
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall variations in particle-level interactions lead to large structural heterogeneities in colloidal gels
Deepak
Mangal
 a,
Gonzalo Sanchez
Vera
c,
Stefano
Aime
c and
Safa
Jamali
*ab
a,
Gonzalo Sanchez
Vera
c,
Stefano
Aime
c and
Safa
Jamali
*ab
aDepartment of Mechanical and Industrial Engineering, Northeastern University, Boston, 02115, USA. E-mail: s.jamali@northeastern.edu
bDepartment of Chemical Engineering, Northeastern University, Boston, 02115, USA
cMolecular, Macromolecular Chemistry, and Materials, ESPCI Paris, France
First published on 13th May 2024
Abstract
Colloidal gels typically exhibit mechanical properties akin to a viscoelastic solid, influenced by their underlying particulate network. Hence, the structural and morphological characteristics of the colloidal network have a significant effect on the rigidity of the gel. In this study, we show how seemingly small variations in the particle-level interactions throughout the system result in larger scale structural heterogeneities. While the microscale particle level descriptors of the colloidal network remain largely unaffected by heterogeneous interactions, larger scale properties of a colloidal gel change appreciably. The overall cluster-level mesostructure of a colloidal gel is found to be sensitive to the small variations in the interaction potential at the particle level.
1 Introduction
Colloidal gels are a class of complex materials that occupy a pivotal position at the interface of soft matter physics, materials science, and nanotechnology. They play a crucial role in various natural and industrial settings, from biological tissues to advanced materials.1,2 These gels formed as colloidal particles aggregate in a liquid medium, creating a network structure that exhibits a remarkable interplay of solid and fluid-like properties.3–5 At the macroscopic level, colloidal gels generally exhibit a complex viscoplastic response,4 manifesting in a measurable yield stress below which the fluid does not flow.6–9 Emergence of elasticity in colloidal gels is believed to be directly governed by the characteristics of their underlying particulate structure.10–12 Recent work on the characterization of the mesoscale description of colloidal gels shows that the size and inter-connectivity of clusters play a major role in the load-bearing ability of the overall structure.11 Hence, different factors that change the cluster-level characteristics of the particle network can potentially impact the overall mechanics of the gel.While the overall intricacies of gel preparation and their potential to drastically affect gel mechanics are generally understood, there is little information in the literature on their origin. For instance, it's been established that kinematics of the pre-shear stage before gelation can foundationally change a gel's structure and thus its mechanical properties.13–15 Attractive interaction between colloids can be induced via different mechanisms such as adding salt to screen the surface charges on a particle, or addition of a non-adsorbing polymer to create depletion interactions.2,16 In principle, gel properties can be tailored by adjusting the system's state variables: particle volume fractions, or the strength and range of attraction between the particles.17 However, regardless of the source of attraction between the particle, it is very common to observe large scale structural heterogeneities within the structure of the particle network formed.18,19 What is interesting is that virtually all computational efforts do not reproduce the same scale of structural heterogeneities.15,20–24 While the role of hydrodynamic interactions in large-scale structural heterogeneities and shear-induced structuration of these colloidal networks is quite established,15,24–27 there remains a significant deviation between the gel structure under the quiescent conditions observed in experiments and that resulted from large scale particle simulations.11
Whether the attraction between the particles is a result of polymer depleting agents or addition of salt, it is plausible to presume slight concentration gradients within the bulk of particulate suspensions. This type of uneven distribution can easily be caused by inadequate mixing during gel preparation and can lead to regions with varying concentrations. Even for very homogeneously mixed suspensions, the mobility of gelation agents themselves, and their diffusion within the system can contribute to a non-uniform distribution.28 For example, a relatively faster diffusion in one region may lead to local accumulation, while slower diffusion in another area may result in lower concentrations. Additionally, additives can interact with each other, further contributing to non-uniform concentration profiles within the system.29 On the other hand, the strength of attraction between interacting colloids is a direct consequence of the ion or polymer concentration in their vicinity.30,31 Therefore it is logical to assume that the interactions will not precisely be the same for all particles and throughout the entire suspension of colloids. Nonetheless, in virtually all numerical efforts to date, a constant interaction between the particles is considered with no variation over time or space.
In this work, we explore the impact of a non-uniform distribution of attraction strength in both weak and strong colloidal systems. By comparing colloidal structures formed with [slightly] non-uniform interactions to those with ideally uniform ones, we isolate the influence of spatial interaction variation on the overall gel structure morphology. Previous studies have shown that at the very weak attraction limits of u0 = 3–4kBT, inherent structural heterogeneities of the colloidal gels are most visible;11,26,32,33 however, there also exist inconsistencies between the experimentally observed structures and computationally modeled ones at these limits. Thus, here, we conduct simulations at two average attraction strengths, u0 = 6kBT and 12kBT, representing weak and strong gels. Our results demonstrate that while particle-level descriptions of the system show no sensitivity to the interaction potential non-uniformity, the larger cluster-level gel structure is directly affected.
