
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular behavior of silicone adhesive at buried polymer interface studied by molecular dynamics simulation and sum frequency generation vibrational spectroscopy†
Yuchen
Wu
ab,
Ting
Lin
ab,
Elizabeth
Santos
c,
Dongchan
Ahn
 c,
Ryan
Marson
c,
Pranab
Sarker
de,
Xiaoyun
Chen
c,
Frédéric
Gubbels
c,
Nick E.
Shephard
c,
Carol
Mohler
c,
Tao
Wei
*de,
Tzu-Chi
Kuo
c and
Zhan
Chen
*ab
c,
Ryan
Marson
c,
Pranab
Sarker
de,
Xiaoyun
Chen
c,
Frédéric
Gubbels
c,
Nick E.
Shephard
c,
Carol
Mohler
c,
Tao
Wei
*de,
Tzu-Chi
Kuo
c and
Zhan
Chen
*ab
aDepartment of Chemistry, University of Michigan, MI 48109, USA. E-mail: zhanc@umich.edu
bDepartment of Macromolecular Science and Engineering, University of Michigan, MI 48109, USA
cThe Dow Chemical Company, Midland, MI 48674, USA
dDepartment of Biomedical Engineering, University of South Carolina, Columbia, SC 29208, USA. E-mail: TAOW@mailbox.sc.edu
eDepartment of Chemical Engineering, University of South Carolina, Columbia, SC 29208, USA
First published on 3rd June 2024
Abstract
Silicones have excellent material properties and are used extensively in many applications, ranging from adhesives and lubricants to electrical insulation. To ensure strong adhesion of silicone adhesives to a wide variety of substrates, silane-based adhesion promotors are typically blended into the silicone adhesive formulation. However, little is known at the molecular level about the true silane adhesion promotion mechanism, which limits the ability to develop even more effective adhesion promoters. To understand the adhesion promotion mechanism of silane molecules at the molecular level, this study has used sum frequency generation vibrational spectroscopy (SFG) to determine the behavior of (3-glycidoxypropyl)trimethoxy silane (γ-GPS) at the buried interface between poly(ethylene terephthalate) (PET) and a bulk silicone adhesive. To complement and extend the SFG results, atomistic molecular dynamics (MD) simulations were applied to investigate molecular behavior and interfacial interaction of γ-GPS at the silicone/PET interface. Free energy computations were used to study the γ-GPS interaction in the sample system and determine the γ-GPS interfacial segregation mechanism. Both experiments and simulations consistently show that γ-GPS molecules prefer to segregate at the interface between PET and PDMS. The methoxy groups on γ-GPS molecules orient toward the PDMS polymer phase. The consistent picture of interfacial structure emerging from both simulation and experiment provides enhanced insight on how γ-GPS behaves in the silicone – PET system and illustrates why γ-GPS could improve the adhesion of silicone adhesive, leading to further understanding of silicone adhesion mechanisms useful in the design of silicone adhesives with improved performance.
Introduction
Polydimethylsiloxane (PDMS) is a major constituent of silicone elastomers with excellent mechanical properties, environmental resistance, insulation, and flexibility.1–7 Due to these remarkable properties, PDMS is widely used in many applications ranging from construction and electronics, to automotive manufacturing.4,8–11 Compared with other adhesives, PDMS-based adhesives have significantly higher thermal stability while retaining flexibility at lower temperatures, leading to their use in many outdoor applications under extreme conditions.12,13Despite its extensive applications, it can be challenging to use PDMS-based adhesives to adhere to all the possible types of substrates. For example, some PDMS elastomers may not bond well to polar thermoplastic substrates like poly(ethylene terephthalate) unless they are formulated appropriately. To improve the adhesion performance of PDMS elastomers, additives such as adhesion promoters (e.g., silane molecules) are added to the formulation.14,15 These silane molecules typically have head groups and end groups that may react with other materials. Typically, the head groups of a silane molecule consist of three methoxy, ethoxy, or chloride groups. Such head groups can react with hydroxyl groups at the interface to enhance adhesion. Depending on the application requirement, the silane tail group can be versatile, examples of which include amine, maleic anhydride, or epoxy groups. Among all the silane molecules, (3-glycidyloxypropyl)trimethoxysilane (γ-GPS) has been widely used in perhaps the greatest variety of addition curing silicone adhesive systems to enhance the adhesion to various substrates.16–20
We have applied a nonlinear optical spectroscopic technique, sum frequency generation (SFG) vibrational spectroscopy, to study the molecular behavior of γ-GPS at various interfaces in situ.16–23 Earlier studies have shown that the methoxy groups are ordered at the PET/neat γ-GPS interface.21,22 The addition of hydroxy-terminated dimethyl co-methylvinyl siloxane (DMMVS) to the γ-GPS bulk enhanced the interfacial ordering of γ-GPS at the PET/γ-GPS-DMMVS mixture interface.20 When a small amount of γ-GPS is added to PDMS matrix, the γ-GPS molecules can segregate to the PET/PDMS interface with ordered methoxy groups, while a small amount of DMMVS molecules in the PDMS matrix can enhance such segregation/ordering.23 It was found that the interfacial segregation and ordering of methoxy groups of γ-GPS at the PET/PDMS interface is important for adhesion.19 In addition to SFG, we carried out additional experiments to measure adhesion at the PET/PDMS adhesive interfaces,19 and concluded that the strong adhesion between PET and PDMS is partially caused by the chemical reactions between the ordered methoxy groups of γ-GPS and the PET hydroxyl end groups at the PET/PDMS interface. Therefore, the presence, either inherent or induced by interfacial segregation, and any ordering of the γ-GPS methoxy groups at the interface should occur before curing. After curing, the SFG signal intensity of methoxy groups decreased, because the interfacial γ-GPS methoxy groups were consumed.19 Similar conclusions can be obtained from the studies on PET/PDMS interfaces as well as nylon/PDMS interfaces.16,17
While our previous research studied the γ-GPS segregation to the PDMS/PET interface in silicone systems with and without silica fillers, but the orientation of γ-GPS molecule at the interface is still unknown. The orientation of the γ-GPS molecules at the interface may play a critical role in promoting adhesion, because the molecular orientation could influence the interfacial chemical reactions. In this study, SFG was utilized to deduce the absolute orientation of γ-GPS molecules at the PET/PDMS interface. To further interpret SFG results and provide more fundamental understanding on the mechanism regarding the molecular behavior of adhesion promoters such as γ-GPS in silicone adhesives, atomistic molecular dynamics (MD) simulations24–31 were applied to study γ-GPS in the PET/PDMS adhesive system. The diffusion of the γ-GPS silane molecules in the PDMS matrix to the buried PET/PDMS interface was investigated. The potential of mean force (PFM) profile32,33 was calculated to probe the interactions between the adhesion promotor γ-GPS and the two contacting bulk materials, PET and PDMS. With this approach, the reason why γ-GPS molecules segregate to the interface and prefer to stay at the interface can be addressed. The solvation free energies34–36 of the system at different γ-GPS methoxy group orientations were computed to deduce the preferred absolute orientation of the methoxy groups at the interface. The results obtained from computer simulation can be correlated to and validate those determined from SFG results.
Methods
Sum frequency generation vibrational spectroscopy
SFG is a second-order non-linear optical spectroscopic technique, which can probe various surfaces and buried interfaces in situ.25,37–47 According to the selection rules of SFG, SFG signal can only be generated from the surfaces and interfaces without bulk inversion symmetry,37–42i.e. the bulk materials cannot generate resonant SFG signal for most cases. In the SFG experiment, a fixed frequency visible laser beam and a frequency tunable IR laser beam are spatially and temporally overlapped at the buried interface of interest. When the IR frequency matches a resonant vibration frequency of the molecules, the SFG signal can be enhanced. The intensity of the SFG signals is determined by the orientation, ordering, and interfacial coverage of the molecules. SFG is sensitive to the monolayer of the molecules at the buried interface, providing unique molecular information on adhesion.Sample preparation
In the SFG experiment, silica windows (Altos Photonics, Inc, Bozeman, MT) were used as substrates. A 100 nm TiO2 film was coated onto each silica window by an electron-beam evaporator (EvoVac, Angstrom Engineering, USA). Aliphatic chain-deuterated poly(ethylene terephthalate) (d4-PET, Mn = 72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) was purchased from Polymer Source, Inc. (Quebec, Canada), and used to prepare a 1.5 wt% solution in 2-chlorophenol. Then a d4-PET film was made by dripping the d4-PET solution onto the window and spin-coating at 2000 rpm by a spin-coater from Specialty Coating Systems P-6000 (Speedline Technologies). The d4-PET coated silica window was placed on a hot plate at 90 °C for 2 h in air to ensure that all the solvent was evaporated. Similar d4-PET films coated on silica windows without deposited TiO2 film were also prepared using the same method. Unless otherwise noted, measurements were performed at ambient conditions in a climate-controlled laboratory (typical laboratory conditions 21 ± 2 °C and 40 ± 5% relative humidity).
