
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Methods of changing low molecular weight gel properties through gelation kinetics
Rebecca E.
Ginesi
 * and
Emily R.
Draper
*
* and
Emily R.
Draper
*
School of Chemistry, University of Glasgow, Glasgow, UK G12 8QQ, UK. E-mail: emily.draper@glasgow.ac.uk
First published on 26th April 2024
Abstract
Low molecular weight gels continue to attract notable interest, with many potential applications. However, there are still significant gaps in our understanding of these systems and the correlation between the pre-gel and final gel states. The kinetics of the gelation process plays a crucial role in the bulk properties of the hydrogel and presents an opportunity to fine-tune these systems to meet the requirements of the chosen application. Therefore, it is possible to use a single gelator for multiple applications. This review discusses four ways to modify the pre-gelled structures before triggering gelation. Such modifications can enhance the material's intended performance, which may result in significant advancements in high-tech areas, such as drug delivery, cell culturing, electronics, and tissue engineering.
 Rebecca E. Ginesi | Rebecca Ginesi received her MSci degree from the University of Glasgow, UK in 2020, where she is currently carrying out her PhD under the supervision of Dr Emily Draper. Her research focuses on tuning the self-assembly of small organic molecules for use in organic electronic devices. |
 Emily R. Draper | Emily Draper received her PhD in Chemistry from the University of Liverpool, UK in 2015. She started a Leverhulme Trust Early Career Fellowship in 2017 before becoming a Lecturer at the University of Glasgow in 2018. She then became a UKRI Future Leaders Fellow in 2021 and a Senior Lecturer in 2022. Her work focuses on the assembly of small organic molecules for applications such as electronics, chromics and cell culture. |
Introduction
Low molecular weight gelators (LMWGs) can self-assemble to form entangled gel networks governed by various non-covalent interactions.1–5 Compared to polymeric hydrogels, LMWGs possess discrete molecular components and well-defined chemical structures.6 As such, it is possible to tune these materials at the molecular level and thus control the resulting gel properties.7–9 Such control is crucial since the gel properties determine the applications for which the gel is suitable. There are many potential applications for these materials, including cell growth, drug delivery and waste management.10–15 Furthermore, some LMWGs can actively contribute to the material's functionality, such as electronics, chromics, water-splitting, and sensing.16–21However, one disadvantage of LMWGs is that designing and synthesizing these materials is difficult, as it can be challenging to predict whether a molecule will gel.22 Recently, computational models have been used to identify dipeptide gelators that can successfully form gels.23,24 However, some predictors require synthesis of a library of materials and screening, making them time- and labour-intensive. Furthermore, using such models to predict gel properties is limited because of the pathway dependence of gelation.25–27 Therefore, there is a need to better understand the assembly process and correlate the precursor, assembly conditions, and self-assembled structures.28
Many hydrogelators are relatively hydrophobic which drives gelation.29 Therefore, self-assembly in a gel is considered a non-equilibrium process in which the system moves from a “highly soluble” to a “less soluble” state and so gelation is regarded as kinetically dependent.30,31 The self-assembly is also an energetically downhill process, allowing the gels to form under thermodynamic equilibrium.25,32–35 The competition between the thermodynamic and kinetic pathways presents an opportunity to switch from thermodynamic to kinetic control, allowing the system to exist as a kinetically trapped species.36 Therefore, materials with different properties can be prepared from the same precursor depending on the assembly pathway kinetics.25,32,33,35–37 The gelation kinetics can be monitored using various techniques such as rheology,38,39 monitoring pH change,40 NMR,41,42 absorption and fluorescence spectroscopy,43,44 and X-ray and neutron scattering.45–47 There are extensive studies on energy landscapes and kinetically trapped states in the literature,6,26,48–50 which is why it will not be the focus of this review. However, as the gelation kinetics play such a crucial role in the final gel properties, it is important to acknowledge this.
While altering the final gel properties by changing the gelation trigger, changing the solvent mixtures,51,52 and controlling the gelation kinetics has been extensively studied,52–57 changing the pre-gelled solution before triggering gelation is rarely discussed. There are many advantages of altering the pre-gelled solution to alter the gel properties. One limitation of changing gelation trigger to change the gel properties is that some gelation triggers may not be suitable for the final applications. For example, DMSO and high temperatures can be detrimental for cells.3,58 Another advantage is that due to their hydrophobicity, the pre-gelled state may contain micellar aggregates (such as spherical, cylindrical, and worm-like micelles) above the critical micelle concentration and Krafft temperature.59–62 How these molecules pack is dependent on their size and shape, but also on the non-covalent interactions present.60,61 Owing to their weak nature, these interactions can be tuned by parameters such as temperature, ionic strength, or pH, thus impacting the bulk properties of the pre-gelled solution.33 These changes in solution-phase properties could potentially be translated into the resulting gels. For example, it has been shown in functionalized dipeptide-based gelators that the structures formed in the gel state can be templated by the micellar state.63 Varying the micellar aggregate presents an opportunity to control the “apparent” pKa value of the aggregate (the pKa value associated with the self-assembled structures as opposed to the single molecule), changing the pH at which the gel forms and the properties of the gel.29 By tuning the gel, this creates new applications for the material. Currently, the properties of gels (such as stiffness) are controlled by varying the concentration of the gelator.64 However, this method also tends to lead to other changes in properties. Thus, the opportunity to selectively control the properties of hydrogels is extremely desirable.
In this review, we discuss four different methods used to tune the properties of pre-gelled LMWG solutions. We focus our discussion on hydrogels formed primarily by peptide LMWGs. Peptide-based hydrogels have been identified as important tools in drug delivery, tissue regeneration, cell culturing and imaging.65–68 We address how this approach can be used to prepare gels with controlled and specific properties. By modifying the gelation kinetics, one can use materials which are already well-studied and tailor the molecule to suit the chosen application. We hope to shine light upon an area we believe can be heavily exploited to save time, money, and resources.
Modification of pre-gel structures
Using heat–cool cycles
When LMWGs are heated, this increases their solubility and therefore, the molecules are more dispersed in solution due to reduced intermolecular interactions.69 As a result, these molecules may reassemble differently after cooling as they are molecularly dissolved. Thus, the kinetics will be affected by both the temperature the solution is heated to and the cooling rate, directly influencing the structures formed. Recently, Zhou et al. have demonstrated the modulation of the self-assembly pathway in peptide hydrogels.70 By controlling the initial temperature at the liquid–liquid phase separation stage, structurally distinct phase-separated droplets formed, resulting in different self-assembly pathways. As a result, the morphology of the hydrogel network could be tuned to alter the mechanical strength and recovery performance of the resulting hydrogel.At elevated temperatures, some charged amphiphilic molecules can form structures that template alignment of supramolecular fibrils.71 The Stupp group have utilized this behaviour to form supramolecular noodles, which when mixed with cells at physiological temperature, formed monodomain gels of aligned cells and filaments.71 A peptide amphiphile with an alkyl tail was studied, which self-assembles into 1D nanofibers in aqueous solution and can form gels. Alignment of 1D nanostructures has potential applications from cell culturing to organic electronics.72–75 Gel noodles were formed by dispensing the self-assembled amphiphile solution from a pipette into a CaCl2 solution to trigger gelation. Upon heating the solution to 80 °C and cooling to 25 °C, alignment of the nanostructures parallel to the long direction of the gel noodles occurred, which was attributed to a heat-induced change in the self-assembled structure. In comparison, non-heat-cooled solutions could not form mechanically stable noodles. Transmission electron microscopy (TEM) showed that the solutions formed thin plaque-like structures after a heat–cool treatment. Heat–cool cycles also resulted in a threefold increase in viscosity. It was postulated that the formation of plaque-like structures and increase in viscosity meant that the shear force experienced by the solution when pipetted aligned the nanostructures. Small-angle X-ray scattering (SAXS) suggested that the local packing was not changed by heating and cooling, but instead the aggregates were dehydrated when heated, leading to filaments with larger diameters. Microscopy highlighted large birefringent domains in the resulting gel noodles. Such birefringence suggests alignment along the noodle axis, which was shown to control the orientation of cells in 3D cultures. These findings offer a route to develop therapies which require directed cell migration or cell growth.
Another example of modifying pre-gelled solutions using heat–cool cycles comes from Draper et al., who have reported on the change in the physical properties of a naphthalene dipeptide-based gelator (2NapFF, where Nap = naphthalene and F = phenylalanine) solutions.76 These gelators can form worm-like micelles at high pH due to their hydrophobicity.77,78 Upon a heat–cool cycle, the viscosity of the samples at low shear significantly increased. SAXS showed that heat-cooling resulted in an increase in the length of the hollow structures formed due to dehydration of the aggregate core. This increase in length resulted in an increase in extensional viscosity (Fig. 1a), making it a potential candidate for electrospinning.79 When CaCl2 was used to trigger gelation (Fig. 1b), the gels formed from heat-cooled solutions were significantly stiffer than those from pre-heated solutions (storage modulus, G′, values of 122.7 ± 4.1 kPa for gels formed from heat-cooled solutions and 18.9 ± 3.4 kPa for gels formed from pre-heated solutions). Other gelators were also tested to prove this behaviour was not just observed with 2NapFF. This work again highlights a method to tune the properties of hydrogels by simply changing how the solution is prepared to offer more potential applications from a single gelator.
