
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A simple approach to produce hydrophobic biobased coatings using methylcellulose and organosolv lignin†
Kourosh
Mobredi
 ,
Isaac Y.
Miranda-Valdez
,
Tero
Mäkinen
*,
Juha
Koivisto
and
Mikko J.
Alava
,
Isaac Y.
Miranda-Valdez
,
Tero
Mäkinen
*,
Juha
Koivisto
and
Mikko J.
Alava
Complex Systems and Materials, Department of Applied Physics, Aalto University, P.O. Box 15600, FI-00076 Aalto, Espoo, Finland. E-mail: tero.j.makinen@aalto.fi
First published on 4th July 2024
Abstract
Substituting plastics with circular and sustainable alternatives has increasingly become a priority. Protective coatings, crucial components in numerous industries, are now in demand for biodegradable options to replace their plastic-based counterparts. Being one of nature's most abundant components, lignin remains underutilized, and this study focuses on investigating its potential for the production of biobased coatings. The method used here involved formulating coating suspensions by mixing methylcellulose and organosolv lignin powders and adding water to the mixture. Glass wafers were coated with the formulated suspensions using spin-coating. The morphology of the coated surfaces was assessed using optical and scanning electron microscopy. In addition, the wettability of the surfaces was examined through water contact angle experiments, and a numerical model was introduced to predict the water contact angle evolution over time. The results revealed that the sample coated with a 2.5 wt% lignin suspension exhibited the highest initial contact angle (114°), with a decreasing trend as the lignin fraction increases. Moreover, coatings with 3.5 wt% lignin and above exhibited lower surface coverage due to lignin particle aggregation and surface defects. By approximating the water droplet on the surface as a spherical cap, the introduced numerical model successfully predicted the time-dependent evolution of the water contact angle by showing strong alignment with experimental results. Taken altogether, we have showcased here a method for modifying coating properties—in a practical sense from water-absorbent to splash-proof—using readily available forest-based materials. This advancement is paving the way for sustainable protective packaging, aiming to replace styrofoam in the electronics and food industries.
1 Introduction
The demand for plastics has surged in recent decades due to their advantageous properties, and this upward trend is expected to continue.1 Plastics serve limitless applications, from consumer products to value-added items such as coatings and adhesives.2–4 However, concerns about plastic waste management and environmental pollution have prompted scientists to explore more sustainable alternatives.5–9 One prominent alternative to traditional plastics is biobased materials, which have gained popularity owing to their sustainable nature.10–16 Sourced primarily from biomass, a rich source of biopolymers, these materials offer the potential to manufacture sustainable and biodegradable products, providing alternatives to conventional plastics. Lignocellulosic biomass is an excellent choice for producing biobased materials, whose main constituents are cellulose, hemicellulose, and lignin.17 Discovering more effective ways to produce and utilize this biomass has the potential to result in the creation of more sustainable products and structures. This paper focuses on finding a sustainable application for lignin.Lignin is the second most abundant complex organic substance found on Earth18,19 and the only large volume renewable feedstock that comprises aromatics.20,21 Nevertheless, the complex nature of lignin structure22 and the limited range of effective uses and methods for incorporating lignin as a renewable chemical feedstock23 result in its continued underutilization in the production of biomaterials. Currently, lignin is sourced from two main sectors: the paper and pulp industry and the biorefinery industry. Each year, a total of 100 million tonnes of lignin are isolated from biomass, with the paper and pulp industry contributing to 70% of this production.24 Furthermore, lignin production forecasts increasing substantially to reach 225 million tonnes annually by 2030, primarily due to the growing adoption of cellulosic biorefineries.25 However, there are several factors, including non-uniform structure, unique chemical reactivity, and the presence of various impurities, that have limited lignin widespread use in producing value-added products.26 Currently, only a small lignin fraction, approximately 1–2% of the annual lignin production, is employed for creating value-added products, whereas the remainder is utilized as a low-energy source for energy generation in power plants.27 Therefore, exploring strategies is essential for overcoming the constraints associated with lignin utilization and discovering innovative ways to derive more value from lignin.
Numerous investigations have been undertaken to discover alternative uses for lignin that offer greater value than its conventional use as a low-energy fuel. Lignin has been found to enhance mechanical properties of composite materials,28,29 as well as being useful in biomedical applications,30–35 fire retardancy,36,37 and more. Due to its hydrophobic nature, lignin has found valuable applications in situations requiring hydrophobic coatings. Several research studies have explored the wettability of coatings based on lignin. These investigations have revealed that various types of lignin exhibit varying levels of hydrophobicity.38 Furthermore, researchers have employed different modification methods, such as esterification,39 acetylation,40 silylation,40 and methylation,41 on raw lignin to assess their impact on the hydrophobic properties of coatings using modified lignin. In addition, Souza Jr et al.42 investigated the effects of UV and plasma treatments on coatings using raw lignin, resulting in the successful creation of super-hydrophobic coatings. One limitation of utilizing lignin to create hydrophobic coatings is its insolubility in water. Alwadani et al.41 investigated the influence of solvents on the hydrophobic properties of lignin-based coatings and determined that using pure water as a solvent for lignin is unsuitable for coating formation. Consequently, additional organic solvents such as acetone and dimethyl sulfoxide must be introduced to water to generate a lignin suspension.43 This reliance on organic solvents to create lignin suspensions prevents these coatings from being entirely derived from renewable sources. Moreover, Hambardzumyan et al.44 studied the interaction between organosolv lignin and cellulose nanocrystals by creating thin films from a suspension containing both components, resulting in water-resistant thin films.
