
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel non-helical antimicrobial peptides insert into and fuse lipid model membranes†
Saheli
Mitra
 a,
Bhairavi
Chandersekhar
a,
Yunshu
Li
a,
Mark
Coopershlyak
a,
Margot E.
Mahoney
a,
Brandt
Evans
a,
Rachel
Koenig
a,
Stephen C. L.
Hall
b,
Beate
Klösgen
c,
Frank
Heinrich
ad,
Berthony
Deslouches
e and
Stephanie
Tristram-Nagle
*a
a,
Bhairavi
Chandersekhar
a,
Yunshu
Li
a,
Mark
Coopershlyak
a,
Margot E.
Mahoney
a,
Brandt
Evans
a,
Rachel
Koenig
a,
Stephen C. L.
Hall
b,
Beate
Klösgen
c,
Frank
Heinrich
ad,
Berthony
Deslouches
e and
Stephanie
Tristram-Nagle
*a
aBiological Physics Group, Physics Department, Carnegie Mellon University, Pittsburgh, PA 15213, USA. E-mail: stn@cmu.edu
bISIS Neutron and Muon Source, Rutherford Appleton Laboratory, Harwell Campus, Didcot, Oxfordshire OX11 0QX, UK
cUniversity of Southern Denmark, Dept. Physics, Chemistry & Pharmacy, PhyLife, Campusvej 55, Odense M5230, Denmark
dCenter for Neutron Research, National Institute of Standards and Technology, Gaithersburg, MD 20899, USA
eDepartment of Environmental and Occupational Health, University of Pittsburgh, Pittsburgh, PA 15261, USA
First published on 29th April 2024
Abstract
This research addresses the growing menace of antibiotic resistance by exploring antimicrobial peptides (AMPs) as alternatives to conventional antibiotics. Specifically, we investigate two linear amphipathic AMPs, LE-53 (12-mer) and LE-55 (16-mer), finding that the shorter LE-53 exhibits greater bactericidal activity against both Gram-negative (G(−)) and Gram-positive (G(+)) bacteria. Remarkably, both AMPs are non-toxic to eukaryotic cells. The heightened effectiveness of LE-53 is attributed to its increased hydrophobicity (H) compared to LE-55. Circular dichroism (CD) reveals that LE-53 and LE-55 both adopt β-sheet and random coil structures in lipid model membranes (LMMs) mimicking G(−) and G(+) bacteria, so secondary structure is not the cause of the potency difference. X-ray diffuse scattering (XDS) reveals increased lipid chain order in LE-53, a potential key distinction. Additionally, XDS study uncovers a significant link between LE-53's upper hydrocarbon location in G(−) and G(+) LMMs and its efficacy. Neutron reflectometry (NR) confirms the AMP locations determined using XDS. Solution small angle X-ray scattering (SAXS) demonstrates LE-53's ability to induce vesicle fusion in bacterial LMMs without affecting eukaryotic LMMs, offering a promising strategy to combat antibiotic-resistant strains while preserving human cell integrity, whereas LE-55 has a smaller ability to induce fusion.
1 Introduction
Since their discovery almost a century ago, antibiotics have been hailed as a revolutionary treatment for bacterial infections.1 Unfortunately, the effectiveness of antibiotics has been compromised by their excessive use, leading to the development of antimicrobial resistance in a growing number of bacterial strains.2–4 This has warranted the exploration of new therapeutic options to combat resistant bacteria, including antimicrobial peptides (AMPs). AMPs, a diverse group of bioactive small proteins, are part of the body's first line of defense for pathogen inactivation. They work by disrupting bacterial cell membranes, modulating the immune response, and regulating inflammation.5,6 AMPs being amphipathic (comprising both hydrophilic and hydrophobic parts) readily attach to lipid bilayers characterized by a hydrophilic headgroup and hydrophobic interior. Conventional antibiotics function through diverse mechanisms, such as impeding bacterial cell wall synthesis,7–9 DNA replication,10 protein synthesis,11 or folic acid metabolism.12 By contrast, AMPs kill bacteria by perturbing their membrane in a non-specific manner, which delays onset of resistance by several weeks.13 Therefore, AMPs could offer an important alternative to traditional antibiotics.14,15 It has been shown that the negatively charged phosphate group on lipid A, when modified with phosphoethanolamine (pEtN), renders it neutral. Consequently, the positively charged AMP is no longer attracted to the modified lipid A.16–18 This rapid membrane-lytic mechanism grants AMPs a wide range of antimicrobial success, combating susceptible and multi-drug-resistant bacteria. The way AMPs disrupt membranes can vary. This disruption can involve creating pores in the membrane,19–21 which can take shapes like “barrel-stave”22 or “toroidal”,15,23 or it can involve actions like interfacial activity,24 thinning the membrane,25–27 segregating lipid domains,28 or solvation (known as the “carpet” model).29 Additionally, Chen et al. suggested the “membrane discrimination model,” where the composition of membrane lipids determines how AMPs act; for example, a eukaryotic membrane might cause the same helical AMP to act like a barrel-stave, but a bacterial membrane would facilitate a carpet mechanism.30 Bacterial membranes have more negatively charged lipids compared to mammalian cell membranes, so cationic AMPs interact selectively with them.31,32While membrane disruption is often seen as the primary mechanism of action for AMPs, other processes may also contribute to their antibacterial efficacy.5 For example, Buforin II, which is structurally similar to the pore forming peptide magainin 2 kills bacteria without cell lysis and has a strong affinity for DNA and RNA, suggesting that Buforin II's target is intracellular nucleic acids, not cell membranes.33–35 Other processes such as inhibition of nucleic-acid synthesis,36 protein synthesis37–39 and enzymatic activity40,41 also contribute to antimicrobial peptide effectiveness. Further, several homopolypeptides, such as poly-L-lysine and poly-L-arginine, that belong to the class of cell-penetrating peptides (CPPs), have antibacterial properties.42–47 CPPs penetrate the cell membrane through two independent mechanisms: one involving endocytosis and the other membrane translocation,48–52 often leaving the membrane intact. However, the precise mechanism of the antibacterial action of CPPs remain incompletely understood.53
Recent advancements have demonstrated that we can modify cationic AMPs to improve their effectiveness and selectivity. One way is by carefully selecting specific combinations of amino acids. For instance, incorporating positively charged arginine (Arg, R) residues on one side of the helix and hydrophobic valine (Val, V) residues on the other side can enhance selectivity.54 Additionally, extending the peptide chain length and introducing tryptophan (Trp, W) on the hydrophobic side of the peptide can also boost antimicrobial activity.55–57 Tryptophan-rich natural AMPs like tritrpticin and indolicidin have shown enhanced potency against a wide range of bacteria while also being less sensitive to salt and serum.55 For example, the engineered peptide WLBU2, which is rich in W, exhibits broader antimicrobial activity compared to other available AMPs such as LL-37, polymyxin B, and colistin.58 However, a key challenge in designing AMPs is to enhance their antibacterial effects without increasing the risk of harming the host. To address this, we employ a systematic approach to peptide design, adjusting the length and sequence of specific amino acids. Some studies have found that using W exclusively in the hydrophobic domain can enhance both antimicrobial activity and host toxicity due to its high hydrophobicity and bulky indole ring.55,59 Thus, in order to mitigate toxicity to eukaryotic cells we added three, four or five Ws to the remaining Vs in the hydrophobic domain.60,61
Various scientific methods have been used to study AMPs.62–65 For instance, we’ve employed X-ray diffuse scattering (XDS) to observe how colistin affects the elasticity and lipid organization of membranes, hinting at a mechanism involving lipid domains.66 Our research has shown that both WLBU2 and its stereoisomer D8-WLBU2 cause similar changes in membrane elasticity as colistin.67 One notable aspect of AMPs is their ability to adopt different secondary structures (like α-helix, β-sheet, β-turn, or random coil) when interacting with bacterial membranes, which is crucial for their effectiveness.68 We’ve worked to optimize the secondary structure of WLBU2 to form an amphipathic α-helix, enhancing its ability to fight bacteria.69 In our recent study involving peptides rich in R and W, such as E2-35 (16 AAs) and E2-05 (22 AAs), we found that the percentage of α-helicity varies depending on the lipid composition of the membranes: greater in G(−) bacteria inner membranes compared to G(+) bacteria or eukaryotic membranes with 33% cholesterol. Our XDS data revealed that a headgroup location correlates with efficacy, but also with toxicity. The membrane bending modulus KC displayed non-monotonic changes due to increasing concentrations of E2-35 and E2-05 in G(−) and G(+) LMMs, suggesting a bacterial killing mechanism where lipid domain formation causes ion and water leakage.70
The present study aims to compare the antibacterial activity, cytotoxicity and biophysics of two linear amphipathic peptides, LE-53 (12-mer) and LE-55 (16-mer), which are both rich in R and W. We classify them as linear amphipathic due to the linear separation between hydrophilic and hydrophobic residues in their primary sequences (as shown in Table 1). By maintaining this linear separation, formation of a stable helix is prevented. In a helical wheel scheme, hydrophobic and hydrophilic amino acids are intermixed which disrupts the helical structure. The peptide LE-55 has the same amino acid composition as the helical peptide E2-35,70 but is designed to not be helical. The secondary structures of the AMPs were analyzed using circular dichroism (CD) measurements to explore potential correlations with their activity. To investigate the structure of the membranes, XDS was employed to explore the location of AMPs within different lipid model membranes, as well as membrane rigidity and lipid chain order. Neutron reflectometry (NR) experiments served to validate the X-ray findings. Additionally, solution small angle X-ray scattering (SAXS) was utilized to study the fusogenic properties of these peptides. Through in vitro microbiological assays, the antibacterial activity and cytotoxicity of LE-53 and LE-55 were determined.
| Peptide | Peptide primary sequence | #AA | Charge | H |
|---|---|---|---|---|
| LE-53 | RR RR RR WW WW VV | 12 | +6 | 0.448 |
| LE-55 | RR RR RR RR WW WW VV VV | 16 | +8 | 0.362 |
2 Experimental
2.1 Materials
The synthetic lyophilized lipids 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(10-rac-glycerol) sodium salt (POPG), 10,30-bis[1,2-dioleoyl-sn-glycero-3-phospho]-sn-glycerol sodium salt (TOCL, i.e., cardiolipin), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), egg sphingomyelin (ESM), and 1,2-dioleoyl-3-trimeathylammoniumpropane chloride salt (DOTAP) were purchased from Avanti Polar Lipids (Alabaster, AL) and used as received. Cholesterol was from Nu-Chek-Prep (Waterville, MN). HPLC-grade organic solvents were purchased from Sigma-Aldrich (St. Louis, MO). Lipid stock solutions in chloroform were combined to create lipid mixtures in molar ratios mimicking the G(−) Inner Membrane (IM): POPE/POPG/TOCL (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 molar ratio), G(+) membrane: POPG/DOTAP/POPE/TOCL (6:1.5:1.5:1),71 and eukaryotic membrane, Euk33: POPC/ESM/POPE/cholesterol (15:4:1:10) (33 mole % cholesterol).72 Bacterial cation-adjusted Mueller Hinton Broth (MHB2), Test Condition Media, Roswell Park Memorial Institute (RPMI) media, fetal bovine serum (FBS) and phosphate-buffered saline (PBS) were obtained from Millipore Sigma (St Louis, MO). RPMI media contains the reducing agent glutathione as well as biotin, vitamin B12, and para aminobenzoic acid. In addition, RPMI media includes high concentrations of the vitamins inositol and choline. Because RPMI contains no proteins, lipids, or growth factors, it is commonly supplemented with FBS. FBS contains more than 1000 components such as growth factors, hormones, and transport proteins that contribute to cell growth when supplemented into culture media.73 Formaldehyde was obtained from Thermo Fisher (Waltham, MA). The peptides LE-53 (MW: 2610 gm mol−1) and LE-55 (MW: 3313 gm mol−1) were purchased in lyophilized form (10 mg in a 1.5 mL vial) from Genscript (Piscataway, NJ) with HPLC/MS spectra corresponding to each designed primary sequence. The traditional antibiotics and colistin were purchased from Millipore Sigma (St. Louis, MO). Peptides’ compositions and their physical attributes are provided in Table 1.