2 Simulation method
In this study, we simulated a system of monodisperse colloidal particles with radii a = 1 in a cubic box with periodic boundary conditions using the Brownian dynamics method. Hydrodynamic interactions between colloids were ignored. The simulations were conducted at a particle volume fraction of ϕ = 0.1 with 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 colloidal particles. The attraction between colloids was modeled using a Morse potential
000 colloidal particles. The attraction between colloids was modeled using a Morse potential 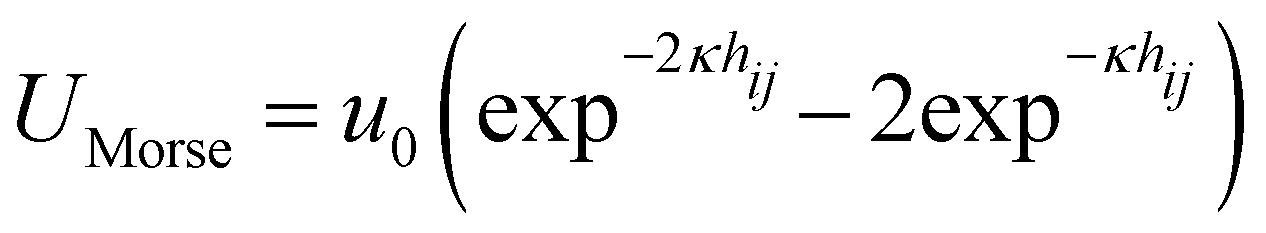 , where u0 and κ−1 are the depth and range of the attraction well. The equation of motion is then described by the following equation:
, where u0 and κ−1 are the depth and range of the attraction well. The equation of motion is then described by the following equation: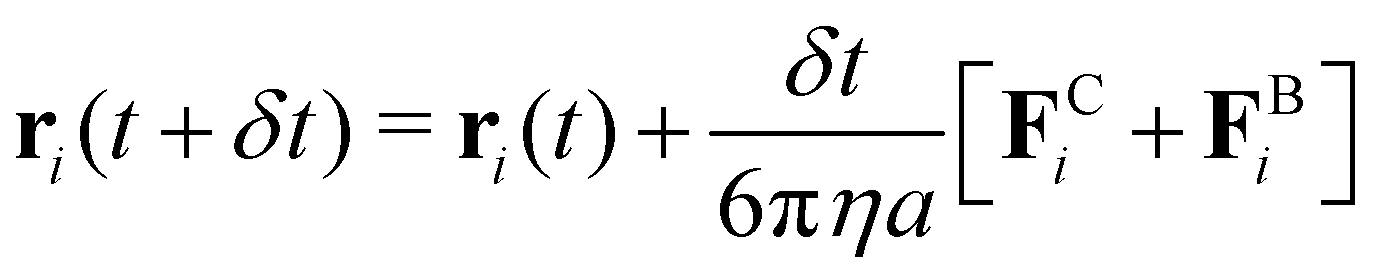 | (1) |
The simulations were performed using a, kBT, and τ = 6πηa3/kBT as the characteristic units for length, energy, and time, respectively. Here, kB is the Boltzmann constant, T is temperature, τ is the diffusive time scale of a single colloidal particle, and η is the dynamic fluid viscosity. The simulations were carried out with the attraction range set to κa = 30, resulting in an attractive interaction range of size 0.1a. To introduce spatially non-uniform attraction within the system, we partitioned the simulation box into sub-cells of size 10a, each assigned an arbitrary well-depth (u0,j). These arbitrary well-depth values u0,j were drawn from both a uniform distribution  and a normal distribution
and a normal distribution 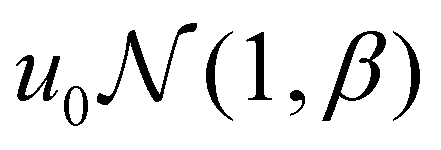 , where u0 was the mean well depth and β was the half-width of the distribution. The study considered two different β values, 0.15 and 0.30, to investigate variations in the distribution of attraction strength. Fig. 1 illustrates a schematic view of the interaction heterogeneities throughout a slice of the simulation box.
, where u0 was the mean well depth and β was the half-width of the distribution. The study considered two different β values, 0.15 and 0.30, to investigate variations in the distribution of attraction strength. Fig. 1 illustrates a schematic view of the interaction heterogeneities throughout a slice of the simulation box.