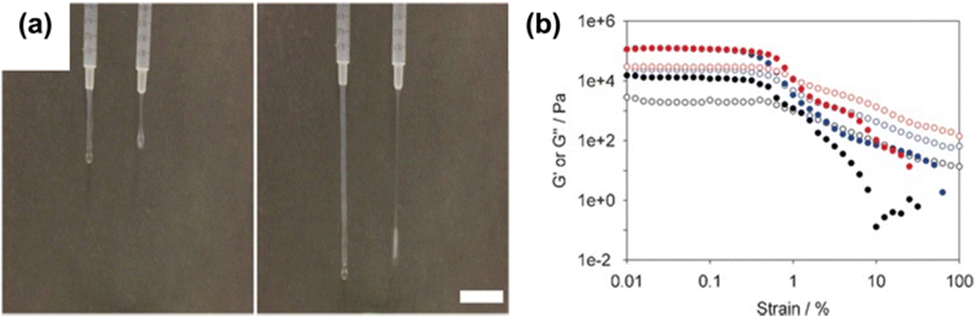 | ||
| Fig. 1 (a) Photographs demonstrating the increase in extensional viscosity upon heat-cooling 2NapFF solutions. Scale bar represents 2 cm. (b) Strain sweeps of gels formed by adding CaCl2 to non-heat-cooled (black), heated (blue), and heat-cooled (red) solutions of 2NapFF. Filled circles represent G′, and empty circles represent G′′. Figure adapted from ref. 74 with permission from Wiley-VCH. | ||
A more recent example of kinetically tuning the behaviour of LMWGs using heat-cooling comes from the Ulijn group, showing that thermal history is a simple method to control the structure and function of supramolecular hydrogels.80 Using the fluorenylmethoxycarbonyl (Fmoc) dipeptide, FmocYL (Fig. 2a), the group showed that tunable gels can form by altering the assembly temperature, with the resulting structures being “locked in” by cooling. The differential self-assembly was tuned by altering the dominant non-covalent interactions present. Using 1H NMR spectroscopy, they found that at higher temperatures (333–363 K (60–90 °C)) π-stacking interactions dominated, whereas at lower temperatures (313–323 K (40–50 °C)), hydrogen bonding was the primary interaction (Fig. 2b). Previously, such modifications were achieved by introducing functional groups to alter the self-assembly.81–84 However, Uljin's group showed that it was possible to obtain a variety of supramolecular structures from a single molecule.80 The balance of non-covalent interactions also influenced the proteolytic degradation of the gel, with gels containing more ordered H-bonding structures having lower degradation rates. Again, upon gelation the hydrogels formed from higher pre-assembly temperatures were mechanically stiffer and showed higher melting temperatures. Atomic force microscopy (AFM) also showed that the morphology of the gel networks had changed, with dried films of gels formed at lower pre-assembly temperatures having shorter and wider fibres. Overall, this work highlights the influence of temperature on the self-assembled structures formed whilst emphasizing that thermodynamic and kinetic considerations must be in place when designing functional nanomaterials.
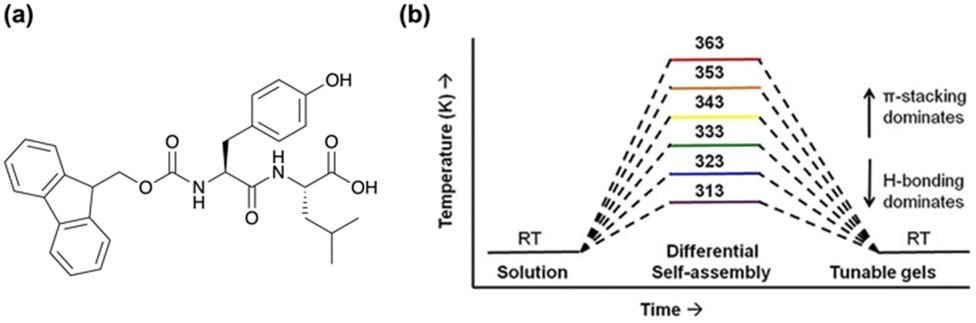 | ||
| Fig. 2 (a) Chemical structure of FmocYL gelator. (b) Cartoon illustrating the pathway-dependent self-assembly of FmocYL and the dominant intermolecular interactions at various temperatures. Figure adapted from ref. 78 with permission from Wiley-VCH. | ||
Using solution pH
LMWGs containing ionizable groups (such as carboxylic acids and amines) are sensitive to changes in pH due to protonation and deprotonation of these groups.85–87 As such, changing the pH of the pre-gelled solution can result in a change in the self-assembly due to differences in solubility.88–90 Therefore, this is another potential way to tune the properties of the resulting hydrogels.In the literature, it is common for solutions to be prepared at high pH, to ensure a LMWG with carboxylic acid groups is completely dissolved, and then adjusting to another pH before triggering gelation. We have recently shown the importance of starting pH and how you get to that pH on the mechanical properties of the resulting hydrogels of amino acid-appended perylene bisimides (PBIs).41 We compared the self-assembly and gelation of solutions of the alanine-appended PBI (PBI-A) at pH 9, where the molecules are more soluble, and at pH 6, which lies between the two “apparent” pKa values of the gelator. The pKa indicates the pH at which these structures change, and in this case is related to the deprotonation of the two carboxylic groups present.91,92 Thus, we expected the structures formed at these two pHs to be different. Using UV-vis absorption spectroscopy, rheology, and small-angle neutron scattering (SANS), we found the solutions at pH 6 were more aggregated and contained worm-like micelles. The lack of scattering and low viscosity observed at pH 9 suggested that the PBI molecules were more dispersed or not forming persistent aggregates. These differences in the solution-phase were translated to the gels, with the starting pH influencing the strength and stiffness of these materials. Furthermore, SANS highlighted differences in the gel fibres, with gels formed from solutions at pH 9 forming more rigid cylindrical fibres. In comparison, the solutions at pH 6 gave more flexible gel fibres. By monitoring the kinetics of the gelation process, we found that the self-assembly is significantly impacted by the starting pH, with the two different starting pHs following different kinetic pathways (Fig. 3). We also found that it was not possible to switch between the aggregated states at the different pHs if the self-assembled structures are already pre-formed, again due to differences in the kinetic pathway. This change was most apparent for gels formed from the two solutions at pH 6, with the resulting gels having more than an order of magnitude in difference in their stiffness. Therefore, we could access three different gels with distinct properties depending on how we prepared the solution. This work provides an opportunity to precisely control the morphology of the network to suit the chosen application without the need to make brand-new materials. Such control could be valuable in cell culturing and tissue engineering, where the morphology of the scaffolds can impact cell proliferation and differentiation.93–95
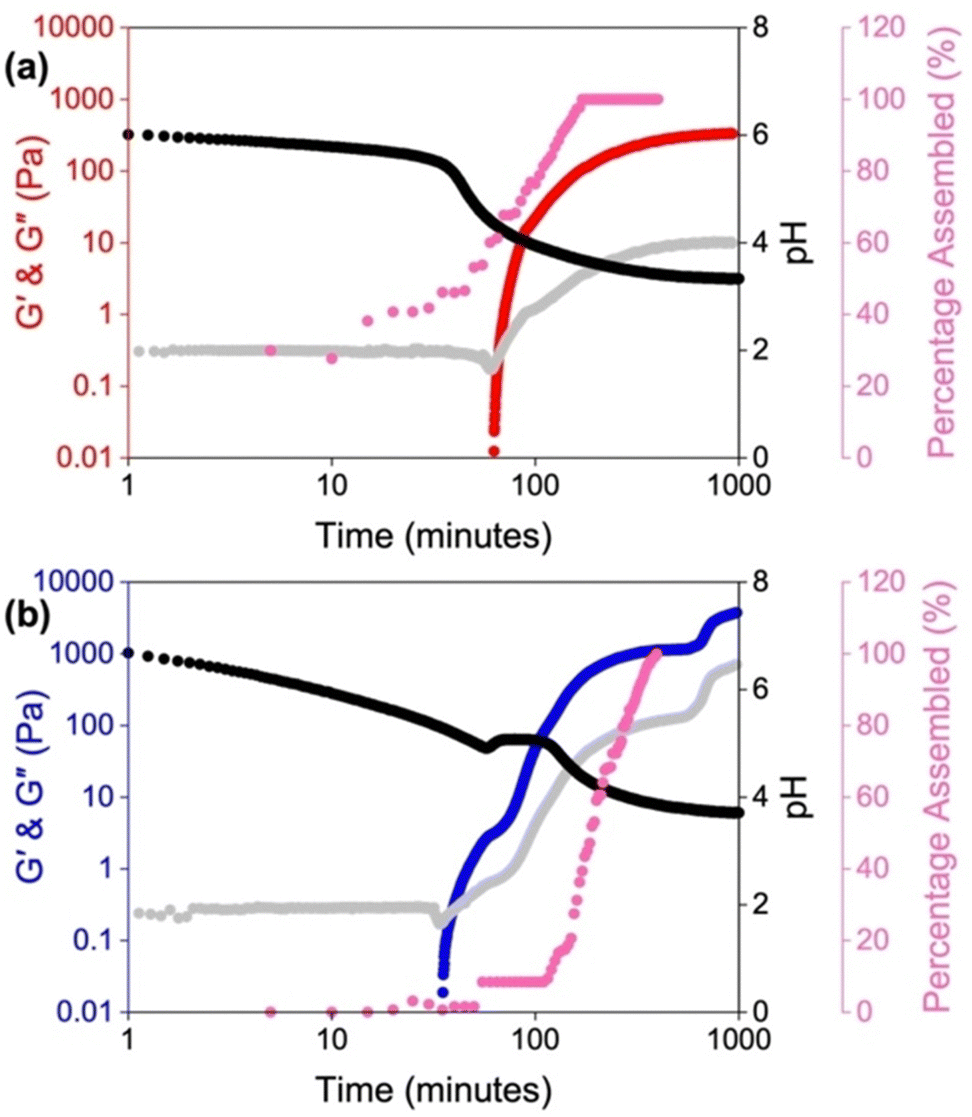 | ||
| Fig. 3 Plots showing the gelation kinetics of PBI-A solutions starting at a pH of (a) 6 and (b) 9. The graphs show the development of G′ (red for a starting pH of 6 and blue for a starting pH of 9) and G′′ (grey) with time and change in pH (black) and the change in percentage assembly (pink). Image reproduced from ref. 41 with permission from Wiley-VCH. | ||
