
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly flexible PEG-LifeAct constructs act as tunable biomimetic actin crosslinkers†
Tyler D.
Jorgenson
 *a,
Kashmeera D.
Baboolall
b,
Cristian
Suarez
c,
David R.
Kovar
cd,
Margaret L.
Gardel
*aefg and
Stuart J.
Rowan
*ah
*a,
Kashmeera D.
Baboolall
b,
Cristian
Suarez
c,
David R.
Kovar
cd,
Margaret L.
Gardel
*aefg and
Stuart J.
Rowan
*ah
aPritzker School of Molecular Engineering, University of Chicago, Chicago, IL, USA. E-mail: gardel@uchicago.edu; stuartrowan@uchicago.edu
bGraduate Program in Biosciences, University of Chicago, Chicago, IL, USA
cDepartment of Molecular Genetics and Cell Biology, University of Chicago, Chicago, IL, USA
dDepartment of Biochemistry and Molecular Biology, University of Chicago, Chicago, IL, USA
eInstitute for Biophysical Dynamics, University of Chicago, Chicago, IL, USA
fJames Franck Institute, University of Chicago, Chicago, IL, USA
gDepartment of Physics, University of Chicago, Chicago, IL, USA
hDepartment of Chemistry, University of Chicago, Chicago, IL, USA
First published on 2nd January 2024
Abstract
In vitro studies of actin filament networks crosslinked with dynamic actin binding proteins provide critical insights into cytoskeletal mechanics as well as inspiration for new adaptive materials design. However, discontinuous variance in the physiochemical properties of actin binding proteins impedes holistic relationships between crosslinker molecular parameters, network structure, and mechanics. Bio-synthetic constructs composed of synthetic polymer backbones and actin binding motifs would enable crosslinkers with engineered physiochemical properties to directly target the desired structure–property relationships. As a proof of concept, bio-synthetic crosslinkers composed of highly flexible polyethylene glycol (PEG) polymers functionalized with the actin binding peptide LifeAct, are explored as actin crosslinkers. Using bulk rheology and fluorescence microscopy, these constructs are shown to modulate actin filament network structure and mechanics in a contour length dependent manner, while maintaining the stress-stiffening behavior inherent to actin filament networks. These results encourage the design of more diverse and complex peptide-polymer crosslinkers to interrogate and control semi-flexible polymer networks.
Introduction
The actin cytoskeleton is a critical regulator of cellular architecture and mechanics. Actin cytoskeleton networks are a composite system composed of actin filaments (F-actin) crosslinked with a variety of actin binding proteins (ABPs).1,2 ABPs transiently crosslink F-actin to form assemblies within the cell such as bundled stress-fibers or filopodia, the isotropic network in the cortex, or branched networks in the lamellipodia.3–6 The physiochemical parameters of the ABP determine the mechanical properties of the resulting network.7–10 However, the physicochemical properties of naturally occurring ABPs involve numerous variables including size, flexibility, binding domain kinetics, and geometric factors. Elucidating the relationships between network structure and mechanics with these ABP physiochemical properties has been a key focus of the biomaterial community. In particular, rheological studies of reconstituted in vitro actin networks have allowed for direct interrogation of these relationships.9,11–13 For instance, changes to the repeat units in the F-actin bundler filamin control the network's non-linear strain-stiffening properties.14 While these studies provided critical insights into how crosslinker properties impact network mechanics, the physiochemical properties probed are inherently coupled, e.g., size and binding kinetics, when relying on naturally existing ABPs.Point-mutations of existing ABPs or construction of chimeric ABPs allow for better control over their physical properties, e.g., length or binding strength, yielding further insights.15,16 However, difficulty in producing these systems prevents facile interrogation of the full physicochemical parameter space. Bio-synthetic crosslinkers provide an engineering handle through which the crosslinker's physiochemical properties can be independently and systematically tuned. Recent work by Lorenz et al. demonstrated that rigid DNA duplexes functionalized with either phalloidin or LifeAct actin binding peptides were able to form robust actin networks with properties akin to networks formed with biologically relevant ABPs.17 While these biomimetic crosslinkers represent a first step towards tunable actin crosslinkers, they are limited to rigid rod geometries given the relatively large persistence length, lp, of double stranded DNA (∼50 nm).18
Harnessing the expansive library of synthetic polymer backbones would enable a continuum of new bio-synthetic crosslinkers with well-defined physiochemical properties to be explored. However, it is an open question as to whether crosslinking constructs with synthetic polymer backbones will mimic their biopolymer counterparts as their persistence length is often significantly smaller than the polymer's contour length, lc, yielding unstructured globular crosslinkers (Fig. 1). Herein the utility of synthetic polymers as biomimetic crosslinker backbones via the development of LifeAct – polyethylene glycol (PEG) constructs is explored. These PEG-LifeAct constructs are demonstrated to be effective actin crosslinkers with contour length dependent network morphology and mechanics despite having equivalent hydrodynamic sizes. Additionally, the generated networks maintained many of the characteristic mechanical properties observed in native actin networks.