The most commonly used technique for assessing the wettability of various surfaces, including solids and thin films, is the measurement of the water contact angle (WCA).45 Among the available methods for measuring WCA, the sessile drop method is particularly popular for thin film structures due to its ease of use and ability to provide valuable information.46 This study utilized the sessile drop method to evaluate surface wettability. Surfaces are classified by their WCA: less than 90° as non-hydrophobic, greater than 90° as hydrophobic, and over 150° as superhydrophobic.
In this study, we employed a method for formulating a lignin suspension which includes mixing methylcellulose (MC) with lignin, allowing for the stable dispersion of lignin in water. This method had previously been utilized in a research study to introduce water resistance to biofoams.14 However, in this study, the focus was placed on preparing thin coatings based on organosolv lignin through spin-coating. The lignin was characterized using nuclear magnetic resonance (NMR) and Gel permeation chromatography (GPC) techniques. Subsequently, glass wafers were coated with suspensions containing different lignin concentrations, and the morphology and wettability of the coated glass wafers were investigated. Finally, based on the experimental results, a numerical approach for determining the contact angle of the coated samples over time is presented. The results of this research can assist in the development of eco-friendly alternatives to current hydrophobic coatings, which rely on unsustainable materials like plastics.
2 Methodology
2.1 Materials
This research employed powdered lignin isolated from wheat straw via organosolv process.14 Wheat straw was cut to 3–5 mm length and mixed with biosolvent, containing formic acid, acetic acid and water, in elevated temperature. After specific reaction time the mixture was cooled down, filtrated and washed with pure biosolvent to obtain all the dissolved material, including hemicellulosic sugars and lignin, in filtrate. Lignin precipitation was done by adding water to the filtrate and precipitated lignin was recovered by filtration. Lignin was washed with water to remove dissolved material. Lignin was air dried. The MC powder, commercially known as Benecel MX-50000, was procured from Ashland Specialties (Belgium). The molecular weight averages of the MC powder were determined in our previous study,47 with the averages calculated as follows: Mn = 353 kg mol−1, Mw = 534 kg mol−1, and Mz = 756 kg mol−1. Additionally, the degree of substitution (DS) for the MC powder was found to be 1.87. Dimethyl sulfoxide-d6 (DMSO-d6), Chromium (3) acetylacetonate, and 5 mm NMR tubes were supplied by Sigma-Aldrich (Germany). Glass wafers were procured from Greatcell Solar Materials Pty Ltd (Australia). All chemicals were used as received without any modifications or alterations.2.2 Lignin characterization
Nuclear magnetic resonance (NMR) spectroscopy was used to analyze the chemical structure of the organosolv lignin utilized in this study. Specifically, a Bruker Avance III 400 MHz spectrometer (USA) was employed to conduct 1H NMR and 2D HSQC NMR experiments on the organosolv lignin. These experiments were carried out at a temperature of 20 °C using a solution containing 120 mg of organosolv lignin dissolved in 0.5 mL of DMSO-d6. Chromium(III) acetylacetonate was added to the solution (0.02 M) as a relaxation agent for all nuclei. For the 2D HSQC NMR, a matrix of 256 × 2048 points was obtained through 24 scans. Data processing was conducted using Bruker TopSpin software. The solvent (DMSO-d6) peak was utilized as the chemical shift reference point for spectrum calibration (δC/δH = 39.51/2.5).48 The S/G ratio of the organosolv lignin was determined by calculating the volume integrals of the S2/6 and G2 contours, then doubling the integral value of the G2 contour.49The molar mass distribution of the organosolv lignin was characterized using an high-performance liquid chromatography (HPLC) system, specifically an Agilent 1100 (USA). A calibration curve was created with polystyrene sulfonate standards (1000–29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 g mol−1), ascorbic acid (176 g mol−1), and NaCl (58 g mol−1; detection with a refractive index detector). Molar masses were determined based on the UV signal at 280 nm. The columns used were Polymer Standards Service MCX, 8 × 300 mm, comprising three columns with pore sizes of 100 Å, 500 Å, and 1000 Å. The flow rate was 0.7 mL min−1, and the injection volume was 50 μL. Samples were dissolved in eluent (0.1 M NaOH) at a concentration of approximately 2 mg mL−1. Both samples and standards were filtered with syringe filters (Pall Acrodisc 0.2 μm) before analysis.
800 g mol−1), ascorbic acid (176 g mol−1), and NaCl (58 g mol−1; detection with a refractive index detector). Molar masses were determined based on the UV signal at 280 nm. The columns used were Polymer Standards Service MCX, 8 × 300 mm, comprising three columns with pore sizes of 100 Å, 500 Å, and 1000 Å. The flow rate was 0.7 mL min−1, and the injection volume was 50 μL. Samples were dissolved in eluent (0.1 M NaOH) at a concentration of approximately 2 mg mL−1. Both samples and standards were filtered with syringe filters (Pall Acrodisc 0.2 μm) before analysis.