:2:1 molar ratio), G(+) membrane: POPG/DOTAP/POPE/TOCL (6:1.5:1.5:1),71 and eukaryotic membrane, Euk33: POPC/ESM/POPE/cholesterol (15:4:1:10) (33 mole % cholesterol).72 Bacterial cation-adjusted Mueller Hinton Broth (MHB2), Test Condition Media, Roswell Park Memorial Institute (RPMI) media, fetal bovine serum (FBS) and phosphate-buffered saline (PBS) were obtained from Millipore Sigma (St Louis, MO). RPMI media contains the reducing agent glutathione as well as biotin, vitamin B12, and para aminobenzoic acid. In addition, RPMI media includes high concentrations of the vitamins inositol and choline. Because RPMI contains no proteins, lipids, or growth factors, it is commonly supplemented with FBS. FBS contains more than 1000 components such as growth factors, hormones, and transport proteins that contribute to cell growth when supplemented into culture media.73 Formaldehyde was obtained from Thermo Fisher (Waltham, MA). The peptides LE-53 (MW: 2610 gm mol−1) and LE-55 (MW: 3313 gm mol−1) were purchased in lyophilized form (10 mg in a 1.5 mL vial) from Genscript (Piscataway, NJ) with HPLC/MS spectra corresponding to each designed primary sequence. The traditional antibiotics and colistin were purchased from Millipore Sigma (St. Louis, MO). Peptides’ compositions and their physical attributes are provided in Table 1.
2.2 Methods
:100 with fresh cation-adjusted MHB2 and incubating for an additional 3–4 h. Bacteria were spun at 3000g for 10 min. The pellet was resuspended in Test Condition Media to determine bacterial turbidity using a Den-1B densitometer (Grant Instruments, Beaver Falls, PA) at 0.5 McFarland units corresponding to 108 CFU mL−1.
To examine antibacterial activity, we used minor modifications of a standard growth inhibition assay endorsed by the Clinical and Laboratory Standards Institute (CLSI), as previously described.74 Bacteria were incubated with each of the indicated peptides in MHB2. The bacterial cells were kept in an incubator for 18 h at 37 °C, which is linked to a robotic system that feeds a plate reader every hour with one of 8 × 96-well plates. The 96-well plates are standard flat-bottom microliter plates purchased from Thermo Fisher (Waltham, MA). This setup allows the collection of growth kinetic data at A570 (absorbance at 570 nm) to examine growth inhibition in real-time (BioTek Instruments, Winooski, VT). We define minimum inhibitory concentration (MIC) as the minimum peptide concentration that completely prevents bacterial growth, demonstrated by a flat (horizontal line) growth curve as a function of hourly determinations for 18 h. at A570 (Fig. S5, ESI†). The assays are typically repeated a second time. If the MIC differs from the first assay, a third experimental trial is performed to confirm the MIC.
Human WBCs RPMI and 10% FBS were incubated with each selected peptide for 1 h at 37 °C. The cells were then immediately washed with PBS at 1000 g for 7 min, while in a round-bottom 96-well plate. After resuspension in PBS, fixable blue live/dead stain from Life Technologies was added according to manufacturer's instructions. The cells were again washed and resuspended in PBS to remove non-specific stain and then fixed with 4% formaldehyde for 1 h. After washing again with PBS, the samples were stored at 4 °C overnight (in the dark) before examination by flow cytometry using the Novocyte flow cytometer (Agilent Technologies, Santa Clara, CA). Peptide-treated cells were compared with untreated cells for dye incorporation, and data were analyzed using the Novocyte analytical software. Dye incorporation was quantified as percent toxicity directly determined by distinguishing live from dead populations,75 which was plotted using GraphPad (Prizm software, San Diego, CA).
:1 and 70:1. Higher molar ratios of lipid:peptide were not possible due to absorption flattening in the UV region. The samples remained at room temperature for ∼16 hours before the CD measurement. Data were collected in 3 mL quartz cuvettes using a Jasco 1500 CD spectrometer at 37 °C in the Center for Nucleic Acids Science and Technology (CNAST) at Carnegie Mellon University. The samples were scanned from 200 to 240 nm 20 times and the results were averaged. The temperature was controlled at 37 °C via a Peltier element and water circulation through the sample compartment. Nitrogen gas was used at a flow rate between 0.56 and 0.71 m3 h−1 to protect the UV bulb. OriginPro 2019 (OriginLab, Northampton, MA) was used to carry out a Levenberg–Marquardt least squares fit of the tryptophan-subtracted ellipticity traces to four secondary structural motifs representing α-helix, β-sheet, β-turn and random coil.25,76 This analysis gives a percentage match of each secondary structural motif to the total sample ellipticity. Instrument ellipticity (ε) was converted to Mean Residue Ellipticity using MRE (deg cm2 dmol−1) = ε × 104/N, where N = # amino acids and peptide concentration was always 10 μM.
:1, v/v) or trifluoroethanol:chloroform (1:1, v/v), were deposited onto a Si wafer (15 mm W × 30 mm L × 1 mm H) inside a fume hood. After rapid evaporation while rocking the substrate, an immobile film formed which was then further dried inside the fume hood for two hours, followed by overnight drying under vacuum to evaporate residual organic solvent. The samples were trimmed to occupy 5 mm W × 30 mm L along the center of the Si substrate. The substrate was fixed to a glass block (5 mm H × 10 mm W × 32 mm L) using heat sink compound (Dow Corning, Freeland, MI). The sample was stored in a refrigerator at 4 °C for several hours. Cold storage prior to transfer into a well-insulated hydration chamber held at 37 °C caused 100% hydration through the vapor within just 10 minutes. This process is faster than our previous method that required a Peltier cooler under the sample.78 Low-angle XDS (LAXS) data from oriented, fully hydrated samples were obtained at the ID7A1 line at Center for High Energy X-ray Sciences (CHEXS, Ithaca, NY) on two separate trips to the Cornell High Energy Synchrotron Source (CHESS) using X-ray wavelengths of 0.8434 Å and 0.8819 Å and sample-to-detector (S)-distances of 410 mm and 390.5 mm, with an Eiger 4M detector. Measurements were carried out in the fluid phase at 37 °C. The flat silicon wafer was rotated from −1 to 6 degrees during the data collection at CHESS to vary the angle of incidence. The background was collected by setting the X-ray angle of incidence to −1.7 degree, where sample scattering does not contribute to the image. For data analysis, backgrounds were first subtracted to remove extraneous air and mylar scattering and the images were laterally symmetrized to increase the signal-to-noise ratio. As the sample nears full hydration, membrane fluctuations occur which produce ‘‘lobes’’ of diffuse X-ray scattering data.79 The fluctuations are quantitated by measuring the fall-off in lobe intensity in the lateral qr direction. The fitting procedure is a non-linear least squares fit that uses the free energy functional from liquid crystal theory,80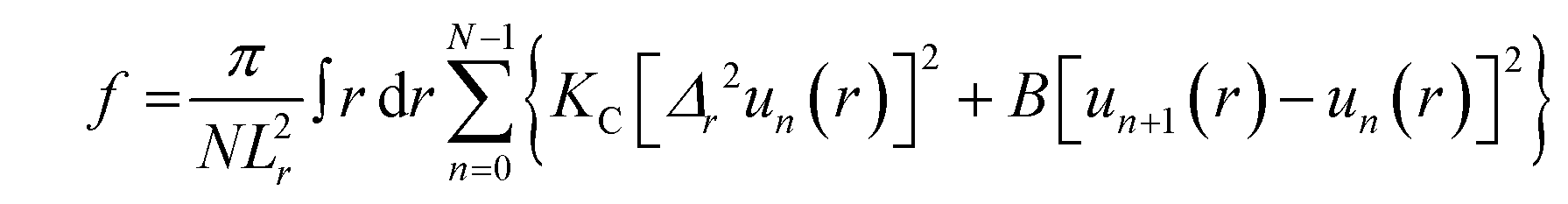 | (1) |
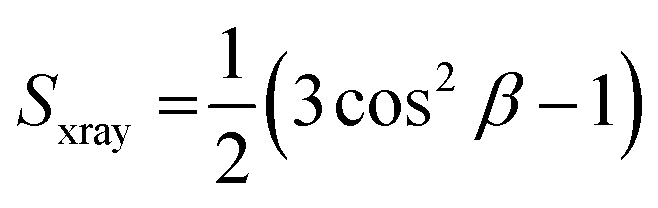 | (2) |
We also obtain the RMSE (root mean square error), which reports the goodness of the fit. An example of LAXS (Fig. S2a, ESI†) and WAXS (Fig. S2b, ESI†) for LE-53 in G(−)IM LMMs at 250:1 molar ratio is added in ESI.†
sin(θ), where λ is the wavelength and 2θ is the scattering angle) was obtained by azimuthally averaging the 2D data. As demonstrated in ref. 83, the absorption coefficient by ULV solution is independent of q over the range studied; hence, no absorption correction was required. Further, a linear intensity corresponding to pure water was subtracted from the acquired scattering intensity I(q).
:30 mol:mol β-mercaptoethanol:HC18 tether solution in ethanol for at least 60 min, leading to the formation of a self-assembled monolayer (SAM) of both molecules at the gold surface.85 SAM-decorated wafers were assembled in the NR cell, and lipid bilayers were completed by fusing vesicles of the desired lipid/peptide mixtures using an osmotic shock procedure.86 NR data were sequentially collected after rinsing the NR cell with ∼6 cell volumes of either D2O or H2O using a syringe. NR datasets collected on stBLMs immersed in isotopically different buffers were analyzed simultaneously (2 datasets per stBLM). One-dimensional structural profiles of the substrate and the lipid bilayer along the interface normal z were parameterized with a model that utilizes continuous volume occupancy distributions of the molecular components.87 Free-form peptide profiles were modeled using Hermite splines with control points on average 15 Å apart.88 The protein extension along the membrane normal determines the number of spline control points and was iteratively refined. A Monte Carlo Markov Chain-based global optimizer was used to determine best-fit parameters and their confidence limits.