Another example of using solution pH to control the gel properties comes from the Banerjee group, who reported on a phenylalanine-based pyrene conjugated LMWG.96 This molecule can form hydrogels across a wide range of phosphate buffers at different pHs (7.46–15.0). Scanning electron microscopy (SEM) showed how pH impacted the morphology of the nanofibers (Fig. 4). Gel structures formed at pH 7.46–12.0 were helical in nature, whereas when the pH was greater than 12, straight, tape-like structures formed. Furthermore, the gels formed at higher pHs had wider gel fibres. It was found that the starting pH had a significant influence on the thixotropic behaviour of the hydrogels, where only the gels formed at pH 7.46 showed full recoverability after high strain was applied. Such behaviour allows these hydrogels to be suitable candidates for 3D printing,97–100 or to be used to encapsulate and release biomolecules over time without the need for heat–cool cycles.96
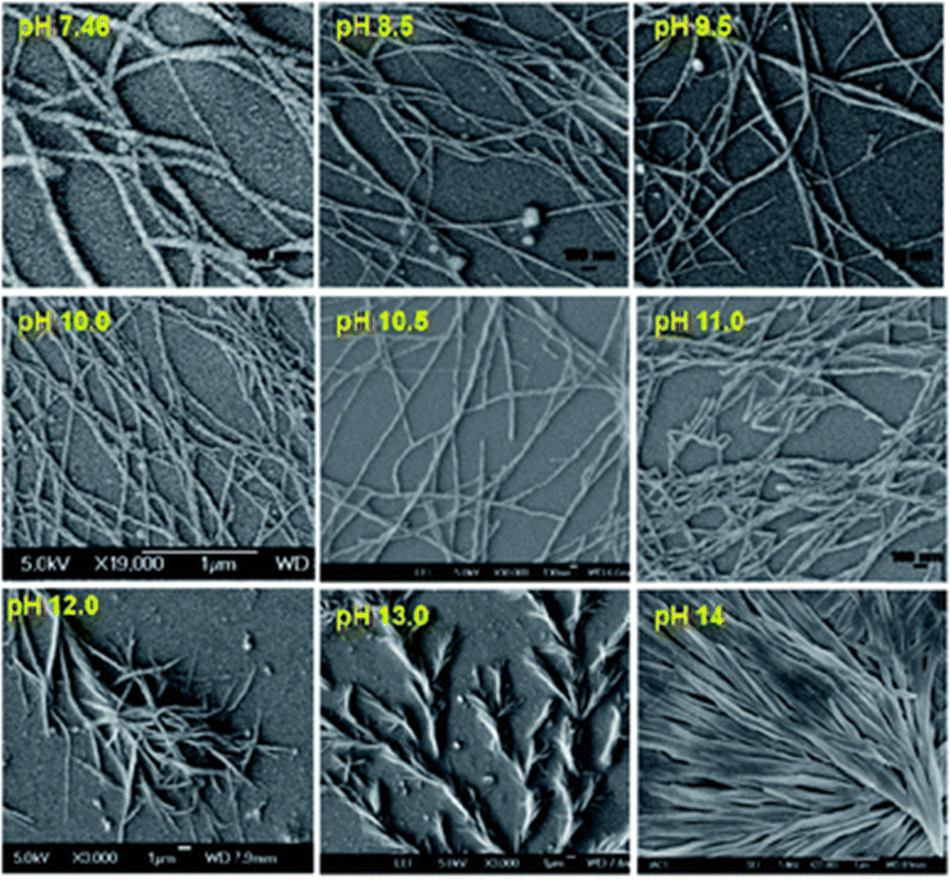 | ||
| Fig. 4 SEM images of the pyrene-based hydrogels formed at different pHs. Figure taken from ref. 94 with permission from the Royal Society of Chemistry. | ||
Similarly, Singh et al. studied the self-assembly and gelation of a Fmoc derivative of 3-nitrotyrosine (3-NT), FNT, in different phosphate buffer solutions ranging from pH 5–8.5 (Fig. 5a).101 This molecule was chosen as the phenolic hydroxyl group is highly sensitive to pH due to the electron-withdrawing nitro group. The gelation kinetics were dependent on the pH of the buffer, with higher pH buffers taking longer to gel. This difference in kinetics was due to the ionic species present in solution impacting the self-assembly behaviour. At lower pH, the carboxylate form of FNT was the dominant ion and at higher pH, the tyrosinate form became the prominent species. The carboxylate resulted in faster gel formation and a higher gelation efficiency, whereas the ionic repulsion between the nitrotyrosinate species meant the gels formed at higher pH took longer to form. As such, the gels at higher pH formed stiffer gels. Furthermore, the colour intensities of the gels increased with increasing pH (Fig. 5b). Overall, this work again highlights the importance of solution pH in controlling the kinetic process of gelation.
 | ||
| Fig. 5 (a) Chemical structure of the FNT gelator. (b) Photographs of FNT hydrogels formed in different pH buffers to show the change in colour intensity with pH. Adapted with permission from ref. 99 with permission from the American Chemical Society. | ||
Using counterions
Salts can also affect aggregation in accordance to the Hofmeister series.29,102–104 The Hofmeister series can affect the stability of secondary and tertiary structures of self-assembled materials.105 Therefore, it should be possible to tune the micellar aggregates formed from a single gelator by changing the cation.29,106–108 McAulay et al. have reported that by changing the size of the cation used to prepare 2NapFF solutions, there was a change in the structure of the micellar aggregates.29 These aggregates showed different behaviours upon heating and cooling and changes in their “apparent” pKa values. Depending on the ability of the salt to influence the solubility in aqueous solutions, counterions can be divided into two categories: “salting-in” or “salting-out”.109 Weakly hydrated cations are more likely to “salt-out”, whereas divalent cations typically “salt-in” (Fig. 6). “Salting-out” describes the increase in surface tension at the fibre-solution interface, leading to an increase in the aggregate stability due to strengthening of the hydrophobic effect.110 By contrast, “salting-in” describes a decrease in surface tension at the fibre-solution interface. As a result, the hydrophobic effect is weakened, and the gelators become more soluble. With this increased solubility, the gelator can more easily interact with water and the stability of the aggregate formed is decreased. These changes in solubility can change the self-assembled structures formed and again impact the gelation kinetics.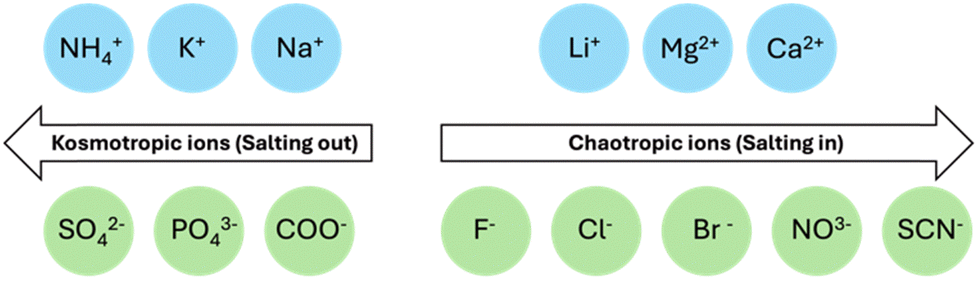 | ||
| Fig. 6 Hofmeister series showing ions which are more likely to “salt-in” on the right and ions more likely to “salt-out” on the left. | ||
Mañas-Torres et al. recently investigated the gelation kinetics of FmocFF in the presence of different metallic cations (Cs+ and Ca2+) using in situ fluorescence lifetime imaging microscopy (FLIM).111 They anticipated that Ca2+ would show “salting-in” behaviour, whilst Cs+ would result in “salting-out”. FLIM showed that fibril formation with Ca2+ ions was faster, whilst Cs+-promoted fibril formation was an order of magnitude slower. The difference in self-assembly mechanism was explained using differential scanning calorimetry (DSC) (Fig. 7a). The more complex DSC profile for Cs+-mediated gelation suggested a multistep assembly process into several increasingly stable intermediate species (a mixture of nanospheres and amorphous fibres). In comparison, only nanofibers were formed when Ca2+ was used. This mixture of fibres and nanospheres for FmocFF with Cs+ was observed using both TEM and FLIM. The difference in the self-assembly process was reflected in the physical properties of the resulting gels. Rheological measurements showed hydrogels formed in the presence of Ca2+ were 500 times stiffer than the analogous Cs+ hydrogels (Fig. 7b). It was postulated that this difference was the result of the different properties of the counterions regarding their coordination abilities and capability to stabilise the water–solute interactions.