000 Da) was purchased from Polymer Source, Inc. (Quebec, Canada), and used to prepare a 1.5 wt% solution in 2-chlorophenol. Then a d4-PET film was made by dripping the d4-PET solution onto the window and spin-coating at 2000 rpm by a spin-coater from Specialty Coating Systems P-6000 (Speedline Technologies). The d4-PET coated silica window was placed on a hot plate at 90 °C for 2 h in air to ensure that all the solvent was evaporated. Similar d4-PET films coated on silica windows without deposited TiO2 film were also prepared using the same method. Unless otherwise noted, measurements were performed at ambient conditions in a climate-controlled laboratory (typical laboratory conditions 21 ± 2 °C and 40 ± 5% relative humidity).
The adhesion promotors γ-GPS and 3-aminopropyl trimethoxysilane (referred to as amino silane below) were obtained from Millipore-Sigma, Inc. (St. Louis, MO). Hydroxy-terminated dimethyl co-methylvinyl siloxane (DMMVS) was supplied by The Dow Chemical Company, along with siloxane (PDMS) polymer. The silicone sample used in the experiments is a two-part platinum-catalyzed addition-cured elastomer. Part A contains vinyl-terminated PDMS matrix polymer (Vi-PDMS60) (99.067 wt%) that has Mn of about 60 kDa and Karstedt's catalyst complex (0.033 wt%). This Karstedt's catalyst loading in Part A yields a total Pt level of about 9 ppm (w/w) for curing of the total formulation when Part A and Part B are mixed at a 1:1 (w/w) ratio. Part B consists of Vi-PDMS60 (91.64 wt%), PDMS cross-linker (8.268 wt%) that is about 40 mol% randomly substituted with methylhydridosiloxane units and has Mn of about 900 Da, and 1-ethylnyl-1-cyclohexanol (Sigma-Aldrich) (0.092 wt%) as a catalyst inhibitor to permit working time before thermal curing. All materials were used as received without further purification. For the PDMS samples with added silane studied in the SFG experiments, the samples were made by mixing the amino silane or epoxy silane and the PDMS polymer, with the concentration of silane in PDMS at 2 wt%. SFG spectra were collected from the interfaces between d4-PET and silane or PDMS with 2 wt% silane (see the SFG sample geometry shown in Fig. 1). The molecular formulas of γ-GPS, amino silane, and DMMVS are shown in Fig. 2a.
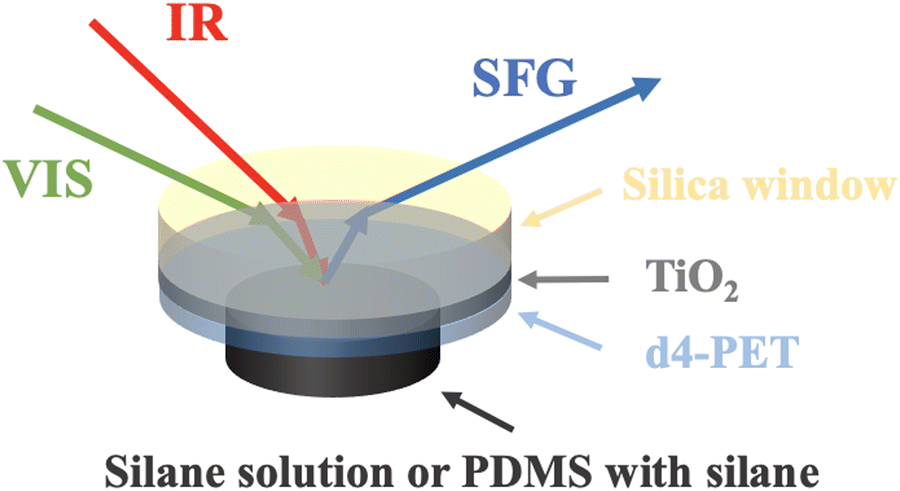 | ||
| Fig. 1 Sample configuration used to collect SFG spectra from the interface between d4-PET and silane or PDMS with 2 wt% silane. SFG spectra were also collected from the similar interfaces without the TiO2 coating on silica window. | ||
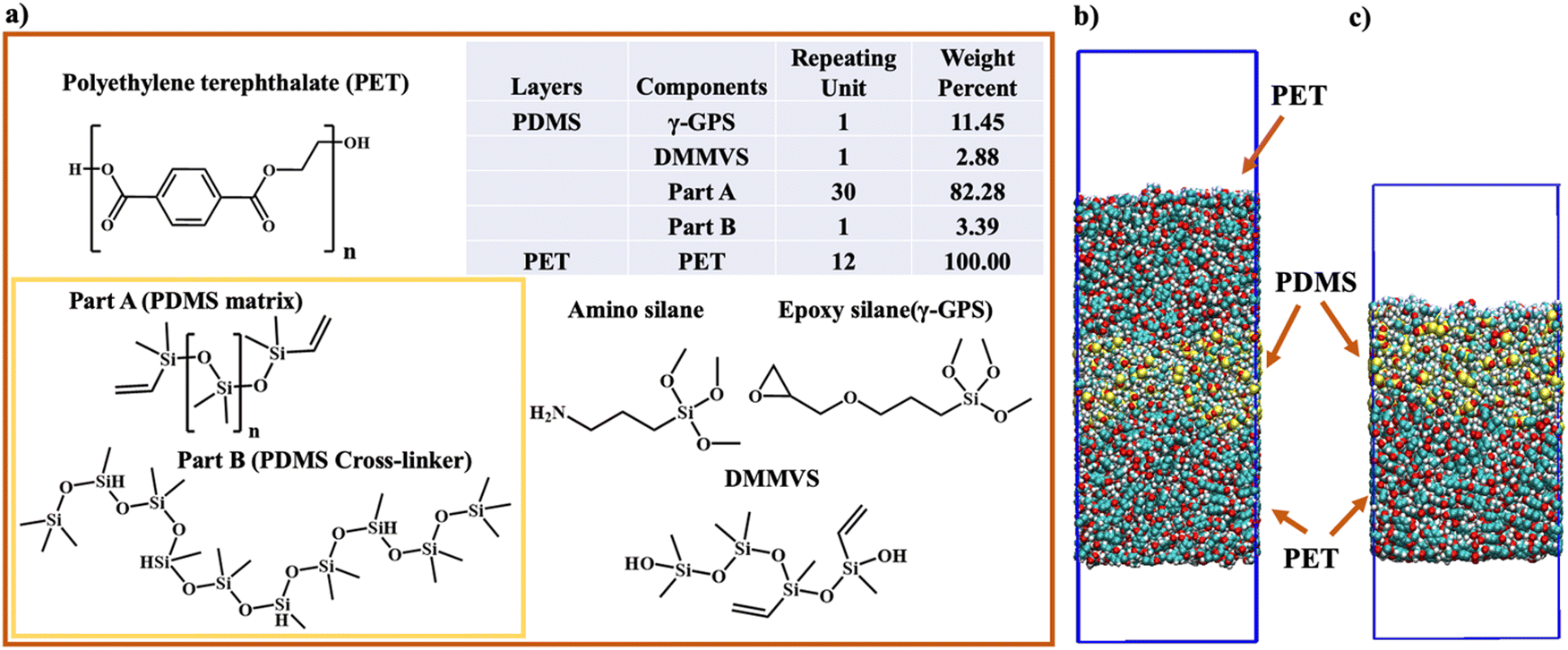 | ||
| Fig. 2 The chemical structures and the ratios of all components in the PET layer and the PDMS layer in computer simulation. (a) The chemical structure, repeating unit, and the weight percent of each component in PET and PDMS layers in computer simulation. The structure of amino silane, which was not included in the simulation but used in SFG experiment, is also displayed. (b) The initial structure of three-layer system. (c) The initial structure of two-layer system. PBC conditions were applied in X, Y and Z directions in both three-layer system and two-layer system. The top layer (6 nm × 6 nm × 16 nm) in Fig. 2(c) comprises PDMS components (Part A, Part B, γ-GPS, and DMMVS), with two boxes of vacuum to eliminate its interaction with the image layer of PET. | ||
Atomistic molecular dynamics simulation and free energy computation
The atomistic MD simulations were performed in this study, utilizing the GROMACS software with the CHARMM36 force field.48,49 For simplicity, we only studied uncured PDMS systems. To investigate the γ-GPS distribution at the PDMS/PET interface, a 3-layer system (Fig. 2b) with dimensions of 6 nm × 6 nm × 20 nm was simulated with periodic boundary conditions (PBC) in the X, Y, and Z directions. The initial system consists of two PET layers and a PDMS layer, which includes a silicone matrix (Part A), a silicone cross-linker (Part B), DMMVS and epoxy silane (γ-GPS) (Fig. 2a). Part A (silicone matrix) has a repeating unit of 30, while Part B (silicone cross-linker) has a repeating unit of 5. Amino silane and epoxy silane are the two silanes used in this research. DMMVS is an additive used with γ-GPS as an adhesion promotor in the silicone system.16,17 The PDMS layer, with a density of 0.97 g cm−3, was randomly packed, and energy minimization was employed at the start of MD simulations to prevent spatial overlap of the molecules. The PET layer, with a repeat unit of 12 and a density of 1.38 g cm−3, consisted solely of PET molecules, randomly packed into the simulation box.The leapfrog algorithm with a time step of 1.0 fs was adopted in simulations. The partial charge of the γ-GPS used in the simulation was obtained from CHARMM,50,51 while the partial charges of the Part A, Part B, PET and DMMVS needed for the MD simulations were obtained following the protocol in our previous work,26,52 utilizing the restrained ESP (RESP) framework implemented in Amber software.53 The structures of γ-GPS and DMMVS used for the RESP calculations were optimized at the density functional theory (DFT) level (see ESI† for details of DFT optimization and RESP calculations). To calculate the long-range electrostatic interactions, the particle mesh Ewald method (PME) was applied in the atomistic MD simulations. The cutoff distance was set to 1.2 nm. For the thermostat at 298 K, the inverse friction constant was 1.0 ps. For an NPT ensemble at constant temperature and pressure, the method of Parrinello–Rahman was applied to keep the pressure at 1 bar.