3 Results and discussion
3.1 Toxicity to bacteria
LE-53 and LE-55 were initially screened for antibacterial potency against an MDR panel of Gram negative (G(−)) and Gram positive (G(+)) bacterial isolates from UPMC. The MIC is measured by a horizontal growth curve taken every hour;61 these MIC values are listed in Table 2. The MICs represent the average of different strains of each type of bacteria. The G(−) bacterial strains are: Pseudomonas aerginosa (PA) (PA14, PA01, PA828), Acinetobacter baumannii (AB) (ABF8, ABF9, AB-A3, AB-A2), Klebsiella pneumoniae (KP) (KPC3, KPC3, KP43816, KPB3, KPA5), Escherichia coli (EC) (YDC748, YDC337, YDC107, 25922) and Enterobacter (Entbac) (EA518, EA596, EC470, EC 560). The G(+) bacterial strains are: Enterococci (Entcoc) (EF23614, EF24670, EF26215, EF26692) and Staphylococcus aureus (SA) (SA49775, MRSA-US300). Notably, the short peptide LE-53 displays broad activity against both G(−) and G(+) bacterial species, outperforming LE-55 and tobramycin (a conventional antibiotic) with the lowest MIC values. The results presented in Table 2 clearly demonstrate a significant variation in MIC values across different bacterial species.| MIC (μM) | % Toxicity | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| G(−) | G(+) | ||||||||||
| PA | AB | KP | EC | Entbac | Average | Entcoc | SA | Average | RBC | WBC | |
| LE-53 | 10.7 | 3.3 | 3.6 | 4.8 | 6.5 | 5.8 ± 1.3 | 14.4 | 2.00 | 8.2 ± 3.0 | 0 | 0 |
| LE-55 | 32.0 | 21.3 | 32.0 | 28.8 | 32.0 | 29.2 ± 2.1 | 28.0 | 24.0 | 26.0 ± 1.3 | 0 | 0 |
| Colistin | 8.4 | 0.5 | 0.7 | 4.3 | 12.1 | 5.2 ± 2.2 | 32.0 | 64.0 | 48.0 ± 23 | — | — |
| Tobramycin | 32.0 | 32.0 | 2.1 | 28.0 | 24.5 | 23.7 ± 3.4 | 25.0 | 13.1 | 19.0 ± 10 | — | — |
The antimicrobial peptides were examined for minimum inhibitory concentrations (MIC) against G(−) and G(+) MDR isolates from UPMC. % RBC lysis at 32 μM and % toxicity at 16 μM against human WBCs are shown. The MICs are the average of different strains of each type of bacteria. Data are representative of 2–3 experimental trials. See text for bacteria names.
In order to compare MIC values to physical attributes, we used the HeliQuest website (https://heliquest.ipmc.cnrs.fr)89,90 to calculate hydrophobicity (H). The two peptides LE-53 and LE-55 differ in length, charge, and H as detailed in Table 1. Two fewer Rs and two fewer Vs in LE-53 compared to LE-55 increased its H considerably. Fig. 1 compares bactericidal efficiency vs. H. Strikingly, a clear correlation between H and bactericidal efficiency was observed. LE-53, characterized by higher values of H, demonstrated significantly lower MICs than LE-55, indicating hydrophobicity could be the cause of higher efficacy. A similar trend was observed by Rosenfeld et al. with a group of 12-mer peptides composed of D,L-amino acids and their fatty acid conjugates.91 Further, linear amphipathic LE-55 has a similar hydrophobicity with our previously published amphipathic α-helical peptide, E2-35.70 However, LE-55 demonstrated inferior antibacterial activity compared to E2-35. Consequently, we believe that altering the primary sequence does indeed impact bactericidal efficiency, while hydrophobicity may play a secondary role.
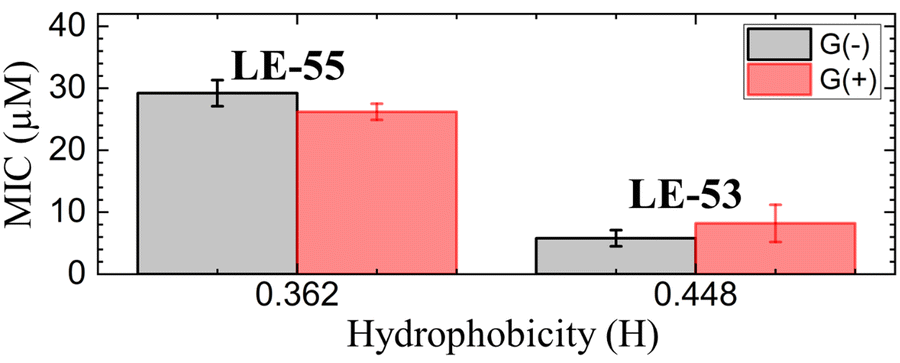 | ||
| Fig. 1 MIC vs. hydrophobicity (H) of LE-peptides in G(−) and G(+) bacteria. Errors bars correspond to standard error of the mean values, σ = Std. dev/√N. Std. dev are calculated by combining the standard deviations for each bacterial species, σAve = √((σA)2 + (σB)2 + (σC)2…)/N. | ||
It is important to note that this trend cannot be universally generalized. Many studies have reported conflicting or inconclusive trends regarding the relationship between H and bactericidal efficiency. In a study by Chen et al. MIC displayed a “sweet spot” of hydrophobicity vs. efficacy.30 In that study, peptide aggregation decreased antibacterial activity as hydrophobicity increased beyond an ideal value.30 In another study, antibacterial activity increased above a threshold value of hydrophobicity.92 Recently Mitra et al. reported poorer efficacy with increasing hydrophobicity for R and W rich helical peptides.70 A similar trend was also reported by Jakkampudi et al. in their recent study with SPLUNC1 (short palate lung and nasal epithelial clone 1) AMP derivatives.93
3.2 Toxicity to eukaryotic cells
The lytic activity of all peptides was systematically examined to determine their potential toxicity to eukaryotic cells, specifically RBCs and WBCs. The obtained data, as summarized in Table 2, shows that neither of the peptides exhibits any discernible toxic effects on eukaryotic cells, highlighting their promising potential for therapeutic applications. This contrasts to helical forming peptides, which usually display some toxicity.703.3 Secondary structure
CD results of LE-53 and LE-55 in three distinct LMM ULVs are shown in Fig. 2. Fig. S1 (ESI†) presents the MRE data for lipid/peptide molar ratios. A comprehensive summary of the percentage of all four secondary structural motifs observed in the peptides can be found in Tables S1–S6 (ESI†). Results indicate that both LE-53 and LE-55 displayed approximately 55–60% random coil and 40–45% β-sheet structures in their pure form, as well as when interacting with G(−)IM, G(+), and Euk33 LMMs. Despite similar secondary structure, LE-53 exhibited notable bactericidal activity against both G(−) and G(+) bacteria, as demonstrated in Table 2, while LE-55 was less effective.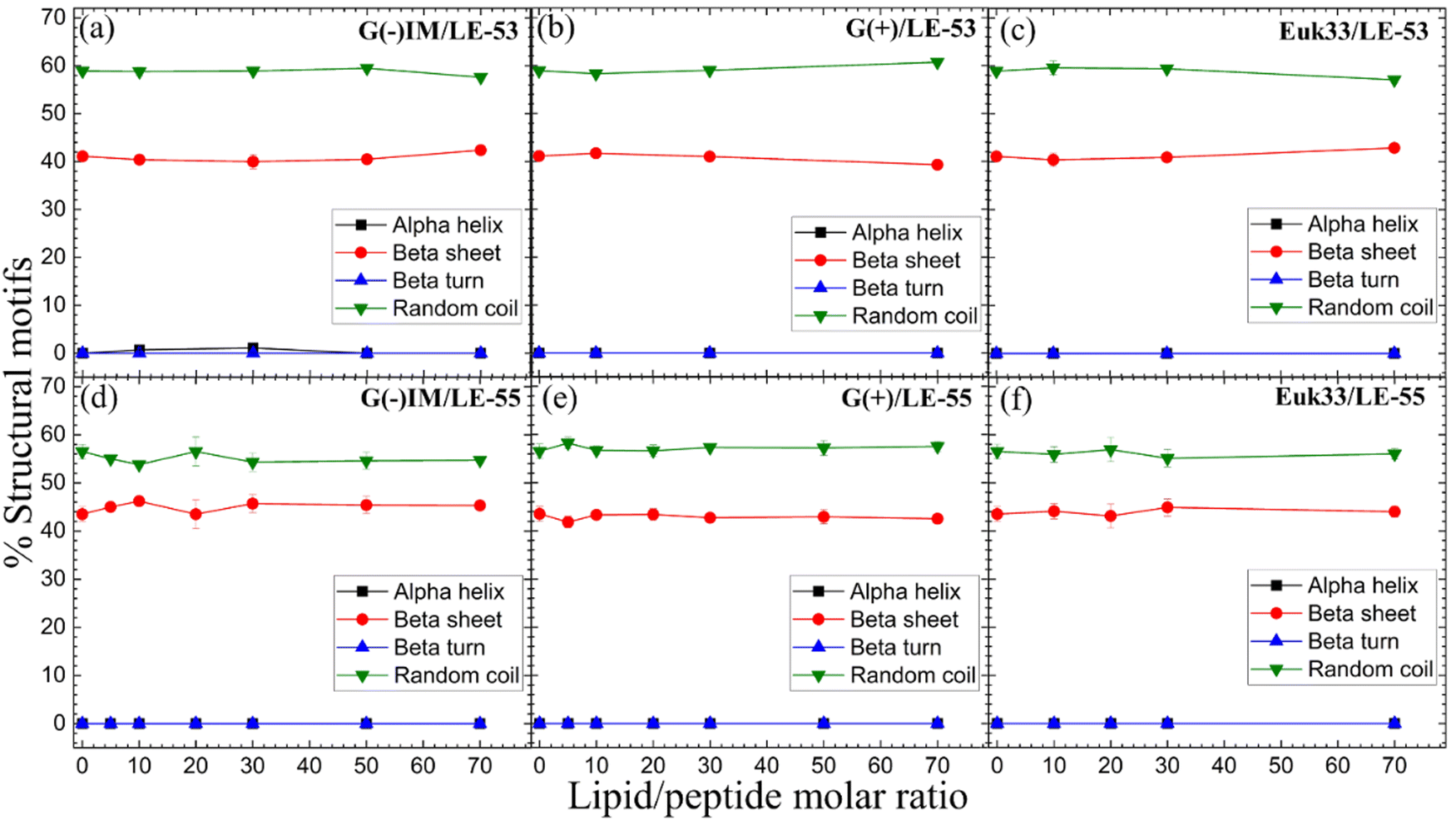 | ||
| Fig. 2 %Structural motifs vs. lipid/peptide molar ratio of LE-53 (a)–(c) and LE-55 (d)–(f) in three different LMM ULVs. Standard deviations represent 3–4 fitting results using shape analysis. | ||
Antimicrobial peptides are typically short in length, consisting of fewer than 100 amino acids. They possess amphipathic properties due to the presence of cationic and hydrophobic residues. While sharing these common characteristics, AMPs exhibit significant diversity in their primary, secondary, and tertiary structures.94,95 The α-helical secondary structure, characterized by hydrophilic residues aligned on one face and hydrophobic residues on the opposite face, facilitates optimal peptide–membrane interactions.96,97 For example, peptides derived from Magainin 2, which have higher α-helicity, promote antibacterial activity.98 In our recent study α-helicity was found to be LMM dependent, showing a higher helicity in G(−) > G(+) > Euk33 LMMs.70 Two peptides, E2-35 and E2-05, rich in R and W residues, exhibited predominantly helical structures in G(−) IM (85–90%) and G(+) LMMs (50–60%), while substitution of R with K (E2-35 → E2-35K) reduced helicity.70 The helicity observed in E2-35 and E2-05 showed a strong correlation with their antibacterial efficiency compared to E2-35K.70 While α-helicity is often associated with higher efficacy, this is not always the case. For instance, the D8 form of WLBU2, containing 8 valines as the D-enantiomer, displayed a random coil structure in G(−) LMMs, in contrast to WLBU2's predominantly helical structure. Surprisingly, both AMPs exhibited similar efficacy in killing bacteria.25,99 Different secondary structures such as β-sheet have also been shown to be efficacious challenging the notion that helicity is crucial for antimicrobial function.100
Therefore, based on our results and the broader literature, it appears that the relationship between secondary structure and antibacterial efficacy of AMPs is complex and multifaceted. Factors beyond secondary structure, such as peptides’ physical attributes and location in lipid membranes may play significant roles in determining antimicrobial activity.