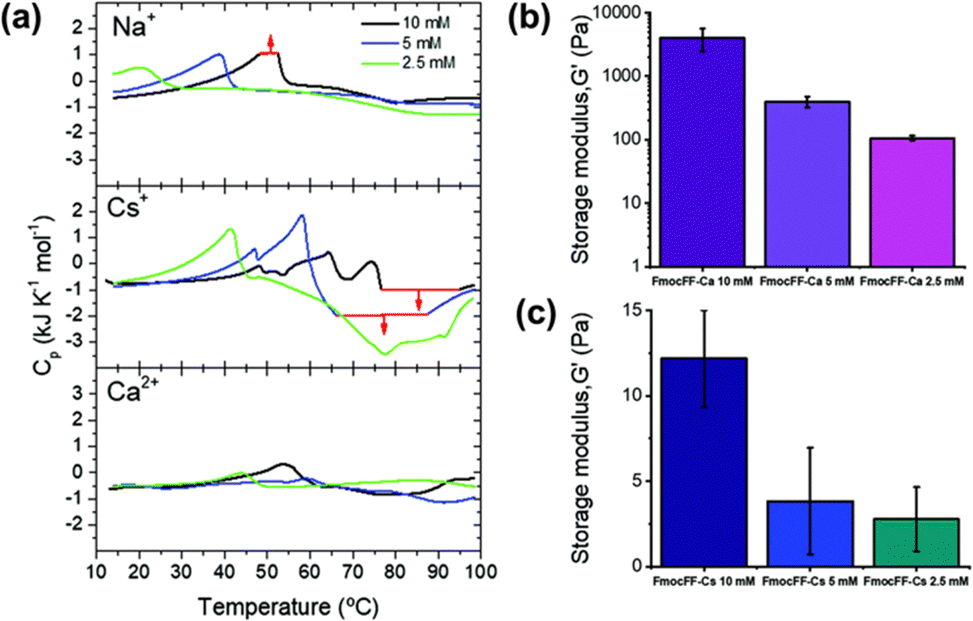 | ||
| Fig. 7 (a) DSC scans of FmocFF at a concentration of 10 mM (black), 5 mM (blue), and 2.5 mM (green) in the presence of Na+, Cs+, and Ca2+. G′ values of hydrogels formed at different FmocFF concentrations in the present of (b) Ca2+ and (c) Cs+ ions. Figure adapted from ref. 109 with permission from the Royal Society of Chemistry. | ||
Overall, this work shows the influence of different metallic ions on the mechanism of nucleation and growth of dipeptides, and thus presents another method to tune the properties of hydrogels.
Similarly, the Adams groups have reported the impact of different counterions on the assembly of 2NapFF.62 Both metal (Li+, Na+, K+, Rb+, and Cs+) and non-coordinating organic (tetra-n-butylammonium (TBA) and benzyltrimethylammonium (BTMA)) ions were used to access different micellar structures at high pH. Differences in the micellar aggregation were evident from viscosity measurements. They found that the viscosity increased with increasing size of the metallic counterions, with Li+ ions giving the lowest viscosity and Rb+ and Cs+ the highest. This increase in viscosity was thought to be the result of the more labile and soft Rb+ and Cs+ ions causing a more viscous micellar aggregation of 2NapFF. The organic ions showed a significant increase in viscosity compared to the metal ions. TBA had a slightly higher viscosity, which was attributed to its larger size and greater hydrophobicity compared to BTMA. Hydrogels were formed by cross-linking the dipeptides with Ca2+ to replace the counterions with this ion in the gel-state. As such, any differences in the gel's network and properties would be due to the distinct self-assembled structures formed in the pre-gelled form. Rheology showed that the gels became weaker with increasing size of metal ion. Furthermore, the gels formed from the organic salts were stiffer than the Li+ and Na+ salts. Upon preparation of gel noodles from metal–salt solutions, the resulting noodles were more rigid than those formed from the organic salts (Fig. 8a), which were too fragile to be measured or manipulated. The noodles from the metal salts could resist external perturbation and deformation. However, the organic salts formed significantly fragile noodles which broke when slight strain was applied or when they were shaken. Nanoindentation (Fig. 8b) and tensile testing experiments showed that the stiffness of the metal–salt noodles varied with the different ions. It was found that Cs+ ions produced the stiffest noodles. This work highlights that the variation of micellar arrangement in the pre-gel state can be translated to the hierarchical networks.
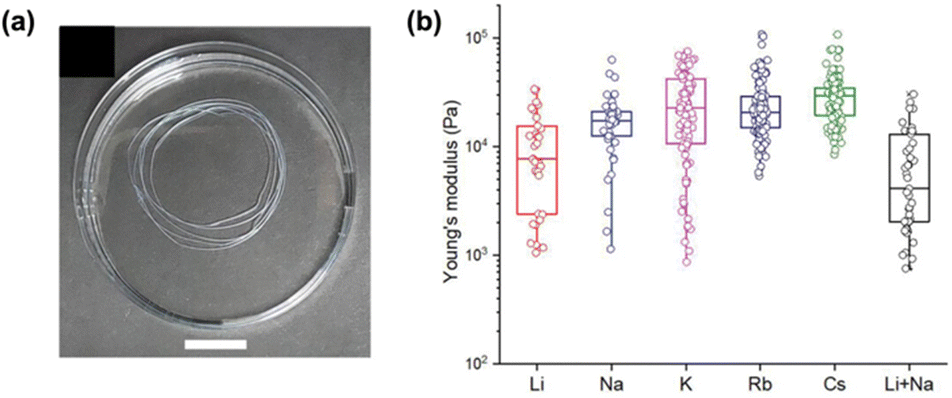 | ||
| Fig. 8 (a) Photograph of gel noodles obtained from 2NapFF (20 mg mL−1) and Li+. (b) Statistical bar plot of nanoindentation data of gel noodles obtained from different 2NapFF salts and counterions. Figure adapted from ref. 61 with permission from Wiley-VCH. | ||
Uljin and co-workers also exploited the Hofmeister series to tune the self-assembly of various Fmoc-dipeptide hydrogels.112 The efficiency of the anions in promoting the hydrophobic interactions, and thus self-assembly, was monitored by fluorescence spectroscopy. The ratio of the emission intensity of the excimer/monomer suggested that strongly hydrated ions (also known as kosmotropes) promoted stacking of fluorenyl groups, aided by increased hydrogen bonding. In comparison, weakly hydrated ions (referred to as chaotropes) resulted in weaker hydrophobic interactions. Such differences were demonstrated in the AFM results, which showed gels produced in the presence of kosmotropes formed dense, fibrous networks (Fig. 9). However, when chaotropes were present, spherical aggregates instead formed. The dense fibrous networks resulted in mechanically stiffer gels when hydrogels were formed with kosmotropes. Overall, this study demonstrates that salts have a dramatic effect on the hydrophobic interactions of dipeptides, resulting in differential order and supramolecular chirality. Therefore, the salts directly impact the mechanical properties of the resulting gels. In summary, ionic composition is another important parameter to consider when designing supramolecular hydrogels.
