. Plotkin, Antibiotics, 2023, 12, 315 CrossRef CAS.
- J. Li, S. Liu, J.-J. Koh, H. Zou, R. Lakshminarayanan, Y. Bai, K. Pervushin, L. Zhou, C. Verma and R. W. Beuerman, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 1023–1031 CrossRef CAS PubMed.
- W. Liu, L. Yuan and S. Wang, J. Med. Chem., 2020, 63, 12429–12459 CrossRef CAS.
- H. B. Konuk and B. Ergüden, Folia Microbiol., 2020, 65, 775–783 CrossRef CAS.
- A. I. P. M. de Kroon, P. J. Rijken and C. H. De Smet, Prog. Lipid Res., 2013, 52, 374–394 CrossRef CAS PubMed.
- L. Klug and G. Daum, FEMS Yeast Res., 2014, 14, 369–388 CrossRef CAS.
- S. E. Horvath, A. Wagner, E. Steyrer and G. Daum, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 1811, 2011, 1030–1037 Search PubMed.
- J. Malinsky, W. Tanner and M. Opekarova, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 1861, 2016, 806–811 Search PubMed.
- Y. Zasshi, T. Maeda, M. Takase, A. Ishibashi, T. Yamamoto, K. Sasaki, T. Arika, M. Yokoo and K. Amemiya, J. Pharm. Soc. Jpn., 1991, 111, 126–137 CrossRef PubMed.
- A. M. G. Porras, B. S. Terra, T. C. Braga, T. F. F. Magalhães, C. V. B. Martins, D. L. da Silva, L. M. Baltazar, L. F. Gouveia, G. J. C. de Freitas, D. A. Santos, M. A. Resende-Stoianoff, B. B. Fuchs, E. Mylonakis, R. P. de Freitas and Â. de Fátima, J. Adv. Res., 2018, 14, 81–91 CrossRef CAS PubMed.
- K. Lipponen, S. K. Wiedmer and M.-L. Riekkola, J. Chromatogr. Open, 2021, 1, 100020 CrossRef.
- F. Duša, J. Witos, E. Karjalainen, T. Viitala, H. Tenhu and S. K. Wiedmer, Electrophoresis, 2016, 37, 363–371 CrossRef.
- D. Regan, J. Williams, P. Borri and W. Langbein, Langmuir, 2019, 35, 13805–13814 CrossRef CAS PubMed.
- J. Witos, G. Russo, S.-K. Ruokonen and S. K. Wiedmer, Langmuir, 2017, 33, 1066–1076 CrossRef CAS PubMed.
- Q. Yang, X.-Y. Liu, J. Miyake and H. Toyotama, Supramol. Sci., 1998, 5, 769–772 CrossRef CAS.
- E. Ornskov, S. Ullsten, L. Soderberg, K. E. Markides and S. Folestad, Electrophoresis, 2002, 23, 3381–3384 CrossRef CAS.
- J. T. Hautala, M. V. Linden, S. K. Wiedmer, S. J. Ryhanen, M. J. Saily, P. K. J. Kinnunen and M. L. Riekkola, J. Chromatogr. A, 2003, 1004, 81–90 CrossRef CAS PubMed.
- J. Lokajová, H. Tiala, T. Viitala, M.-L. Riekkola and S. K. Wiedmer, Soft Matter, 2011, 7, 6041–6050 RSC.
- S. D. Clas, C. R. Dalton and B. C. Hancock, Pharm. Sci. Technol. Today, 1999, 2, 311–320 CrossRef CAS.
- T.-T. Wei, B.-B. Cao, X.-L. Hao, J.-Y. Gu and R.-G. Wu, Thermochim. Acta, 2021, 703, 178993 CrossRef CAS.
- R. G. Wu, J. D. Dai, F. G. Wu, X. H. Zhang, W. F. Li and Y. R. Wang, Int. J. Pharm., 2012, 438, 91–97 CrossRef CAS PubMed.