3.4 Membrane elasticity and lipid chain order parameter
Fig. 3(a) and (b) show the elastic bending modulus parameter (KC) of G(−) IM (black), G(+) (red) and Euk33 LMMs (blue) in the presence of two AMPs. The KC is greatest for Euk33 LMM because it has 33% cholesterol. Research from our lab suggests that cholesterol primarily interacts with the saturated palmitoyl chain in POPC and POPE, which makes these membranes stiffer and organizes the lipid acyl chains.101–103 The greater KC value for the G(−) control compared to the G(+) control is because of the higher content of PE, as demonstrated by Dupuy et al.66 A general softening was observed for LE-53 in G(+) and Euk33 LMMs and for LE-55 in all three LMMs, indicating that softening behavior was not significantly different for these peptides and may be unrelated to their different bactericidal efficiency.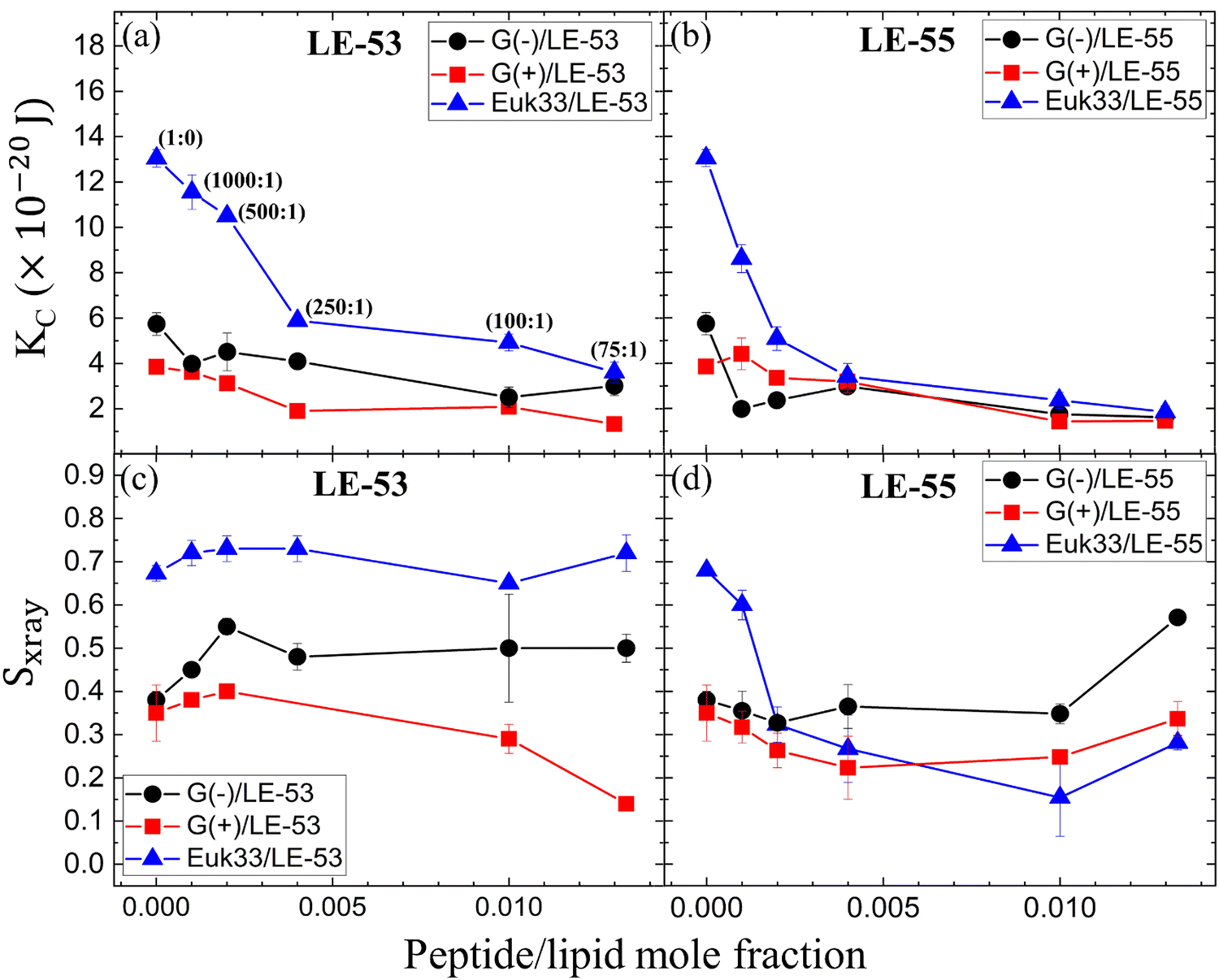 | ||
| Fig. 3 (a) and (b) Bending modulus (KC) of G(−) IM (black circles), G(+) (red squares) and Euk33 (blue triangles) LMMs interacting with two AMPs as shown. (c) and (d) Chain order parameter (Sxray) of two AMPs with LMMs (colors as in a and b). In (a), lipid-to-peptide molar ratios are denoted within parentheses, and this notation applies to all the subsequent plots (b)–(d). The standard deviations are from duplicate or triplicate samples. | ||
What might be more significant is their distinct acyl chain order parameters (Sxray), as illustrated in Fig. 3(c) and (d). Higher Sxray values indicate well-organized lipid acyl chains, while lower values indicate disorganized chains. The Euk33 control, which contains 33 mol% cholesterol, exhibited the most ordered chains. In contrast, LE-53 initially increased ordering at low concentrations, but at higher concentrations, there was either no change or a decrease in ordering across all three LMMs. This behavior is different from LE-55 which caused initial disordering and some degree of ordering at higher concentraions in all three LMMs. This implies that enhancing the order of lipid chains at a low peptide concentration could play a crucial role in the mechanism of bacterial cell death.
3.5 Membrane structural results
In this part, we’re investigating the connections between where peptides are located in bilayers and the structural changes of the LMMs in relation to the effectiveness of two peptides in killing bacteria. Fig. S3 (ESI†) shows form factors |F(qz)|, while Fig. 4 presents electron density profiles (EDPs) of three LMMs containing a lipid/peptide molar ratio of 75:1 for either LE-53 or LE-55. These were determined by fitting XDS form factor data using the scattering density profile (SDP) program.104 This program accounts for the volumes of lipids, peptides, and their component groups in the bilayer, along with the number of electrons each component contributes. We fitted the form factors by placing a Gaussian envelope for the peptide in three potential locations: the headgroup, hydrocarbon region, or a combination of both, then assessed the fit quality using chi-square. Generally, the SDP bilayer model fit the XDS form factor data well (Fig. S3, ESI†), resulting in EDPs typical of fully hydrated membranes. The various component groups in EDPs are Phos (phosphate plus outer headgroup), CG (carbonyl/glycerol), CH2 (methylene hydrocarbon region containing CH groups), CH3 (terminal methyl group), Water (fills volumes around other groups to maintain a total probability of one), and Total (sum of all component groups). Key measures derived from these EDPs include the combined peak-to-peak distance of Phos and CG (DHH), and the full-width at half-maximal of the hydrocarbon region (2DC), both of which indicate membrane thickness. The EDP also determines the area per lipid molecule (AL) using lipid and peptide volumes. A summary of the XDS structural results for the three LMMs used in this study interacting with LE-53 and LE-55 is shown in Table 3.
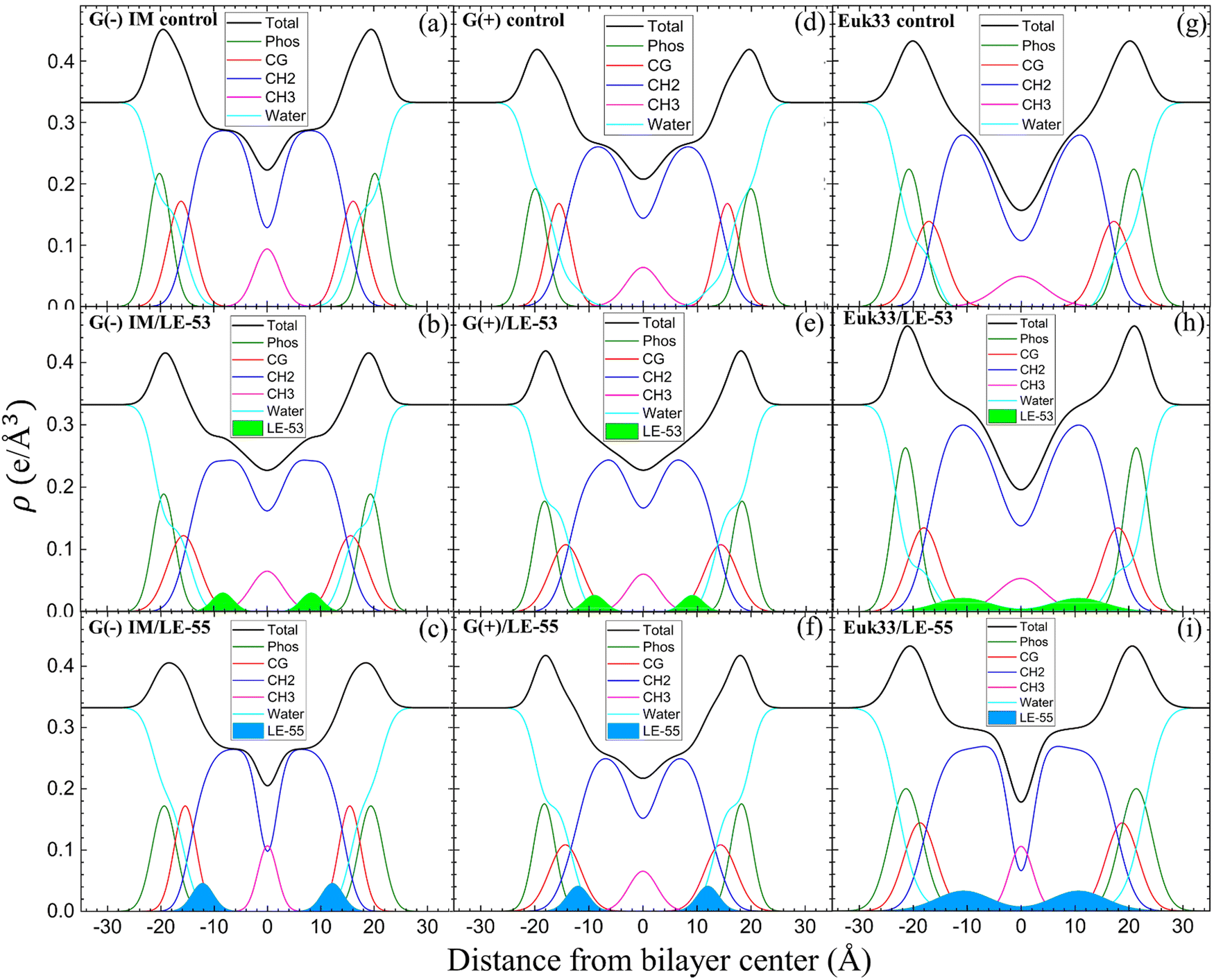 | ||
| Fig. 4 EDPs for G(−) IM LMMs (a)–(c), G(+) LMMs (d)–(f) and Euk33 LMMs (g)–(i) in the presence of LE-53 and LE-55. Component groups in EDPs: phosphate + external headgroup (Phos, green), carbonyl-glycerol (CG, red), CH2 (dark blue), CH3 (magenta), water (cyan), total (black), LE-53 (filled lime green), LE-55 (filled sky blue). Lipid/peptide molar ratio is 75:1. | ||
| Sample (lipid/peptide (75:1)) |
Area/lipid AL [Å2] (±1.0) | D HH [Å] (±0.5) | 2DC [Å] (±0.5) | Net charge/residue |
|---|---|---|---|---|
| G(−) IM/control | 70.8 | 39.2 | 29.1 | — |
| G(−) IM/LE-53 | 73.0 | 38.1 | 28.9 | −0.24 |
| G(−) IM/LE-55 | 74.6 | 36.9 | 28.6 | −0.18 |
| G(+)/control | 73.4 | 38.5 | 28.9 | — |
| G(+)/LE-53 | 82.5 | 36.1 | 26.3 | −0.28 |
| G(+)/LE-55 | 83.5 | 36.1 | 26.3 | −0.21 |
| Euk33/control | 64.0 | 40.3 | 32.0 | — |