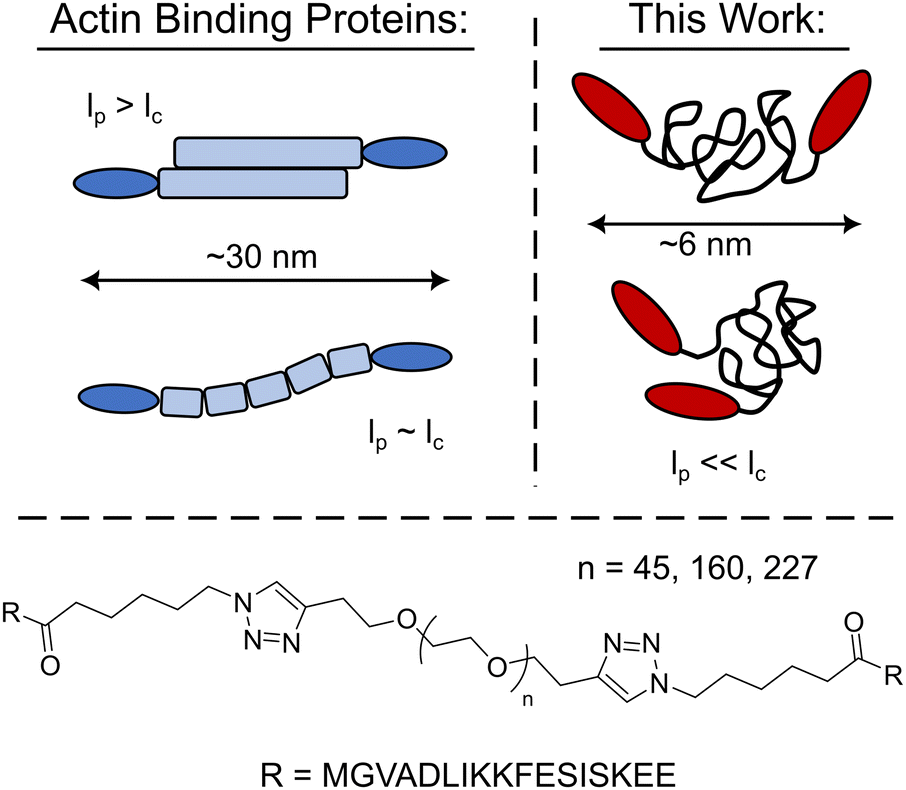 | ||
| Fig. 1 Schematic comparison of the PEG-LifeAct constructs to that of typical biopolymer actin binding proteins and the chemical structure of the PEG-LifeAct construct's core. | ||
Results and discussion
With the goal of exploring highly flexible synthetic actin crosslinkers, a series of telechelic PEG polymers end-capped with the known actin binding peptide, LifeAct, were targeted. As schematized in Fig. 1, copper assisted azide–alkyne click chemistry was used to conjugate ditopic alkyne end-capped polyethylene glycol polymers (PEGs) with two azide-functionalized LifeAct peptides. LifeAct is a short peptide fragment from yeast ABP140 that binds to one monomer within F-actin with a KD of ∼2 μM.19 To explore the impact of construct contour length, three molecular weights of PEG were used, 2, 7 and 10 kDa. As compiled in Table 1, the hydrodynamic diameter of these PEG-LifeAct constructs were all approximately 5–6 nm, although contour lengths vary from approximately 13 to 64 nm. Commonly studied proteinaceous crosslinkers like α-actinin and DdFilamin have approximate contour lengths, lc, of 30–36 nm.20 The synthesized PEG-LifeAct (abbreviated as PEG-X-LA, where X is the molecular weight of the PEG) constructs systematically probe below and above this length regime while maintaining a consistent backbone chemistry and actin binding domain. The consistent hydrodynamic size of these constructs highlights the key difference between PEG-LA constructs and proteinaceous crosslinkers: the highly flexible nature of the PEG backbone. On account of the extremely small persistence length, lp, of PEG (∼0.37 nm), the presented bio-synthetic constructs should be viewed as isotropic spheres with the two actin binding domains presented randomly at the surface (Fig. 1). As the effective spring constant of a flexible polymer chain is inversely dependent on the contour length of the chain (see Note S1, ESI†), the synthesized constructs sweep effective crosslinker stiffnesses of ∼1000 pN μm−1 to ∼200 pN μm−1. These spring constants make the constructs potentially extensible under strain. The physical properties of the PEG-LA constructs are summarized in Table 1.| Name | Contour Length, lc (nm) | Spring Constant (pN μm−1) | Diameter (nm) |
|---|---|---|---|
| Contour length was calculated assuming a repeat distance of 0.28 nm and the number average molecular weight of the polymer. The spring constant was calculated assuming an entropic spring with the given contour length and a persistence length of 0.37 nm (see Note S1 for details). The diameter was measured via dynamic light scattering (see Fig. S4). | |||
| PEG-2k-LA | 13 | 1300 | 4.7 ± 1.7 |
| PEG-7k-LA | 45 | 370 | 6.2 ± 1.5 |
| PEG-10k-LA | 64 | 260 | 5.6 ± 0.7 |
To test whether the synthesized PEG-LA constructs can restructure F-actin networks, fluorescently labeled G-actin was spontaneously assembled in the presence of the PEG-LA constructs and visualized via fluorescence microscopy. PEG-LA crosslinkers can restructure the F-actin network as can be seen in TIRF (top row) and confocal (bottom row) images presented in Fig. 2a. Bundling is observed for PEG-7k-LA and PEG-10k-LA at a relative concentration, Rc, of 33 mol% (top) and 15 mol% (bottom), while PEG-2k-LA shows no bundling. Time-lapse imaging of the bundling process suggests that G-actin is almost immediately organized into larger bundles of multiple filaments in the presence of PEG-7k-LA and PEG-10k-LA. The network morphology produced by either PEG-7k-LA or PEG-10k-LA features branched and curved bundles, reminiscent of network architectures generated by filamin.13 To quantify the differences in crosslinker bundling affinity, the percent of bundling, i.e., the fractional length of filaments contained within a bundle, was calculated from the TIRF images. The presented bundling affinities in Fig. 2b, suggest an increase in the PEG contour length improves the apparent bundling kinetics of the crosslinker despite having comparable hydrodynamic sizes.