- M. Di Foggia, S. Bonora, A. Tinti and V. Tugnoli, J. Therm. Anal. Calorim., 2016, 127, 1407–1417 CrossRef.
- T. P. W. McMullen and R. N. McElhaney, Biochim. Biophys. Acta, Biomembr., 1995, 1234, 90–98 CrossRef.
- S. Jaikishan, A. Bjorkbom and J. P. Slotte, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 1615–1622 CrossRef CAS.
- B. D. Ladbrooke, R. M. Williams and D. Chapman, Biochim. Biophys. Acta, Biomembr., 1968, 150, 333–340 CrossRef CAS.
- J. A. Urbina, S. Pekerar, H. B. Le, J. Patterson, B. Montez and E. Oldfield, Biochim. Biophys. Acta, Biomembr., 1995, 1238, 163–176 CrossRef PubMed.
- E. Oldfield, M. Meadows, D. Rice and R. Jacobs, Biochemistry, 1978, 17, 2727–2740 CrossRef CAS PubMed.
- M. Alwarawrah, J. Dai and J. Huang, J. Phys. Chem. B, 2010, 114, 7516–7523 CrossRef CAS PubMed.
- S. Jaikishan, A. Bjorkbom and J. P. Slotte, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 1987–1994 CrossRef CAS PubMed.
- M. R. Vist and J. H. Davis, Biochemistry, 1990, 29, 451–464 CrossRef CAS PubMed.
- T. K. M. Nyholm, S. Jaikishan, O. Engberg, V. Hautala and J. P. Slotte, Biophys. J., 2019, 116, 296–307 CrossRef CAS PubMed.
- S. Jaikishan and J. P. Slotte, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 1940–1945 CrossRef CAS.
- J. N. van der Veen, J. P. Kennelly, S. Wan, J. E. Vance, D. E. Vance and R. L. Jacobs, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1558–1572 CrossRef CAS.
- C. H. Huang, Lipids, 2001, 36, 1077–1097 CrossRef CAS.
- R. N. McElhaney, Chem. Phys. Lipids, 1982, 30, 229–259 CrossRef CAS.
- H. Farzaneh, M. Ebrahimi Nik, M. Mashreghi, Z. Saberi, M. R. Jaafari and M. Teymouri, Int. J. Pharm., 2018, 551, 300–308 CrossRef CAS.
- A. C. Alves, D. Ribeiro, M. Horta, J. L. F. C. Lima, C. Nunes and S. Reis, J. R. Soc., Interface, 2017, 14, 20170408 CrossRef.
- J. A. Svetlovics, S. A. Wheaten and P. F. Almeida, Biophys. J., 2012, 102, 2526–2535 CrossRef CAS.
- M. Alwarawrah, F. Hussain and J. Huang, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 253–263 CrossRef CAS.
- H. Martinez-Seara, T. Róg, M. Pasenkiewicz-Gierula, I. Vattulainen, M. Karttunen and R. Reigada, Biophys. J., 2008, 95, 3295–3305 CrossRef CAS.
- D. A. Mannock, M. Y. T. Lee, R. N. A. H. Lewis and R. N. McElhaney, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 2191–2202 CrossRef CAS PubMed.
- S. Béni, M. Budai, B. Noszál and P. Gróf, Eur. J. Pharm. Sci., 2006, 27, 205–211 CrossRef PubMed.
- L. Zhao, S. S. Feng, N. Kocherginsky and I. Kostetski, Int. J. Pharm., 2007, 338, 258–266 CrossRef CAS.
- M. P. Mingeot-Leclercq, X. Gallet, C. Flore, F. Van Bambeke, J. Peuvot and R. Brasseur, Antimicrob. Agents Chemother., 2001, 45, 3347–3354 CrossRef CAS.
- S. Li, F. Huang, T. Xia, Y. Shi and T. Yue, Langmuir, 2023, 39, 5995–6005 CrossRef CAS PubMed.
- H. H. Huang, J. Jia, L. Ren, S. Wang, T. Yue, B. Yan and Y. H. Chu, J. Hazard. Mater., 2023, 453, 131430 CrossRef CAS.