2.3 Thin film deposition
Two distinct groups of suspensions were prepared. In the first group, the primary component was lignin and tap water. We used tap water due to the industrial scalability of the process; the water analysis was previously published elsewhere.50 In contrast, the second group of suspensions featured a mixture of lignin and MC as the main components, with tap water added to this mixture. In both sets of suspensions, one-third of the water was heated to 80 °C, while the remaining two-thirds were kept cold with an ice bath (4 °C). The hot water was added to the powder, and the resulting mixture was stirred for 5 minutes at a rate of 800 rpm using a magnetic stirrer. Following this, the cold water was gradually introduced into the suspension while stirring, and this mixture was stirred for an additional 30 minutes. A total of 14 suspensions were prepared, with their lignin and MC wt% summarized in Table 1. The constant 1 wt% of MC in the set 2 suspensions provided the optimal consistency for the film-forming process.| No. | Suspension | Lignin wt% | MC wt% | |
|---|---|---|---|---|
| Set 1 | 1 | 0.5Lig–0MC | 0.5 | 0 |
| 2 | 1Lig–0MC | 1 | 0 | |
| 3 | 1.5Lig–0MC | 1.5 | 0 | |
| 4 | 2Lig–0MC | 2 | 0 | |
| Set 2 | 5 | 0.5Lig–1MC | 0.5 | 1 |
| 6 | 1Lig–1MC | 1 | 1 | |
| 7 | 1.5Lig–1MC | 1.5 | 1 | |
| 8 | 2Lig–1MC | 2 | 1 | |
| 9 | 2.5Lig–1MC | 2.5 | 1 | |
| 10 | 3Lig–1MC | 3 | 1 | |
| 11 | 3.5Lig–1MC | 3.5 | 1 | |
| 12 | 4Lig–1MC | 4 | 1 | |
| 13 | 4.5Lig–1MC | 4.5 | 1 | |
| 14 | 5Lig–1MC | 5 | 1 | |
Glass wafers (1.5 × 2.5 cm2) underwent a preparation process for spin-coating. This involved washing the wafers with tap water, followed by rinsing them with deionized Milli-Q-water, and finally, drying them with nitrogen gas. The spin-coating procedure was performed using a WS-650SX-6NPP/LITE spin coater from Laurell Technologies Corporation (USA), at a speed of 1500 rpm for 2 minutes. In cases where multiple layers were to be applied to the substrate, the coated substrate was left to thoroughly dry in room conditions before applying the next layer.
To assess the efficacy of blending MC with lignin in the process of preparing coating suspensions, four glass wafers were coated with suspensions from the set 1 (Table 1) for a single layer. An additional four glass wafers were then coated with suspensions from the set 2 (Table 1), maintaining the same lignin weight percentage as the first set of suspensions. Upon examination of the coated glass wafers, the suspensions from the set 2, where MC and lignin were combined, were selected for further coatings. Furthermore, determining the optimal number of layers for enhanced water resistance of the coated substrate involved an initial spin-coating experiment. This experiment utilized the 1.5Lig–1MC suspension to coat three glass wafers with 1, 2, and 3 coating layers, followed by conducting water contact angle tests on each. A comparison between the water contact angles of these samples is presented in Fig. S4 (ESI†). Finally, equipped with knowledge of the effective suspension preparation method and the ideal number of coating layers, suspensions in the set 2 were used to coat glass wafers with two layers of coating at various lignin concentration. The coating process was carried out on three glass wafers using each suspension, leading to a total of 30 glass wafers coated with 10 different coating suspensions, each applied in two layers.
2.4 Surface characterization
Field emission scanning electron microscopy (FE-SEM) with a Zeiss Sigma VP (Germany) instrument was employed to investigate the morphology of the coated surfaces. The electric potential was consistently maintained at 5 kV, and the SE2 signal was utilized to generate the images. To improve the conductivity of the coated glass wafers, they were securely mounted on carbon tape and then coated with a thin layer (20 nm) of an 80/20 Au/Pd mixture. This coating process was performed using a Q150R sputter coater manufactured by Quorum Technologies Ltd (United Kingdom). Furthermore, the surface of the coated substrates was examined by optical microscopy using a combination of Olympus BX53M and DP74 (Japan).2.5 Contact angle analysis
A goniometer, provided by Biolin Scientific (Finland), was utilized to determine the water contact angle on the surface of the specimens. The experimental procedure followed the methodology outlined in a research paper by Huhtamäki et al.51 In summary, 10 μL of Milli-Q water was deposited on the surface of the coated sample at a controlled rate of 2 μL s−1, and the water droplet image was recorded using a high-speed camera at a rate of 1.4 frames per second. Subsequently, using the goniometer software (OneAttension) provided by Biolin Scientific, the contact angle of the droplet as well as the baseline length of the droplet was calculated at each frame. Each test was conducted for 30 minutes, and for each coating suspension, the test was repeated three times, with each repetition being carried out on one of the three glass wafers coated with the respective coating suspension.The droplet is approximated as a spherical cap with volume
 | (1) |
 gives
gives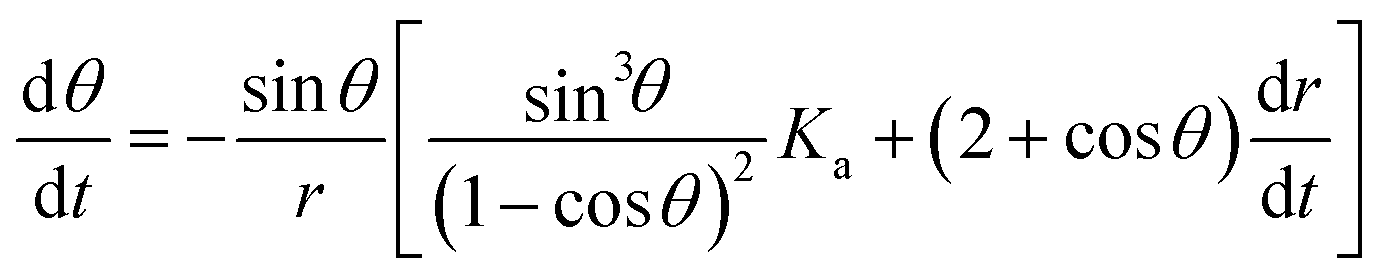 | (2) |
 i.e. the rate of volume change of the droplet per area of the droplet base. The contact angle can then be solved by numerically integrating eqn (2) as
i.e. the rate of volume change of the droplet per area of the droplet base. The contact angle can then be solved by numerically integrating eqn (2) as  . Taking the time-evolution of r from the experimental data leaves exactly two fitting parameters: θ0 and Ka (which we take to be constant for each lignin wt%).
. Taking the time-evolution of r from the experimental data leaves exactly two fitting parameters: θ0 and Ka (which we take to be constant for each lignin wt%).