To establish a 3-layer system (Fig. 2b), the PET layer underwent a heating/annealing protocol in the NVT ensemble (at constant temperature and volume), gradually reaching 600 K, which is 100 K above its melting point. During heating (298–600 K), a 30-ns MD simulation was run every 50 K, followed by a 700 ns MD simulation at 600 K to relax the PET chains. The cooling process mirrored the heating, with a 30 ns MD simulation at every 50 K from 600 K to room temperature. This cycle was repeated twice for further PET layer relaxation. Before combining the PET/PDMS layers, the annealing process for the PDMS layer was omitted to prevent γ-GPS movement. The construction of the three-layer system involved placing a PDMS layer between two identical PET layers. Simulations comprised a 200 ns NVT ensemble run at room temperature, followed by an NPT ensemble run of heating/annealing relaxation. No significant changes occurred during the initial 200 ns MD simulation in the NVT ensemble. In the NPT process, the temperature gradually increased from 298.15 K to 500 K, with 30 ns MD simulations at every 50 K during heating. After reaching 500 K, an 800 ns MD simulation in the NPT ensemble was performed. The system's temperature was then gradually decreased to room temperature with 30 ns NPT simulations at every 50-degree interval.
To quantify γ-GPS interactions with PET and PDMS layers, the free energy was estimated using both methods of umbrella sampling32 and alchemical free energy perturbation.34,35 To reduce computational load, a two-layer (Fig. 2c) system was established using a similar heating/annealing protocol as the 3-layer system. The PMF profile for γ-GPS was computed to examine its interactions with polymer layers, shedding light on the distribution of γ-GPS. Nonequilibrium-steered molecular dynamics (SMD) simulations54,55 and weighted histogram analysis (WHAM) were used in the computation of PMF32 (see ESI† for details) to approximate the free energy profile of the interactions between a γ-GPS molecule and polymer layers.
The orientation-dependent solvation free energies (ΔF) for γ-GPS at the PET/PDMS interface were also calculated using FEP computations with a two-layer system. A γ-GPS molecule was rotated at the PET/PDMS interface, while three randomly chosen atoms from both the ends and the middle of the molecule were fixed during the simulation to maintain orientation stability. The free energy ΔF was computed as the molecule rotated (0° to 180°). Before the free energy calculation, the system's temperature was increased to 550 K, followed by a 200 ns MD simulation for complete relaxation, and then the temperature was gradually decreased to room temperature. In the computation of FEP, the two end states of a transformation with Hamiltonian H1 (in polymers: with coupling with the polymers) and H0 (in the vacuum: without coupling with the polymers) are coupled by a parameter λ,
| H(λ) = λH1 + (1 − λ)H0 | (1) |
The free energy difference ΔF between both states can be estimated using discrete values of λ,
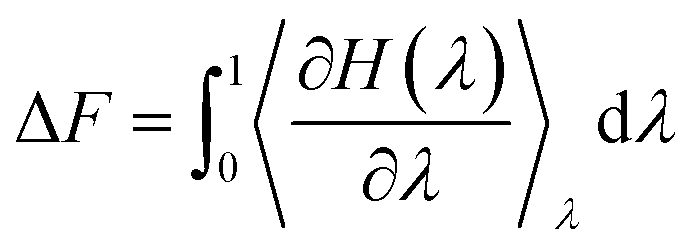 | (2) |
In this work, we used twenty different discrete values of λ (see ESI† for details of FEP calculations).
As a justification of the use of the two-layer system, we compared the interfacial segregation for both three-layer and two-layer systems at a given orientation. For both cases, we obtained the similar interfacial segregation, and thus we adopted two-layer system to calculate free energies at different orientations to reduce the computational load.
Results and discussion
Orientation study of the silane molecules at the PET/PDMS interface by SFG
To study the absolute orientation of γ-GPS at the PET/PDMS interface, we carried out SFG experiments. We first studied PET/silane interfaces without PDMS. Fig. 3a shows the SFG spectrum collected from the d4-PET/amino silane interface and the interface between d4-PET (coated on a thin TiO2 film on silica window) and amino silane. At the room temperature, amino silane is a liquid, thus the spectrum was collected from a solid/liquid interface. The peak at 2840 cm−1 in Fig. 3a can be assigned to the symmetric C–H stretching of the methoxy groups. Here we use this peak to monitor the interfacial orientation of the silane methoxy groups at the interface. The thin TiO2 film was coated on silica to create a non-resonant SFG signal to interfere with the SFG methoxy symmetric C–H stretching signal to determine the relative phase of this stretching mode vs. the TiO2 non-resonant signal. The constructive or destructive interferences can be deduced from the SFG signal intensity increase or decrease. With TiO2 coating, the intensity of the methoxy symmetric C–H stretching signal intensity increased in comparison to that collected without TiO2 coating. This means that the non-resonant signal from TiO2 coating has a constructive interference with the methoxy symmetric C–H stretching signal at the d4-PET/amino silane interface, showing that they have the same phase. This result is the same as our previously published paper.21 Since the hydrogen bond formed between the NH2 groups on amino silane and the oxygen atoms on PET, the NH2 groups point to the PET. Therefore, the methoxy groups of amino silanes at the d4-PET/amino silane interface face to the bulk silane. Based on this knowledge, the amino silane can be used as a reference to deduce the orientation of the epoxy silane γ-GPS. Fig. 3b shows the SFG spectrum collected from the d4-PET/epoxy silane γ-GPS interface and d4-PET (on TiO2 coating on silica)/epoxy silane interface. For the sample without TiO2 coating, the SFG signal from the methoxy groups at 2840 cm−1 was detected but has a very weak intensity. We hypothesize this is because the methoxy groups of the epoxy silane are not very well aligned with preferred orientation at the PET surface. From the d4-PET (on TiO2 coating on silica)/epoxy silane interface, a distinct negative peak at 2840 cm−1 was observed, showing a “destructive” interference or a different phase of the non-resonant SFG signal from TiO2 coating with the methoxy symmetric C–H stretching of the epoxy silane. Therefore, the methoxy groups of epoxy silane adopt a different absolute orientation compared to the methoxy groups of the amino silane at the d4-PET/silane interface. For epoxy silane γ-GPS, the methoxy groups face the PET at the d4-PET interface, while the epoxy groups face the silane bulk.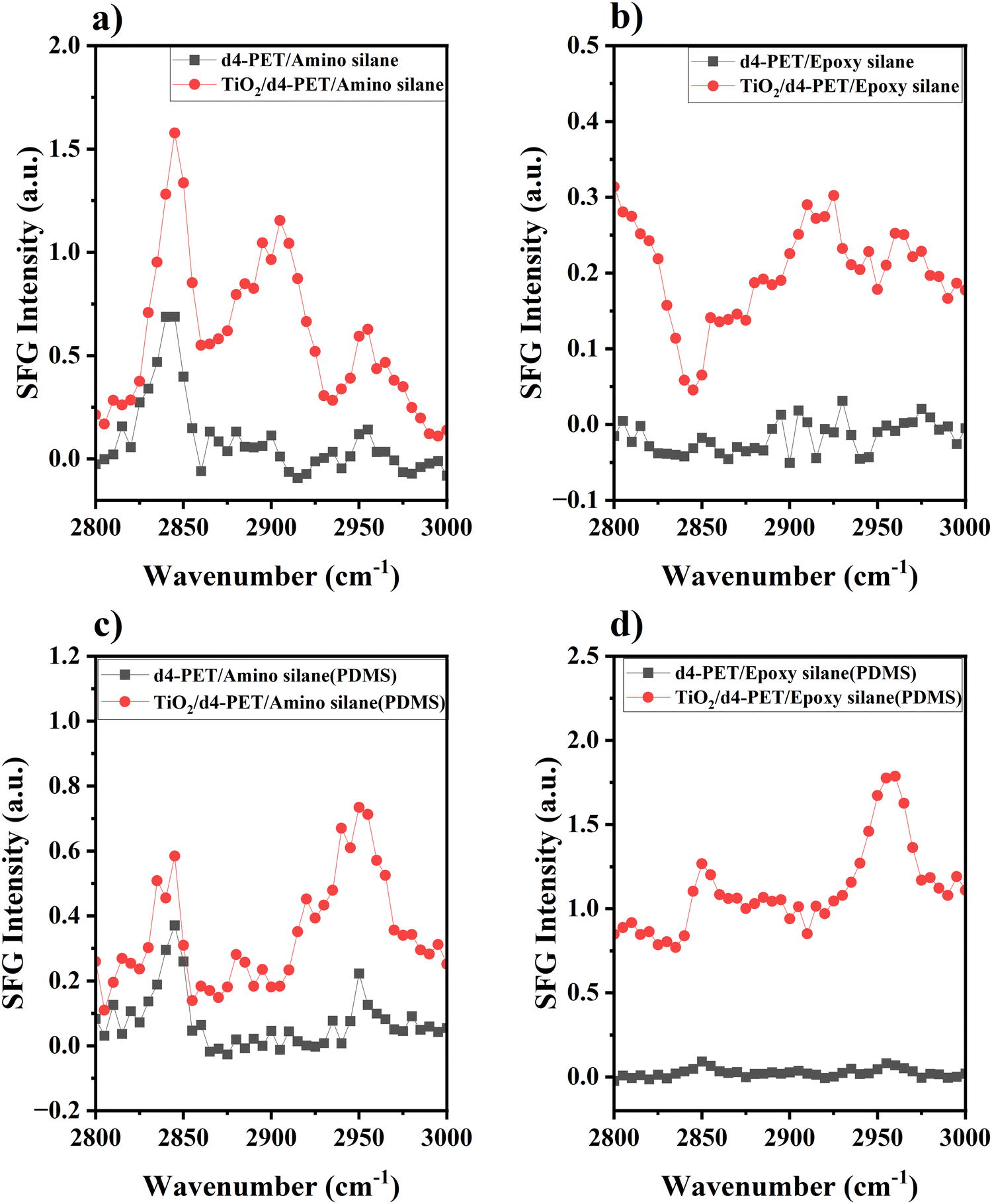 | ||