| Euk33/LE-53 | 63.1 | 42.2 | 33.8 | +0.01 |
| Euk33/LE-55 | 61.8 | 41.2 | 35.1 | +0.01 |
XDS data reveal that LE-53 locates in the hydrocarbon region, close to the CG headgroup Gaussian in both G(−) and G(+) LMMs (Fig. 4(b) and (e)). LE-55 also locates in the hydrocarbon region of both bacterial LMMs, but closer to the CG headgroup than LE-53 (Fig. 4(c) and (f)). Therefore, LE-53's upper hydrocarbon location correlates with efficient bacterial destabilization. The cause of shallower insertion of LE-55 (8 Rs) in the hydrocarbon region could be its lower hydrophobicity. In addition LE-55 has higher R content compared to LE-53 (6 Rs). The amino acid R contains two extra nitrogens, which allows the guanidinium part of the molecule to form up to six hydrogen bonds.105 This unique feature of R enables it to interact with phosphate groups in various ways, forming complexes. A simulation study conducted by Allolio et al. discovered that the charged side chains of nona-arginine (R9) tend to attach to lipid headgroups, particularly those with negatively charged phosphates.106 In our recent study we observed similar behavior with R-rich peptides E2-35 and E2-05.70 Therefore, it's more probable for the R-rich peptide LE-55 to be attached to the phosphate groups and negatively charged headgroups of lipids (such as POPG and TOCL) in both Gram-negative and Gram-positive lipid multilayer membranes. The smaller size and higher hydrophobicity of LE-53 (Table 1) might explain its tendency to reside in the upper hydrocarbon region. In order to verify the locations of the peptides, we conducted NR on LE-53 and LE-55. The LMMs were attached to the gold-coated silicon substrate by a lipid tether. NR curves are shown in Fig. S4 (ESI†). Fig. 5 provides a graphical summary of the membrane location of both LE-53 and LE-55 in all three LMMs from NR measurements. These NR results are quantified in Table S7 (ESI†). As illustrated in Fig. 5, the peptides’ locations found by NR are in agreement with the locations determined using XDS.
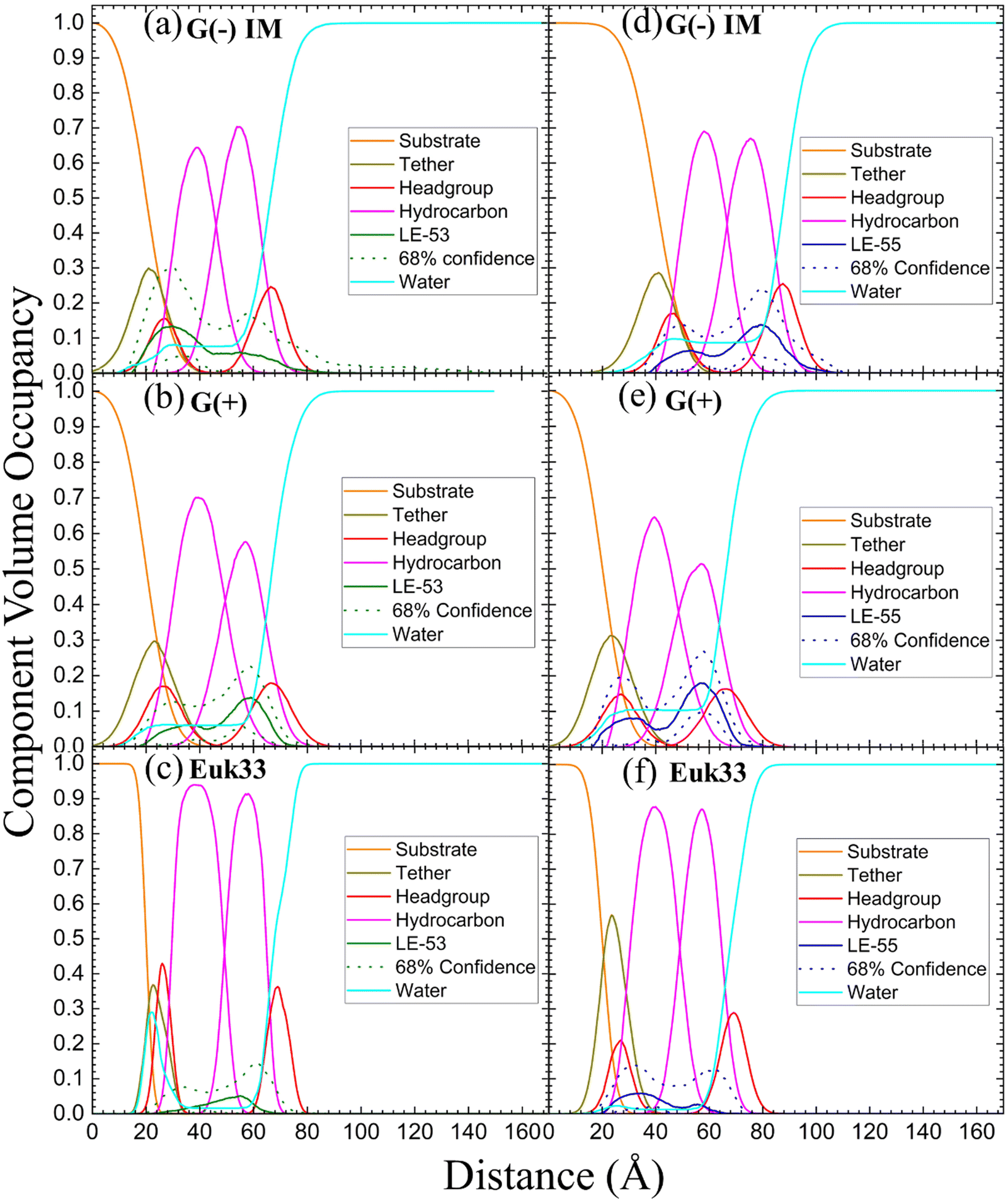 | ||
| Fig. 5 Neutron reflectivity component volume occupancy of LE-53 (a)–(c) and LE-55 (d)–(f) in a single tethered bilayer of G(−) IM, G(+) and Euk33. Component volumes: gold-covered silicon substrate (orange), tether (olive), headgroups (red), hydrocarbons (magenta), LE-53 (green), LE-55 (blue), water (cyan). The dotted lines represent the 68% confidence limit of the composition-space fit. | ||
As indicated in Table 3, both peptides decrease the thickness of the membrane (measured by 2DC and DHH) in both G(−) and G(+) membranes, regardless of their position within the bilayer. Likewise, the peptides increase the area per lipid (AL) in both types of membranes. Thus, this suggests that changes in membrane thickness and area per lipid are not related to the effectiveness of killing bacteria. Interestingly, LE-53 and LE-55 peptides do not exhibit toxicity towards eukaryotic cells where both peptides localize in the hydrocarbon region of eukaryotic LMMs (Euk33). In our recent studies involving helical peptides rich in Arg and Trp we have observed that peptides which exhibit a certain level of toxicity tend to localize in the headgroup region of the Euk33 bilayer.70 Conversely, the non-toxic peptide E2-35K prefers to locate within the hydrocarbon region of the lipid bilayer.70 This suggests a correlation between the peptides’ lack of toxicity and their location within the hydrocarbon region of the bilayer.
3.6 Fusogenicity of LE peptides
While numerous reports have discussed lipid vesicle fusion induced by peptides,107–112 only a limited number of studies have investigated the fusogenicity of AMPs.113–115 In this work, we examine the correlation between antibacterial activity and fusogenicity of LE peptides by conducting solution SAXS measurements on G(−)IM, G(+), and Euk33 ULVs in the presence of these peptides.The SAXS data (Fig. 6(a)–(c)) obtained from control samples (G(−)IM, G(+), and Euk33) LMM ULVs showed a diffuse modulation of scattered intensity, which is indicative of positionally uncorrelated bilayers commonly found in ULV dispersions. In the presence of LE-53, the ULVs underwent a significant structural transition and exhibited characteristic Bragg, lamellar orders at q values of 0.123 Å−1, 0.246 Å−1 and 0.369 Å−1 with D spacing of 51.1 Å for both G(−) and G(+). With the addition of LE-55, only small Bragg orders became visible in the first lobe. This pattern suggests the formation of multilamellar vesicles (MLVs) with lamellar structure. Using the Scherrer equation,116,117 LE-53 produces lamellarity of 86 (G(−)) and 107 (G(+)) bilayers while LE-55 produces 30 (G(−)) and 20 (G(+)) bilayers. The transition from ULV to MLV implies vesicle fusion. Free energy changes associated with this process are illustrated in Fig. 7.