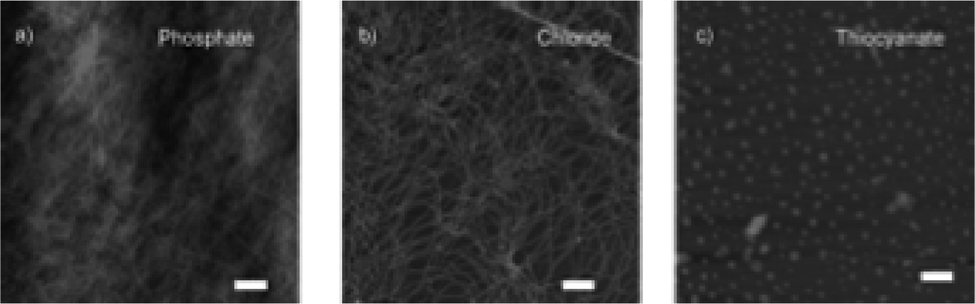 | ||
| Fig. 9 AFM images of FmocYL gels in the presence of (a) phosphate, (b) chloride, and (c) thiocyanate salts of sodium. Scale bar represents 500 nm. Figure adapted from ref. 110 with permission from Wiley-VCH. | ||
Using polymer additives
Polymer additives have generated interest due to the ability to enhance the desired properties of a material without the need for synthesis.113 Such additives have been reported to both impede and promote gelation.4,114–116 These polymers can modify the nucleation and growth process during gelation,117 facilitate aggregation,118 or increase fibre branching.119 As such, the addition of additives could impact the gelation kinetics and thus change the resulting gel properties.Recently, the Durand group investigated the impact of dextran on the gel kinetics and properties of two L-lysine-based gelators (A and B, Fig. 10).120 Samples were prepared with varying dextran quantities ranging from 0 to 240 mg. Increasing the dextran concentration resulted in weaker gels, thought to be due to the polymer reducing the topological interactions between fibre-like aggregates. Such behaviour has also been reported by the Adams group.121,122 Increasing the dextran concentration also resulted in a decrease in the gelation kinetics, suggesting gel formation was more difficult. This behaviour suggests that the polymer increased the viscosity of the pre-gelled solution, resulting in diffusion-limited self-assembly. It was also hypothesized that there were no interactions between the dextran macromolecules and the LMWG, and the polymer was instead sterically hindering aggregate formation and growth. In summary, these studies show that careful consideration must be given when choosing polymer additives, as they can directly impact the self-assembly process to alter the properties of the gel.
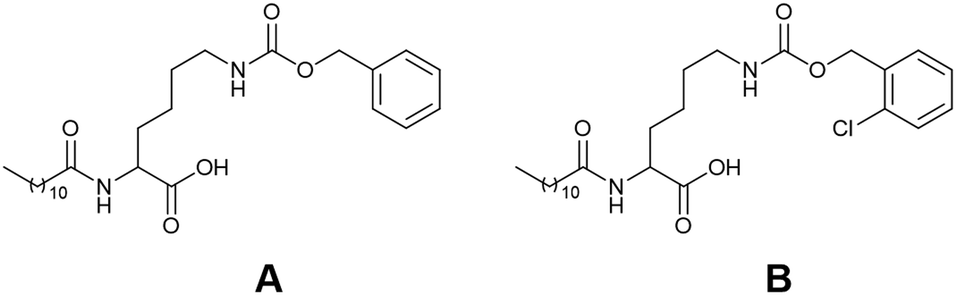 | ||
| Fig. 10 Chemical structures of the two L-lysine-based gelators (A and B) used by Durand and co-workers. Image reproduced from ref. 118 with permission from MDPI. | ||
Chakraborty et al. have also used polymer additives with the aim to improve the desired mechanical properties of tripeptide-based gels and their durability in cell culture media.123 This work focused on composite hydrogels formed from Fmoc-RGD and chitosan (Fig. 11a). Gels formed from Fmoc-RGD alone were too weak to be used for cell culturing. The composite Fmoc-RGD/chitosan hydrogels showed a significant increase in the storage modulus (G′ values of 529 Pa and 3436 Pa for Fmoc-RGD and Fmoc-RGD/chitosan, respectively). TEM showed that the fibre diameter of the composite gel had decreased, resulting in a higher aspect ratio of the fibres (Fig. 11b). These higher aspect ratio fibres entrapped the solvent more tightly, explaining the increase in G′ from the rheology. Furthermore, chitosan provided additional nucleation sites during gelation, enhancing the number of fibres, and consequently, the fibre network density. This was further suggested when monitoring the gelation over time, with the composite gels forming much faster than the Fmoc-RGD gels (239 minutes versus 46 minutes for Fmoc-RGD and Fmoc-RGD/chitosan, respectively). When placed in cell media, the Fmoc-RGD gels completely dissolved after 30 minutes. However, the Fmoc-RGD/chitosan gels were stable in media for several months. Washing with cell media made these composite gels ideal for cell adherence. This work demonstrates the use of polymers to tailor the properties of supramolecular gels by forming a gel with attributes from both the gelator and the polymer to make them suitable for multiple applications.
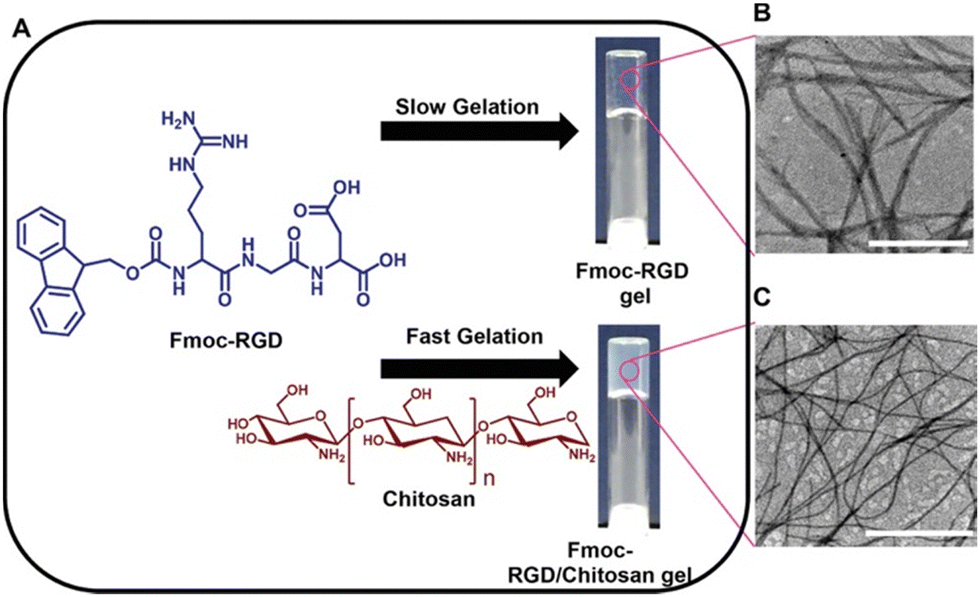 | ||
| Fig. 11 (A) Chemical structures of the Fmoc-RGD gelator and chitosan additive and images of the resulting hydrogels. TEM micrographs of (B) Fmoc-RGD and (C) Fmoc-RGD/chitosan hydrogels. Scale bars represent 1 μm. Images reproduced from ref. 121 with permission from Wiley-VCH. | ||
When using polymer additives, the molecular weight, concentration, and order of mixing can all impact the self-assembly process. The Thordarson group focused on how these factors influenced the gels formed from the dipeptide FmocFF.124 The gelator was dissolved in various polyethylene glycol (PEG)/water mixtures, and the gelation compared. Furthermore, the group explored the gelation behaviour in different molecular weight PEGs. The viscosity significantly increased upon increasing the polymer weight from PEG 400 to PEG 800, after which the viscosity only slightly increased. In the resulting gels, G′ was found to increase with increasing molecular weight of PEG from PEG 200 to PEG 400 (Fig. 12a). However, for gels formed with PEG 400 to PEG 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, there was very little change in G′, suggesting that the storage modulus of these gels cannot be correlated with the viscosity of PEG. This behaviour differs from that reported by Adams and co-workers, who showed that upon the addition of water/dextran mixtures to FmocFF in DMSO, the storage modulus showed a negative correlation with the viscosity of the water/polymer mixtures.122 Thordarson's group also found that the ratio of PEG 400/water significantly impacts the properties of the resulting gel (Fig. 12b). Between concentrations of 0% and 60% (v/v) PEG 400, a gel can be formed, and an increase in the G′ values is observed with increasing PEG concentration. However, above concentrations of 60% (v/v) PEG 400, gels could no longer form. It was hypothesized that this increase in G′ and the inability to form gels above 60% (v/v) PEG was the result of macromolecular crowding effects, which provides additional gel stability, resulting in stiffer gels. However, there is an optimal ratio for PEG-to-water interactions, which the authors think could explain why gels do not form above 60% PEG 400. Finally, the group also performed gelation experiments where the order of mixing was changed. They compared gels where the FmocFF was dissolved in the chosen PEG, followed by the addition of water, to those that were first dissolved in basic water before the polymer was added. Rheology showed that when the polymer was added first, the resulting gels were much stronger, due to better dissolution of the FmocFF in PEG 400 than water. This work emphasizes the importance of controlling experimental conditions when preparing hydrogels, as even the order in which one mixes the components can impact the resulting gels.