| Fig. 3 SFG spectra collected from the PET/silane interfaces and the PET/PDMS (with 2 wt% silane) interfaces. SFG spectra collected from (a) d4-PET/amino silane interface and TiO2/d4-PET/amino silane interface. (b) d4-PET/epoxy silane (γ-GPS) interface and TiO2/d4-PET/epoxy silane (γ-GPS) interface. (c) d4-PET/PDMS (with 2 wt% amino silane) interface and TiO2/d4-PET/PDMS (with 2 wt% amino silane) interface. (d) d4-PET/PDMS (with 2 wt% epoxy silane (γ-GPS)) interface and TiO2/d4-PET/PDMS (with 2 wt% epoxy silane (γ-GPS)) interface. | ||
Fig. 3c displays the SFG spectrum collected from the d4-PET/PDMS (with 2 wt% amino silane) interface and d4-PET (on TiO2 coating)/PDMS (with 2 wt% amino silane) interface. For the sample without TiO2 coating, the strong methoxy symmetric C–H stretching peak can be observed at 2840 cm−1, indicating that the amino silane molecules migrate to the d4-PET/PDMS interface and are ordered at the interface. With the TiO2 coating, the intensity of the methoxy symmetric C–H stretching signal intensity slightly increased, indicating its constructive interference with the TiO2 non-resonant signal. This suggests for amino silane, the methoxy groups face the PDMS side at the PET/PDMS interface, which may result from the formation of hydrogen bonds between amino silane NH2 groups and the oxygen atoms of PET at this interface. Fig. 3d shows the SFG spectrum collected from the d4-PET/PDMS (with 2 wt% epoxy silane γ-GPS) interface and d4-PET (on TiO2)/PDMS (with 2 wt% epoxy silane) interface. For the uncoated sample, some γ-GPS molecules segregated to the interface and were ordered there with preferred orientation, so the methoxy symmetric C–H stretching peak (2840 cm−1) could be observed at the interface. With TiO2 layer on silica window, the intensity of the methoxy symmetric C–H stretching signal intensity increased, indicating its constructive interference with the TiO2 non-resonant signal and that it has the same phase as the TiO2 non-resonant signal. This is consistent with the hypothesis that the methoxy groups of the epoxy silane γ-GPS at the d4-PET interface have the same absolute orientation as those of amino silanes. Therefore, for γ-GPS, the methoxy groups at the d4-PET/PDMS interface face the PDMS side, while the epoxy groups face the PET side. This is the same absolute orientation which we found from our simulations, which will be presented below.
As mentioned above, the PDMS system studied here is uncured system. In the past, our SFG results indicate that there are two requirements for good silicone adhesion to PET: (1) interfacial segregation of silane methoxy groups at interface before curing, evidenced by strong SFG methoxy signal; (2) chemical reaction between silane methoxy groups and PET hydroxyl end groups during the curing process, shown by SFG methoxy signal intensity decrease after curing.16 Here we show that interfacial methoxy groups in the uncured system face the PDMS side. We believe that during the curing process, methoxy groups change orientation to face to PET which enables an interfacial reaction. The cross-linking involved during thermal curing is beyond the scope of the current simulation study.
Atomistic simulations of adhesion promotor in the silicone adhesive system: three-layer system
We performed atomistic MD simulation and free energy analyses to better interpret SFG results and provide more insights into the interfacial interactions of γ-GPS. Fig. 4a and b display the final configuration after the heating/annealing relaxation. After the initial 300 ns of simulation in the NPT ensemble at 500 K, the γ-GPS molecules segregated to the PET/PDMS interface and remained there. At the same time, a crystalline structure gradually formed in the PET bulk and is discussed in more detail in the ESI.† After 800 ns, most γ-GPS molecules were segregated to the PET/PDMS interfaces, and it is likely that they could not move across the PET bulk. The density of PET, PDMS and γ-GPS in the last configuration of the three-layer system is shown in Fig. 4c, which demonstrated that the γ-GPS molecules were segregated at the interface after the system was equilibrated. A small number of γ-GPS molecules remained or were trapped in the PET and PDMS layers, after the system was gradually cooled down to room temperature. To verify the reproducibility and reliability of the result, two additional atomistic MD simulations with different random seeds were conducted. Both used the same NPT simulations discussed above and went through the heating/annealing processes. During these processes, the segregation of the γ-GPS molecules to the PET/PDMS interfaces and the formation of the crystalline structures in the PET bulk were observed, which is the same as the results presented above. Such results support the SFG results that γ-GPS molecules prefer to segregate to the PET/PDMS and remain there.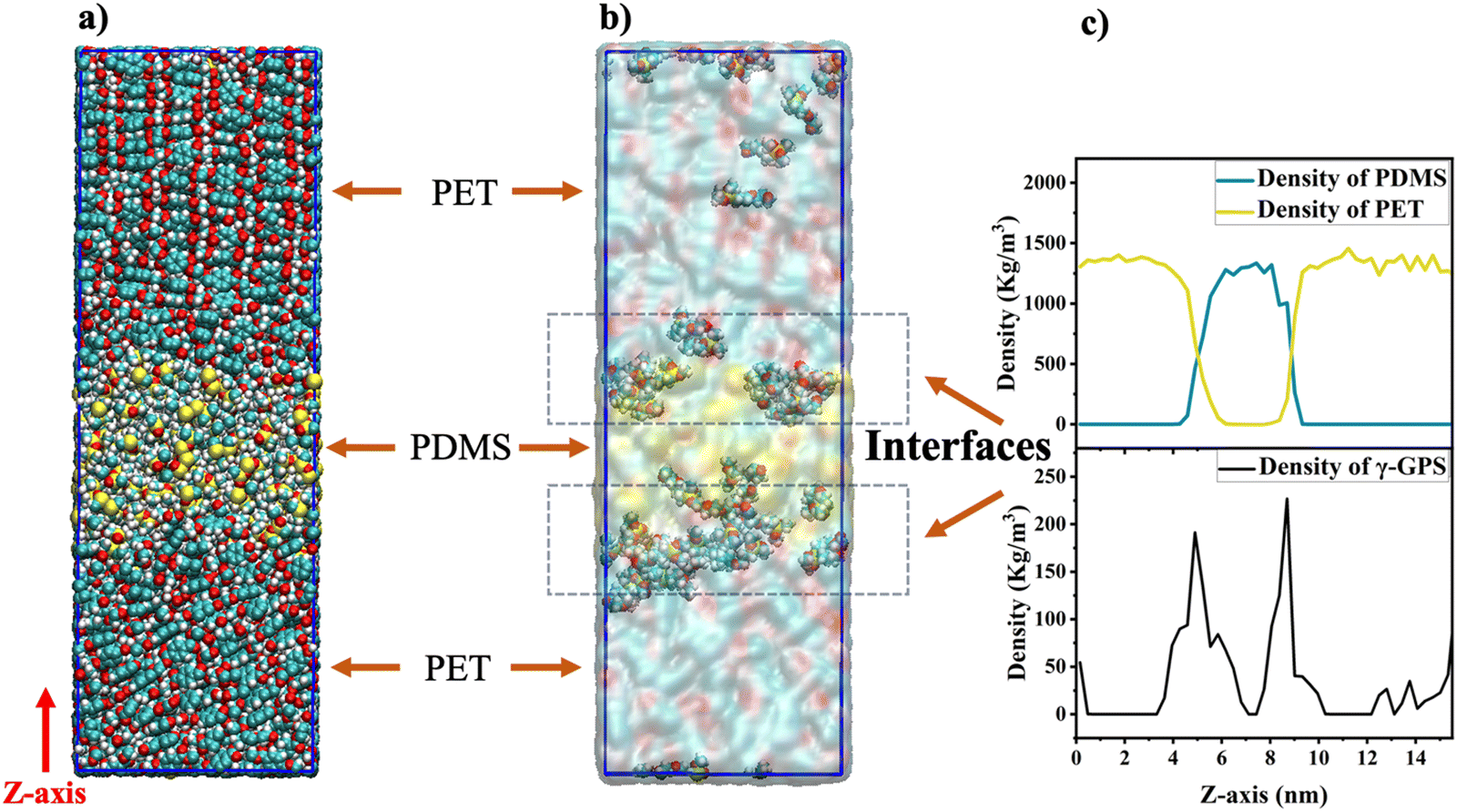 | ||
| Fig. 4 Segregation of adhesion promoter γ-GPS in silicone to the interface studied by atomistic MD simulations of the three-layer system with the PBC condition in X, Y and Z directions: (a) the final snapshot of the three-layer system after equilibrium; (b) the final snapshot of the three-layer system after equilibrium (only the γ-GPS molecules are shown in detail. The red, blue, yellow, and white balls indicate oxygen, carbon, silicon, and hydrogen atoms); (c) the density of PET, PDMS and γ-GPS in the sandwich structure system. | ||
To study how temperature affects the results and whether a correct temperature (500 K) in the heating/annealing process was chosen, the same system using the same NPT ensemble was also calculated to compare different temperatures: 450 K and 500 K. We believe that both temperatures are very close to the actual temperature in our previously published paper. Both temperatures are between the glass transition temperature Tg (350 K) and the melting temperature Tm (533.1 K) of PET. In the 450 K system, γ-GPS molecules were observed to gradually segregate to the interface and remain there after equilibrium, which is similar to the results of the 500 K MD simulation. The only difference is that the 450 K system requires more time to reach equilibrium.