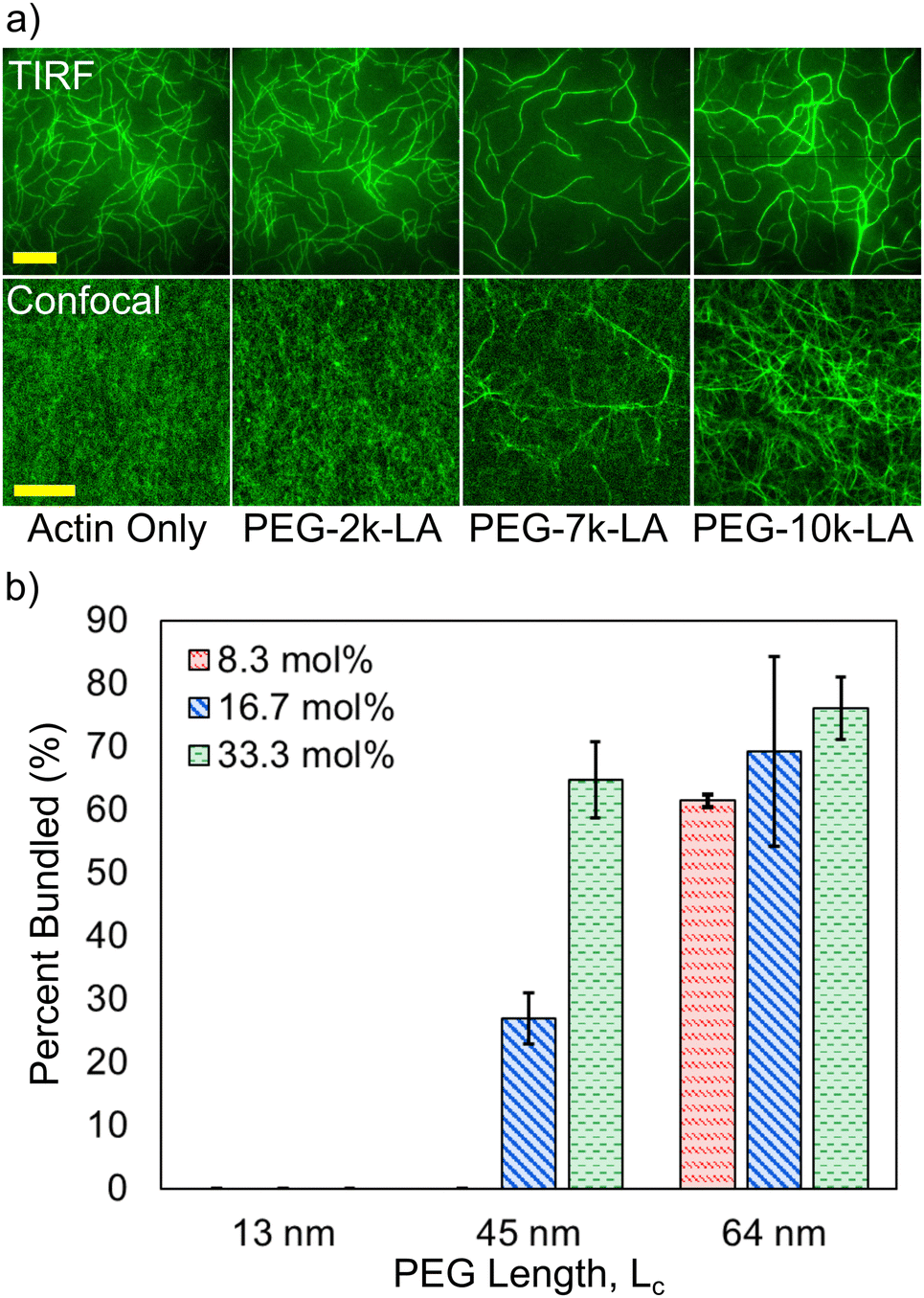 | ||
| Fig. 2 (a) Top row: TIRF microscopy images of actin networks in the presence of PEG-LifeAct crosslinker at Rc = 33.3 mol% and CA = 1.5 μM. Bottom row: Confocal microscopy of actin networks in the presence of PEG-LifeAct crosslinker at Rc = 15 mol% and CA = 11.9 μM. Scale bar represents 20 μm for confocal and TIRF images. (b) Percent of the network that is bundled for each crosslinker at different relative concentrations as a function of the PEG contour length. Percentages were determined from TIRF microscopy images. Errors are standard sample deviations with N = 3. | ||
The observed differences in crosslinker bundling ability and resulting actin network microstructures shown in Fig. 2a should translate into noticeable differences in network mechanics. To investigate such effects, the crosslinker dependent network mechanics were probed using bulk rheological measurements. Characteristic frequency sweeps of actin networks crosslinked with 15 mol% PEG-LA are shown in Fig. 3a. As may be intuited from microstructures presented in Fig. 2a, the storage modulus, G′, of actin networks crosslinked with PEG-10k-LA is higher than that of PEG-7k-LA and PEG-2k-LA networks at 15 mol%. In contrast to PEG-7k-LA and PEG-10k-LA, PEG-2k-LA crosslinked networks are more viscoelastic. To interrogate the impact of construct contour length on the linear mechanics of the actin networks, the loss factor, G′′/G′ = tan(δ), and the frequency dependence of G′ were analyzed as a function of Rc up to 20 mol% as shown in Fig. 3b. The tan(δ) (measured at 10 mHz) transitions from a value of ∼0.3 to ∼0.2 over the tested concentration regime for both PEG-2k-LA and PEG-7k-LA. Despite being the largest and most flexible crosslinker the loss factor remained below 0.2 for all PEG-10k-LA concentrations tested. Fitting the storage modulus from 0.01 to 1 Hz with a power law, G′(f) ∼ fS, reveals the moduli's frequency dependence (Fig. S8, ESI†). For PEG-10k-LA this steepness, S, is low for the entire tested concentration regime with an average value of 0.08. PEG-7k-LA networks have a similar steepness for Rc above 5 mol%. In contrast, PEG-2k-LA networks exhibit a much greater dependence between S and Rc. Only at a Rc of 20 mol% did PEG-2k-LA networks reach a value analogous to the longer PEG-LA crosslinked networks.
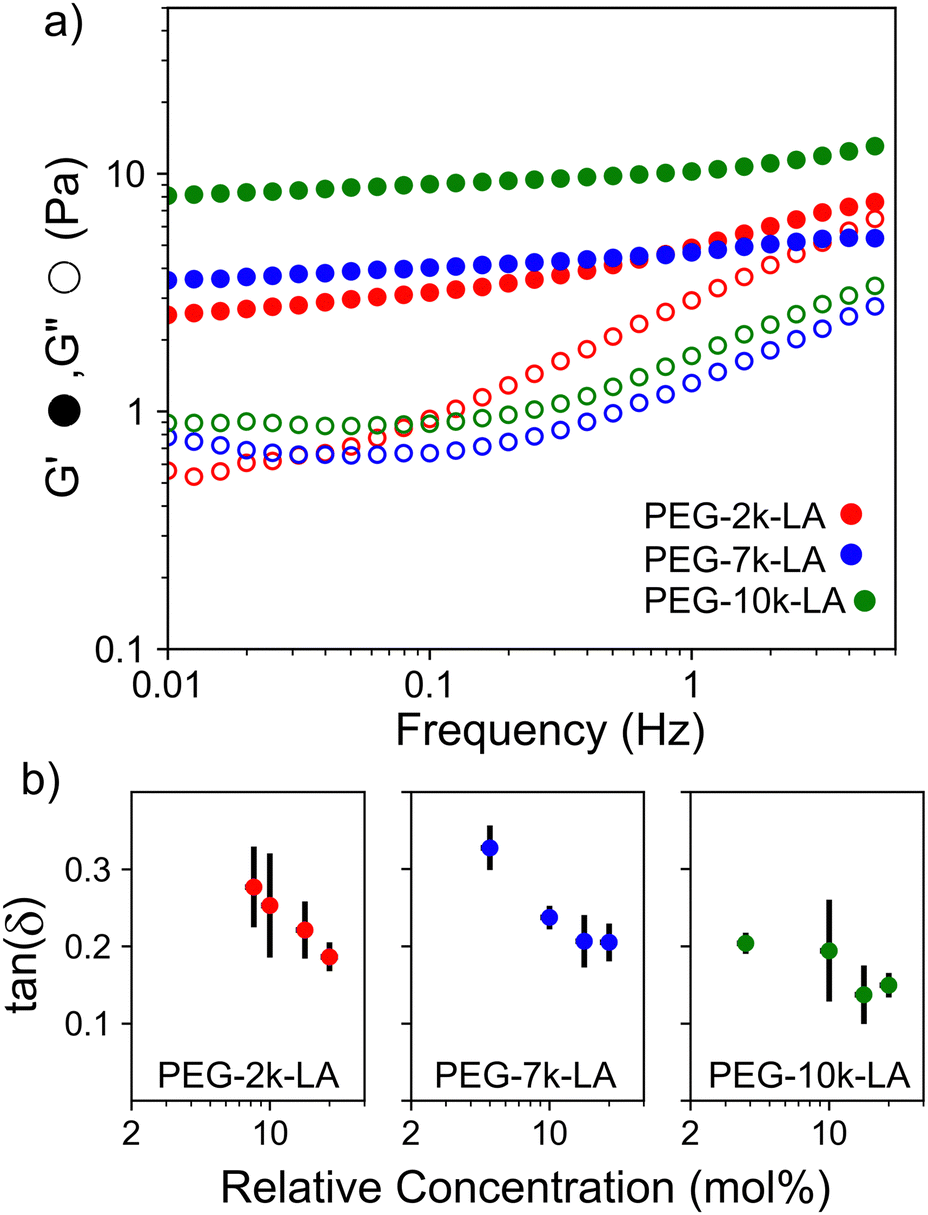 | ||
| Fig. 3 (a) Representative frequency sweeps of networks of 23.8 μM actin crosslinked with 15 mol% of PEG-2k-LA, PEG-7k-LA, and PEG-10k-LA (b). Loss factor, tan(δ), is measured at 10 mHz. Y-Axis error bars are for N = 3. X-Axis error bars are propagated from concentration errors based on UV measurements of the actin and crosslinkers. | ||
The lower tan(δ) and S for PEG-7k-LA and PEG-10k-LA compared to the PEG-2k-LA likely arises from differences in the bundling that is observed in Fig. 2. Bundled filaments have a higher bending modulus resulting in a more elastic network. Additionally, these results further support the observed differences in effective binding affinity suggested by the microstructure analysis presented in Fig. 2b. Elastic insensitivity to frequency occurs when the concentration of crosslinker is high enough to ensure filaments are always crosslinked, thus, arresting non-bending filament motion. This condition appears to be satisfied for concentrations of PEG-7k-LA and PEG-10k-LA above 4 mol% while higher concentrations of PEG-2k-LA are needed to reach such an arrested state. To better understand these observed differences for PEG-LA crosslinkers, the pseudo-plateau modulus, G0, of the crosslinked actin networks measured at 10 mHz was determined as a function of Rc. As shown in Fig. 4a, a plateau region exists until a critical concentration of crosslinker, 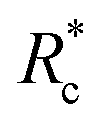 , at which point G0 increases substantially in a log-linear manner for each crosslinker. The observed