000, there was very little change in G′, suggesting that the storage modulus of these gels cannot be correlated with the viscosity of PEG. This behaviour differs from that reported by Adams and co-workers, who showed that upon the addition of water/dextran mixtures to FmocFF in DMSO, the storage modulus showed a negative correlation with the viscosity of the water/polymer mixtures.122 Thordarson's group also found that the ratio of PEG 400/water significantly impacts the properties of the resulting gel (Fig. 12b). Between concentrations of 0% and 60% (v/v) PEG 400, a gel can be formed, and an increase in the G′ values is observed with increasing PEG concentration. However, above concentrations of 60% (v/v) PEG 400, gels could no longer form. It was hypothesized that this increase in G′ and the inability to form gels above 60% (v/v) PEG was the result of macromolecular crowding effects, which provides additional gel stability, resulting in stiffer gels. However, there is an optimal ratio for PEG-to-water interactions, which the authors think could explain why gels do not form above 60% PEG 400. Finally, the group also performed gelation experiments where the order of mixing was changed. They compared gels where the FmocFF was dissolved in the chosen PEG, followed by the addition of water, to those that were first dissolved in basic water before the polymer was added. Rheology showed that when the polymer was added first, the resulting gels were much stronger, due to better dissolution of the FmocFF in PEG 400 than water. This work emphasizes the importance of controlling experimental conditions when preparing hydrogels, as even the order in which one mixes the components can impact the resulting gels.
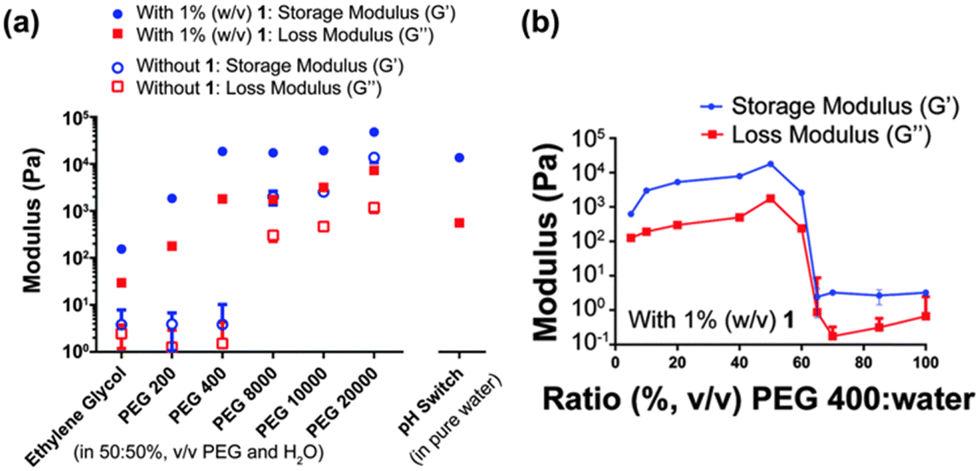 | ||
| Fig. 12 (a) Variation of G′ (blue) and G′′ (red) of gels formed with different molecular weight PEGs in water (50:50%, v/v) with 1% FmocFF (w/v) present (filled markers) and without any gelator (hollow markers). A gel of FmocFF formed using glucono-δ-lactone is also shown on the right. (b) G′ (blue) and G′′ (red) of FmocFF gels formed in various PEG 400:water mixtures. Image adapted from ref. 122 with permission from the Royal Society of Chemistry. | ||
Summary and outlook
This review discusses using pre-processing as a method to fine-tune the properties of low molecular weight hydrogels. Four different parameters used to control the formation of diverse self-assembled structures are summarised. All methods mentioned here focus on changing the pre-gelled precursor solution. However, we stress that differences in gel properties are likely due to changes in the kinetics during the gelation process. How the kinetics are changed depends on the pre-processing conditions. Using heat–cool cycles, pH and salts all change the solubility of the gelator and thus the kinetics. In comparison, polymer additives only interfere with the kinetics.The challenges that arise from adjusting the process of assembly is that it can be difficult to know where to begin, as there are no patterns to which method will give the desired properties in the end material. How a material is impacted by the different pre-processing methods discussed here is dependent on each system. As we have shown, there are many factors that must be considered and controlled when preparing materials for the given application. However, such control is not always applied in academic labs. Therefore, future work should also focus on studies into why gelation has occurred to help better understand the relationship between the pre-gelled and final gel states. A deeper understanding of this very complex process will allow us to further improve prediction models to correlate the micellar structures to the bulk gel properties. Such understanding will result in significant advances in areas such as organic electronics and regenerative medicine. Overall, this work provides new opportunities to increase the number of applications these materials are suitable for without the need to design new gelators.
Author contributions
Conceptualisation: R. G. and E. D.; funding acquisition: E. D.; supervision: E. D.; visualisation: R. G. and E. D.; writing – original draft: R. G. and E. D.; writing – review and editing: R. G. and E. D.Conflicts of interest
There are no conflicts to declare.Acknowledgements
For the purpose of open access, the authors have applied a Creative Commons Attribution (CC BY) license to any Author Accepted Manuscript version arising from this submission. We would like to thank the UKRI (MR/V021087/1) and the EPSRC (EP/S032673/1) and (EP/R513222/1) for funding.Notes and references
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CAS.
- J. Omar, D. Ponsford, C. A. Dreiss, T. Lee and X. J. Loh, Chem. – Asian J., 2022, 17, e202200081 CrossRef CAS PubMed.
- S. Panja and D. J. Adams, Chem. Soc. Rev., 2021, 50, 5165–5200 RSC.
- R. K. Mishra, S. Das, B. Vedhanarayanan, G. Das, V. K. Praveen and A. Ajayaghosh, Molecular Gels: Structure and Dynamics, Royal Society of Chemistry, Cambridge, 2018, pp. 190–226 Search PubMed.
- G. Deen and X. Loh, Gels, 2018, 4, 13 CrossRef PubMed.
- P. R. A. Chivers and D. K. Smith, Nat. Rev. Mater., 2019, 4, 463–478 CrossRef CAS.
- R. Jain and S. Roy, ACS Biomater. Sci. Eng., 2020, 6, 2832–2846 CrossRef CAS PubMed.
- V. I. B. Castro, A. R. Araújo, F. Duarte, A. Sousa-Franco, R. L. Reis, I. Pashkuleva and R. A. Pires, ACS Appl. Mater. Interfaces, 2023, 15, 29998–30007 CrossRef CAS PubMed.
- A. K. Patterson and D. K. Smith, Chem. Commun., 2020, 56, 11046–11049 RSC.
- G. Zhang, J. Li, W. Cai, S. Li, Y. Kong and Z.-Z. Yin, Microchem. J., 2023, 193, 109160 CrossRef CAS.
- J. Y. C. Lim, S. S. Goh, S. S. Liow, K. Xue and X. J. Loh, J. Mater. Chem. A, 2019, 7, 18759–18791 RSC.
- N. Hou, R. Wang, F. Wang, J. Bai, J. Zhou, L. Zhang, J. Hu, S. Liu and T. Jiao, ACS Omega, 2020, 5, 5470–5479 CrossRef CAS PubMed.
- D. McDowall, B. J. Greeves, R. Clowes, K. McAulay, A. M. Fuentes-Caparrós, L. Thomson, N. Khunti, N. Cowieson, M. C. Nolan, M. Wallace, A. I. Cooper, E. R. Draper, A. J. Cowan and D. J. Adams, Adv. Energy Mater., 2020, 10, 2002469 CrossRef CAS.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014, 6, 964–970 CrossRef CAS PubMed.
- T. Yoshii, S. Onogi, H. Shigemitsu and I. Hamachi, J. Am. Chem. Soc., 2015, 137, 3360–3365 CrossRef CAS PubMed.
- A. Döring, W. Birnbaum and D. Kuckling, Chem. Soc. Rev., 2013, 42, 7391 RSC.
- V. K. Praveen, B. Vedhanarayanan, A. Mal, R. K. Mishra and A. Ajayaghosh, Acc. Chem. Res., 2020, 53, 496–507 CrossRef CAS PubMed.
- G. L. Eakins, R. Pandey, J. P. Wojciechowski, H. Y. Zheng, J. E. A. Webb, C. Valéry, P. Thordarson, N. O. V. Plank, J. A. Gerrard and J. M. Hodgkiss, Adv. Funct. Mater., 2015, 25, 5640–5649 CrossRef CAS.
- D. J. Adams, J. Am. Chem. Soc., 2022, 144, 11047–11053 CrossRef CAS PubMed.
- J. K. Gupta, D. J. Adams and N. G. Berry, Chem. Sci., 2016, 7, 4713–4719 RSC.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- J. Raeburn, A. Zamith Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143 RSC.