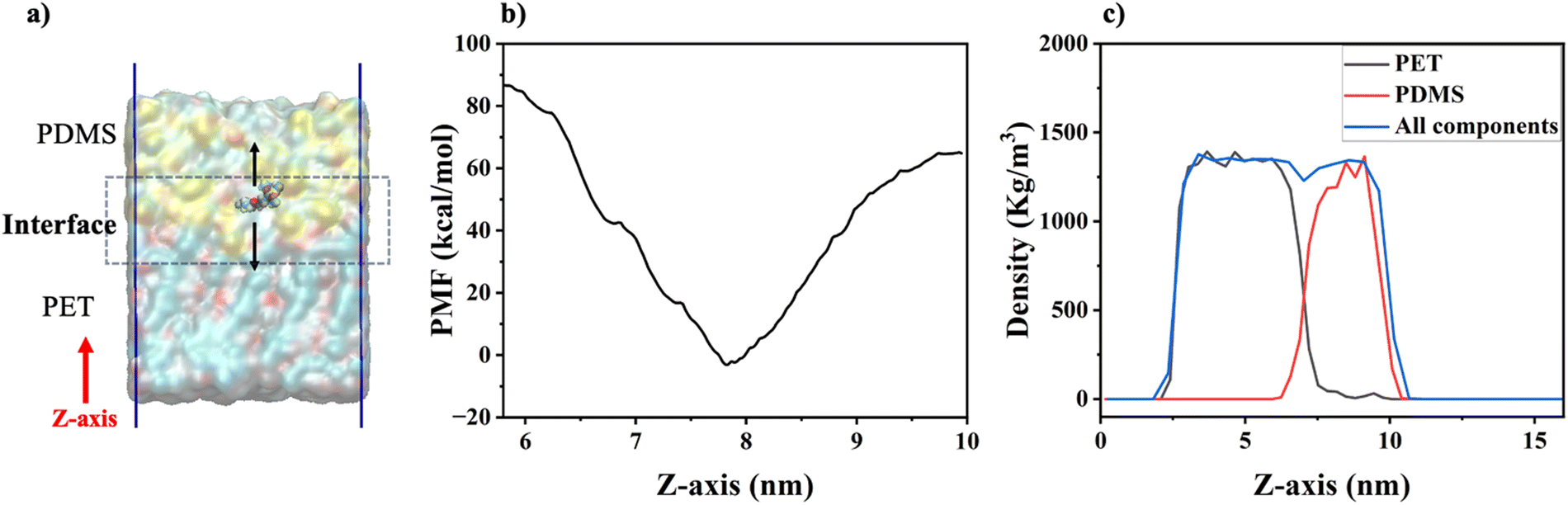 | ||
| Fig. 5 A schematic of the γ-GPS-pulling, the calculated PMF, and the density profiles of the simulation sample system: (a) the schematic of the pulling of the γ-GPS molecule in the steered MD simulations in the 2-layer system; (b) the PMF profile of the γ-GPS molecule at different locations of the box, calculated with umbrella sampling; (c) density profiles of overall system, the PET and the PDMS. Note: The Z-axis in the figure is the distance between the current γ-GPS position and the zero position on the Z-axis (or the bottom position of the simulation box). A γ-GPS molecule randomly chosen at the interface was pulled during the pulling process. | ||
It is interesting to observe that the γ-GPS molecule has a larger PMF in the PET phase than that of the PDMS phase. This may be due to the lower solubility of γ-GPS in PET compared to PDMS. As shown in Table 1, the large differences in Hildebrand solubility parameter at 20 °C reported previously16,57 between γ-GPS and PET, and between γ-GPS and PDMS, are consistent with lower solubility of γ-GPS in both PET and PDMS.
| Molecule | Solubility parameter (at 20 °C) (cal cm−3)0.5 |
|---|---|
| γ-GPS | 8.49 |
| PET | 10.26 |
| PDMS | 7.38 |
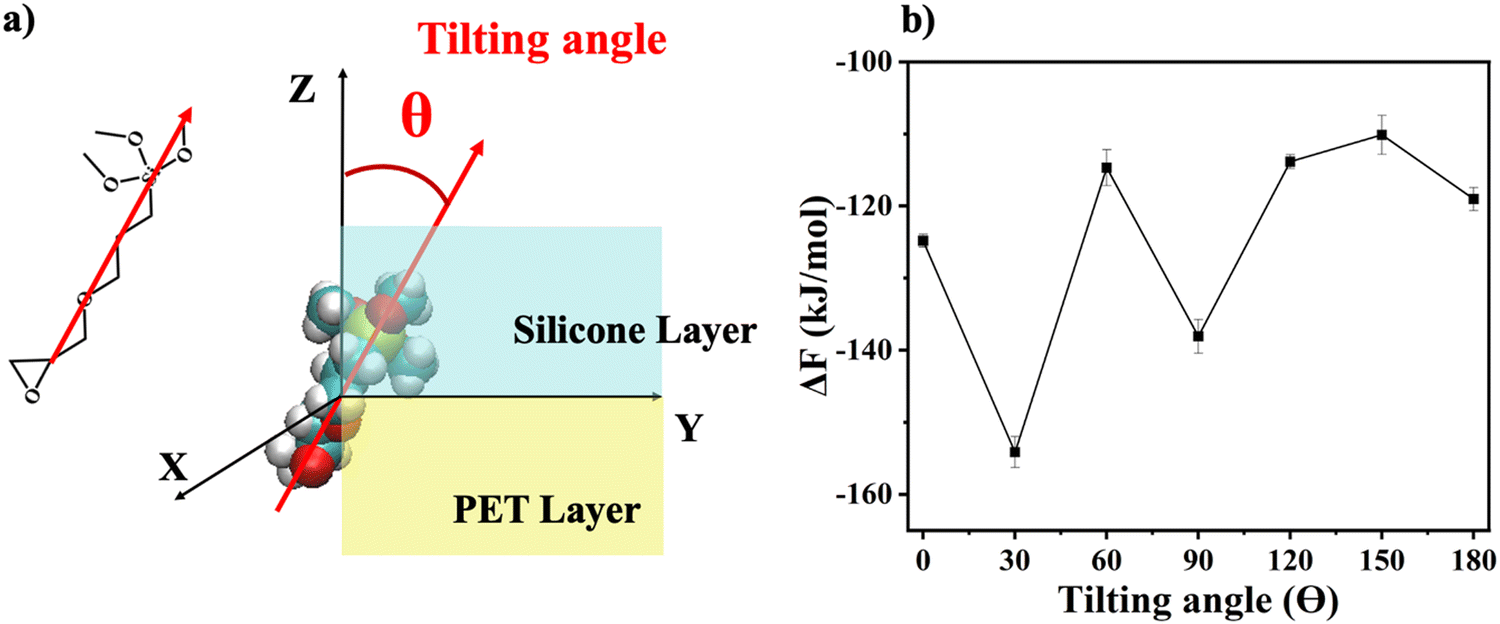 | ||
| Fig. 6 Orientation definition and orientation-dependent free energy (ΔF) profile: (a) the definition of the orientation angle (θ); (b) the calculated solvation free energy ΔF results of the system with different γ-GPS orientations. | ||
To further investigate the orientation distribution of γ-GPS at the PET/PDMS interface, the local chemical environment around the γ-GPS molecule was calculated at different orientations. Fig. 7 shows that the local Si concentration around the Si atom of the silane. Both silicone matrix and the γ-GPS molecules have Si atoms, but the system has very few γ-GPS molecules. Therefore, the Si atoms around the Si atom in silane are from the silicone matrix, which means that the local Si concentration presented in Fig. 7 represents the local silicone matrix concentration around the chosen γ-GPS molecule. In Fig. 7, the 30°-sample has a strong silicon peak at 4.8 Å, which is closest to the chosen γ-GPS molecule. For the 90°-sample, it has a broad and strong silicon peak at 6 Å, which is the second closest to the chosen γ-GPS molecule among all the samples. For the silicon peaks of the samples with orientation angles of 0°, 60°, 120°, 150° and 180°, they are farther and weaker than those of the 30°- and 90°-samples. This trend matches the profile of ΔF (Fig. 6b), which means that when the chosen γ-GPS molecule is closer to the silicone matrix, the entire system will be more stable, with a lower free energy. The high stabilities at 30° and 90° orientations of γ-GPS can be attributed to the solubility parameter difference between γ-GPS and silicone/PET. From the above simulations, it can be concluded that the methoxy groups of the γ-GPS molecule prefer to face toward the silicone as it is more stable. A schematic showing the interfacial segregation process of γ-GPS is shown in Fig. 8.