, at which point G0 increases substantially in a log-linear manner for each crosslinker. The observed 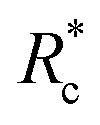 depends on the contour length of the crosslinker, with
depends on the contour length of the crosslinker, with 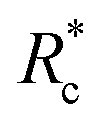 values of approximately 8, 5, and 2 mol% for PEG-2k-LA, PEG-7k-LA, and PEG-10k-LA, respectively (Fig. 4a). The lowering of
values of approximately 8, 5, and 2 mol% for PEG-2k-LA, PEG-7k-LA, and PEG-10k-LA, respectively (Fig. 4a). The lowering of 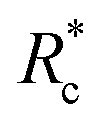 with increasing contour length further confirms differences in effective crosslinking kinetics. The mechanical effectiveness of the crosslinkers, G0 ∼ Rxc, appears to vary inversely with the contour length of the construct. The scaling value, x, decreases from 1.5 ± 0.3 to 0.8 ± 0.1 for an increase in PEG contour length of 13 to 64 nm.
with increasing contour length further confirms differences in effective crosslinking kinetics. The mechanical effectiveness of the crosslinkers, G0 ∼ Rxc, appears to vary inversely with the contour length of the construct. The scaling value, x, decreases from 1.5 ± 0.3 to 0.8 ± 0.1 for an increase in PEG contour length of 13 to 64 nm.
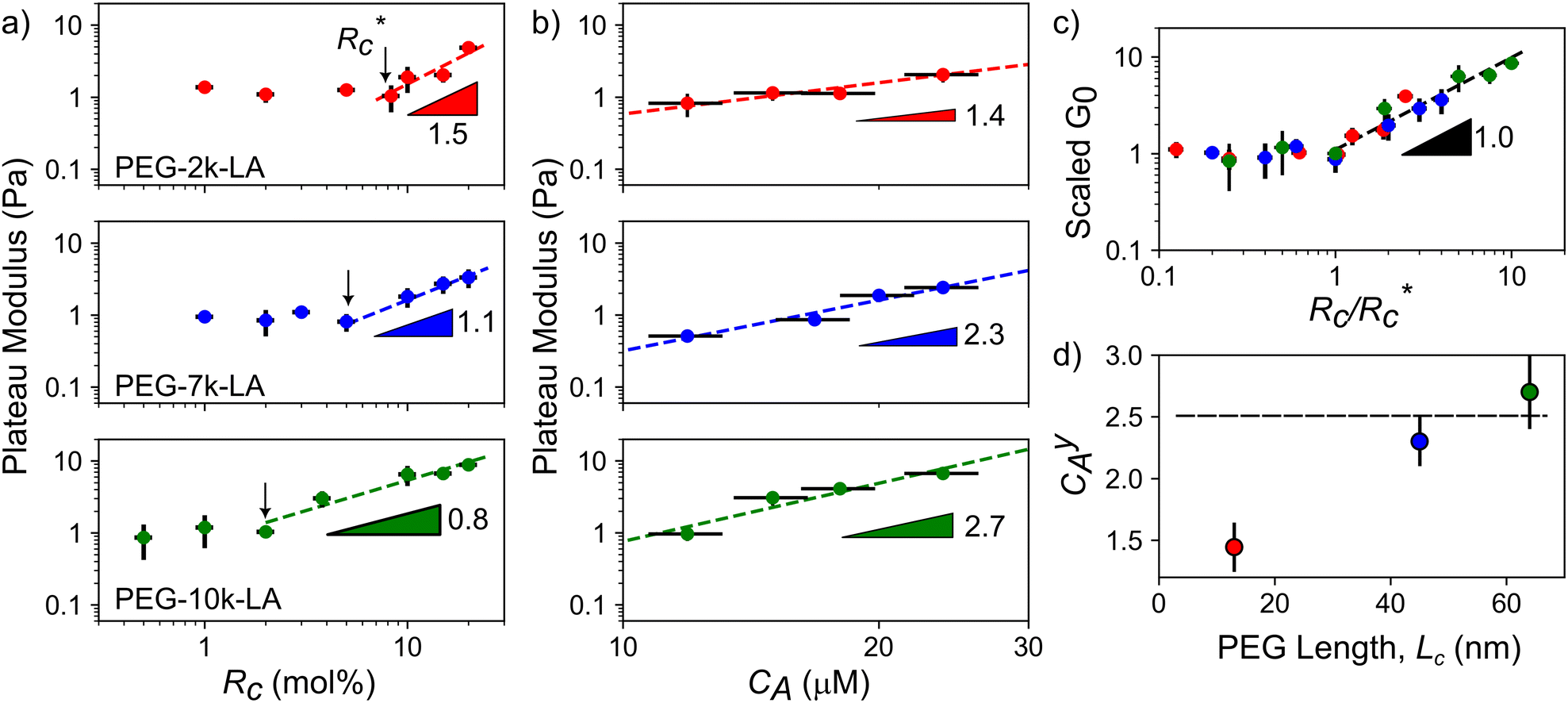 | ||
Fig. 4 Scaling of plateau modulus with relative concentration (a) and actin concentration (b). Errors in G0 are standard deviations over N = 3 while errors in Rc and CA are propagated from UV-vis concentration calibrations curves. (c) Scaled G0vs. Rc data from (a) revealing a master curve with a single Rxc. (d) Relationship between actin concentration scaling and crosslinker contour length. Errors for scaling values are determined from least squares fitting of a power model to G0vs. Rc above 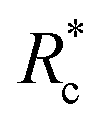 . . | ||
Interestingly, a similar relationship between crosslinker size and G0 ≈ Rxc scaling has been reported for semi-flexible DdFilamin-like biopolymer constructs composed of hisactophilin actin binding domains separated by a variable number of an IgG fold protein spacer.15 Specifically, a dimer of hisactophilin scaled with R1.2c while the largest construct studied scaled with R0.5c. The authors suggested the decrease in scaling of G0 with Rc was related to a reduced bundling propensity of the larger crosslinkers. Our results suggest that bundling propensity is not directly correlated with Rxc as the larger PEG-LA crosslinkers yield higher bundling affinity but lower elastic sensitivity to Rc. In a study of synthetic actin crosslinkers comprised of rigid 20 nm DNA duplexes functionalized with either phalloidin or LifeAct peptides, Lorenz et al. reported the phalloidin and LifeAct crosslinkers yield power law dependences of R0.4c and R1.1c, respectively.17 The authors concluded that the order of magnitude lower KD of phalloidin led to an increased partitioning of crosslinker into actin bundles leading to fewer elastically active inter-bundle crosslinks. The observed PEG-LA contour length dependent crosslinking kinetics may lead to a similar partitioning of crosslinker within the networks. Interestingly, when mechanical data presented in Fig. 4a are scaled by the low concentration G0 and 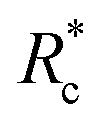 , a master curve is generated with a scaling of 1.0 ± 0.1 (Fig. 4c). This result suggests that crosslinking kinetics plays a central role in determining a construct's mechanical effectiveness when all other crosslinker properties are constant.