On long timescales the baseline radius saturates to a constant value r∞ and eqn (2) can be approximated as
 | (3) |
3 Results and discussion
3.1 Lignin characterization
Nuclear magnetic resonance (NMR) spectroscopy was utilized to examine the chemical structure of the organosolv lignin used in this study. Fig. S1 (ESI†) displays the 1H NMR spectra of the organosolv lignin, where proton functional groups are identified based on the chemical shifts of the peaks.52,53 However, the 1H NMR spectra alone cannot identify all the functional groups in the lignin, highlighting the need for 2D NMR spectra. The 2D HSQC NMR spectra, which can identify many more functional groups of the organosolv lignin, are shown in Fig. S2 (ESI†), with the full data from the NMR software (Bruker TopSpin) provided as ESI.† An important feature of different types of lignin is the syringyl to guaiacyl (S/G) ratios, which can be accurately determined using the 2D HSQC NMR spectra.49Fig. 1 presents a section of the 2D HSQC NMR spectra of the organosolv lignin, with the blue contours indicating the peaks in the spectra, and the S2/6 and G2 contours labeled.54,55 For the organosolv lignin in this study, the S/G ratio was found to be 0.9. Additionally, the molar mass distribution of the organosolv lignin was analyzed using an HPLC system, as illustrated in Fig. S3 (ESI†). The number-averaged molar mass (Mn), mass-averaged molar mass (Mw), and Z-averaged molar mass (Mz) were determined to be 1.46 kg mol−1, 5.65 kg mol−1, and 14.41 kg mol−1, respectively.
 | ||
| Fig. 1 2D HSQC NMR spectra of the organosolv lignin dissolved in DMSO-d6. The full spectra is illustrated in Fig. S2 (ESI†), while here a section of the spectra is shown to visualise the two S2/6 and G2 contours and their corresponding peaks. | ||
3.2 SEM and optical microscopy
In our earlier investigations,14,15 we observed significant synergy between methylcellulose and various types of lignin when combined to create foams. Additionally, the fact that methylcellulose is an edible compound introduces a potential advantage in addressing the safety concerns associated with the coatings. So, we assessed the efficiency of mixing MC and lignin as the primary powder for the coating suspension, as opposed to using lignin only, in terms of surface coverage. Glass wafers were coated with one layer of each type of suspension and imaged using an optical microscope at 10× magnification. The images obtained from the two glass wafers are displayed in Fig. 2. The main difference between both images is the presence of 1.0 wt% MC next to 1.0 wt% lignin in the coating suspension (Fig. 2(a) and (b)), while the other coating suspension contains only lignin at 1.0 wt%. When the suspension contained MC and lignin was used, a noticeable enhancement in coating coverage became evident. In simpler terms, the presence of MC in the coating suspension led to a nearly uniform distribution of lignin particles across the surface, resulting in complete coverage, as depicted in Fig. 2(a). On the other hand, when only lignin was used in the suspension, it led to the aggregation of lignin particles on the surface, leaving a substantial portion uncovered, as shown in Fig. 2(b). As a result, it can be concluded that using MC promoted the even spread of lignin particles when applying the coating, significantly improving the surface coverage. | ||
| Fig. 2 Optical microscope images of two glass wafers coated with (a) 1Lig–1MC, and (b) 1Lig–0MC suspensions. | ||
SEM was employed to analyze the morphology of glass wafers coated with suspensions containing 1.0 wt% MC and varying wt% lignin. Fig. 3(a)–(j) displays images captured at a 500× magnification. It is evident that as the wt% of lignin increased in the coating suspension, a greater portion of the surface was covered. As illustrated in Fig. 3(g), complete coverage was achieved at 3.5 wt% lignin suspension. In other words, an increase in the wt% of lignin in the coating suspension led to a higher number of lignin particles in a specific area. However, surpassing the 3.5 wt% threshold did not result in improved coverage; instead, lignin particles began to aggregate. This phenomenon exhibited its most notable impact when the lignin wt% were at 4.5 and 5 wt% in the coating suspension. At these levels, the aggregation of lignin particles had a significant impact on surface coverage. This can be clearly observed by comparing Fig. 3(e) and (j). In the case of the sample with higher lignin concentration, particles start to aggregate, leading to a decrease in coverage compared to samples with lower lignin wt% in their coating suspension. Fig. 4(a) illustrates this phenomenon, showcasing the sample coated with suspension containing 5 wt% lignin. The image, captured at 500× magnification, reveals the aggregation of lignin particles in the top-left corner, with a noticeable area of the surface remaining uncovered. This can be attributed to the fact that the wt% of MC in the coating suspensions remained consistent for these samples. As the lignin wt% exceeded 3.5 wt% in the coating suspension, the quantity of MC in the coating suspension became insufficient to prevent the aggregation of lignin particles. Consequently, this led to the aggregation of lignin particles at higher lignin concentration and a reduced surface coverage.