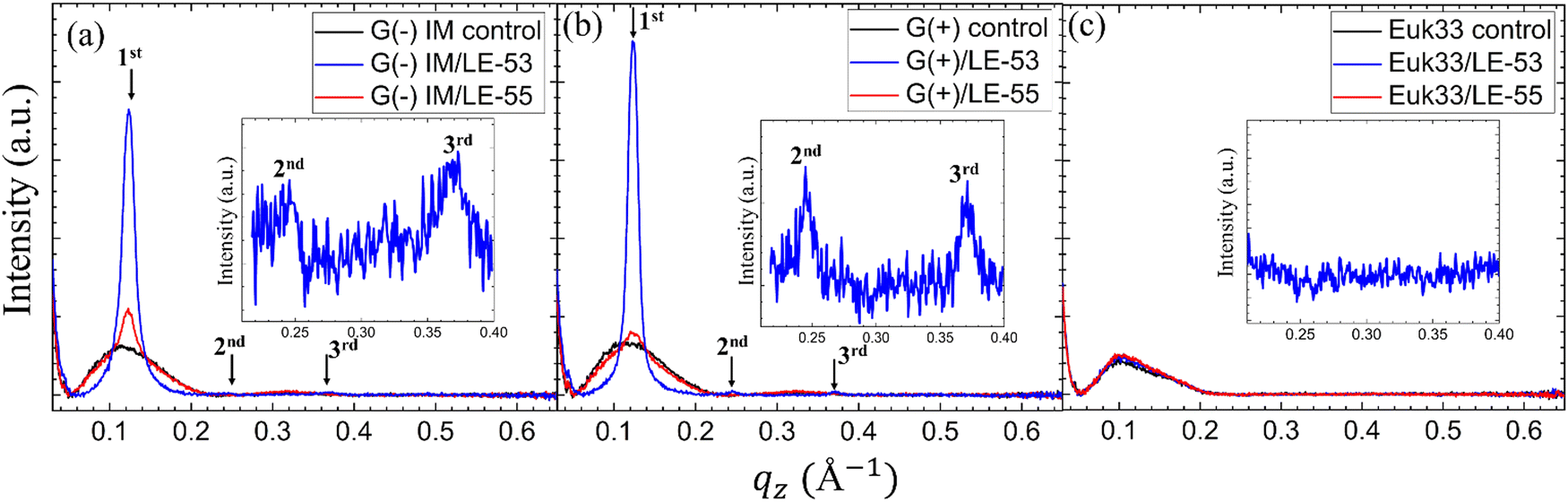 | ||
| Fig. 6 Small angle X-ray scattering profiles of (a) G(−)IM, (b) G(+) and (c) Euk33 ULVs (black) in the presence of LE-53 (blue) and LE-55 (red) AMPs. The arrows indicate Bragg orders. Lipid/peptide molar ratio is 75:1. Additionally, (a) and (b) contain insets highlighting the 2nd and 3rd Bragg orders observed in G(−) and G(+) ULVs, and (c) contains inset highlighting no Bragg orders in Euk33 ULVs when incorporated with LE-53. | ||
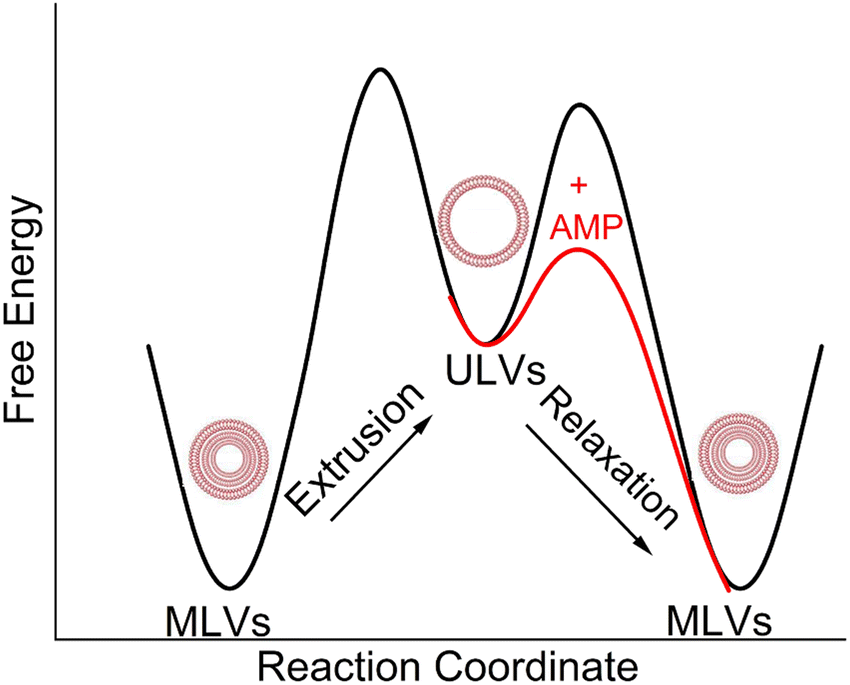 | ||
| Fig. 7 A schematic diagram of free energy vs. reaction coordinate, which illustrates the transition of MLVs from a low energy configuration to ULVs in a high energy configuration through extrusion. The diagram also depicts the subsequent transition of ULVs back to MLVs with the addition of AMPs. | ||
Membrane fusion plays a crucial role in various biological processes such as the entry of enveloped viruses into host cells, fertilization, cell division, and synaptic transmission.113 This process is facilitated by fusogenic peptide sequences, typically found within larger proteins.113,118 Additionally, certain membrane-active peptides, including cell-penetrating peptides (CPPs), possess the capability to induce fusion of membranes and vesicles.113,119,120 In interactions between CPPs and complex lipid mixtures such as phosphatidylcholine/PE/bis(monoacylglycerol), not only fusion and leakage, but also leaky fusion (simultaneous fusion and leakage) have been observed.111,121 Recently, Allolio et al. have shown that arginine-rich CPPs enter vesicles by inducing membrane multilamellarity and fusion.106 A combination of magainin 2 and PGLa leads to morphological changes in the membrane that resemble fusion, which could potentially account for their ability to cause leakage.96 Leakage activity through leaky vesicle fusion has also been reported as a likely mechanism of membrane permeabilization by an antimicrobial polycation (poly-NM) according to Shi et al.122 For fusion between vesicles to happen, two obstacles must be surmounted. Firstly, both membranes must be brought into close proximity,123,124 a task aided by the neutralization of negative charges on lipid headgroups through the interaction with positively charged peptides.123,124 Secondly, the creation of a fusion stalk demands high curvatures of the lipid layers. Various lipid types exhibit different tendencies for forming fusion stalks.125 The inherent negative curvatures of POPE126,127 and TOCL,128 coupled with their well-documented inclination to form non-lamellar structures, contribute to the process of membrane fusion.125,126,128–131 This lipid-intrinsic tendency can be further enhanced by the binding and insertion of amphiphiles, such as AMPs.114,132 In the present study, it is noteworthy that membrane fusion is more pronounced in the presence of the shorter LE-53 peptide, which exhibits higher efficiency as an antibacterial agent compared to LE-55. This finding raises the possibility of vesicle fusion playing a role in the antimicrobial activity of the peptides. It suggests that induction of curvature may help to disrupt the bacterial membrane. In contrast, it is noteworthy that both LE-55 and LE-53, which are non-toxic to eukaryotic cells, did not induce fusion in the eukaryotic ULVs, as exemplified in Fig. 6(c).
Experiments utilizing X-ray diffuse scattering on lipid bilayer stacks have shed light on the fusion mediation mechanism by observing a significant reduction in membrane bending rigidity upon the addition of small molar fractions of fusion peptides such as FP23,133 which localizes in the upper hydrocarbon region.134 In addition, studies have highlighted the hydrophobic nature of most fusion peptides, which has been proposed to contribute to the fusion process.135–137 Remarkably, the behavior of LE-53, being more hydrophobic than LE-55 and localizing in the upper hydrocarbon region, is similar to these findings.
4 Conclusions
This research demonstrates a systematic examination of a possible response to the growing threat of antibiotic resistance. By exploring the potential of AMPs as effective alternatives, the study highlights LE-53 (12 AAs) as a potent solution against both G(−) and G(+) bacteria, all the while ensuring the safety of human cells. The efficacy of LE-53 can be attributed to its elevated hydrophobicity, resulting in notably lower MICs compared to its counterpart, LE-55 (16 AAs), suggesting that physical attributes drive enhanced efficacy. Increased order of lipid acyl chains surfaces as a second determinant for bacterial destabilization. Thirdly, the study unveils the pivotal connection between LE-53's upper hydrocarbon location and its efficacy, shedding light on its mode of action. Fourthly, the most remarkable revelation lies in LE-53's distinctive capacity to trigger vesicle fusion in bacterial model membranes, while remaining inert in eukaryotic LMMs, underscoring a targeted disruption strategy. CD study reveals that LE-53 and LE-55 both maintain random coil and β-sheet structures when in contact with G(−) and G(+) LMMs, which suggests that secondary structure does not distinguish these two AMPs. These exceptional characteristics of LE-53 hold significant promise in addressing antibiotic-resistant strains without compromising human cell integrity.Author contributions
Investigation (SM, BC, YL, MC, MM, BE, RK, SH, BK, FH, STN), data curation and formal analysis (SM, BC, YL, MC, MM, BE, RK, FH, STN), conceptualization (SM, STN), resources (BD,STN), funding acquisition (BD, STN), supervision (SM, STN), methodology (FH, STN), Project administration (BD, STN), writing – original draft (SM, STN), writing – review & editing (BK, SH, FH, BD, RK, MC, BE, YL, SM, STN).Conflicts of interest
The authors declare no conflict of interest in this work.Acknowledgements
This work is based upon research conducted at the Center for High Energy X-ray Sciences (CHEXS at CHESS), which is supported by the National Science Foundation under award DMR-1829070, and the Macromolecular Diffraction at CHESS (MacCHESS) facility, which is supported by award 1-P30-GM124166-01A1 from the National Institute of General Medical Sciences, National Institutes of Health, and by New York State's Empire State Development Corporation (NYSTAR). Additional support for this work was from the National Science Foundation (NSF) MCB-2115790 (S. M., S. T-N.), National Institute of Allergy and Infectious Diseases (NIAID) 1R01AI172861-01A1 (B. D., S. T-N.). Bhairavi Chandersekhar was supported by Carnegie Mellon University SURF program, and Yunshu Li by the Robert Kramer Award at CMU Physics. F. H. was supported by the U.S. Department of Commerce (award 70NANB17H299). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. B. K. was supported by the Danish Agency for Science, Technology and Innovation (DanScatt). The authors would like to acknowledge the ISIS facility at Rutherford Appleton Laboratory, Harwell Campus, U.K, doi:10.5286/ISIS.E.RB2310027, and the Materials Characterization Facility Xenocs Xeuss 3.0, Carnegie Mellon University, funded by the NSF Major Research Instrumentation (MRI) award CMMI-2117523. The authors would also like to acknowledge Dr Richard Gillilan for his help at CHEXS (CHESS).References
- B. A. Wilson and B. T. Ho, Revenge of the Microbes: How Bacterial Resistance is Undermining the Antibiotic Miracle, John Wiley & Sons, 111 River St, Hoboken, New Jersey, 07030-5773, USA, 2023 Search PubMed.
- C. Årdal, M. Balasegaram, R. Laxminarayan, D. McAdams, K. Outterson, J. H. Rex and N. Sumpradit, Nat. Rev. Microbiol., 2020, 18, 267–274 CrossRef.
- N. A. Rizk, R. Moghnieh, N. Haddad, M.-C. Rebeiz, R. M. Zeenny, J.-R. Hindy, G. Orlando and S. S. Kanj, Antibiotics, 2021, 10, 1320 CrossRef CAS PubMed.
- C. J. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. R. Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao and E. Wool, Lancet, 2022, 399, 629–655 CrossRef CAS.
- M. Magana, M. Pushpanathan, A. L. Santos, L. Leanse, M. Fernandez, A. Ioannidis, M. A. Giulianotti, Y. Apidianakis, S. Bradfute and A. L. Ferguson, Lancet Infect. Dis., 2020, 20, e216–e230 CrossRef CAS PubMed.
- N. Mookherjee and R. Hancock, Cell. Mol. Life Sci., 2007, 64, 922–933 CrossRef CAS.
- Y. Liu and E. Breukink, Antibiotics, 2016, 5, 28 CrossRef.
- P. Sarkar, V. Yarlagadda, C. Ghosh and J. Haldar, MedChemComm, 2017, 8, 516–533 RSC.
- S. Sibinelli-Sousa, J. T. Hespanhol and E. Bayer-Santos, J. Bacteriol., 2021, 203, 00478, DOI:10.1016/j.celrep.2020.107813.