- S. Panettieri and R. V. Ulijn, Curr. Opin. Struct. Biol., 2018, 51, 9–18 CrossRef CAS PubMed.
- E. R. Draper and D. J. Adams, Langmuir, 2019, 35, 6506–6521 CrossRef CAS PubMed.
- E. R. Draper and D. J. Adams, Supramolecular Nanotechnology: Advanced Design of Self-Assembled Functional Materials, VCH, Weinheim, 2023, pp. 619–639 Search PubMed.
- K. McAulay, P. A. Ucha, H. Wang, A. M. Fuentes-Caparrós, L. Thomson, O. Maklad, N. Khunti, N. Cowieson, M. Wallace, H. Cui, R. J. Poole, A. Seddon and D. J. Adams, Chem. Commun., 2020, 56, 4094–4097 RSC.
- S. Panja and D. J. Adams, Chem. – Eur. J., 2020, 26, 6130–6135 CrossRef CAS PubMed.
- A. Arango-Restrepo, J. M. Rubi and D. Barragán, J. Phys. Chem. B, 2018, 122, 4937–4945 CrossRef CAS PubMed.
- S. Panja, K. Boháčová, B. Dietrich and D. J. Adams, Nanoscale, 2020, 12, 12840–12848 RSC.
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
- F. Tantakitti, J. Boekhoven, X. Wang, R. V. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. O. De La Cruz, G. C. Schatz and S. I. Stupp, Nat. Mater., 2016, 15, 469–476 CrossRef CAS PubMed.
- Y. Li and Y. Cao, Chin. J. Polym. Sci., 2018, 36, 366–378 CrossRef CAS.
- Y. Yan, J. Huang and B. Z. Tang, Chem. Commun., 2016, 52, 11870–11884 RSC.
- N. A. Dudukovic, B. C. Hudson, A. K. Paravastu and C. F. Zukoski, Nanoscale, 2018, 10, 1508–1516 RSC.
- S. M. Hashemnejad and S. Kundu, Langmuir, 2017, 33, 7769–7779 CrossRef CAS PubMed.
- S. Bianco, S. Panja and D. J. Adams, Gels, 2022, 8, 132 CrossRef CAS PubMed.
- P. Ravarino, S. Panja and D. J. Adams, Macromol. Rapid Commun., 2022, 43, 2200606 CrossRef CAS PubMed.
- R. E. Ginesi, N. R. Murray, R. M. Dalgliesh, J. Doutch and E. R. Draper, Chem. – Eur. J., 2023, 43, e202301042 CrossRef PubMed.
- A. M. Castilla, E. R. Draper, M. C. Nolan, C. Brasnett, A. Seddon, L. L. E. Mears, N. Cowieson and D. J. Adams, Sci. Rep., 2017, 7, 8380 CrossRef PubMed.
- L. Chen, S. Revel, K. Morris and D. J. Adams, Chem. Commun., 2010, 46, 4267 RSC.
- O. El Hamoui, K. Gaudin, S. Battu, P. Barthélémy, G. Lespes and B. Alies, Langmuir, 2021, 37, 297–310 CrossRef CAS PubMed.
- D. McDowall, D. J. Adams and A. M. Seddon, Soft Matter, 2022, 18, 1577–1590 RSC.
- K. McAulay, L. Thomson, L. Porcar, R. Schweins, N. Mahmoudi, D. J. Adams and E. R. Draper, Org. Mater., 2020, 02, 108–115 CrossRef CAS.
- E. R. Draper, B. Dietrich, K. McAulay, C. Brasnett, H. Abdizadeh, I. Patmanidis, S. J. Marrink, H. Su, H. Cui, R. Schweins, A. Seddon and D. J. Adams, Matter, 2020, 2, 764–778 CrossRef.
- F. Tantakitti, J. Boekhoven, X. Wang, R. V. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. O. De La Cruz, G. C. Schatz and S. I. Stupp, Nat. Mater., 2016, 15, 469–476 CrossRef CAS PubMed.
- S. Panja and D. J. Adams, Chem. Commun., 2019, 55, 10154–10157 RSC.
- S. Panja and D. J. Adams, Giant, 2021, 5, 10041 CrossRef.
- C. Yuan, R. Xing, J. Cui, W. Fan, J. Li and X. Yan, CCS Chem., 2024, 6, 255–265 CrossRef CAS.
- J. Raeburn, C. Mendoza-Cuenca, B. N. Cattoz, M. A. Little, A. E. Terry, A. Zamith Cardoso, P. C. Griffiths and D. J. Adams, Soft Matter, 2015, 11, 927–935 RSC.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856 RSC.
- V. Lakshminarayanan, C. Chockalingam, E. Mendes and J. H. van Esch, ChemPhysChem, 2021, 22, 2256–2261 CrossRef CAS PubMed.
- Z. Huang, T. Jiang, J. Wang, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2021, 60, 2855–2860 CrossRef CAS PubMed.
- J. S. Foster, J. M. Zurek, N. M. S. Almeida, W. E. Hendriksen, V. A. A. Le Sage, V. Lakshminarayanan, A. L. Thompson, R. Banerjee, R. Eelkema, H. Mulvana, M. J. Paterson, J. H. Van Esch and G. O. Lloyd, J. Am. Chem. Soc., 2015, 137, 14236–14239 CrossRef CAS PubMed.
- A. Aufderhorst-Roberts, W. J. Frith and A. M. Donald, Soft Matter, 2012, 8, 5940–5946 RSC.
- X. Chen and S. Thibeault, 2013 35th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), 2013, 6228–6231.
- F. E.-T. Heakal and A. E. Elkholy, J. Mol. Liq., 2017, 230, 395–407 CrossRef CAS.
- C. A. Dreiss, Wormlike Micelles: Advances in Systems, Characterisation and Applications, Royal Society of Chemistry, Cambridge, 2017, pp. 1–8 Search PubMed.
- C. A. Dreiss, Soft Matter, 2007, 3, 956 RSC.
- D. Ghosh, L. J. Marshall, G. Ciccone, W. Liu, A. Squires, A. Seddon, M. Vassalli and D. J. Adams, Macromol. Mater. Eng., 2023, 308, 2300082 CrossRef CAS.
- K. McAulay, B. Dietrich, H. Su, M. T. Scott, S. Rogers, Y. K. Al-Hilaly, H. Cui, L. C. Serpell, A. M. Seddon, E. R. Draper and D. J. Adams, Chem. Sci., 2019, 10, 7801–7806 RSC.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- N. Kulkarni, P. Rao, G. S. Jadhav, B. Kulkarni, N. Kanakavalli, S. Kirad, S. Salunke, V. Tanpure and B. Sahu, ACS Omega, 2023, 8, 3551–3570 CrossRef CAS PubMed.
- N. Yadav, M. K. Chauhan and V. S. Chauhan, Biomater. Sci., 2020, 8, 84–100 RSC.
- R. Binaymotlagh, L. Chronopoulou, F. H. Haghighi, I. Fratoddi and C. Palocci, Materials, 2022, 15, 5871 CrossRef CAS PubMed.
- N. Kulkarni, S. D. Shinde, G. S. Jadhav, D. R. Adsare, K. Rao, M. Kachhia, M. Maingle, S. P. Patil, N. Arya and B. Sahu, Bioconjugate Chem., 2021, 32, 448–465 CrossRef CAS PubMed.
- K. M. Wolfe, M. Mooney, C. Crep, S. Rondeau-Gagné and G. C. Welch, Flexible Printed Electron., 2022, 7, 044007 CrossRef CAS.
- P. Zhou, R. Xing, Q. Li, J. Li, C. Yuan and X. Yan, Matter, 2023, 6, 1945–1963 CrossRef CAS.
- S. Zhang, M. A. Greenfield, A. Mata, L. C. Palmer, R. Bitton, J. R. Mantei, C. Aparicio, M. O. de la Cruz and S. I. Stupp, Nat. Mater., 2010, 9, 594–601 CrossRef CAS PubMed.
- D. McDowall, M. Walker, M. Vassalli, M. Cantini, N. Khunti, C. J. C. Edwards-Gayle, N. Cowieson and D. J. Adams, Chem. Commun., 2021, 57, 8782–8785 RSC.
- B. D. Wall, S. R. Diegelmann, S. Zhang, T. J. Dawidczyk, W. L. Wilson, H. E. Katz, H. Mao and J. D. Tovar, Adv. Mater., 2011, 23, 5009–5014 CrossRef CAS PubMed.
- A. Chalard, L. Vaysse, P. Joseph, L. Malaquin, S. Souleille, B. Lonetti, J.-C. Sol, I. Loubinoux and J. Fitremann, ACS Appl. Mater. Interfaces, 2018, 10, 17004–17017 CrossRef CAS PubMed.
- G. Liu, D. Zhang and C. Feng, Angew. Chem., Int. Ed., 2014, 53, 7789–7793 CrossRef CAS PubMed.