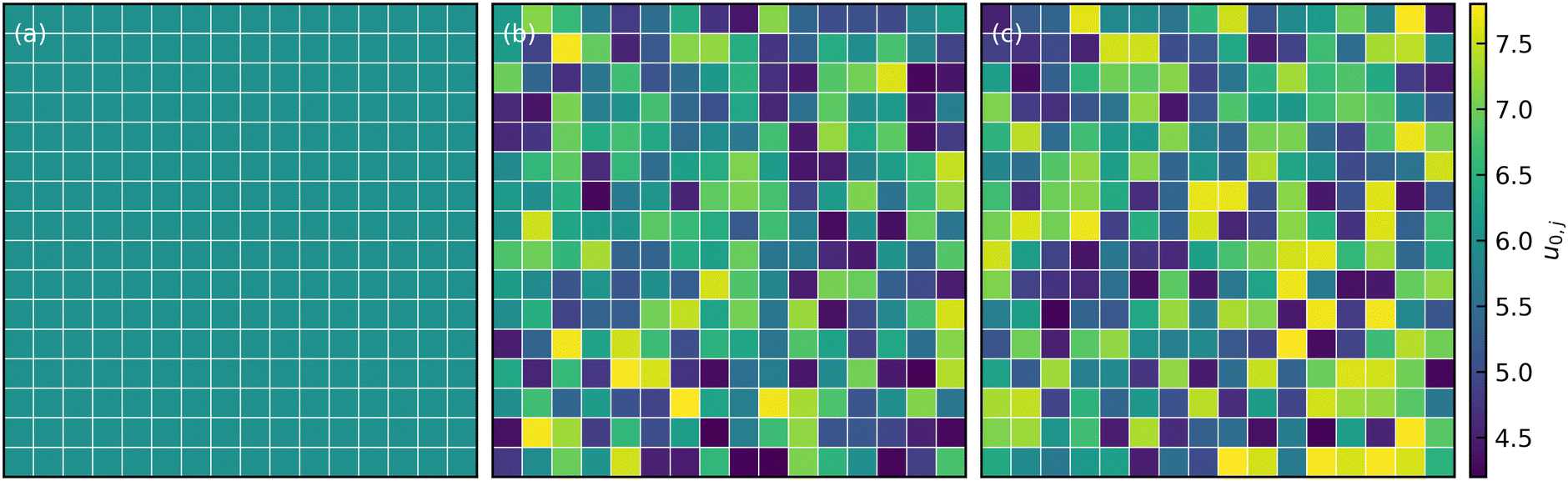 | ||
| Fig. 1 Attraction strength distribution in the x − y plane in (a) constant (b) normally distributed, and (c) uniformly-distributed interaction systems with β = 0.3 and mean well-depth u0 = 6. | ||
The simulations consisted of two distinct steps. In the first step, particles evolved under short-range hard-sphere repulsion without any attractive interactions to eliminate initial unphysical overlaps and generate a disordered initial configuration. Then, in the second step, attractive interactions were added to induce gel formation. During this latter step, we ensured that a quasi-steady structure was attained with minimal alterations to the microstructure. The presented results correspond to a total simulation time of 104τ, where τ is the Brownian diffusion time of a single particle. All simulations were executed using HOOMD-blue, an open-source molecular dynamics simulation toolkit.34
3 Results and discussion
Snapshots of the final morphologies for various attractive colloidal systems are presented in Fig. 2. The top row corresponds to the weakly attractive 6kT structures, while the bottom row corresponds to the strongly attractive 12kT structures. Generally, stronger attractions tend to produce more fractal-like structures compared to the coarser domains observed for weaker attractions. This is evident in the thicker particle strands and larger interstitial void pores observed in all weakly attractive systems (Fig. 2). More importantly, regardless of attraction strength, introducing spatial non-uniformity in the interaction leads to a visible coarsening of the structure and increased structural heterogeneity. | ||
| Fig. 2 Final gel structures for systems with (a) constant (b) normally distributed with β = 0.3, and (c) uniformly distributed with β = 0.3, attractive interactions throughout the calculation box as depicted in Fig. 1. The top row corresponds to weakly attractive colloids with u0 = 6, while the bottom row corresponds to strongly attractive colloids with u0 = 12. Particles are color-coded using a blue-yellow scheme based on their coordination number. | ||
While visual inspection of structures in Fig. 2 suggests that variations within the interaction potential leads to relatively more heterogeneous network structures, quantification across different length scales is necessary. First, we examine the global [percolated] network formation at the largest scale by computing the number of independent clusters and the fraction of particles in the largest connected component (LCC) within the system. Remarkably, gelation at a higher attraction strength (u0 = 12) occurs nearly two orders of magnitude faster than at a lower attraction strength (u0 = 6). However, the details of interaction uniformity (or lack thereof) show no significant effects on the gelation kinetic for a given attraction strength (Fig. 3). This suggests that in a macroscopic system with a large number of particles, local variations due to non-uniform attractions are statistically averaged out, and the overall system behavior is determined by the average attraction strength.
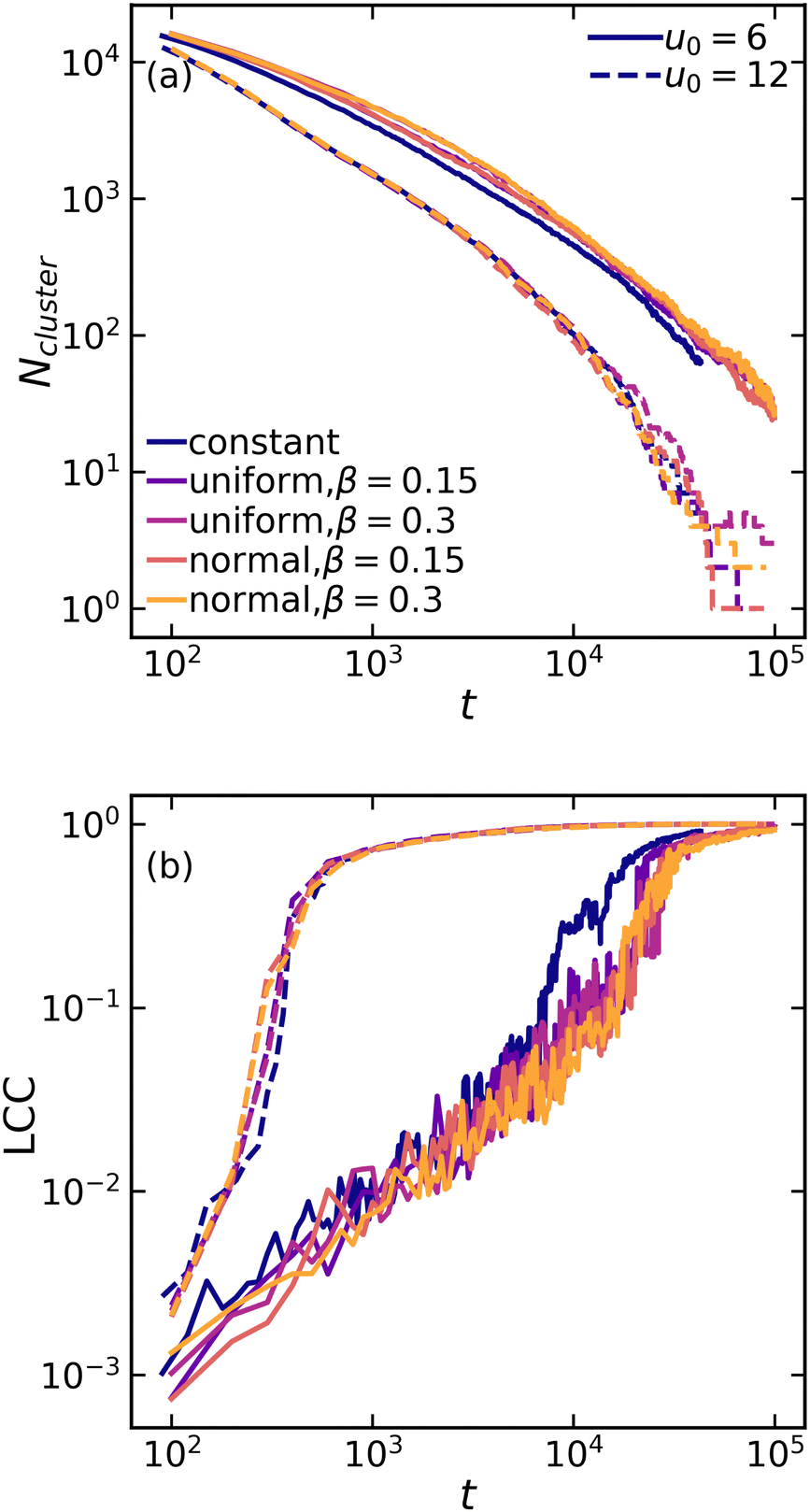 | ||
| Fig. 3 Evolution of (a) the number of independent clusters and (b) the fraction of particles in the largest connected component (LCC) as a function of time for various attraction systems at mean values of u0 = 6 (solid lines) and u0 = 12 (dashed lines). | ||