- C. Martinotti, L. Ruiz-Perez, E. Deplazes and R. L. Mancera, ChemPhysChem., 2020, 21, 1486–1514 CrossRef CAS PubMed.
- J. A. Jackman, A. Rahim Ferhan and N.-J. Cho, Chem. Soc. Rev., 2017, 46, 3615–3660 RSC.
- G. Russo, J. Witos, A. H. Rantamäki and S. K. Wiedmer, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 2361–2372 CrossRef CAS PubMed.
- R. Frost, S. Svedhem, C. Langhammer and B. Kasemo, Langmuir, 2016, 32, 2708–2717 CrossRef CAS.
- F. Dusa, W. Chen, J. Witos, A. H. Rantamaki, A. W. T. King, E. Sklavounos, M. Roth and S. K. Wiedmer, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183115 CrossRef CAS.
- A. R. Ferhan, B. K. Yoon, W. Y. Jeon and N. J. Cho, Nanoscale Adv., 2020, 2, 3103–3114 RSC.
- S. Jaikishan, M. Lavainne and S. K. Wiedmer, Sens. Actuators Rep., 2024, 7, 100192 CrossRef.
- A. R. Ferhan, J. A. Jackman and N. J. Cho, Phys. Chem. Chem. Phys., 2017, 19, 2131 RSC.
- M. Dacic, J. A. Jackman, S. Yorulmaz, V. P. Zhdanov, B. Kasemo and N. J. Cho, Langmuir, 2016, 32, 6486–6495 CrossRef CAS PubMed.
- N. B. Leite, A. Aufderhorst-Roberts, M. S. Palma, S. D. Connell, J. Ruggiero Neto and P. A. Beales, Biophys. J., 2015, 109, 936–947 CrossRef CAS.
- E. Calzada, O. Onguka and S. M. Claypool, Int. Rev. Cell Mol. Biol., 2016, 321, 29–88 CAS.
- H. Ohvo-Rekila, B. Ramstedt, P. Leppimaki and J. P. Slotte, Prog. Lipid Res., 2002, 41, 66–97 CrossRef CAS PubMed.
- H. I. Ingólfsson, M. N. Melo, F. J. van Eerden, C. Arnarez, C. A. Lopez, T. A. Wassenaar, X. Periole, A. H. de Vries, D. P. Tieleman and S. J. Marrink, J. Am. Chem. Soc., 2014, 136, 14554–14559 CrossRef PubMed.
- Q. Chen, S. Xu, Q. Liu, J. Masliyah and Z. Xu, Adv. Colloid Interface Sci., 2016, 233, 94–114 CrossRef CAS PubMed.
- N.-J. Cho, C. W. Frank, B. Kasemo and F. Hook, Nat. Protoc., 2010, 5, 1096–1106 CrossRef CAS.
- N.-Y. Lu, K. Yang, J.-L. Li, B. Yuan and Y.-Q. Ma, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 1918–1925 CrossRef CAS.
- F. Duša, S.-K. Ruokonen, J. Petrovaj, T. Viitala and S. K. Wiedmer, Colloids Surf., B, 2015, 136, 496–505 CrossRef.
- P. Losada-Pérez, M. Khorshid and F. U. Renner, PLoS One, 2016, 11, e0163518 CrossRef.
- S. Moon, B. K. Yoon and J. A. Jackman, Langmuir, 2022, 38, 4606–4616 CrossRef CAS PubMed.
- A. Benedetto, Biophys. Rev., 2023, 15, 1909–1939 CrossRef CAS PubMed.
- J. A. Jackman, S. Yorulmaz Avsar, A. R. Ferhan, D. Li, J. H. Park, V. P. Zhdanov and N.-J. Cho, Anal. Chem., 2017, 89, 1102–1109 CrossRef CAS.
- F. Duša, W. Chen, J. Witos and S. K. Wiedmer, Front. Mater., 2019, 6, 23 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00648h |
| This journal is © The Royal Society of Chemistry 2024 |