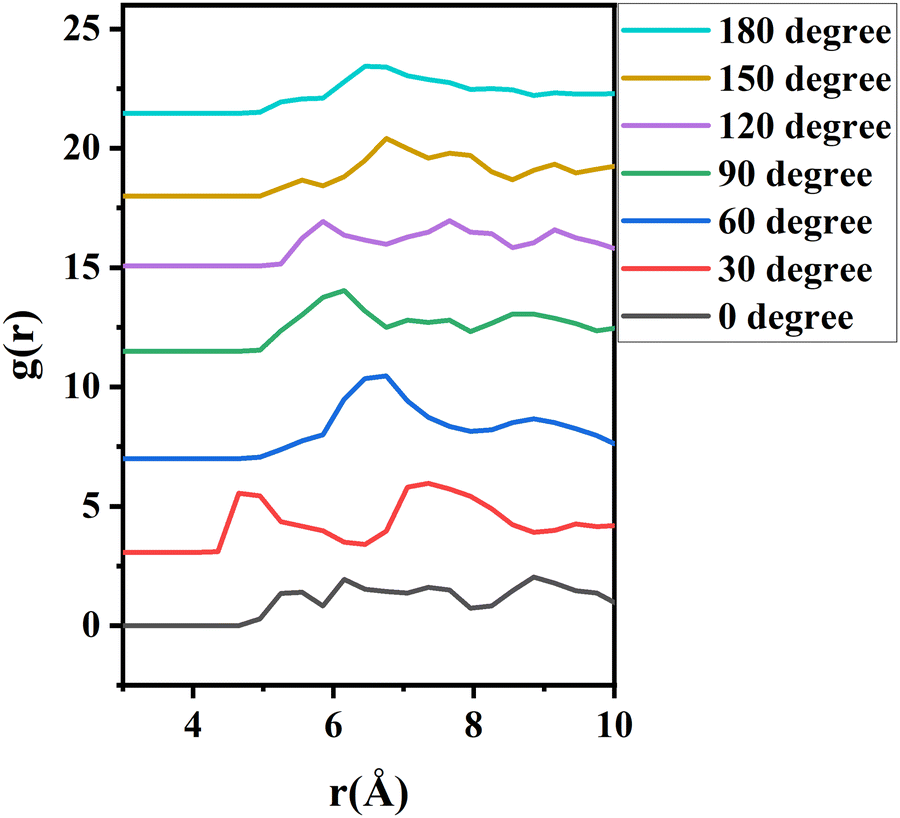 | ||
| Fig. 7 The radial distribution function (g(r)) of silicon atoms around the silicon atom of γ-GPS. The curves are offset. | ||
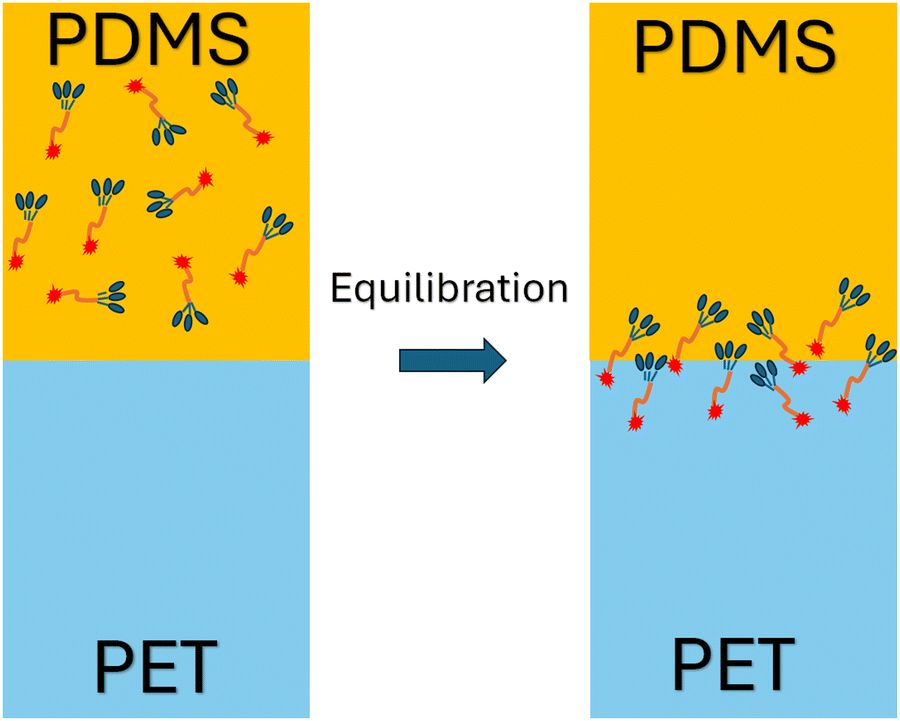 | ||
| Fig. 8 Schematic showing the interfacial segregation process of γ-GPS. | ||
Conclusions
In this research, SFG experimental measurements, atomistic MD simulation and free energy analyses were applied to study interfacial segregation of the adhesion promoter γ-GPS in the PDMS – PET system. In the SFG experiments, TiO2 coating layer was applied to silica used as PET substrate. This introduces an SFG non-resonance background signal to interfere with the SFG resonant signal from interfacial methoxy groups, helping to determine the absolute orientation of the γ-GPS molecule at the interface. Using the orientation of the methoxy groups of amino silane molecules at interface as a standard, the methoxy groups of epoxy silane molecules were found to face the PDMS side at the PET/PDMS interface. The SFG results supported the conclusion obtained from simulations. Since the interfacial methoxy groups should react with the hydroxyl end groups on PET during the curing process, they must have changed their absolute orientations to face the PET side while curing.Consistent with the experimental SFG measurement, atomistic MD simulation shows that the γ-GPS molecules diffuse to the PET/PDMS interface from the PDMS bulk. They prefer to stay at the interface, instead of remaining in the polymer bulk after the system was equilibrated. The interfacial segregation behavior of γ-GPS is consistent with its PMF profile, indicating that the PDMS/PET interface is the location with the lowest PMF. This observation also aligns with the solubility analyses that reveal significant solubility parameter differences between γ-GPS and PET, as well as between γ-GPS and PDMS.
Solvation free energies of the system were calculated for seven different γ-GPS molecule methoxy group orientations at the PDMS/PET interface. When the methoxy groups of the γ-GPS molecule face towards PDMS with 30-degree tilting, the free energy was calculated to be −154 ± 2 kJ mol−1, which is the lowest among all seven cases. This is related to the radial distribution function of silicon atoms (from PDMS) around the silicon atom of γ-GPS. For the chosen γ-GPS molecule with 30-degree orientation, the radial distribution function has a strong silicon peak at 4.8 Å, which is closest to this γ-GPS molecule, leading to the lowest free energy. This is because the interaction between γ-GPS and PET is larger than that between γ-GPS and PDMS, which is caused by the larger solubility parameter difference between γ-GPS and PET than that between γ-GPS and PDMS. For the 90-degree orientation, it has a broad and strong silicon peak at 6 Å, which is the second closest to the chosen γ-GPS molecule among all the samples, leading to the second lowest free energy.