- J. A. Santos and M. H. Lamers, Antibiotics, 2020, 9, 776 CrossRef CAS.
- G. Kapoor, S. Saigal and A. Elongavan., J. Anaesthesiol., Clin. Pharmacol., 2017, 33, 300–305 CrossRef CAS PubMed.
- D. Fernandez-Villa, M. R. Aguilar and L. Rojo, Int. J. Mol. Sci., 2019, 20, 4996 CrossRef CAS PubMed.
- B. Deslouches, J. D. Steckbeck, J. K. Craigo, Y. Doi, J. L. Burns and R. C. Montelaro, Antimicrob. Agents Chemother., 2015, 59, 1329–1333 CrossRef CAS PubMed.
- Y. Zhu, W. Hao, X. Wang, J. Ouyang, X. Deng, H. Yu and Y. Wang, Med. Res. Rev., 2022, 42, 1377–1422 CrossRef CAS.
- K. Matsuzaki, Antimicrob. Pept., 2019, 9–16 CAS.
- S. Ittig, B. Lindner, M. Stenta, P. Manfredi, E. Zdorovenko, Y. A. Knirel, M. Dal Peraro, G. R. Cornelis and U. Zähringer, PLoS Pathog., 2012, 8, e1002667 CrossRef CAS.
- A. Silipo, L. Sturiale, D. Garozzo, G. Erbs, T. T. Jensen, R. Lanzetta, J. M. Dow, M. Parrilli, M. A. Newman and A. Molinaro, ChemBioChem, 2008, 9, 896–904 CrossRef CAS PubMed.
- M. R. Pelletier, L. G. Casella, J. W. Jones, M. D. Adams, D. V. Zurawski, K. R. Hazlett, Y. Doi and R. K. Ernst, Antimicrob. Agents Chemother., 2013, 57, 4831–4840 CrossRef CAS.
- L. Yang, T. A. Harroun, T. M. Weiss, L. Ding and H. W. Huang, Biophys. J., 2001, 81, 1475–1485 CrossRef CAS PubMed.
- J. L. Velasco-Bolom and R. Garduno-Juarez, J. Biomol. Struct. Dyn., 2022, 40, 5060–5068 CrossRef CAS PubMed.
- N. Uematsu and K. Matsuzaki, Biophys. J., 2000, 79, 2075–2083 CrossRef CAS PubMed.
- G. A. Woolley and B. A. Wallace, J. Membr. Biol., 1992, 129, 109–136 CAS.
- K. J. Hallock, D. K. Lee and A. Ramamoorthy, Biophys. J., 2003, 84, 3052–3060 CrossRef CAS PubMed.
- R. Rathinakumar and W. C. Wimley, J. Am. Chem. Soc., 2008, 130, 9849–9858 CrossRef CAS PubMed.
- F. Heinrich, A. Salyapongse, A. Kumagai, F. G. Dupuy, K. Shukla, A. Penk, D. Huster, R. K. Ernst, A. Pavlova and J. C. Gumbart, Chem. – Eur. J., 2020, 26, 6247–6256 CrossRef CAS PubMed.
- F. Y. Chen, M. T. Lee and H. W. Huang, Biophys. J., 2003, 84, 3751–3758 CrossRef CAS PubMed.
- C. Aisenbrey, A. Marquette and B. Bechinger, in Antimicrobial Peptides Basics for Clinical Application, Advances in Experimental Medicine and Biology, ed. K. Matsuzaki, Springer Nature, Singapore, Singapore, 2019, vol. 1117, ch. 4, pp.33–64 Search PubMed.
- R. M. Epand and R. F. Epand, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 289–294 CrossRef CAS PubMed.
- Y. Shai and Z. Oren, Peptides, 2001, 22, 1629–1641 CrossRef CAS.
- Y. X. Chen, M. T. Guarnieri, A. I. Vasil, M. L. Vasil, C. T. Mant and R. S. Hodges, Antimicrob. Agents Chemother., 2007, 51, 1398–1406 CrossRef CAS.
- H.-K. Kang, C. Kim, C. H. Seo and Y. Park, J. Microbiol., 2017, 55, 1–12 CrossRef CAS PubMed.
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS.
- C. B. Park, K.-S. Yi, K. Matsuzaki, M. S. Kim and S. C. Kim, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8245–8250 CrossRef CAS PubMed.
- C. B. Park, M. S. Kim and S. C. Kim, Biochem. Biophys. Res. Commun., 1996, 218, 408–413 CrossRef CAS PubMed.
- C. B. Park, H. S. Kim and S. C. Kim, Biochem. Biophys. Res. Commun., 1998, 244, 253–257 CrossRef CAS.
- A. Patrzykat, C. L. Friedrich, L. Zhang, V. Mendoza and R. E. Hancock, Antimicrob. Agents Chemother., 2002, 46, 605–614 CrossRef CAS PubMed.
- H. G. Boman, B. Agerberth and A. Boman, Infect. Immun., 1993, 61, 2978–2984 CrossRef CAS PubMed.
- R. Roy, M. Tiwari, G. Donelli and V. Tiwari, Virulence, 2018, 9, 522–554 CrossRef CAS PubMed.
- R. Lehrer, A. Barton, K. A. Daher, S. Harwig, T. Ganz and M. E. Selsted, J. Clin. Invest., 1989, 84, 553–561 CrossRef CAS.
- D. Andreu and L. Rivas, Pept. Sci., 1998, 47, 415–433 CrossRef CAS.
- L.-j Zhang and R. L. Gallo, Curr. Biol., 2016, 26, R14–R19 CrossRef CAS.
- A. Mutschler, L. Tallet, M. Rabineau, C. Dollinger, M.-H. Metz-Boutigue, F. Schneider, B. Senger, N. E. Vrana, P. Schaaf and P. Lavalle, Chem. Mater., 2016, 28, 8700–8709 CrossRef CAS.
- A. Mutschler, C. Betscha, V. Ball, B. Senger, N. E. Vrana, F. Boulmedais, A. Schroder, P. Schaaf and P. Lavalle, Chem. Mater., 2017, 29, 3195–3201 CrossRef CAS.
- L. Vidal, V. Thuault, A. Mangas, R. Coveñas, A. Thienpont and M. Geffard, J. Amino Acids, 2014, 2014, 672367 Search PubMed.
- M. Nishikawa and K. i Ogawa, Antimicrob. Agents Chemother., 2004, 48, 229–235 CrossRef CAS.
- L. Eloïse, L. Petit, Y. Nominé, B. Heurtault, I. B. H. Kaddour, B. Senger, J. R. Fores, N. E. Vrana, F. Barbault and P. Lavalle, Eur. J. Med. Chem., 2024, 116224 CrossRef.
- S. S. Shah, N. Casanova, G. Antuono and D. Sabatino, Front. Chem., 2020, 8, 218 CrossRef CAS PubMed.
- D. J. Mitchell, L. Steinman, D. Kim, C. Fathman and J. Rothbard, J. Pept. Res., 2000, 56, 318–325 CrossRef CAS.
- M. Aróstica, R. Rojas, L. F. Aguilar, P. Carvajal-Rondanelli, F. Albericio, F. Guzmán and C. Cárdenas, Membranes, 2022, 12, 1180 CrossRef.
- H. Derakhshankhah and S. Jafari, Biomed. Pharmacother., 2018, 108, 1090–1096 CrossRef CAS PubMed.
- N. Schmidt, A. Mishra, G. H. Lai and G. C. Wong, FEBS Lett., 2010, 584, 1806–1813 CrossRef CAS PubMed.
- I. Ruseska and A. Zimmer, Beilstein J. Nanotechnol., 2020, 11, 101–123 CrossRef CAS PubMed.
- F. Madani, S. Lindberg, Ü. Langel, S. Futaki and A. Gräslund, J. Biophys., 2011, 2011, 414729 Search PubMed.
- P. Tan, H. Fu and X. Ma, Nano Today, 2021, 39, 101229 CrossRef CAS.
- B. Deslouches, S. M. Phadke, V. Lazarevic, M. Cascio, K. Islam, R. C. Montelaro and T. A. Mietzner, Antimicrob. Agents Chemother., 2005, 49, 316–322 CrossRef CAS PubMed.
- A. A. Stromstedt, L. Ringstad, A. Schmidtchen and M. Malmsten, Curr. Opin. Colloid Interface Sci., 2010, 15, 467–478 CrossRef.
- H. J. Vogel, D. J. Schibli, W. Jing, E. M. Lohmeier-Vogel, R. F. Epand and R. M. Epand, Biochem. Cell Biol., 2002, 80, 49–63 CrossRef CAS PubMed.
- M. Skinner, A. Kiselev, C. Isaacs, T. Mietzner, R. Montelaro and M. Lampe, Antimicrob. Agents Chemother., 2010, 54, 627–636 CrossRef CAS PubMed.
- J. B. Mandell, J. A. Koch, B. Deslouches and K. L. Urish, J. Orthop. Res., 2020, 38, 2657–2663 CrossRef CAS PubMed.
- W. C. Wimley and S. H. White, Nat. Struct. Biol., 1996, 3, 842–848 CrossRef CAS PubMed.
- W. Xiang, P. Clemenza, J. Klousnitzer, J. Chen, W. Qin, S. Tristram-Nagle, Y. Doi, Y. P. Di and B. Deslouches, Front. Microbiol., 2022, 13, 889791 CrossRef PubMed.
- V. Booth, D. E. Warschawski, N. P. Santisteban, M. Laadhari and I. Marcotte, Biochim. Biophys. Acta, Proteins Proteomics, 2017, 1865, 1500–1511 CrossRef CAS.
- A. Marquette and B. Bechinger, Biomolecules, 2018, 8, 18 CrossRef PubMed.
- R. Leber, M. Pachler, I. Kabelka, I. Svoboda, D. Enkoller, R. Vacha, K. Lohner and G. Pabst, Biophys. J., 2018, 114, 1945–1954 CrossRef CAS PubMed.
- A. K. Buck, D. E. Elmore and L. E. O. Darling, Future Med. Chem., 2019, 11, 2447–2460 CrossRef PubMed.
- F. G. Dupuy, I. Pagano, K. Andenoro, M. F. Peralta, Y. Elhady, F. Heinrich and S. Tristram-Nagle, Biophys. J., 2018, 114, 919–928 CrossRef CAS PubMed.
- A. Kumagai, F. G. Dupuy, Z. Arsov, Y. Elhady, D. Moody, R. K. Ernst, B. Deslouches, R. C. Montelaro, Y. P. Di and S. Tristram-Nagle, Soft Matter, 2019, 15, 1860–1868 RSC.
- G. Pandit, N. Chowdhury, S. A. Mohid, A. P. Bidkar, A. Bhunia and S. Chatterjee, ChemMedChem, 2021, 16, 355–367 CrossRef CAS PubMed.
- B. Deslouches, I. A. Gonzalez, D. DeAlmeida, K. Islam, C. Steele, R. C. Montelaro and T. A. Mietzner, J. Antimicrob. Chemother., 2007, 60, 669–672 CrossRef CAS PubMed.
- S. Mitra, M. Coopershlyak, Y. Li, B. Chandersekhar, R. Koenig, M.-T. Chen, B. Evans, F. Heinrich, B. Deslouches and S. Tristram-Nagle, Adv. NanoBiomed Res., 2023, 3, 2300013 CrossRef CAS PubMed.
- S. G. Wilkinson, Microbial Lipids, Academic Press, San Diego, CA, 1988 Search PubMed.