We then analyze the microscopic particle-level structure by examining the coordination number  and its distribution with a fixed cutoff distance of rc = 0.1. It's important to note that since the attraction range is the same for all systems studied, adjusting the cutoff distance won't change our overall findings. One should however note that variations in the range of attraction, especially in depletion gels, is another plausible source of heterogeneities, since there may exist an inherent size polydispersity in the polymer depleting agents. Nonetheless, if all variations are within the short-range limit and do not become comparable with the particle size, the overall effect will arguably be negligible. We observed that gelation kinetics, where percolation is faster for strongly attractive systems, is indirectly evident in the evolution of the average coordination number (Fig. 4(a)). However, weakly attractive systems tend to have a slightly higher steady-state average coordination number, indicating that thicker strands within their structures that allow for each particle to find more neighbours.22 These observations are also reflected in the coordination number distribution (Fig. 4(b)). While all systems studied show a clear peak at
and its distribution with a fixed cutoff distance of rc = 0.1. It's important to note that since the attraction range is the same for all systems studied, adjusting the cutoff distance won't change our overall findings. One should however note that variations in the range of attraction, especially in depletion gels, is another plausible source of heterogeneities, since there may exist an inherent size polydispersity in the polymer depleting agents. Nonetheless, if all variations are within the short-range limit and do not become comparable with the particle size, the overall effect will arguably be negligible. We observed that gelation kinetics, where percolation is faster for strongly attractive systems, is indirectly evident in the evolution of the average coordination number (Fig. 4(a)). However, weakly attractive systems tend to have a slightly higher steady-state average coordination number, indicating that thicker strands within their structures that allow for each particle to find more neighbours.22 These observations are also reflected in the coordination number distribution (Fig. 4(b)). While all systems studied show a clear peak at 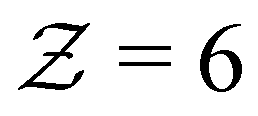 , indicating overall isostatic rigidity, weakly attractive systems exhibit relatively higher probability densities of large coordination numbers (
, indicating overall isostatic rigidity, weakly attractive systems exhibit relatively higher probability densities of large coordination numbers (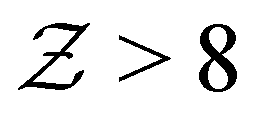 ) compared to the strongly attractive ones. Interestingly, regardless of the attraction strength, all heterogeneous attraction systems reach the same asymptotic
) compared to the strongly attractive ones. Interestingly, regardless of the attraction strength, all heterogeneous attraction systems reach the same asymptotic 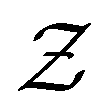 values. While there are minor differences in probability distributions for non-uniform interactions at different coordination numbers, the overall shape remains consistent across all systems. This suggests that at the particle-level, spatial variations in the interaction potential do not significantly affect colloidal assembly structure.
values. While there are minor differences in probability distributions for non-uniform interactions at different coordination numbers, the overall shape remains consistent across all systems. This suggests that at the particle-level, spatial variations in the interaction potential do not significantly affect colloidal assembly structure.