, a master curve is generated with a scaling of 1.0 ± 0.1 (Fig. 4c). This result suggests that crosslinking kinetics plays a central role in determining a construct's mechanical effectiveness when all other crosslinker properties are constant.
While the scaling of G0 ≈ Rxc depends on the physiochemical properties of the crosslinker, the relationship between G0 and actin concentration, CA, has been shown to track closely with expected scaling laws derived from polymer physics.11,21–23 To determine if the expected scaling holds for the PEG-LA constructs, G0 was probed at a constant Rc of 15 mol% to ensure robust networks. The expected log-linear increase in G0 with increasing CA was observed for all crosslinkers (Fig. 4b). PEG-7k-LA and PEG-10k-LA networks scale with C2.3 ± 0.3A and C2.7 ± 0.2A, respectively (Fig. 4d). These observed values are within error of the expected scaling for a densely crosslinked (C2.5A) network when assuming a mechanical bending model of semi-flexible polymers.23 Surprisingly, the modulus of PEG-2k-LA networks is relatively insensitive to changes in actin concentration with scaling of G0 ∼ C1.4 ± 0.2A. While the limited data range used to determine the scaling warrants caution of over-analysis, the observed insensitivity to CA may arise from the previously discussed differences in linear network mechanics. Networks composed of PEG-2k-LA exhibit higher frequency dependence suggesting larger extents of filament mobility. For the tested range of CA and Rc of 15 mol%, the frequency dependence, S, is approximately 2× higher for PEG-2k-LA networks than the analogous PEG-7k-LA and PEG-10k-LA networks. The additional mobility in these networks indicates modes of stress dissipation other than the assumed bending modes that lead to the expected C2.5A scaling.
Collectively, the results presented in Fig. 2–4 demonstrate that highly flexible (lp ≪ lc) synthetic PEG constructs that have a lack of geometry can indeed act as effective actin crosslinkers with apparent contour length dependent control over network mechanics and structure. The observed contour length dependent network mechanics and structure can be attributed to the apparent crosslinking kinetic difference of the PEG-LA constructs. We posit that augmented crosslinking kinetics with increasing contour length results from improved positioning of the LifeAct binding domains. In aqueous conditions, PEG is well described as a freely jointed chain.24 As the LifeAct peptides are connected to the PEG chain ends, the average end-to-end distance, re, is a decent proxy for the LifeAct-LifeAct distance in the crosslinker. For PEG-2k-LA, the re is ∼50% of the hydrodynamic diameter suggesting a large fraction of constructs have the LifeAct functionalized chain-ends in close proximity, which inhibits effective crosslinking through the formation of singly bound constructs or doubly bound “loops”. In contrast, for PEG-7k-LA and PEG-10k-LA the re is ∼70% and 94% of the hydrodynamic diameter, respectively, suggesting the LifeAct domains are better positioned for inter-filament crosslinking (see Note S2 and Fig. S9, ESI†). The contour length dependent 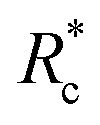 presented in Fig. 4a supports this physical view of PEG-2k-LA as higher concentrations are needed to overcome the fraction of constructs with inefficiently positioned LifeAct units. Moreover, these elastically ineffective bound PEG-2k-LA constructs would similarly lead to the observed inability of PEG-2k-LA to restructure the F-actin network's morphology and limit filament mobility.
presented in Fig. 4a supports this physical view of PEG-2k-LA as higher concentrations are needed to overcome the fraction of constructs with inefficiently positioned LifeAct units. Moreover, these elastically ineffective bound PEG-2k-LA constructs would similarly lead to the observed inability of PEG-2k-LA to restructure the F-actin network's morphology and limit filament mobility.