- E. R. Draper, H. Su, C. Brasnett, R. J. Poole, S. Rogers, H. Cui, A. Seddon and D. J. Adams, Angew. Chem., Int. Ed., 2017, 56, 10467–10470 CrossRef CAS PubMed.
- L. Chen, T. O. McDonald and D. J. Adams, RSC Adv., 2013, 3, 8714–8720 RSC.
- E. R. Draper, M. Wallace, R. Schweins, R. J. Poole and D. J. Adams, Langmuir, 2017, 33, 2387–2395 CrossRef CAS PubMed.
- J. H. Yu, S. V. Fridrikh and G. C. Rutledge, Polymer, 2006, 47, 4789–4797 CrossRef CAS.
- S. Debnath, S. Roy, Y. M. Abul-Haija, P. W. J. M. Frederix, S. M. Ramalhete, A. R. Hirst, N. Javid, N. T. Hunt, S. M. Kelly, J. Angulo, Y. Z. Khimyak and R. V. Ulijn, Chem. – Eur. J., 2019, 25, 7881–7887 CrossRef CAS PubMed.
- C. G. Pappas, Y. M. Abul-Haija, A. Flack, P. W. J. M. Frederix and R. V. Ulijn, Chem. Commun., 2014, 50, 10630–10633 RSC.
- D. M. Ryan, T. M. Doran, S. B. Anderson and B. L. Nilsson, Langmuir, 2011, 27, 4029–4039 CrossRef CAS PubMed.
- D. M. Ryan, S. B. Anderson and B. L. Nilsson, Soft Matter, 2010, 6, 3220 RSC.
- W. Liyanage and B. L. Nilsson, Langmuir, 2016, 32, 787–799 CrossRef CAS PubMed.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS PubMed.
- S. Wan, M. Jiang and G. Zhang, Macromolecules, 2007, 40, 5552–5558 CrossRef CAS.
- S. Panja and D. J. Adams, Chem. Commun., 2019, 55, 47–50 RSC.
- E. R. Draper, J. J. Walsh, T. O. McDonald, M. A. Zwijnenburg, P. J. Cameron, A. J. Cowan and D. J. Adams, J. Mater. Chem. C, 2014, 2, 5570–5575 RSC.
- M. C. Nolan, J. J. Walsh, L. L. E. Mears, E. R. Draper, M. Wallace, M. Barrow, B. Dietrich, S. M. King, A. J. Cowan and D. J. Adams, J. Mater. Chem. A, 2017, 5, 7555–7563 RSC.
- B. A. K. Kriebisch, C. M. E. Kriebisch, A. M. Bergmann, C. Wanzke, M. Tena-Solsona and J. Boekhoven, ChemSystemsChem, 2023, 5, e202200035 CrossRef CAS.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS PubMed.
- D. J. Adams, L. M. Mullen, M. Berta, L. Chen and W. J. Frith, Soft Matter, 2010, 6, 1971 RSC.
- A. J. Rufaihah, S. Cheyyatraivendran, M. D. M. Mazlan, K. Lim, M. S. K. Chong, C. N. Z. Mattar, J. K. Y. Chan, T. Kofidis and D. Seliktar, Front. Physiol., 2018, 9, 1555 CrossRef PubMed.
- W. Wang, G. Caetano, W. Ambler, J. Blaker, M. Frade, P. Mandal, C. Diver and P. Bártolo, Materials, 2016, 9, 992 CrossRef PubMed.
- L. Ghasemi-Mobarakeh, World J. Stem Cells, 2015, 7, 728 CrossRef PubMed.
- J. Nanda, A. Biswas and A. Banerjee, Soft Matter, 2013, 9, 4198 RSC.
- R. Pugliese, F. Fontana, A. Marchini and F. Gelain, Acta Biomater., 2018, 66, 258–271 CrossRef CAS PubMed.
- C. B. Highley, C. B. Rodell and J. A. Burdick, Adv. Mater., 2015, 27, 5075–5079 CrossRef CAS PubMed.
- L. Latxague, M. A. Ramin, A. Appavoo, P. Berto, M. Maisani, C. Ehret, O. Chassande and P. Barthélémy, Angew. Chem., Int. Ed., 2015, 54, 4517–4521 CrossRef CAS PubMed.
- K. J. Skilling, B. Kellam, M. Ashford, T. D. Bradshaw and M. Marlow, Soft Matter, 2016, 12, 8950–8957 RSC.
- P. Singh, S. Misra, A. Das, S. Roy, P. Datta, G. Bhattacharjee, B. Satpati and J. Nanda, ACS Appl. Bio Mater., 2019, 2, 4881–4891 CrossRef CAS PubMed.
- S. Roy, N. Javid, J. Sefcik, P. J. Halling and R. V. Ulijn, Langmuir, 2012, 28, 16664–16670 CrossRef CAS PubMed.
- K. Aoki, K. Shiraki and T. Hattori, Phys. Chem. Chem. Phys., 2016, 18, 15060–15069 RSC.
- B. Kang, H. Tang, Z. Zhao and S. Song, ACS Omega, 2020, 5, 6229–6239 CrossRef CAS PubMed.
- Y. Li, T. Zhao, C. Wang, Z. Lin, G. Huang, B. D. Sumer and J. Gao, Nat. Commun., 2016, 7, 13214 CrossRef PubMed.
- C. Liu, Y. Wang, Y. Gao, Y. Zhang, L. Zhao, B. Xu and L. S. Romsted, Phys. Chem. Chem. Phys., 2019, 21, 8633–8644 RSC.
- N. Vlachy, M. Drechsler, J.-M. Verbavatz, D. Touraud and W. Kunz, J. Colloid Interface Sci., 2008, 319, 542–548 CrossRef CAS PubMed.
- G. O. Lloyd and J. W. Steed, Nat. Chem., 2009, 1, 437–442 CrossRef CAS PubMed.
- A. Klaus, G. J. T. Tiddy, R. Rachel, A. P. Trinh, E. Maurer, D. Touraud and W. Kunz, Langmuir, 2011, 27, 4403–4411 CrossRef CAS PubMed.
- A. M. Hyde, S. L. Zultanski, J. H. Waldman, Y.-L. Zhong, M. Shevlin and F. Peng, Org. Process Res. Dev., 2017, 21, 1355–1370 CrossRef CAS.
- M. C. Mañas-Torres, C. Gila-Vilchez, J. A. González-Vera, F. Conejero-Lara, V. Blanco, J. M. Cuerva, M. T. Lopez-Lopez, A. Orte and L. Álvarez de Cienfuegos, Mater. Chem. Front., 2021, 5, 5452–5462 RSC.
- S. Roy, N. Javid, P. W. J. M. Frederix, D. A. Lamprou, A. J. Urquhart, N. T. Hunt, P. J. Halling and R. V. Ulijn, Chem. – Eur. J., 2012, 18, 11723–11731 CrossRef CAS PubMed.
- H. K. Can and S. Parvizikhosroshahi, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 680–689 CrossRef CAS PubMed.
- J. L. Li, X. Y. Liu, C. S. Strom and J. Y. Xiong, Adv. Mater., 2006, 18, 2574–2578 CrossRef CAS.
- J. Li and X. Liu, Adv. Funct. Mater., 2010, 20, 3196–3216 CrossRef CAS.
- Y. J. Adhia, T. H. Schloemer, M. T. Perez and A. J. McNeil, Soft Matter, 2012, 8, 430–434 RSC.
- J.-L. Li, B. Yuan, X.-Y. Liu, X.-G. Wang and R.-Y. Wang, Cryst. Growth Des., 2011, 11, 3227–3234 CrossRef CAS.
- M. Numata, K. Sugiyasu, T. Kishida, S. Haraguchi, N. Fujita, S. M. Park, Y. J. Yun, B. H. Kim and S. Shinkai, Org. Biomol. Chem., 2008, 6, 712 RSC.
- J.-L. Li and X.-Y. Liu, J. Phys. Chem. B, 2009, 113, 15467–15472 CrossRef CAS PubMed.
- G. Rangel Euzcateguy, C. Parajua-Sejil, P. Marchal, D. Chapron, M.-C. Averlant-Petit, L. Stefan, G. Pickaert and A. Durand, Colloids Surf., A, 2021, 625, 126908 CrossRef CAS.
- L. Chen, S. Revel, K. Morris, D. G. Spiller, L. C. Serpell and D. J. Adams, Chem. Commun., 2010, 46, 6738 RSC.
- G. Pont, L. Chen, D. G. Spiller and D. J. Adams, Soft Matter, 2012, 8, 7797 RSC.
- P. Chakraborty, M. Ghosh, L. Schnaider, N. Adadi, W. Ji, D. Bychenko, T. Dvir, L. Adler-Abramovich and E. Gazit, Macromol. Rapid Commun., 2019, 40, 1900175 CrossRef PubMed.
- Md. M. Hassan, A. D. Martin and P. Thordarson, J. Mater. Chem. B, 2015, 3, 9269–9276 RSC.
| This journal is © The Royal Society of Chemistry 2024 |