 | ||
Fig. 4 (a) Evolution of ensemble-averaged coordination number 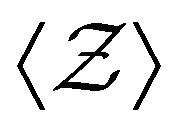 , and (b) distribution of coordination number , and (b) distribution of coordination number 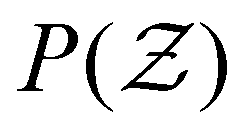 at final configuration for various attractive colloidal systems with strengths of mean values u0 = 6 (solid lines) and u0 = 12 (dashed lines). at final configuration for various attractive colloidal systems with strengths of mean values u0 = 6 (solid lines) and u0 = 12 (dashed lines). | ||
At the cluster-level mesoscale, particulate structure can be characterized through several methods. Firstly, differences in the large-scale cluster level structures can be clearly isolated by examining the static structure factor, S(q), of the resulting networks at low q values, defined as follows:35
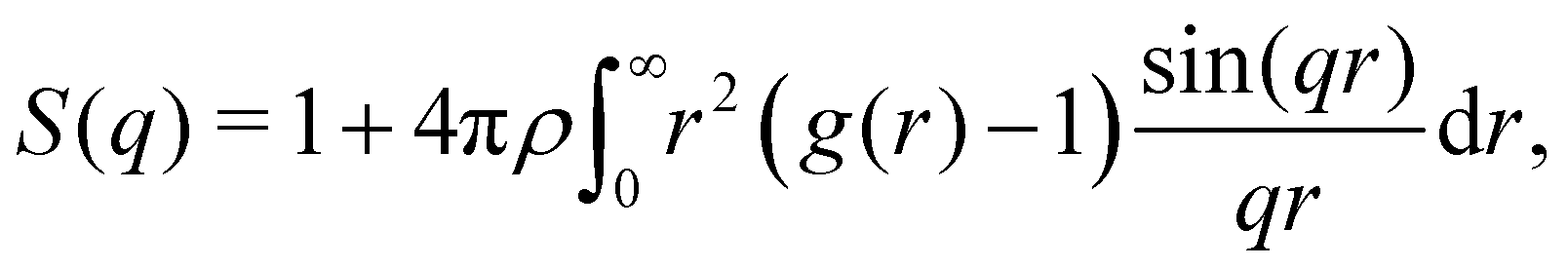 | (2) |
 | ||
| Fig. 5 Structure factor S(q) as a function of q for various attractive colloidal systems at mean values of u0 = 6 (solid lines) and u0 = 12 (dashed lines). | ||
As the colloidal structures construct a network and coarsen, distinguishable voids/pores form as well. To complement the particulate structure characterization via the structure factor, we measure the distribution of interstitial void sizes within the final structure using a method introduced by Gubbins and colleagues.36 This method involves selecting an arbitrary point in the void space and then determining the largest possible radius of a sphere that can encompass that point without overlapping with any particles. We found that in both weak and strong gels, the average pore size increases when spatial variation in the interaction potential is introduced (Fig. 6). This difference is particularly evident when examining the tail end of the pore size distribution. The largest pores within the system with heterogeneous interactions can be appreciably larger than those in systems with constant interactions. Moreover, the pore size distribution in strong gels shows greater sensitivity to interaction heterogeneities compared to weak gels. It should be noted that while the distribution broadens generally for non-constant interactions, the peak pore size remains rather unchanged for all systems.
 | ||
| Fig. 6 Pore size distribution for various attraction systems at mean values u0 = 6 (solid lines) and u0 = 12 (dashed lines). | ||
4 Experimental verification
In order to validate the results of our simulations, we compare those with experiments on colloidal gels prepared by aggregating silica nanoparticles (Ludox HS40, from Sigma Aldrich, diameter a = 8 nm37) at a volume fraction of ϕ = 0.11. The colloidal suspension is initially stabilized by the negative charges decorating the silica–water interface at the suspension's basic pH. To aggregate the colloids, we dilute the mother suspension of concentrated silica particles (at volume fraction ϕ0 = 0.22) with an aqueous solution of sodium chloride (NaCl), to increase the ionic strength I to a prescribed value, ranging between 0.15 and 0.55 mol l−1. DLVO theory indicates that under these conditions the range of the interaction between colloids is about 0.1a, matching that of numerical simulations. Samples are mixed on a stirring plate for 1 min at 100 rpm and then loaded in a rheometer (ARES-G2, from TA instruments) equipped with cone-plate geometry to monitor the time evolution of the suspension rheology. For I < 0.2 mol l−1, we find that the suspension remains liquid on timescales exceeding one day. For I ≥ 0.2 mol l−1, we observe a well-defined transition from a liquid suspension to a gel, after a waiting time, tgel, strongly decreasing with increasing I. For I ≥ 0.55 mol l−1, we find that gelation takes place before the rheology experiment starts, about 1 minute after sample mixing. It is worth noting that, at this large volume fraction, the expected gel time in the limit of fully screened electrostatic repulsion, associated with diffusion-limited cluster aggregation (DLCA) kinetics, is about 1 microsecond.38 The necessity to load the suspension before gelation restricts our experiments to much longer gel times, in the reaction-limited cluster aggregation (RLCA) regime.39As a first quantification of large-scale structural heterogeneities in the gels, we measure the turbidity of fully formed gels, 24 h after sample preparation. Subsequent measurements a few days later don't show significant time evolution of the results. Turbidity measurements are performed by preparing the sample in sealed rectangular cuvettes and measuring the attenuation of a laser beam with beam size w = 1 mm and wavelength λ = 633 nm across an optical path of 1 cm. To estimate experimental uncertainty, we repeat the measurement on ≈ 10 different locations in the sample. We find that turbidity strongly increases with salt concentration, indicating that sample heterogeneity increases with increasing I, as shown in Fig. 7. We interpret this as the result of the interplay between incipient particle aggregation and sample mixing: when I is large, colloidal clusters form before the salt has been fully mixed, and therefore in a locally heterogeneous environment, resulting in an increasingly heterogeneous gel. To test the role of mixing on the large-scale heterogeneities of the gels, we aggregate the same suspension by gradually increasing the ionic strength of the solvent after sample mixing and loading. We achieve this by means of an enzyme-catalyzed chemical reaction: the hydrolysis of urea, catalyzed by urease.40 In this case, we prepare a water suspension of silica nanoparticles at volume fraction ϕ = 0.11, urea at concentration MUrea = 2 Mol l−1 and urease (U1500-20KU, from Sigma Aldrich) at 10 mg ml−1. Right after mixing, the suspension is stable and can be safely handled without perturbing the aggregation pathway. As urea hydrolysis produces charged species, the ionic strength of the solvent gradually increases, eventually exceeding that of the most concentrated salt solutions, as monitored by conductivity measurements. Yet, we find that the turbidity of the resulting gel remains small, rather comparable to that of samples with the smallest ionic strengths, as shown by the dashed line in Fig. 7.