 | ||
| Fig. 3 SEM images of the surface of the glass wafers coated with (a) 0.5Lig–1MC, (b) 1Lig–1MC, (c) 1.5Lig–1MC, (d) 2Lig–1MC, (e) 2.5Lig–1MC, (f) 3Lig–1MC, (g) 3.5Lig–1MC%, (h) 4Lig–1MC, (i) 4.5Lig–1MC, and (j) 5Lig–1MC suspensions. | ||
 | ||
| Fig. 4 SEM images of the (a) particles aggregation at 5 wt% lignin concentration, (b) local detachment of coating from substrate at 4.5 wt% lignin concentration, (c) microscopic linear detachment of coating from substrate at 4 wt% lignin concentration, and (d) local fracture of coating layer at 3.5 wt% lignin concentration. | ||
Apart from the aggregation of lignin particles at high lignin concentration (Fig. 4(a)), other types of defects were also identified at the microscopic scale in samples coated with suspensions containing 3.5 wt% lignin or higher. These defects are depicted in Fig. 4(a)–(d). Firstly, some local micro-detachments in the shape of semi-spheres were observed on these samples, resulting in small points of coating where lignin had shifted towards the edges of these spherical structures. This is shown in Fig. 4(b), taken from the sample coated with 4.5Lig–1MC suspension at 1750× magnification. In addition, the coating showed linear detachment from the substrate, as illustrated in Fig. 4(b), which was obtained from the sample coated with 4Lig–1MC suspension at 60× magnification. Lastly, tiny cracks in the coating layer were observed in these samples, causing the coating layer to split into two pieces, as illustrated in Fig. 4(d), which was captured from the sample coated with 3.5Lig–1MC suspension at 2500× magnification. When examining the surfaces of samples coated with suspensions containing 0.5 to 3.0 wt% lignin, no significant defects were observed.
3.3 Surface wettability
Fig. 5 depicts the water contact angle of glass wafers coated with different wt% of lignin suspensions at various time intervals. For each time point, a second-degree polynomial curve is fitted to the WCA values corresponding to samples coated with suspensions containing lignin concentration ranging from 0.5 to 4 wt%, and these curves are represented with dashed lines. Detailed WCA values and their associated standard deviations can be found in Table 2. The data suggests that initially, all samples treated with varying lignin wt% in their coating suspensions exhibited hydrophobic properties (WCA > 90°). In contrast to earlier studies that reported an initial contact angle of 50° for glass slides coated with unmodified kraft lignin dissolved in Ammonium hydroxide41 and an initial angle of 130° for paper sheets coated with esterified kraft lignin dissolved in acetone,39 our findings appear promising. This is particularly remarkable as water was employed as the solvent, and no modifications were made to the lignin in our investigation. However, as time progressed, the WCA decreased due to interfacial interactions between the coating and water, resulting in the dissolution of the coating in the water droplet. Fig. 6 displays the water droplet and its associated contact angle on the glass wafer coated with the 2Lig–1MC suspension at four distinct time points. It's noteworthy to mention that the majority of previous research in this particular field has placed primary emphasis on the initial contact angle, neglecting to adequately address the evolution of the contact angle over time. In contrast, our study not only explores the initial contact angle but also specifically investigates how the contact angle changes over time. | ||
| Fig. 5 WCA of coated glass wafers at different time points. A curve was fitted to WCA data for samples coated with 1 wt% MC and 0.5 to 5 wt% lignin suspensions at each time point. | ||
| Lignin wt% | WCA | |||||
|---|---|---|---|---|---|---|
| t = 0 s | t = 10 s | t = 60 s | t = 300 s | t = 900 s | t = 1800 s | |
| 0.5 | 100° ± 2° | 98° ± 2° | 96° ± 2° | 81° ± 5° | 55° ± 7° | 42° ± 4° |
| 1 | 101° ± 1° | 98° ± 1° | 92° ± 1° | 83° ± 3° | 70° ± 4° | 55° ± 3° |
| 1.5 | 106° ± 5° | 102° ± 6° | 97° ± 7° | 89° ± 3° | 80° ± 6° | 64° ± 7° |
| 2 | 110° ± 3° | 108° ± 4° | 100° ± 6° | 95° ± 6° | 85° ± 5° | 74° ± 6° |
| 2.5 | 114° ± 5° | 113° ± 4° | 108° ± 2° | 102° ± 1° | 94° ± 2° | 82° ± 3° |
| 3 | 114° ± 5° | 112° ± 5° | 107° ± 5° | 102° ± 7° | 95° ± 6° | 83° ± 7° |
| 3.5 | 112° ± 3° | 108° ± 2° | 104° ± 2° | 99° ± 4° | 90° ± 4° | 74° ± 4 |
| 4 | 111° ± 2° | 106° ± 1° | 97° ± 1° | 88° ± 3° | 78° ± 3° | 63° ± 5° |
| 4.5 | 110° ± 3° | 103° ± 3° | 92° ± 3° | 68° ± 4° | 0 | 0 |
| 5 | 108° ± 2° | 99° ± 3° | 84° ± 3° | 51° ± 2° | 0 | 0 |
 | ||
| Fig. 6 Deposited water droplet on the glass wafer coated with the 2Lig–1MC suspension with corresponding contact angles at four different times. | ||
Based on the predefined criteria, after 15 minutes, only two samples coated with suspension containing 2.5 wt% and 3.0 wt% lignin remained hydrophobic, and even these two transitioned to hydrophilic surfaces after 30 minutes. Additionally, Fig. 5 reveals that the WCA of the coatings did not follow a linear trend with increasing lignin wt% in the suspension. In simpler terms, throughout the observation period, samples coated with suspensions containing 2.5 wt% or 3.0 wt% lignin consistently exhibited the highest WCA values. Beyond these concentration levels, as the lignin wt% in the coating suspension increased, the WCA began to decrease. It is evident that samples coated with 4.5Lig–1MC and 5Lig–1MC suspensions became completely wet in less than 15 minutes (WCA = 0°). This trend can be attributed to the surface properties observed during SEM imaging. It was observed that in samples coated with suspensions containing lignin at wt% ranging from 3.5 and above, various types of surface defects were present, and these defects became more pronounced in the samples coated with 4.5Lig–1MC and 5Lig–1MC suspensions. In these cases, lignin aggregation occurred, resulting in a significant portion of the samples remaining incompletely covered. In other words, the defects in samples with higher lignin concentration led to decreased hydrophobicity. With an increase in lignin concentration in the suspension from 3.5 wt%, the aggregation of lignin particles intensified, causing samples coated with 4.5Lig–1MC and 5Lig–1MC suspensions to become completely wet in less than 15 minutes. The stability of the prepared suspension with a lignin concentration greater than 3.5 wt% can be improved by employing natural cross-linking agents. For instance, El Bouchtaoui et al.56 proposed using citric acid as a cross-linker to develop methylcellulose/lignin biocomposites that exhibit hydrophobicity and barrier properties. Our future research will focus on incorporating cross-linkers such as citric acid into our suspension preparation method.