The results of computer simulations and SFG experiments reported here demonstrate the interfacial segregation and ordering of γ-GPS methoxy groups at the buried PET/PDMS interface, indicating that they play significant roles in enhancing the PDMS adhesion to PET. These results on γ-GPS behavior and methoxy interfacial segregation/ordering offer important insights for consideration in optimizing the design of elastomeric silicone adhesive systems in the future.
Author contributions
Conceptualization: YW, ES, DA, XC, CM, TW, TCK, ZC; funding acquisition: ZC, CM; investigation: YW, TL, RM, PS, TW; methodology: all the authors; project administration: CM, TW, TCK, ZC; software: YW, TL, RM, PS, TW; writing – original draft: YW, ZC; writing – review & editing: all the authors.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This research is supported by The Dow Chemical Company.References
- I. Miranda, A. Souza, P. Sousa, J. Ribeiro, E. M. S. Castanheira, R. Lima and G. Minas, Properties and Applications of PDMS for Biomedical Engineering: A Review, J. Funct. Biomater., 2021, 13(1), 2, DOI:10.3390/jfb13010002.
- K. Raj M and S. Chakraborty, PDMS Microfluidics: A Mini Review, J. Appl. Polym. Sci., 2020, 137(27), 48958, DOI:10.1002/app.48958.
- S. Apolónia, A. Victor, J. Ribeiro and F. F. Araújo, Portugal Study of PDMS Characterization and Its Applications in Biomedicine: A Review, J. Mech. Eng. Biomech., 2019, 4(1), 1–9, DOI:10.24243/JMEB/4.1.163.
- M. P. Wolf, G. B. Salieb-Beugelaar and P. Hunziker, PDMS with Designer Functionalities—Properties, Modifications Strategies, and Applications, Prog. Polym. Sci., 2018, 83, 97–134, DOI:10.1016/j.progpolymsci.2018.06.001.
- A. Mata, A. J. Fleischman and S. Roy, Characterization of Polydimethylsiloxane (PDMS) Properties for Biomedical Micro/Nanosystems, Biomed. Microdevices, 2005, 7(4), 281–293, DOI:10.1007/s10544-005-6070-2.
- T. K. Kim, J. K. Kim and O. C. Jeong, Measurement of Nonlinear Mechanical Properties of PDMS Elastomer, Microelectron. Eng., 2011, 88(8), 1982–1985, DOI:10.1016/j.mee.2010.12.108.
- Q. Li, C. Wen, J. Yang, X. Zhou, Y. Zhu, J. Zheng, G. Cheng, J. Bai, T. Xu, J. Ji, S. Jiang, L. Zhang and P. Zhang, Zwitterionic Biomaterials, Chem. Rev., 2022, 122(23), 17073–17154, DOI:10.1021/acs.chemrev.2c00344.
- P. Pan, Z. Bian, X. Song and X. Zhou, Properties of Porous PDMS and Stretchability of Flexible Electronics in Moist Environment, J. Appl. Mech., 2020, 87(10), 101009, DOI:10.1115/1.4047775.
- J. Chen, J. Zheng, Q. Gao, J. Zhang, J. Zhang, O. Omisore, L. Wang and H. Li, Polydimethylsiloxane (PDMS)-Based Flexible Resistive Strain Sensors for Wearable Applications, Appl. Sci., 2018, 8(3), 345, DOI:10.3390/app8030345.
- D.-P. Wang, Z.-H. Zhao, C.-H. Li and J.-L. Zuo, An Ultrafast Self-Healing Polydimethylsiloxane Elastomer with Persistent Sealing Performance, Mater. Chem. Front., 2019, 3(7), 1411–1421, 10.1039/C9QM00115H.
- V. Anoop, S. Subramani, S. N. Jaisankar, C. Sohini and N. L. Mary, Mechanical, Dielectric, and Thermal Properties of Polydimethylsiloxane/Polysilsesquioxane Nanocomposite for Sealant Application, J. Appl. Polym. Sci., 2019, 136(12), 47228, DOI:10.1002/app.47228.
- A. Syafiq, B. Vengadaesvaran, N. Abd Rahim, A. K. Pandey, A. R. Bushroa, K. Ramesh and S. Ramesh, Transparent Self-Cleaning Coating of Modified Polydimethylsiloxane (PDMS) for Real Outdoor Application, Prog. Org. Coat., 2019, 131, 232–239, DOI:10.1016/j.porgcoat.2019.02.020.
- D. T. Vaimakis-Tsogkas, D. G. Bekas, T. Giannakopoulou, N. Todorova, A. S. Paipetis and N.-M. Barkoula, Effect of TiO2 Addition/Coating on the Performance of Polydimethylsiloxane-Based Silicone Elastomers for Outdoor Applications, Mater. Chem. Phys., 2019, 223, 366–373, DOI:10.1016/j.matchemphys.2018.11.011.
- L. Kersey, V. Ebacher, V. Bazargan, R. Wang and B. Stoeber, The Effect of Adhesion Promoter on the Adhesion of PDMS to Different Substrate Materials, Lab Chip, 2009, 9(7), 1002–1004, 10.1039/B813757A.
- M. Tang, Z. Jiang, Z. Wang, Y. Qin, Y. Jiang, L. Wu and Z. Li, High-Adhesion PDMS/Ag Conductive Composites for Flexible Hybrid Integration, Chem. Eng. J., 2023, 451, 138730, DOI:10.1016/j.cej.2022.138730.
- T. Lin, Y. Wu, E. Santos, X. Chen, D. Ahn, C. Mohler and Z. Chen, Molecular Insights into Adhesion at a Buried Silica-Filled Silicone/Polyethylene Terephthalate Interface, Langmuir, 2020, 36(49), 15128–15140, DOI:10.1021/acs.langmuir.0c02719.
- T. Lin, Y. Wu, E. Santos, X. Chen, J. Kelleher-Ferguson, C. Tucker, D. Ahn, C. Mohler and Z. Chen, Probing Covalent Interactions at a Silicone Adhesive/Nylon Interface, Langmuir, 2022, 38(8), 2590–2600, DOI:10.1021/acs.langmuir.1c03218.
- A. V. Vázquez, A. P. Boughton, N. E. Shephard, S. M. Rhodes and Z. Chen, Molecular Structures of the Buried Interfaces between Silicone Elastomer and Silane Adhesion Promoters Probed by Sum Frequency Generation Vibrational Spectroscopy and Molecular Dynamics Simulations, ACS Appl. Mater. Interfaces, 2010, 2(1), 96–103, DOI:10.1021/am900612r.
- C. Zhang, N. E. Shephard, S. M. Rhodes and Z. Chen, Headgroup Effect on Silane Structures at Buried Polymer/Silane and Polymer/Polymer Interfaces and Their Relations to Adhesion, Langmuir, 2012, 28(14), 6052 CrossRef CAS PubMed.
- C. L. Loch, D. Ahn and Z. Chen, Sum Frequency Generation Vibrational Spectroscopic Studies on a Silane Adhesion-Promoting Mixture at a Polymer Interface, J. Phys. Chem. B, 2006, 110(2), 914–918, DOI:10.1021/jp055377b.
- C. L. Loch, D. Ahn, C. Chen and J. Wang, Sum Frequency Generation Studies at Poly(Ethylene Terephthalate)/Silane Interfaces: Hydrogen Bond Formation and Molecular Conformation Determination, Langmuir, 2004, 20(13), 5467–5473, DOI:10.1021/la0494526.
- C. L. Loch, D. Ahn, C. Chen and Z. Chen, Polymer-Silane Interactions Probed by Sum Frequency Generation Vibrational Spectroscopy, J. Adhes., 2005, 81(3–4), 319–345, DOI:10.1080/00218460590944620.
- A. V. Vázquez, N. E. Shephard, C. L. Steinecker, D. Ahn, S. Spanninga and Z. Chen, Understanding Molecular Structures of Silanes at Buried Polymer Interfaces Using Sum Frequency Generation Vibrational Spectroscopy and Relating Interfacial Structures to Polymer Adhesion, J. Colloid Interface Sci., 2009, 331(2), 408–416, DOI:10.1016/j.jcis.2008.11.065.
- T. Wei and C. Ren, Theoretical Simulation Approaches to Polymer Research, Polymer Science and Innovative Applications, Elsevier, 2020, pp. 207–228 DOI:10.1016/B978-0-12-816808-0.00006-8.
- W. Guo, T. Lu, R. Crisci, S. Nagao, T. Wei and Z. Chen, Determination of Protein Conformation and Orientation at Buried Solid/Liquid Interfaces, Chem. Sci., 2023, 14(11), 2999–3009, 10.1039/D2SC06958J.
- H. Huang, C. Zhang, R. Crisci, T. Lu, H.-C. Hung, M. S. J. Sajib, P. Sarker, J. Ma, T. Wei, S. Jiang and Z. Chen, Strong Surface Hydration and Salt Resistant Mechanism of a New Nonfouling Zwitterionic Polymer Based on Protein Stabilizer TMAO, J. Am. Chem. Soc., 2021, 143(40), 16786–16795, DOI:10.1021/jacs.1c08280.