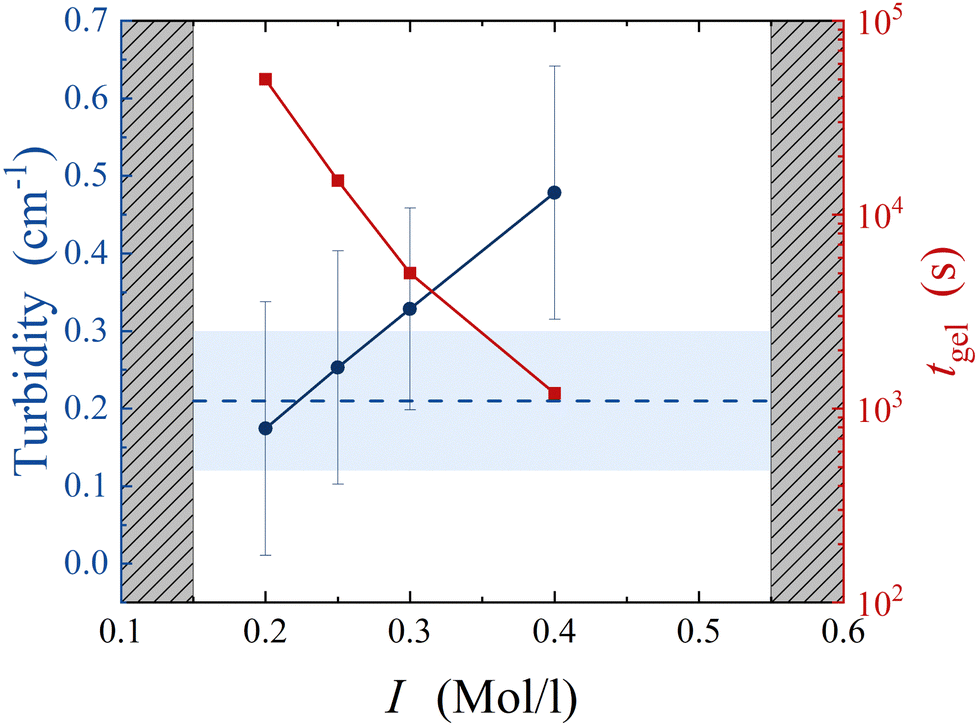 | ||
| Fig. 7 Blue circles: turbidity of colloidal gels at ϕ = 0.11 as a function of the ionic strength of the solvent, I. Red squares: gel time, measured after adding salt to the colloidal suspension. Grey shaded areas mark the boundaries of the experimentally accessible region. Blue dashed line: turbidity of colloidal gel aggregated by the gradual hydrolysis of urea. Blue shaded region denotes experimental uncertainty. | ||
To further detail the difference between gels undergoing fast aggregation due to concentrated salt suspensions and gels undergoing slow aggregation due to urea hydrolysis, we complement these results by optical microscopy measurements. Because of the limited spatial resolution of optical microscopy, we increase the characteristic length scale of the gel microstructure by using larger colloidal particles (Ludox TM50, from Sigma Aldrich, with diameter a = 14 nm37) and by reducing the volume fraction to ϕmicro = 0.01. The gel structure is measured using an upright optical microscope (LEITZ DM RXE equipped with a ×64 oil objective from Leica Microsystems, numerical aperture NA = 1.32), by injecting the liquid suspension into a channel with a thickness of 125 μm, and by taking snapshots of its mid-plane every 10 s until gelation. Under the same Koehler's illumination conditions, we find that the gel aggregated using concentrated salt solutions exhibits significantly enhanced intensity fluctuations in the microscopy images, as shown in the bottom row of Fig. 8. Inspecting the suspension 1 min after injection, we also notice that the gel aggregated with salt at a large concentration already exhibits heterogeneities that are absent in the sample aggregated by hydrolyzing urea, as shown in the top row of Fig. 8. While a quantitative analysis of such differences goes beyond the scope of this paper, this result confirms that colloidal gel aggregation can be strongly affected by mixing, which results in an increased large-scale heterogeneity of the gel microstructure.