Given how the extensible nature of PEG based constructs affected linear network mechanics, it was important to explore the impact on the actin network's non-linear mechanical response. A characteristic feature of actin networks is the pronounced stress-stiffening beyond a critical stress which represents a transition between bending to stretching dominated stress responses. Within this stiffening regime, the differential elastic modulus, K′, increases with applied pre-stress as K′ ≈ σ1.5.11 When networks crosslinked with the compliant ABP Filamin are stressed further, a scaling of unity with σ is observed, indicating a significant amount of stress is mediated through the crosslinker.25,26 To determine whether the highly compliant PEG-LA crosslinkers mediate stress rather than the actin filaments, K′ was measured as a function of applied pre-stress for networks with 15 mol% crosslinker at a variety of actin concentrations. As shown in Fig. 5, all tested PEG-LA networks stress-stiffened and could be collapsed by the zero-stress differential modulus, K0, and the critical stress, σc, to a master curve with a single stiffening regime. The scaling within this regime is well described by the expected power law σ1.5 suggesting the networks were still dominated by the stretching of the actin filaments rather than the extension of the PEG crosslinkers. Previously published coarse-grained simulations of two-dimensional actin networks suggested that crosslinker stiffnesses less than ∼100 pN μm−1 are needed for strain energy to be stored primarily in the crosslinker.27 Our experimental results bolster the validity of this mechanical threshold as the most compliant crosslinker tested herein has an approximate spring constant of ∼260 pN μm−1.
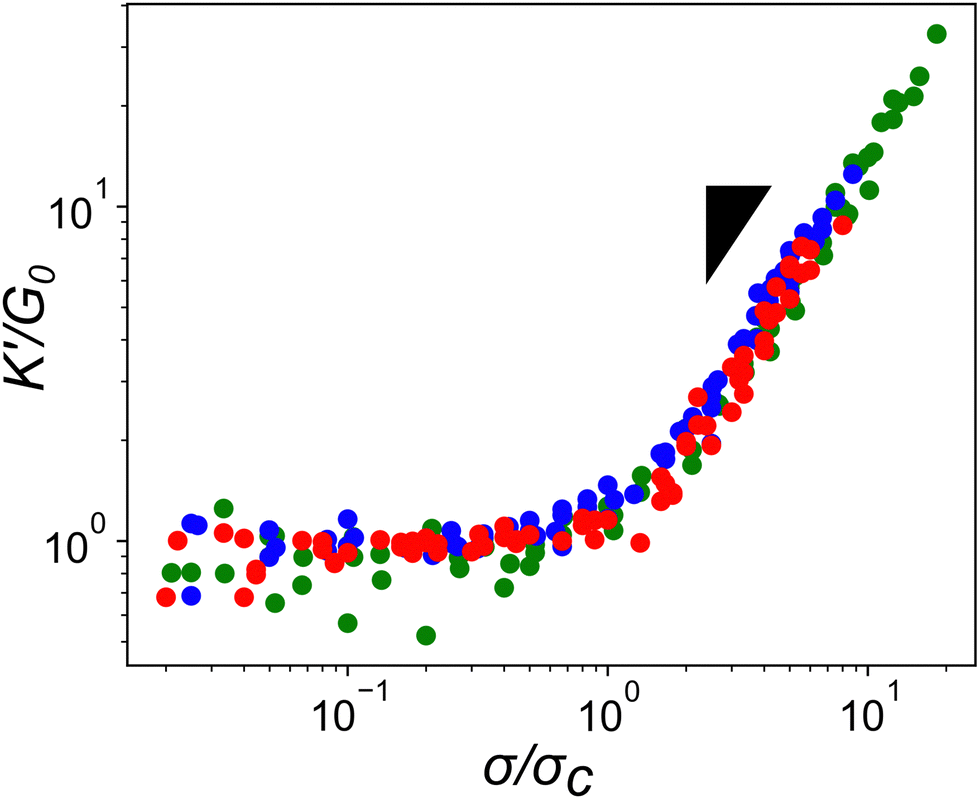 | ||
| Fig. 5 Scaled stress-stiffening response of actin networks crosslinked with PEG-LifeAct of differing molecular weights. Networks are crosslinked with 15 mol% PEG-LifeAct with actin concentrations ranging from 11.9 to 23.8 μM. Red circles are PEG-2k-LA, blue circles are PEG-7k-LA, and green circles are PEG-10k-LA. Inset triangle represents a power law of σ1.5. | ||
Experimental
Actin purification and LifeAct synthesis
Monomeric actin (G-actin) was purified from chicken skeletal muscle acetone powder using a previously published procedure and stored in calcium buffer G at −80 °C.28 Azide functionalized LifeAct peptides were synthesized via solid-phase synthesis (Microwave Peptide Synthesizer Liberty Blue 2.0) using rink amide resin, N,N′-diisopropylcarbodiimide (DIC) and ethyl cyanohydroxyiminoacetate (oxyma) in DMF. Deprotection and cleavage of the peptide was achieved by mixing the reacted resin with a solution of 95% trifluoroacetic acid (TFA), 2.5% triisopropyl silane (TIPS), and 2.5% water for 2 hours. Crude peptide was precipitated in cold diethyl ether and sedimented by centrifugation. Peptide molecular weight was validated using LC-MS and subsequently purified using reverse-phase HPLC using a gradient of acetonitrile to water with 0.1% TFA. Fractions containing the pure peptide were combined and lyophilized. Stock solutions of peptide were made by dissolving peptide in 1X PBS, pH 7.4. Peptide concentration was determined via UV absorbance at 260 nm using the phenylalanine molar extinction coefficient of 190 M−1 cm−1.Polyethylene glycol-LifeAct construct synthesis
Alkyne functionalized polyethylene glycol (PEG) polymers (Creative PEGworks, Mw 2 kDa, 5 kDa, & 10 kDa) were characterized using NMR before use. The molecular weight of the nominally 5 kDa polymer was determined to be approximately 7.1 kDa. PEG-LifeAct constructs were prepared via copper catalyzed azide–alkyne click chemistry using a protocol adapted from Presolski et al.29 Briefly, appropriate volumes of PEG-alkyne stock solutions (10 mg mL−1 in deionized water) were added to an aliquot of azide functionalized LifeAct peptide in PBS buffer (pH 7.4) for a final molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 azide to alkyne groups. Separately, a mixture of copper(II) sulfate (20 mM in DI water) and tris(3-hydroxypropyltriazolyl-methyl)amine ligand (THPTA, 50 mM in DI water) with a ligand to copper ratio of 5:1 was prepared and allowed to incubate for at least 15 minutes. Enough copper-ligand solution was added to the PEG/LifeAct mixture to achieve a final concentration of 0.1 mM copper and 0.5 mM THPTA ligand. Initiation of the reaction was achieved by the addition of freshly prepared sodium ascorbate solution to give a final reaction concentration of 5 mM. The reaction was allowed to proceed under constant agitation at room temperature for 6 hours. The reaction was purified via 6 rounds centrifugal filtration (Amicon Ultra-4 centrifugal filters MWCO 3 kDa) using deionized water. The product was collected, flash frozen, and lyophilized for further characterization. LifeAct functionality was confirmed using NMR with pure D2O as solvent (see Fig. S2 and S3, ESI†). Pure PEG-LifeAct constructs were aliquoted with calcium buffer and stored at −80 °C for further use. Concentration of PEG-LifeAct constructs was determined using the peptide backbone UV absorbance at 220 nm (see Fig. S1, ESI†).
:1 azide to alkyne groups. Separately, a mixture of copper(II) sulfate (20 mM in DI water) and tris(3-hydroxypropyltriazolyl-methyl)amine ligand (THPTA, 50 mM in DI water) with a ligand to copper ratio of 5:1 was prepared and allowed to incubate for at least 15 minutes. Enough copper-ligand solution was added to the PEG/LifeAct mixture to achieve a final concentration of 0.1 mM copper and 0.5 mM THPTA ligand. Initiation of the reaction was achieved by the addition of freshly prepared sodium ascorbate solution to give a final reaction concentration of 5 mM. The reaction was allowed to proceed under constant agitation at room temperature for 6 hours. The reaction was purified via 6 rounds centrifugal filtration (Amicon Ultra-4 centrifugal filters MWCO 3 kDa) using deionized water. The product was collected, flash frozen, and lyophilized for further characterization. LifeAct functionality was confirmed using NMR with pure D2O as solvent (see Fig. S2 and S3, ESI†). Pure PEG-LifeAct constructs were aliquoted with calcium buffer and stored at −80 °C for further use. Concentration of PEG-LifeAct constructs was determined using the peptide backbone UV absorbance at 220 nm (see Fig. S1, ESI†).
Total-internal reflection fluorescence microscopy (TIRFM)
Cross-linkers of interest were initially added to a polymerization mix (9.5 mM imidazole (pH 7.0), 47.6 mM KCl, 952 μM MgCl2, 952 μM EGTA, 95.2 mM DTT, 190 μM ATP, 66.7 μg mL−1 catalase, 333 μg mL−1 glucose oxidase, 14.3 mM glucose, 0.48% methyl cellulose at 400 cp). The cross-linker/polymerization mix was then added to 1.5 μM of Mg-ATP-actin (10% labelled with Alexa Fluor 488) to induce the spontaneous assembly of F-actin in the presence of the cross-linkers of interest. The mixture was flown in a TIRF chamber made from a PEG-Silane coated coverslip and glass slide taped by double-sided TIRF tape. TIRFM movies were obtained at 5 s intervals and at room temperature using an Olympus IX-71 microscope with a 100× TIRF objective, an iXon EMCCD camera (Andor Technology) and a 488 nm laser line (12 mW) connected via a cellTIRF 4Line system (Olympus).Bundling assays
For each experiment, an area containing similar actin filament densities (between 1750 μM and 2000 μM total filament length) was selected for the bundling quantification. The total actin filament length within the area was measured manually by creating ROIs (regions of interest) for every filament and obtaining the total actin filament length using FIJI.30–32 Bundled segments were identified by looking at the history of the TIRF movies and determining that two filaments were associated and had been associated for at least three consecutive frames. ROIs for every segment in a bundle were then created and the total length of bundled filament was measured.Network preparation and bulk rheology
In vitro networks are prepared by adding G-actin and desired PEG-LifeAct crosslinker into calcium buffer while kept on ice. Crosslinker concentration is reported as a ratio Rc = [crosslinker]/[G-actin]. Actin is primed for polymerization through the exchange of Ca2+ ions with Mg2+ ions through the addition of enough 10× magnesium buffer (0.5 mM MgCl2, 2 mM EDTA) to achieve a final concentration of 1×. After 1 minute of incubation, polymerization is initiated with the addition of 1/10th the final volume of 10× polymerization buffer (500 mM KCl, 10 mM MgCl2, 2 mM EGTA, 100 mM Imidazole). Immediately after pipette mixing, 60 μL of the solution was added to the rheometer stage. All rheological measurements were performed on a TA Instruments Discovery Hybrid Rheometer 30 with the Peltier plate stage and a 20 mm parallel plate. All measurements were performed at 22 °C. Network formation was monitored using a small 1 Hz oscillatory strain of 5%. The network is considered to reach a mechanical steady state once both G′ and G′′ plateau, typically 1.5 hours. Frequency sweeps from 0.01 Hz to 10 Hz were performed using a strain of 5%. On account of inertial effects, frequency sweeps were reliably compared below ∼1 Hz. The stress-stiffening properties of the network were determined by applying a pre-stress to the network while simultaneously measuring the differential modulus using a super-imposed small oscillatory stress. The oscillatory stress was always less than or equal to 10% of the applied pre-stress.Conclusions
In this experimental study, bio-synthetic crosslinkers comprised of polyethylene glycol (PEG) chains end functionalized with the actin binding peptide LifeAct were demonstrated to be effective F-actin crosslinkers. Fluorescence microscopy experiments highlighted the role of crosslinker contour length on bundling propensity. Specifically, the larger flexible crosslinkers have an augmented bundling propensity while little to no bundles are observed with small flexible crosslinkers. Bulk rheological studies revealed that longer constructs resulted in more robust F-actin networks that better suppressed filament mobility. Determination of G0 as a function of Rc revealed an inverse correlation between the mechanical effectiveness (G0 ∼ Rxc) and the crosslinker's contour length mirroring previous reports on biopolymer ABPs. Scaling the data by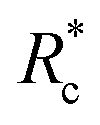 eliminated this contour length dependence suggesting crosslinking kinetics is critical in determining the mechanical effectiveness of the PEG-LA constructs. While the longer crosslinkers exhibited the expected power law dependence on actin concentration, CA, for a densely crosslinked networks (G0 ∼ C2.5A), the shorter crosslinker exhibited an insensitivity to actin likely due to additional filament mobility in these networks. Despite the differences in linear network mechanics, the non-linear stress-stiffening properties were indistinguishable, and the stress dependent stiffening was well described by the expected scaling of σ1.5 for entropic stretching of the F-actin filaments.
eliminated this contour length dependence suggesting crosslinking kinetics is critical in determining the mechanical effectiveness of the PEG-LA constructs. While the longer crosslinkers exhibited the expected power law dependence on actin concentration, CA, for a densely crosslinked networks (G0 ∼ C2.5A), the shorter crosslinker exhibited an insensitivity to actin likely due to additional filament mobility in these networks. Despite the differences in linear network mechanics, the non-linear stress-stiffening properties were indistinguishable, and the stress dependent stiffening was well described by the expected scaling of σ1.5 for entropic stretching of the F-actin filaments.
Collectively, the presented results show that crosslinkers composed of highly flexible synthetic polymer backbones (lp ≪ lc) have contour length dependent properties despite having indistinguishable hydrodynamic sizes. Moreover, the main impact of increasing contour length was an augmented effective crosslinking kinetics arising from improved geometric positioning of the LifeAct domains. It is hypothesized this improved positioning is a consequence of conformational statistics of an ideal chain. The increased crosslinking kinetics leads to improved bundling of filaments as well as more robust F-actin gels that behave analogously to native networks. For short contour length constructs, a larger fraction of conformations orient the LifeAct domains in close proximity resulting in the formation of elastically ineffective bound species. Such a phenomena would limit the construct's ability to crosslink filaments, hinder filament mobility, and reconstruct the network's structure. More broadly, the presented results highlight the potential of synthetic polymers as scaffolds for biomimetic actin crosslinkers. Future work could specifically tune polymer backbone flexibility to better probe the role of crosslinker stiffness on the network's stress-stiffening properties, as well as explore non-native geometries such as multi-armed crosslinkers or mixed binding domain systems. Such synthetic systems promise to provide greater insight into cytoskeletal functions as well as provide specific design rules for adaptive materials design.
Author contributions
SJR and MLG conceived the project. TDJ, KB, and CS synthesized materials and performed the experiments. TDJ, SJR, DRK, and MLG conceived of the experiments. TDJ wrote the manuscript with contributions from all other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Chicago Materials Research Science and Engineering Center, which is funded by National Science Foundation under award number DMR-2011854. M. L. G. acknowledges support from NSF DMR-2215605. D. R. K. acknowledges support from National Institutes of Health Grant R01 GM079265. This work made use of the shared facilities at the University of Chicago Materials Research Science and Engineering Center, supported by National Science Foundation under award number DMR-2011854. Parts of this work were carried out at the Soft Matter Characterization Facility of the University of Chicago. We would like to acknowledge Prof. Aaron Esser-Khan and Dr Adam Weiss for supplying access and expertise with peptide synthesis and purification.Notes and references
- F. Huber, J. Schnauß, S. Rönicke, P. Rauch, K. Müller, C. Fütterer and J. Käs, Adv. Phys., 2013, 62, 1–112 CrossRef CAS PubMed.
- S. Banerjee, M. L. Gardel and U. S. Schwarz, Annu. Rev. Condens. Matter Phys., 2020, 11, 421–439 CrossRef PubMed.
- D. Vignjevic, S.-i Kojima, Y. Aratyn, O. Danciu, T. Svitkina and G. G. Borisy, J. Cell Biol., 2006, 174, 863–875 CrossRef CAS PubMed.
- R. D. Mullins, J. A. Heuser and T. D. Pollard, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6181–6186 CrossRef CAS PubMed.
- E. S. Chhabra and H. N. Higgs, Nat. Cell Biol., 2007, 9, 1110–1121 CrossRef CAS PubMed.
- J. Stricker, T. Falzone and M. L. Gardel, J. Biomech., 2010, 43, 9–14 CrossRef PubMed.
- D. H. Wachsstock, W. Schwarz and T. Pollard, Biophys. J., 1994, 66, 801–809 CrossRef CAS PubMed.
- D. H. Wachsstock, W. Schwartz and T. D. Pollard, Biophys. J., 1993, 65, 205–214 CrossRef CAS PubMed.
- Y. Tseng, E. Fedorov, J. M. McCaffery, S. C. Almo and D. Wirtz, J. Mol. Biol., 2001, 310, 351–366 CrossRef CAS PubMed.
- A. J. Ehrlicher, R. Krishnan, M. Guo, C. M. Bidan, D. A. Weitz and M. R. Pollak, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6619–6624 CrossRef CAS PubMed.
- M. Gardel, J. H. Shin, F. MacKintosh, L. Mahadevan, P. Matsudaira and D. A. Weitz, Science, 2004, 304, 1301–1305 CrossRef CAS PubMed.
- O. Esue, Y. Tseng and D. Wirtz, PLoS One, 2009, 4, e4411 CrossRef PubMed.
- K. Schmoller, O. Lieleg and A. Bausch, Phys. Rev. Lett., 2008, 101, 118102 CrossRef CAS PubMed.
- M. L. Gardel, F. Nakamura, J. H. Hartwig, J. C. Crocker, T. P. Stossel and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1762–1767 CrossRef CAS PubMed.
- B. Wagner, R. Tharmann, I. Haase, M. Fischer and A. R. Bausch, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13974–13978 CrossRef CAS PubMed.
- Y. Mulla, M. J. Avellaneda, A. Roland, L. Baldauf, W. Jung, T. Kim, S. J. Tans and G. H. Koenderink, Nat. Mater., 2022, 21, 1019–1023 CrossRef CAS PubMed.
- J. S. Lorenz, J. Schnauß, M. Glaser, M. Sajfutdinow, C. Schuldt, J. A. Käs and D. M. Smith, Adv. Mater., 2018, 30, 1706092 CrossRef PubMed.
- P. J. Hagerman, Annu. Rev. Biophys. Biophys. Chem., 1988, 17, 265–286 CrossRef CAS PubMed.
- J. Riedl, A. H. Crevenna, K. Kessenbrock, J. H. Yu, D. Neukirchen, M. Bista, F. Bradke, D. Jenne, T. A. Holak and Z. Werb, Nat. Methods, 2008, 5, 605–607 CrossRef CAS PubMed.
- J. Condeelis, M. Vahey, J. M. Carboni, J. DeMey and S. Ogihara, J. Cell Biol., 1984, 99, 119s–126s CrossRef CAS PubMed.
- R. Tharmann, M. M. Claessens and A. R. Bausch, Phys. Rev. Lett., 2007, 98, 088103 CrossRef CAS PubMed.
- R. Tharmann, M. Claessens and A. Bausch, Biophys. J., 2006, 90, 2622–2627 CrossRef CAS PubMed.
- F. MacKintosh, J. Käs and P. Janmey, Phys. Rev. Lett., 1995, 75, 4425 CrossRef CAS PubMed.
- F. Oesterhelt, M. Rief and H. Gaub, New J. Phys., 1999, 1, 6 CrossRef.
- K. Kasza, G. Koenderink, Y. Lin, C. Broedersz, W. Messner, F. Nakamura, T. Stossel, F. MacKintosh and D. Weitz, Phys. Rev. E, 2009, 79, 041928 CrossRef CAS PubMed.
- C. Broedersz, C. Storm and F. MacKintosh, Phys. Rev. Lett., 2008, 101, 118103 CrossRef CAS PubMed.
- S. L. Freedman, S. Banerjee, G. M. Hocky and A. R. Dinner, Biophys. J., 2017, 113, 448–460 CrossRef CAS PubMed.
- J. A. Spudich and S. Watt, J. Biol. Chem., 1971, 246, 4866–4871 CrossRef CAS PubMed.
- S. I. Presolski, V. P. Hong and M. Finn, Curr. Protoc. Chem. Biol., 2011, 3, 153–162 CrossRef PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld and B. Schmid, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- J. R. Christensen, K. E. Homa, A. N. Morganthaler, R. R. Brown, C. Suarez, A. J. Harker, M. E. O'Connell and D. R. Kovar, eLife, 2019, 8, e47279 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm01341c |
| This journal is © The Royal Society of Chemistry 2024 |