- E. L. Gottfried, J. Lipid Res., 1967, 8, 321–327 CrossRef CAS.
- I. Branzoi, M. Iordoc, F. Branzoi, R. Vasilescu-Mirea and G. Sbarcea, Surf. Interface Anal., 2010, 42, 502–509 CrossRef CAS.
- B. Deslouches, J. D. Steckbeck, J. K. Craigo, Y. Doi, T. A. Mietzner and R. C. Montelaro, Antimicrob. Agents Chemother., 2013, 57, 2511–2521 CrossRef CAS PubMed.
- B. Deslouches, M. L. Hasek, J. K. Craigo, J. D. Steckbeck and R. C. Montelaro, J. Med. Microbiol., 2016, 65, 554 CrossRef CAS PubMed.
- S. Brahms and J. Brahms, J. Mol. Biol., 1980, 138, 149–178 CrossRef CAS PubMed.
- S. A. Tristram-Nagle, Methods Mol. Biol., 2007, 400, 63–75 CrossRef CAS PubMed.
- N. Kučerka, Y. Liu, N. Chu, H. I. Petrache, S. Tristram-Nagle and J. F. Nagle, Biophys. J., 2005, 88, 2626–2637 CrossRef PubMed.
- Y. Lyatskaya, Y. Liu, S. Tristram-Nagle, J. Katsaras and J. F. Nagle, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2000, 63, 011907 CrossRef PubMed.
- P.-G. De Gennes and J. Prost, The physics of liquid crystals, Oxford university press, 1993 Search PubMed.
- T. T. Mills, G. E. Toombes, S. Tristram-Nagle, D.-M. Smilgies, G. W. Feigenson and J. F. Nagle, Biophys. J., 2008, 95, 669–681 CrossRef CAS PubMed.
- M. Inc, 2022.
- N. Kučerka, M.-P. Nieh and J. Katsaras, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2761–2771 CrossRef PubMed.
- R. Dalgliesh, S. Langridge, J. Plomp, V. De Haan and A. Van Well, Phys. B, 2011, 406, 2346–2349 CrossRef CAS.
- R. Budvytyte, G. Valincius, G. Niaura, V. Voiciuk, M. Mickevicius, H. Chapman, H.-Z. Goh, P. Shekhar, F. Heinrich and S. Shenoy, Langmuir, 2013, 29, 8645–8656 CrossRef CAS PubMed.
- R. Eells, D. P. Hoogerheide, P. A. Kienzle, M. Lösche, C. F. Majkrzak and F. Heinrich, in Characterization of Biological Membranes: Structure and Dynamics, ed. M.-P. Nieh, F. A. Heberle and J. Katsaras, 2019, pp.87–130 Search PubMed.
- P. Shekhar, H. Nanda, M. Lösche and F. Heinrich, J. Appl. Phys., 2011, 110, 102216 CrossRef PubMed.
- F. Heinrich and M. Lösche, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 2341–2349 CrossRef CAS PubMed.
- J. L. Fauchere and V. Pliska, Eur. J. Med. Chem., 1983, 18, 369–375 CAS.
- D. Eisenberg, R. M. Weiss and T. C. Terwilliger, Nature, 1982, 299, 371–374 CrossRef CAS PubMed.
- Y. Rosenfeld, N. Lev and Y. Shai, Biochemistry, 2010, 49, 853–861 CrossRef CAS PubMed.
- M. Stark, L. P. Liu and C. M. Deber, Antimicrob. Agents Chemother., 2002, 46, 3585–3590 CrossRef CAS PubMed.
- T. Jakkampudi, Q. Lin, S. Mitra, A. Vijai, W. Qin, A. Kang, J. Chen, E. Ryan, R. Wang and Y. Gong, Biomacromolecules, 2023, 24, 2804–2815 CrossRef CAS PubMed.
- J. Koehbach and D. J. Craik, Trends Pharmacol. Sci., 2019, 40, 517–528 CrossRef CAS PubMed.
- X. T. Mai, J. F. Huang, J. J. Tan, Y. B. Huang and Y. X. Chen, J. Pept. Sci., 2015, 21, 561–568 CrossRef CAS PubMed.
- I. Kabelka and R. Vacha, Acc. Chem. Res., 2021, 54, 2196–2204 CrossRef CAS PubMed.
- B. Mojsoska and H. Jenssen, Pharmaceuticals, 2015, 8, 366–415 CrossRef CAS PubMed.
- X. Xu and R. Lai, Chem. Rev., 2015, 115, 1760–1846 CrossRef CAS.
- Y. Di, Q. Lin, C. Chen, R. Montelaro, Y. Doi and B. Deslouches, Sci. Adv., 2020, 6, eaay6817 CrossRef CAS PubMed.
- J.-P. S. Powers and R. E. Hancock, Peptides, 2003, 24, 1681–1691 CrossRef CAS PubMed.
- J. Pan, T. T. Mills, S. Tristram-Nagle and J. F. Nagle, Phys. Rev. Lett., 2008, 100, 198103 CrossRef PubMed.
- J. Pan, S. Tristram-Nagle and J. F. Nagle, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2009, 80, 021931 CrossRef PubMed.
- T. T. Mills, S. Tristram-Nagle, F. A. Heberle, N. F. Morales, J. Zhao, J. Wu, G. E. S. Toombes, J. F. Nagle and G. W. Feigenson, Biophys. J., 2008, 95, 682–690 CrossRef CAS.
- N. Kucerka, J. F. Nagle, J. N. Sachs, S. E. Feller, J. Pencer, A. Jackson and J. Katsaras, Biophys. J., 2008, 95, 2356–2367 CrossRef CAS PubMed.
- K. Hristova and W. C. Wimley, J. Membr. Biol., 2011, 239, 49–56 CrossRef CAS PubMed.
- C. Allolio, A. Magarkar, P. Jurkiewicz, K. Baxova, M. Javanainen, P. E. Mason, R. Sachl, M. Cebecauer, M. Hof, D. Horinek, V. Heinz, R. Rachel, C. M. Ziegler, A. Schrofel and P. Jungwirth, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11923–11928 CrossRef CAS PubMed.
- T. M. Domingues, K. R. Perez and K. A. Riske, Langmuir, 2020, 36, 5145–5155 CrossRef CAS.
- L. Marx, E. F. Semeraro, J. Mandl, J. Kremser, M. P. Frewein, N. Malanovic, K. Lohner and G. Pabst, Front. Med. Technol., 2021, 3, 625975 CrossRef PubMed.
- L. Sun, K. Hristova and W. C. Wimley, Nanoscale, 2021, 13, 12185–12197 RSC.
- D. J. Brock, H. Kondow-McConaghy, J. Allen, Z. Brkljača, L. Kustigian, M. Jiang, J. Zhang, H. Rye, M. Vazdar and J.-P. Pellois, Cell Chem. Biol., 2020, 27, 1296–1307 CrossRef CAS PubMed.
- S.-T. Yang, E. Zaitseva, L. V. Chernomordik and K. Melikov, Biophys. J., 2010, 99, 2525–2533 CrossRef CAS PubMed.
- S. Shi, A. M. Markl, Z. Lu, R. Liu and M. Hoernke, Langmuir, 2022, 38, 2379–2391 CrossRef CAS PubMed.
- A. R. Ferreira, M. Ferreira, C. Nunes, S. Reis, C. Teixeira, P. Gomes and P. Gameiro, Membranes, 2023, 13, 138 CrossRef CAS PubMed.
- K. Beck, J. Nandy and M. Hoernke, Soft Matter, 2023, 19, 2919–2931 RSC.
- J. Sun, Y. Xia, D. Li, Q. Du and D. Liang, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 2985–2993 CrossRef CAS PubMed.
- P. Scherrer, Nachr. Ges. Wiss. Gottingen, 1918, 2, 8–100 Search PubMed.
- A. Patterson, Phys. Rev., 1939, 56, 978 CrossRef CAS.
- L. V. Chernomordik and M. M. Kozlov, Nat. Struct. Mol. Biol., 2008, 15, 675–683 CrossRef CAS PubMed.
- A. Joardar, G. P. Pattnaik and H. Chakraborty, J. Membr. Biol., 2022, 255, 211–224 CrossRef CAS PubMed.
- P. Wadhwani, J. Reichert, J. Bürck and A. S. Ulrich, Eur. Biophys. J., 2012, 41, 177–187 CrossRef CAS.
- D. J. Brock, H. Kondow-McConaghy, J. Allen, Z. Brkljača, L. Kustigian, M. Jiang, J. Zhang, H. Rye, M. Vazdar and J.-P. Pellois, Cell Chem. Biol., 2020, 27, 1296–1307 CrossRef CAS PubMed.
- S. Shi, H. Fan and M. Hoernke, Nanoscale Adv., 2022, 4, 5109–5122 RSC.
- S. L. Leikin, M. M. Kozlov, L. V. Chernomordik, V. S. Markin and Y. A. Chizmadzhev, J. Theor. Biol., 1987, 129, 411–425 CrossRef CAS PubMed.
- A. Witkowska, L. P. Heinz, H. Grubmüller and R. Jahn, Nat. Commun., 2021, 12, 3606 CrossRef CAS PubMed.
- C. S. Poojari, K. C. Scherer and J. S. Hub, Nat. Commun., 2021, 12, 6594 CrossRef CAS PubMed.
- B. Kollmitzer, P. Heftberger, M. Rappolt and G. Pabst, Soft Matter, 2013, 9, 10877–10884 RSC.
- G. Meher and H. Chakraborty, J. Membr. Biol., 2019, 252, 261–272 CrossRef CAS PubMed.
- Y.-F. Chen, K.-Y. Tsang, W.-F. Chang and Z.-A. Fan, Soft Matter, 2015, 11, 4041–4053 RSC.
- B. P. Navas, K. Lohner, G. Deutsch, E. Sevcsik, K. Riske, R. Dimova, P. Garidel and G. Pabst, Biochim. Biophys. Acta, Biomembr., 2005, 1716, 40–48 CrossRef PubMed.
- Y. G. Smirnova, H. J. Risselada and M. Müller, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2571–2576 CrossRef CAS PubMed.
- D. Koller and K. Lohner, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 2250–2259 CrossRef CAS PubMed.
- A. Mishra, G. H. Lai, N. W. Schmidt, V. Z. Sun, A. R. Rodriguez, R. Tong, L. Tang, J. Cheng, T. J. Deming and D. T. Kamei, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16883–16888 CrossRef CAS PubMed.
- S. Tristram-Nagle and J. F. Nagle, Biophys. J., 2007, 93, 2048–2055 CrossRef CAS PubMed.
- S. Tristram-Nagle, R. Chan, E. Kooijman, P. Uppamoochikkal, W. Qiang, D. P. Weliky and J. F. Nagle, J. Mol. Biol., 2010, 402, 139–153 CrossRef CAS PubMed.
- P. Shchelokovskyy, S. Tristram-Nagle and R. Dimova, New J. Phys., 2011, 13, 025004 CrossRef CAS PubMed.
- R. Brasseur, M. Vandenbranden, B. Cornet, A. Burny and J.-M. Ruysschaert, Biochim. Biophys. Acta, Biomembr., 1990, 1029, 267–273 CrossRef CAS PubMed.
- A. Colotto, I. Martin, J.-M. Ruysschaert, A. Sen, S. Hui and R. Epand, Biochemistry, 1996, 35, 980–989 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00220b |
| This journal is © The Royal Society of Chemistry 2024 |