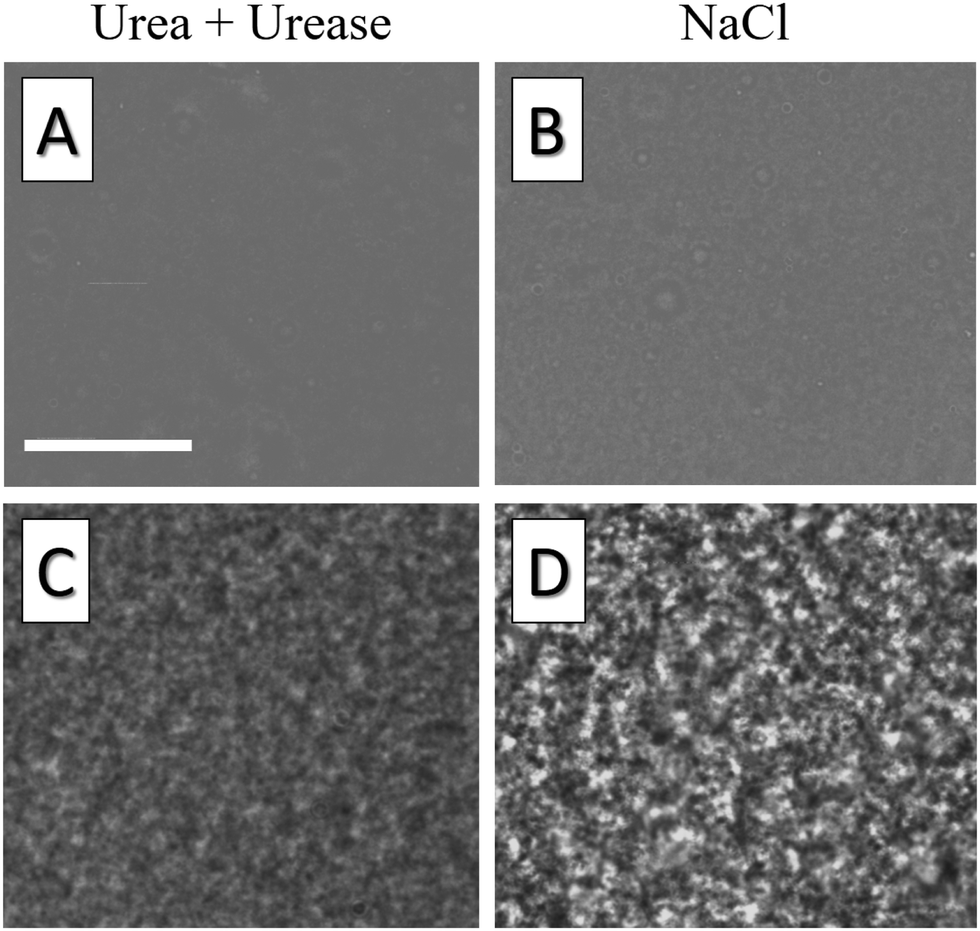 | ||
| Fig. 8 Optical microscopy images of colloidal gels aggregated using urea/urease (c) and NaCl (d). Stronger fluctuations observed in NaCl samples under equal imaging conditions reflect enhanced structural heterogeneities, already present in the liquid suspension, 1 minute after injection (a) and (b). Scale bar: 32 μm. | ||
5 Conclusions
This study explores the role of spatial variations in the particle-level interactions on the particulate structure across different length scales in both weak and strong colloidal gels. While the largest [macro]scale and the smallest [micro]scale characteristics of the structure show no sensitivity to the variations in attraction throughout the system, intermediate cluster-level [meso]scale features are significantly affected. Introducing spatial variation in the interaction potential leads to coarsening of the structure and enhanced structural heterogeneities within the gel network. These large-scale cluster-level structures exhibit clear signals at low wave vectors in the static structure factor, indicating structural heterogeneity. This heterogeneity is associated with a visible broadening of the pore size distribution, where much larger voids/pores are observed. Our results unequivocally show that while some measures of a colloidal gel structure may not reflect the intricate details of the interactions between the particles, mesoscale structures—critical for understanding gel mechanics—are greatly influenced by these variations. Arguably, such spatial variations, common in many experimental setups, are often overlooked or averaged-out in numerical simulations focusing on structure-mechanics coupling in soft glassy materials. To truly recover experimentally relevant structures, one must understand and consider interaction-variations between the individual particles in space and perhaps over time.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (CBET-2118962 and CBET-2104869). Computational resources were generously provided by the Massachusetts Green High-Performance Computing Center in Holyoke, MA.References
- E. Dickinson, An introduction to food colloids, Oxford University Press, 1992 Search PubMed.
- E. Zaccarelli, J. Phys.: Condens. Matter, 2007, 19, 323101 CrossRef.
- Y. M. Joshi, Annu. Rev. Chem. Biomol. Eng., 2014, 5, 181–202 CrossRef CAS PubMed.
- D. Bonn, M. M. Denn, L. Berthier, T. Divoux and S. Manneville, Rev. Mod. Phys., 2017, 89, 035005 CrossRef.
- A. Y. Malkin, S. R. Derkach and V. G. Kulichikhin, Gels, 2023, 9, 715 CrossRef CAS PubMed.