- S. Krishna, I. Sreedhar and C. M. Patel, Molecular Dynamics Simulation of Polyamide-Based Materials – A Review, Comput. Mater. Sci., 2021, 200, 110853, DOI:10.1016/j.commatsci.2021.110853.
- A.-T. Kuo, S. Urata, R. Koguchi, K. Yamamoto and M. Tanaka, Analyses of Equilibrium Water Content and Blood Compatibility for Poly(2-Methoxyethyl Acrylate) by Molecular Dynamics Simulation, Polymer, 2019, 170, 76–84, DOI:10.1016/j.polymer.2019.03.001.
- S. Yamamoto, R. Kuwahara and K. Tanaka, Dynamic Behaviour of Water Molecules in Heterogeneous Free Space Formed in an Epoxy Resin, Soft Matter, 2021, 17(25), 6073–6080, 10.1039/D1SM00529D.
- S. Yamamoto and K. Tanaka, Entropy-Driven Segregation in Epoxy-Amine Systems at a Copper Interface, Soft Matter, 2021, 17(5), 1359–1367, 10.1039/D0SM01600D.
- Y. Tang, Y. Liu, D. Zhang and J. Zheng, Perspectives on Theoretical Models and Molecular Simulations of Polymer Brushes, Langmuir, 2024, 40(2), 1487–1502, DOI:10.1021/acs.langmuir.3c03253.
- T. Wei, Md. S. J. Sajib, M. Samieegohar, H. Ma and K. Shing, Self-Assembled Monolayers of an Azobenzene Derivative on Silica and Their Interactions with Lysozyme, Langmuir, 2015, 31(50), 13543–13552, DOI:10.1021/acs.langmuir.5b03603.
- S. Park and K. Schulten, Calculating Potentials of Mean Force from Steered Molecular Dynamics Simulations, J. Chem. Phys., 2004, 120(13), 5946–5961, DOI:10.1063/1.1651473.
- M. R. Shirts and V. S. Pande, Solvation Free Energies of Amino Acid Side Chain Analogs for Common Molecular Mechanics Water Models, J. Chem. Phys., 2005, 122(13), 134508, DOI:10.1063/1.1877132.
- Z. Yuan, P. McMullen, S. Luozhong, P. Sarker, C. Tang, T. Wei and S. Jiang, Hidden Hydrophobicity Impacts Polymer Immunogenicity, Chem. Sci., 2023, 14(8), 2033–2039, 10.1039/D2SC07047B.
- S.-H. Chong, C. Lee, G. Kang, M. Park and S. Ham, Structural and Thermodynamic Investigations on the Aggregation and Folding of Acylphosphatase by Molecular Dynamics Simulations and Solvation Free Energy Analysis, J. Am. Chem. Soc., 2011, 133(18), 7075–7083, DOI:10.1021/ja1116233.
- Y. Wu, T. Wang, J. Gao, L. Zhang, J. D. B. Fay, S. Hirth, J. Hankett and Z. Chen, Molecular Behavior of 1K Polyurethane Adhesive at Buried Interfaces: Plasma Treatment, Annealing, and Adhesion, Langmuir, 2023, 39(9), 3273–3285, DOI:10.1021/acs.langmuir.2c03084.
- J. Gao, M. R. Khan, Y. Wu, D. D. Hawker, K. E. Gutowski, R. Konradi, L. Mayr, J. M. Hankett, M. Kellermeier and Z. Chen, Probing Interfacial Behavior and Antifouling Activity of Adsorbed Copolymers at Solid/Liquid Interfaces, Langmuir, 2023, 39(13), 4557–4570, DOI:10.1021/acs.langmuir.2c03056.
- J. Gao, P. Stengel, T. Lu, Y. Wu, D. D. Hawker, K. E. Gutowski, J. M. Hankett, M. Kellermeier and Z. Chen, Antiadhesive Copolymers at Solid/Liquid Interfaces: Complementary Characterization of Polymer Adsorption and Protein Fouling by Sum Frequency Generation Vibrational Spectroscopy and Quartz-Crystal Microbalance Measurements with Dissipation Monitoring, Langmuir, 2023, 39(34), 12270–12282, DOI:10.1021/acs.langmuir.3c01759.
- W. Guo, T. Lu, Z. Gandhi and Z. Chen, Probing Orientations and Conformations of Peptides and Proteins at Buried Interfaces, J. Phys. Chem. Lett., 2021, 12(41), 10144–10155, DOI:10.1021/acs.jpclett.1c02956.
- X. Lu, C. Zhang, N. Ulrich, M. Xiao, Y.-H. Ma and Z. Chen, Studying Polymer Surfaces and Interfaces with Sum Frequency Generation Vibrational Spectroscopy, Anal. Chem., 2017, 89(1), 466–489, DOI:10.1021/acs.analchem.6b04320.
- Z. Chen, Investigating Buried Polymer Interfaces Using Sum Frequency Generation Vibrational Spectroscopy, Prog. Polym. Sci., 2010, 35(11), 1376–1402, DOI:10.1016/j.progpolymsci.2010.07.003.
- X. Li and X. Lu, Interfacial Irreversibly and Loosely Adsorbed Layers Abide by Different Evolution Dynamics, ACS Macro Lett., 2019, 8(11), 1426–1431, DOI:10.1021/acsmacrolett.9b00472.
- C. Zhang, W. Chen, Y. Hong and X. Wang, Surface Activity and Structure of Temperature-Responsive Polymer Surfactants Based on PNIPAm at the Air/Solution Interface, Langmuir, 2021, 37(15), 4632–4638, DOI:10.1021/acs.langmuir.1c00320.
- Y. Hong, J. He, C. Zhang and X. Wang, Probing the Structure of Water at the Interface with Graphene Oxide Using Sum Frequency Generation Vibrational Spectroscopy, J. Phys. Chem. C, 2022, 126(3), 1471–1480, DOI:10.1021/acs.jpcc.1c08328.
- R. Harada, D. Kawaguchi, S. Yamamoto and K. Tanaka, Change in Local Conformation of Polymer Chains at Film Surface Attached to Solid Surface, Soft Matter, 2022, 18(17), 3304–3307, 10.1039/D1SM01833G.
- D. Kawaguchi, K. Yamamoto, T. Abe, N. Jiang, T. Koga, S. Yamamoto and K. Tanaka, Local Orientation of Chains at Crystal/Amorphous Interfaces Buried in Isotactic Polypropylene Thin Films, Phys. Chem. Chem. Phys., 2021, 23(41), 23466–23472, 10.1039/D1CP03959H.
- J. Huang and A. D. MacKerell, CHARMM36 All-Atom Additive Protein Force Field: Validation Based on Comparison to NMR Data, J. Comput. Chem., 2013, 34(25), 2135–2145, DOI:10.1002/jcc.23354.
- R. M. Venable, F. L. H. Brown and R. W. Pastor, Mechanical Properties of Lipid Bilayers from Molecular Dynamics Simulation, Chem. Phys. Lipids, 2015, 192, 60–74, DOI:10.1016/j.chemphyslip.2015.07.014.
- T. A. Beu and A. Farcaş, CHARMM Force Field and Molecular Dynamics Simulations of Protonated Polyethylenimine, J. Comput. Chem., 2017, 38(27), 2335–2348, DOI:10.1002/jcc.24890.
- A. Terteci-Popescu and T. A. Beu, Branched Polyethyleneimine: CHARMM Force Field and Molecular Dynamics Simulations, J. Comput. Chem., 2022, 43(31), 2072–2083, DOI:10.1002/jcc.27005.
- P. Sarker, T. Lu, D. Liu, G. Wu, H. Chen, M. S. Jahan Sajib, S. Jiang, Z. Chen and T. Wei, Hydration Behaviors of Nonfouling Zwitterionic Materials, Chem. Sci., 2023, 14(27), 7500–7511, 10.1039/D3SC01977B.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model, J. Phys. Chem., 1993, 97(40), 10269–10280, DOI:10.1021/j100142a004.
- B. Isralewitz, M. Gao and K. Schulten, Steered Molecular Dynamics and Mechanical Functions of Proteins, Curr. Opin. Struct. Biol., 2001, 11(2), 224–230, DOI:10.1016/S0959-440X(00)00194-9.
- P. Sarker, G. T. Chen, M. S. J. Sajib, N. W. Jones and T. Wei, Hydration and Antibiofouling of TMAO-Derived Zwitterionic Polymers Surfaces Studied with Atomistic Molecular Dynamics Simulations, Colloids Surf. Physicochem. Eng. Asp., 2022, 653, 129943, DOI:10.1016/j.colsurfa.2022.129943.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, THE Weighted Histogram Analysis Method for Free-energy Calculations on Biomolecules. I. The Method, J. Comput. Chem., 1992, 13(8), 1011–1021, DOI:10.1002/jcc.540130812.
- R. F. Fedors, A Method for Estimating Both the Solubility Parameters and Molar Volumes of Liquids, Polym. Eng. Sci., 1974, 14(2), 147–154, DOI:10.1002/pen.760140211.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed methods used in simulations, PET crystal formation observed in simulation. See DOI: https://doi.org/10.1039/d4sm00407h |
| This journal is © The Royal Society of Chemistry 2024 |