The water droplet on the coated film can be treated as a spherical cap, and its volume can be determined using eqn (1). The contact angle of the droplet over time can be obtained through the numerical integration of eqn (2). To achieve this, the numerical integration of eqn (2) was fitted to experimental results employing the orthogonal distance regression (ODR) method. The fitting involved determining fitting parameters, namely the absorption rate (Ka) and initial contact angle (θ0). It was assumed that these parameters would remain constant over time, depending solely on the lignin weight fraction of the samples. Fig. 7 displays the Ka values obtained as fitting parameters using the ODR method. Additionally, a second-degree polynomial was fitted to these values (depicted by the dashed line), and the absorption rate associated with this fit was labeled as  . The sample coated with the 2Lig–1MC suspension shows the lowest absorption rate at 0.1698 mL m−2 s−1. The contact angle experiments revealed that the sample coated with the 2.5Lig–1MC suspension demonstrated the highest initial contact angle. The determined absorption rate for this sample was 0.1716 mL m−2 s−1. Even though the sample coated with the 2Lig–1MC showed the minimum absorption rate, the absorption rate value for the sample coated with the 2.5Lig–1MC is not considerably higher. Taking into consideration the potential errors introduced by the spherical cap approximation and the assumption of a constant absorption rate over time in the ODR fitting, it can be stated that the sample with the highest initial contact angle also exhibited the lowest absorption rate value.
. The sample coated with the 2Lig–1MC suspension shows the lowest absorption rate at 0.1698 mL m−2 s−1. The contact angle experiments revealed that the sample coated with the 2.5Lig–1MC suspension demonstrated the highest initial contact angle. The determined absorption rate for this sample was 0.1716 mL m−2 s−1. Even though the sample coated with the 2Lig–1MC showed the minimum absorption rate, the absorption rate value for the sample coated with the 2.5Lig–1MC is not considerably higher. Taking into consideration the potential errors introduced by the spherical cap approximation and the assumption of a constant absorption rate over time in the ODR fitting, it can be stated that the sample with the highest initial contact angle also exhibited the lowest absorption rate value.
 | ||
Fig. 7 Absorption rate of the samples coated with suspensions containing 1 wt% MC and varying lignin wt%. The values were obtained as a fitting parameter of the numerical model (blue dots) and a second degree polynomial was fitted to these values (dashed line). The specific  values obtained from the fitting are indicated by crosses. values obtained from the fitting are indicated by crosses. | ||
In Fig. 8, the contact angles of samples coated with suspensions containing 1, 2, 3, and 4 wt% of lignin are depicted over time. Utilizing the obtained values of Ka and θ0, numerical integration of eqn (2) was performed. The resulting contact angles for each coated sample over time were determined and presented as black lines in Fig. 8. Additionally, the numerical integration was repeated with the same θ0 values, but employing  obtained from fitting a second-degree polynomial to the Ka values as the absorption rate. The results of this integration are represented as dashed lines in Fig. 8. It is evident that the fitting is effective, and the spherical cap approximation proves capable of calculating the contact angle of surfaces when the necessary inputs are available. The contact angles of other samples, including both experimental and fitted values, are presented in Fig. S5 and S6 (ESI†).
obtained from fitting a second-degree polynomial to the Ka values as the absorption rate. The results of this integration are represented as dashed lines in Fig. 8. It is evident that the fitting is effective, and the spherical cap approximation proves capable of calculating the contact angle of surfaces when the necessary inputs are available. The contact angles of other samples, including both experimental and fitted values, are presented in Fig. S5 and S6 (ESI†).
 | ||
| Fig. 8 Water contact angle of the samples coated with suspensions containing 1 wt% MC and 1, 2, 3, and 4 wt% lignin, respectively. Both experimental (dots) and numerical model (lines) values are presented. | ||
4 Conclusion
This research investigated the feasibility of formulating a coating using a mixture of methylcellulose and organosolv lignin powders, employing water as the solvent. Glass wafers were coated with the suspension, and subsequent analysis demonstrated a uniform distribution of lignin particles, accompanied by observed water resistivity behavior. Additionally, under constant methylcellulose concentration, an optimal lignin concentration was identified, exhibiting the highest initial contact angle. Beyond this point, an elevation in lignin concentration led to a reduction in the initial contact angle and introduced aggregation of lignin particles, resulting in diminished surface coverage as observed in SEM images. Moreover, a numerical model based on the spherical cap approximation was introduced to characterize the time evolution of the water contact angle. The model demonstrated a strong fit with the experimental results. Notably, one of the determined fitting parameters, namely the absorption rate, revealed that the sample with the highest initial contact angle exhibited the lowest absorption rate. We believe the bio-based coating method we employed holds potential for application in industries such as packaging, contributing to a more sustainable and environmentally friendly process. Future research could replicate these methods for different lignin types to explore their impact on coating water resistivity, along with validating the numerical model across diverse lignin types.Author contributions
Conceptualization: All the authors contributed to the conceptualization. Experiments: K. M. and I. M. V. performed the experiments. Formal analysis: K. M., I. M. V., and T. M. contributed to the analysis. Writing draft: K. M. wrote the original draft. Revising draft: All the author revised the manuscript. Supervision: J. K. and M. A. supervised the work. Funding: T. M., J. K., and M. A. acquired the funding for the work.Data availability
The 2D HSQC NMR data of the organosolv lignin has been included as part of the ESI.† The data that support the findings of this study are available from the corresponding author, T. M., upon reasonable request.Conflicts of interest
Patent for manufacturing bio-based protective structures is submitted. No other competing interests.Acknowledgements
The authors thank the Aalto University School of Science “Science-IT” project for the computational resources provided. We express our gratitude to Prof. Robin Ras and his research group for generously sharing the goniometer instrument for our project. We would also want to thank the Department of Bioproducts and Biosystems at Aalto University for providing us access to their test instruments during the project. The authors thank Leena Pitkänen, Jari Koivisto and the School of Chemical Engineering at Aalto University for their help in measuring the molar mass and the NMR spectra of the organosolv lignin. M. J. A. and J. K. acknowledge funding from FinnCERES flagship (151830423), Business Finland (211835), and Future Makers programs. M. J. A., T. M., and I. M. V. acknowledge funding from Business Finland (211909). M. J. A. acknowledges funding from the Finnish Cultural Foundation. I. M. V. acknowledges the Vilho, Yrjö and Kalle Väisälä Foundation of the Finnish Academy of Science and Letters for personal funding.References
- P. Stegmann, V. Daioglou, M. Londo, D. P. van Vuuren and M. Junginger, Nature, 2022, 612, 272–276 CrossRef CAS PubMed.
- I. Vollmer, M. J. Jenks, M. C. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van Der Laan, F. Meirer, J. T. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- K. Zhang, A. H. Hamidian, A. Tubić, Y. Zhang, J. K. Fang, C. Wu and P. K. Lam, Environ. Pollut, 2021, 274, 116554 CrossRef CAS PubMed.
- J. N. Hahladakis, C. A. Velis, R. Weber, E. Iacovidou and P. Purnell, J. Hazard. Mater., 2018, 344, 179–199 CrossRef CAS PubMed.
- S. B. Borrelle, J. Ringma, K. L. Law, C. C. Monnahan, L. Lebreton, A. McGivern, E. Murphy, J. Jambeck, G. H. Leonard and M. A. Hilleary, et al. , Science, 2020, 369, 1515–1518 CrossRef CAS PubMed.
- W. W. Lau, Y. Shiran, R. M. Bailey, E. Cook, M. R. Stuchtey, J. Koskella, C. A. Velis, L. Godfrey, J. Boucher and M. B. Murphy, et al. , Science, 2020, 369, 1455–1461 CrossRef CAS PubMed.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- L. Zhang, Y. Huang, P. Sun, Y. Hai and S. Jiang, Soft Matter, 2021, 17, 5231–5239 RSC.
- X. Qian, N. Mu, X. Zhao, C. Shi, S. Jiang, M. Wan and B. Yu, Soft Matter, 2023, 19, 6097–6107 RSC.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicola, K. Van Aelst and M. Maesen, et al. , Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- T. Mäkinen, J. Koivisto, E. Pääkkönen, J. A. Ketoja and M. J. Alava, Soft Matter, 2020, 16, 6819–6825 RSC.
- M. Reichler, S. Rabensteiner, L. Törnblom, S. Coffeng, L. Viitanen, L. Jannuzzi, T. Mäkinen, J. R. Mac Intyre, J. Koivisto, A. Puisto and M. J. Alava, Sci. Rep., 2021, 11, 24306 CrossRef CAS PubMed.
- I. Y. Miranda-Valdez, S. Coffeng, Y. Zhou, L. Viitanen, X. Hu, L. Jannuzzi, A. Puisto, M. A. Kostiainen, T. Mäkinen, J. Koivisto and M. Alava, Cellulose, 2023, 30, 2253–2266 CrossRef CAS.
- I. Y. Miranda-Valdez, M. R. Yazdani, T. Mäkinen, S. Coffeng, L. Viitanen, J. Koivisto and M. J. Alava, J. Energy Storage, 2023, 73, 109036 CrossRef.
- K. M. Cheung, H. L. Chong, Z. Jiang and T. Ngai, Soft Matter, 2023, 19, 7696–7707 RSC.
- C. G. Yoo, X. Meng, Y. Pu and A. J. Ragauskas, Bioresour. Technol., 2020, 301, 122784 CrossRef CAS PubMed.
- D. Watkins, M. Nuruddin, M. Hosur, A. Tcherbi-Narteh and S. Jeelani, J. Mater. Res. Technol., 2015, 4, 26–32 CrossRef CAS.
- M. Xie, J. Zhang, T. J. Tschaplinski, G. A. Tuskan, J.-G. Chen and W. Muchero, Front. Plant Sci., 2018, 9, 1427 CrossRef PubMed.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- M. M. Campbell and R. R. Sederoff, Plant Physiol., 1996, 110, 3 CrossRef CAS PubMed.
- Q. Liu, T. Kawai, Y. Inukai, D. Aoki, Z. Feng, Y. Xiao, K. Fukushima, X. Lin, W. Shi and W. Busch, et al. , Nat. Commun., 2023, 14, 4866 CrossRef CAS PubMed.
- M. Liu and P. J. Dyson, Nat. Commun., 2023, 14, 2830 CrossRef CAS PubMed.
- H. Luo and M. M. Abu-Omar, Encyclopedia of Sustainable Technologies, 2017, vol. 3, pp. 573–585 Search PubMed.
- D. Bajwa, G. Pourhashem, A. Ullah and S. Bajwa, Ind. Crops Prod., 2019, 139, 111526 CrossRef CAS.
- A. Vishtal and A. Kraslawski, BioResources, 2011, 6, 3547–3568 Search PubMed.
- I. F. Demuner, J. L. Colodette, A. J. Demuner and C. M. Jardim, BioResources, 2019, 14, 7543–7581 Search PubMed.
- I. Klapiszewska, A. Parus, L. Lawniczak, T. Jesionowski, L. Klapiszewski and A. Slosarczyk, Cem. Concr. Compos., 2021, 124, 104250 CrossRef CAS.
- T. Aso, K. Koda, S. Kubo, T. Yamada, I. Nakajima and Y. Uraki, J. Wood Chem. Technol., 2013, 33, 286–298 CrossRef CAS.
- J. Domínguez-Robles, Á. Cárcamo-Martínez, S. A. Stewart, R. F. Donnelly, E. Larrañeta and M. Borrega, Sustainable Chem. Pharm., 2020, 18, 100320 CrossRef.
- R. Liu, L. Dai, C. Xu, K. Wang, C. Zheng and C. Si, ChemSusChem, 2020, 13, 4266–4283 CrossRef CAS PubMed.
- I. Ullah, Z. Chen, Y. Xie, S. S. Khan, S. Singh, C. Yu and G. Cheng, Int. J. Biol. Macromol., 2022, 208, 819–832 CrossRef CAS PubMed.
- Y. Liu, X. Wang, Q. Wu, W. Pei, M. J. Teo, Z. S. Chen and C. Huang, Int. J. Biol. Macromol., 2022, 222, 994–1006 CrossRef CAS PubMed.
- R. Kumar, A. Butreddy, N. Kommineni, P. G. Reddy, N. Bunekar, C. Sarkar, S. Dutt, V. K. Mishra, K. R. Aadil and Y. K. Mishra, et al. , Int. J. Nanomed., 2021, 2419–2441 CrossRef CAS PubMed.
- S. Rai, B. K. Singh, P. Bhartiya, A. Singh, H. Kumar, P. Dutta and G. Mehrotra, J. Lumin., 2017, 190, 492–503 CrossRef CAS.
- L. Liu, M. Qian, P. Song, G. Huang, Y. Yu and S. Fu, ACS Sustainable Chem. Eng., 2016, 4, 2422–2431 CrossRef CAS.
- H. Yang, B. Yu, X. Xu, S. Bourbigot, H. Wang and P. Song, Green Chem., 2020, 22, 2129–2161 RSC.
- M. Borrega, S. Päärnilä, L. G. Greca, A.-S. Jääskeläinen, T. Ohra-Aho, O. J. Rojas and T. Tamminen, Langmuir, 2020, 36, 9675–9684 CrossRef CAS PubMed.
- S. S. Singh, A. Zaitoon, S. Sharma, A. Manickavasagan and L.-T. Lim, Int. J. Biol. Macromol., 2022, 223, 1243–1256 CrossRef CAS PubMed.
- P. Buono, A. Duval, P. Verge, L. Averous and Y. Habibi, ACS Sustainable Chem. Eng., 2016, 4, 5212–5222 CrossRef CAS.
- N. Alwadani, N. Ghavidel and P. Fatehi, Colloids Surf., A, 2021, 609, 125656 CrossRef CAS.
- J. R. Souza Jr, J. R. Araujo, B. S. Archanjo and R. A. Simão, Prog. Org. Coat., 2019, 128, 82–89 CrossRef.
- E. Melro, L. Alves, F. E. Antunes and B. Medronho, J. Mol. Liq., 2018, 265, 578–584 CrossRef CAS.
- A. Hambardzumyan, L. Foulon, N. Bercu, M. Pernes, J.-E. Maigret, M. Molinari, B. Chabbert and V. Aguié-Béghin, Chem. Eng. J., 2015, 264, 780–788 CrossRef CAS.
- S. M. Lößlein, R. Merz, D. W. Müller, M. Kopnarski and F. Mücklich, Sci. Rep., 2022, 12, 19389 CrossRef PubMed.
- M. F. Ismail, M. A. Islam, B. Khorshidi, A. Tehrani-Bagha and M. Sadrzadeh, Adv. Colloid Interface Sci., 2022, 299, 102524 CrossRef CAS PubMed.
- I. Y. Miranda-Valdez, J. G. Puente-Córdova, F. Y. Rentera-Baltiérrez, L. Fliri, M. Hummel, A. Puisto, J. Koivisto and M. J. Alava, Food Hydrocolloids, 2024, 147, 109334 CrossRef CAS.
- M. Solutions, Stable Isotopes for Structural Biomolecular NMR, 2011, 75.
- S. D. Mansfield, H. Kim, F. Lu and J. Ralph, Nat. Protoc., 2012, 7, 1579–1589 CrossRef CAS PubMed.
- I. Y. Miranda-Valdez, L. Viitanen, J. Mac Intyre, A. Puisto, J. Koivisto and M. Alava, Carbohydr. Polym., 2022, 298, 119921 CrossRef CAS PubMed.
- T. Huhtamäki, X. Tian, J. T. Korhonen and R. H. Ras, Nat. Protoc., 2018, 13, 1521–1538 CrossRef PubMed.
- C. Inkrod, M. Raita, V. Champreda and N. Laosiripojana, BioEnergy Res., 2018, 11, 277–290 CrossRef CAS.
- J. Rönnols, E. Danieli, H. Freichels and F. Aldaeus, Holzforschung, 2020, 74, 226–231 Search PubMed.
- S. A. Ralph, J. Ralph, L. Landucci and L. Landucci, US Forest Prod. Lab., 2004, 506 Search PubMed.
- J.-L. Wen, S.-L. Sun, B.-L. Xue and R.-C. Sun, Materials, 2013, 6, 359–391 CrossRef PubMed.
- F.-Z. El Bouchtaoui, E.-H. Ablouh, M. Mhada, I. Kassem, M. H. Salim, S. Mouhib, Z. Kassab, H. Sehaqui and M. El Achaby, Int. J. Biol. Macromol., 2022, 221, 398–415 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00427b |
| This journal is © The Royal Society of Chemistry 2024 |