- M. Laurati, S. Egelhaaf and G. Petekidis, J. Rheol., 2011, 55, 673–706 CrossRef CAS.
- L. C. Hsiao, R. S. Newman, S. C. Glotzer and M. J. Solomon, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 16029–16034 CrossRef CAS PubMed.
- S. Jamali, G. H. McKinley and R. C. Armstrong, Phys. Rev. Lett., 2017, 118, 048003 CrossRef PubMed.
- L. C. Johnson, R. N. Zia, E. Moghimi and G. Petekidis, J. Rheol., 2019, 63, 583–608 CrossRef CAS.
- K. A. Whitaker, Z. Varga, L. C. Hsiao, M. J. Solomon, J. W. Swan and E. M. Furst, Nat. Commun., 2019, 10, 1–8 CrossRef CAS PubMed.
- M. Nabizadeh, F. Nasirian, X. Li, Y. Saraswat, R. Waheibi, L. C. Hsiao, D. Bi, B. Ravandi and S. Jamali, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2316394121 CrossRef CAS PubMed.
- H. Vinutha, F. D. Diaz Ruiz, X. Mao, B. Chakraborty and E. Del Gado, J. Chem. Phys., 2023, 158, 114104 CrossRef CAS PubMed.
- A. Helal, T. Divoux and G. H. McKinley, Phys. Rev. Appl., 2016, 6, 064004 CrossRef.
- N. Koumakis, E. Moghimi, R. Besseling, W. C. Poon, J. F. Brady and G. Petekidis, Soft Matter, 2015, 11, 4640–4648 RSC.
- S. Jamali, R. C. Armstrong and G. H. McKinley, Mater. Today Adv., 2020, 5, 100026 CrossRef.
- J. Ruiz-Franco and E. Zaccarelli, Annu. Rev. Cond. Matter Phys., 2021, 12, 51–70 CrossRef CAS.
- P. J. Lu and D. A. Weitz, Annu. Rev. Condens. Matter Phys., 2013, 4, 217–233 CrossRef CAS.
- A. Duri and L. Cipelletti, Europhys. Lett., 2006, 76, 972 CrossRef CAS.
- E. Zaccarelli, P. J. Lu, F. Ciulla, D. A. Weitz and F. Sciortino, J. Phys.: Condens. Matter, 2008, 20, 494242 CrossRef.
- E. Del Gado and W. Kob, Europhys. Lett., 2005, 72, 1032 CrossRef CAS.
- J. Colombo and E. Del Gado, J. Rheol., 2014, 58, 1089–1116 CrossRef CAS.
- R. N. Zia, B. J. Landrum and W. B. Russel, J. Rheol., 2014, 58, 1121–1157 CrossRef CAS.
- Z. Varga, V. Grenard, S. Pecorario, N. Taberlet, V. Dolique, S. Manneville, T. Divoux, G. H. McKinley and J. W. Swan, Proc. Natl. Acad. Sci., 2019, 116, 12193–12198 CrossRef CAS PubMed.
- H. Tsurusawa, S. Arai and H. Tanaka, Sci. Adv., 2020, 6, eabb8107 CrossRef CAS PubMed.
- A. Mohraz and M. J. Solomon, J. Rheol., 2005, 49, 657–681 CrossRef CAS.
- Z. Varga and J. Swan, Soft Matter, 2016, 12, 7670–7681 RSC.
- S. Jamali, R. C. Armstrong and G. H. McKinley, Phys. Rev. Lett., 2019, 123, 248003 CrossRef PubMed.
- M. Zarif and A. Naji, Phys. Rev. E, 2020, 102, 032613 CrossRef CAS PubMed.
- T. D. Edwards and M. A. Bevan, Langmuir, 2012, 28, 13816–13823 CrossRef CAS PubMed.
- H. N. Lekkerkerker, R. Tuinier, H. N. Lekkerkerker and R. Tuinier, Depletion interaction, Springer, 2011 Search PubMed.
- J. N. Israelachvili, Surface forces, CRC Press, 2022, pp. 793–816 Search PubMed.
- P. J. Lu, E. Zaccarelli, F. Ciulla, A. B. Schofield, F. Sciortino and D. A. Weitz, Nature, 2008, 453, 499–503 CrossRef CAS PubMed.
- A. P. Eberle, N. J. Wagner and R. Castaneda-Priego, Phys. Rev. Lett., 2011, 106, 105704 CrossRef PubMed.
- J. A. Anderson, J. Glaser and S. C. Glotzer, Comput. Mater. Sci., 2020, 173, 109363 CrossRef CAS.
- A. Filipponi, J. Phys.: Condens. Matter, 1994, 6, 8415 CrossRef CAS.
- L. D. Gelb and K. Gubbins, Langmuir, 1999, 15, 305–308 CrossRef CAS.
- L. Goehring, J. Li and P.-C. Kiatkirakajorn, Philos. Trans. R. Soc., A, 2017, 375, 20160161 CrossRef PubMed.
- D. A. Weitz, J. S. Huang, M. Y. Lin and J. Sung, Phys. Rev. Lett., 1984, 53, 4 CrossRef.
- M. Y. Lin, H. M. Lindsay, D. A. Weitz, R. C. Ball, R. Klein and P. Meakin, Nature, 1989, 339, 360–362 CrossRef CAS.
- L. J. Gauckler, T. Graule and F. Baader, Mater. Chem. Phys., 1999, 61, 78–102 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |