
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation of supramolecular structures in various aqueous solutions of an amyloid forming peptide using small-angle X-ray scattering†
Ellen
Brunzell
 a,
Kalle
Sigfridsson
b,
Lars
Gedda
c,
Katarina
Edwards
c and
L. Magnus
Bergström
*a
a,
Kalle
Sigfridsson
b,
Lars
Gedda
c,
Katarina
Edwards
c and
L. Magnus
Bergström
*a
aDepartment of Medicinal Chemistry, Pharmaceutical Physical Chemistry, Uppsala University, Uppsala 751 23, Sweden. E-mail: magnus.bergstrom@ilk.uu.se
bAdvanced Drug Delivery, Pharmaceutical Science, R&D, AstraZeneca, Gothenburg 431 83, Sweden
cDepartment of Chemistry–Ångström, Uppsala University, P.O. Box 573, Uppsala 751 23, Sweden
First published on 14th February 2024
Abstract
Aggregation of peptide molecules into amyloid fibrils is a characteristic feature of several degenerative diseases. However, the details behind amyloid-formation, and other self-assembled peptide aggregates, remain poorly understood. In this study, we have used small-angle X-ray scattering (SAXS), static and dynamic light scattering (SLS and DLS) as well as cryogenic transmission electron microscopy (cryo-TEM) to determine the structural geometry of self-assembled peptide aggregates in various dilute aqueous solutions. Pramlintide was used as a model peptide to assess the aggregation behaviour of an amyloid-forming peptide. The effects of adding sodium chloride (NaCl), sodium thiocyanate (NaSCN), and sodium fluoride (NaF) and the co-solvent dimethyl sulfoxide (DMSO) on the aggregation behaviour were studied. Our scattering data analysis demonstrates that small oligomeric fibrils aggregate to form networks of supramolecular assemblies with fractal dimensions. The choice of anion in small amounts of added salt has a significant impact on the size of the fibrils as well as on the fractal dimensions of supramolecular clusters. In DMSO the fractal dimension decreased with increasing DMSO concentration, indicating the formation of a less compact structure of the supramolecular assemblies.
Introduction
Aggregation of peptides is usually an unwanted event that may appear during peptide synthesis in pharmaceutical formulation development, or in vivo.1 The structural combination of polar and non-polar groups that make up peptides, as well as their inherent flexibility, leaves reactive side groups or hydrophobic areas exposed. These properties make them prone to interact with surfaces, interfaces, and other peptide molecules.2Peptides as active pharmaceutical ingredients are becoming increasingly more common due to their high specificity and potency.1,3 However, production of biological drugs is time-consuming and expensive. Peptides are usually less stable and more likely to undergo aggregation or chemical degradation, compared with small molecules, which makes the manufacturing more complicated and usually reduces the shelf life of peptide products.2,4 The structure and aggregation behaviour of peptides can be affected by, among other things, interfaces, formulation components, sheer force, and/or added ions. However, the self-assembly properties and aggregation behaviour can be an advantage if possible to control.5 For example, aggregation can be utilised to alter pharmacokinetic properties of peptides, as a way to achieve long-acting pharmacokinetic profiles.6,7 In this way the dosage interval can be decreased and patient compliance increased. If the mechanisms behind peptide aggregation were better understood, it would be easier to predict under which circumstances aggregation would occur.8 Increased understanding of peptide aggregation pathways can also facilitate manufacturing and stability predictions of pharmaceutical peptide formulations.9
Amyloid fibrils
The link between deposits of aggregated peptides in organ tissue and degenerative diseases was established in the early 20th century when Opie10 found amyloid deposits in the pancreatic tissue of diabetic patients. Amyloid fibrils are insoluble, unbranched fibrils around 10 nm in diameter, that usually are deposited in the extracellular space in tissues and organs.11 This pathology is shared with other so-called misfolding diseases,12 such as Alzheimer's13 and Parkinson's14 disease, in which the brain is the affected organ.15The cause of in vivo deposition of aggregated peptides in specific tissues is believed to have a genetic component, but further details remain unknown.12,16 Besides deposition of aggregated peptides being a trait for misfolding diseases,17 formation of aggregated peptides in vivo can in many cases be immunogenic, but the mechanisms behind peptide aggregate-caused immune activation are not fully understood.18,19 Several studies have reported that the cytotoxicity of aggregated peptides is heavily linked to the ability to disrupt cell membranes. This effect seems strongest by oligomeric species as opposed to fully formed fibrils20–23 or β-sheet structures.24
Amyloid fibril formation has been investigated for decades, and several mechanisms have been proposed, including nucleation-dependent fibril growth,25–27 as well as off-pathway oligomer formation.28,29 However, the mechanism is highly peptide-dependent, and the properties of the resulting aggregates can vary greatly and is difficult to predict, especially from the off-pathway mechanism.29
Pramlintide
Pramlintide is a 37 amino acid analogue of the endogenous peptide hormone amylin, or human islet amyloid polypeptide (hIAPP). Pramlintide was designed based on the sequence of amylin from rat (rIAPP), which does not form amyloid fibrils like hIAPP.30,31 Pramlintide differs from amylin by three proline substitutions (cf.Fig. 1), which were introduced to decrease the rate of aggregation. However, pramlintide can form amyloid fibrils in in vitro conditions.32 Residues 2 and 7 are connected by a disulphide bond, and the C-terminus is amidated, both of which are important post-translational modifications for the biological activity of hIAPP, as for analogues of amylin.33 Pramlintide holds three positive charges at pH 6 and is soluble in water. As pH is raised (>8), the basic amino acids will become less charged, and the solubility of pramlintide will decrease. | ||
| Fig. 1 Molecular structure of pramlintide. Circled segments indicate the replaced amino acids, next to which are the residues in endogenous amylin. | ||
Following subcutaneous administration, pramlintide shares the effect of endogenous amylin.34 Amylin, along with insulin, is secreted from the β-cells in the pancreas in response to increased plasma glucose levels35 and regulates blood glucose levels by affecting satiety and controlling gastric emptying.36 Patients with diabetes mellitus type II have dysfunctional β-cells which leads to decreased release of insulin and amylin in response to plasma glucose increase, in comparison with healthy humans.37 However, unlike insulin, amylin cannot be used as replacement therapy for treatment of diabetes, due to its high propensity for aggregation.38 Adding pramlintide as an adjunctive treatment to insulin can decrease weight gain and glycated haemoglobin, more than insulin alone.39–42
In the present study, we structurally characterize the meta-stable transparent solution that pramlintide forms in various aqueous solvents at about pH 5–6. Our study includes the effect of ion binding and presence of co-solvent on the structural behaviours.
Experimental
Materials
Pramlintide acetate was provided by AstraZeneca, Gothenburg, Sweden, as a lyophilized powder. The total amount of impurities for the present batch of pramlintide has been determined with RP-HPLC to be less than 4.0% of total peak area. The batch is confirmed stable for many years when stored at <−15 °C. Dimethyl sulfoxide (DMSO) (99.9% ACS reagent), sodium thiocyanate (NaSCN) (≥99.99% trace metals basis), sodium chloride (NaCl) (99.5% BioXtra), and sodium fluoride (NaF) (99% ACS reagent), were purchased from SigmaAldrich, Saint Louis, MO.All samples and solvents were prepared using Type I water from a Millipore Synergy water purification system. Lyophilized pramlintide acetate (henceforth referred to as pramlintide) powder was stored under nitrogen gas at −18 °C and allowed to thaw in desiccator before use. Mixed samples were prepared 4–10 days prior to all measurements (SAXS, SLS/DLS, and cryo-TEM). To slow down the aggregation process, samples that could not be measured within four days were stored in refrigerator (2–5 °C) up until four days prior to measurement, after which they were stored at room temperature. Dynamic light scattering shows that the size of large structures in our samples do not change appreciably from a few hours after preparation to about two weeks ahead. A previous study on pramlintide in solution at different storage temperatures and solution pH shows that pramlintide exhibits high stability, especially when stored in refrigerator.43 All measurements were performed at room temperature unless otherwise specified. All solvents were filtered twice before peptide was dissolved. Syringe filter with Durapore PVDF membrane (pore size 0.1 μm) from Merck, Cork, Ireland was used for aqueous solvents without DMSO. Syringe filter with PTFE membrane (pore size 0.2 μm) from Fisher Scientific, Hampton, New Hampshire was used for DMSO-containing solvents.
Methods
Pramlintide solutions at concentrations 2.5–10 mg cm−3 (0.63–2.5 mM) were prepared in eight different aqueous solvents containing pure water, 1 mM of the three salts NaSCN, NaCl and NaF, as well as different concentrations of DMSO added to water. All investigated samples were studied within 10 days from preparation and, except those with 20 wt% DMSO, appeared transparent.Small-angle X-ray scattering
Small-angle X-ray scattering measurements were performed at the B21 beamline at Diamond Light Source, UK, which uses a bending magnet source and delivers X-rays of wavelength 0.9537 Å with a flux of 4 × 1012 photons per second. The sample-to-detector distance was 3688.3 mm, at which the scattered intensity was detected by an EigerX 4M detector (Dectris, Switzerland). This made it possible to cover a q-range of 4.5 × 10−3 ≤ q ≤ 3.4 × 10−1 Å−1. The SAXS data were set to absolute intensity and solvent subtracted before data analysis with the XSACT 2.4 software (Xenocs, France).Static and dynamic light scattering
Static and dynamic light scattering (SLS and DLS) measurements were performed using an ALV/CGS-3/MD-4 Multi-Detection Goniometer System, covering angles between 30° < θ < 150°, with ALV/LSE-5004 Light Scattering Electronics Correlator (Germany), complete with a 50 mW DPSS Nd-YAG Laser 532 nm and fibril-optical single detecting units with fibril-based beam splitter for 532 nm wavelength. Static light scattering was not measured for concentrations below 10 mg cm−3 due to too low intensity.Cryogenic transmission electron microscopy (cryo-TEM)
Specimens for cryo-TEM investigations were prepared at controlled temperature (25 °C) and high humidity within a custom-built environmental chamber. A small drop of sample (∼1 μL) was deposited on a carbon-sputtered copper grid (300 mesh, Agar scientific) covered with a perforated polymer film. Excess liquid was blotted away with a filter paper, leaving a thin film of the sample solution on the grid. The sample was then vitrified by plunging the grid into liquid ethane held at a temperature just above its freezing point. Thereafter the grid was mounted in a Gatan CT3500 sample holder and transferred to a Zeiss TEM Libra 120 transmission electron microscope (Carl Zeiss AG, Oberkochen, Germany), operating at 80 kV and in zero-loss bright-field mode, for viewing. The sample was kept below −160 °C and protected against atmospheric conditions during the transfer and viewing processes. Analyses were performed with a Zeiss Libra 120 transmission electron microscope (Carl Zeiss AG, Oberkochen, Germany) operating at 80 kV and in zero-loss bright-field mode. Digital images were recorded under low-dose conditions with a BioVision Pro-SM Slow Scan CCD camera (Proscan Elektronische Systeme GmbH, Scheuring, Germany).Data analysis
Small-angle X-ray scattering curves, covering a q-range of 4.5 × 10−3 ≤ q ≤ 3.4 × 10−1 Å−1 on absolute intensity scale, were analysed by means of least-square model fitting analysis. All our data could be fitted with an identical model in which a form factor for a spheroid was combined with a structure factor for particles forming clusters with fractal dimensions. The detailed model is provided in the ESI.† Static light scattering data was not included in the fitting but were added to the SAXS curves to show that no plateau of the scattering intensity was reached at q-value 0.00081 Å−1.Results and discussion
The aggregation process of amyloid-forming peptides, from monomer to mature fibril, is proposed to take place over a series of steps. The intermediate products of this stepwise process include oligomeric species, as suggested by several studies on amyloidogenic peptides.12,29,44 The aggregates formed by pramlintide in solution are not thermodynamically stable but behave as a dispersed system where the size of supramolecular structures slowly increases over time and eventually phase separates. For pramlintide in water, low salt, or low amounts of DMSO this process takes several months and the samples are transparent for months. However, the stability of pramlintide in solution was greatly reduced if the salt concentration exceeded 1 mM. At salt concentrations close to physiological conditions (about 150 mM), pramlintide phase separates immediately forming visible, opaque gel-like structures. Higher concentrations of salt or DMSO could in this case contribute to an accelerated aggregation process, typical for a non-equilibrium dispersed system of charged particles that is stabilized according to the DLVO theory.45,46Small-angle X-ray scattering
The high q-region (q > 0.05 Å−1) appeared similar for all samples of pramlintide in solution, regardless of solvent additives used (cf.Fig. 2 and 3). Differences in scattering behaviour between various samples were mainly seen in the low q-region of the SAXS curves. All scattering curves have a plateau region at about q = 0.1 Å−1, which becomes more or less pronounced depending on solvent additives. The main difference between the different solvents used is seen in the interval 0.004 < q < 0.07 Å−1. The extension of the plateau region in this q-range varied depending on the solvent used. The scattering curve with the most pronounced plateau comes from pramlintide in water, whereas no plateau at all could be observed for pramlintide in 20% DMSO. At q-values below this plateau, the scattering intensity increased with decreasing q, and did not level out within the examined q-range, indicating the formation of very large supramolecular aggregates. Although SLS data were included to cover q-values as low as 0.00081 Å−1, the Guiner regime47 was never reached and the aggregates were too large for their size to be determined from our scattering data.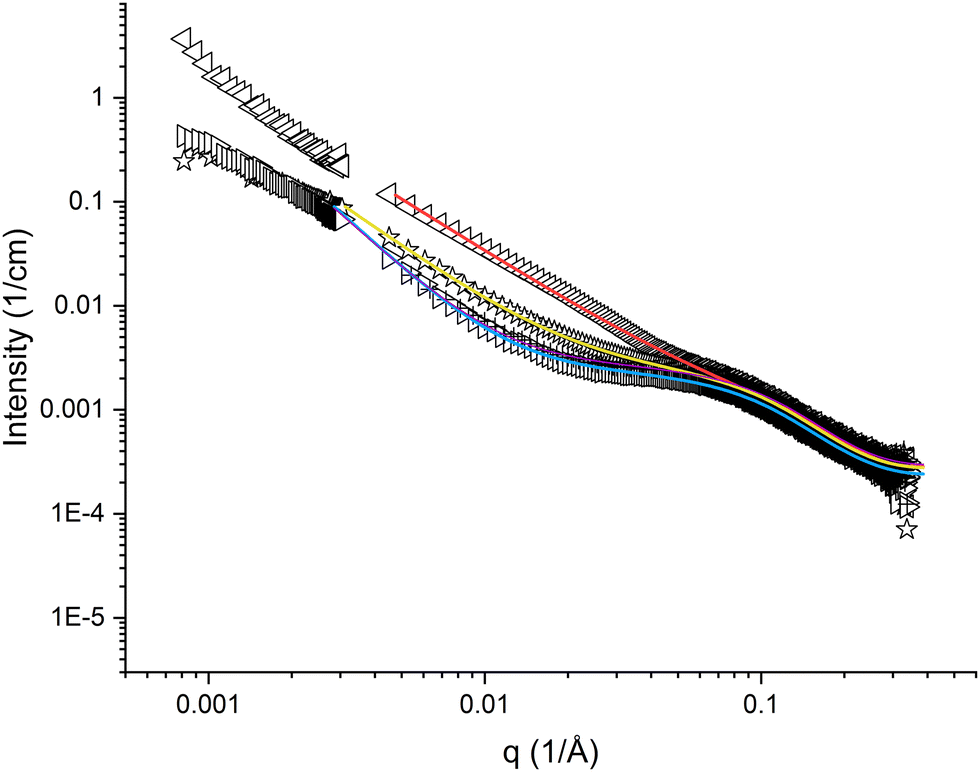 | ||
Fig. 2 SAXS and SLS curves for 10 mg cm−3 pramlintide in 1 mM NaSCN (◁), 1 mM NaCl ( ), water (▷), and 1 mM NaF (+), as hollow symbols, and fitted result as solid lines. ), water (▷), and 1 mM NaF (+), as hollow symbols, and fitted result as solid lines. | ||
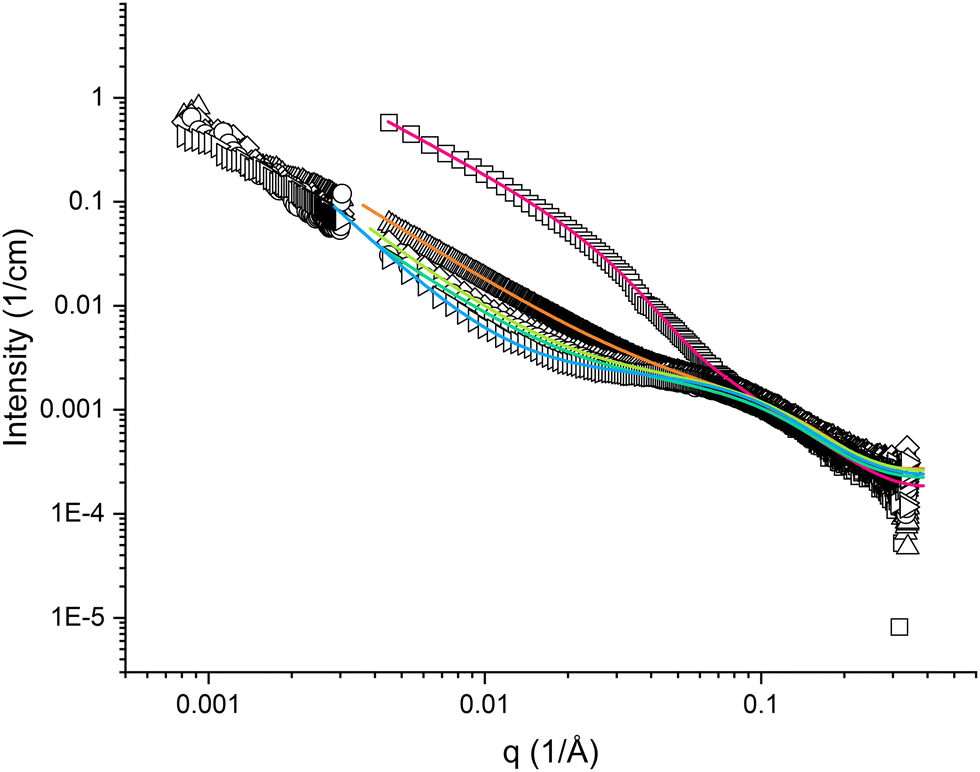 | ||
Fig. 3 SAXS and SLS curves for 10 mg cm−3 pramlintide in 20 wt% DMSO (□), 10 wt% DMSO (△), 5 wt% DMSO (◇), 1 wt% DMSO ( ), and water (▷), as hollow symbols, and fitted result as solid lines. ), and water (▷), as hollow symbols, and fitted result as solid lines. | ||
Scattering data analysis
The high q-region of the SAXS curves was possible to fit with a model for slightly elongated spheroids, in which the dimensions of oligomeric pramlintide fibrils were described by the two semi-axes of a prolate spheroid (cf. Fig. S1, ESI†). However, it was not possible to fit data in the low q-regime assuming individual aggregates, but an indefinite increase in scattering intensity with decreasing q indicates the formation of large aggregates or clusters. Data show a linear behaviour in a log-log plot with the slope varying in the range of −2.5 to −1.4 (cf.Fig. 2 and 3). This means that the peptide does not form compact three-dimensional structures for which the slope is expected to equal −4 according to Porod's law.48 Due to the complex nature of the system, no simple geometrical model could explain the scattering data. The variety of different slopes in the Porod regime is here interpreted as the presence of an open fractal structure of the supramolecular aggregates formed by pramlintide fibrils. Hence, we have included a structure factor to our model that takes into account the aggregation of pramlintide molecules or oligomers into a rather loose network with fractal dimensions. With this model, it was possible to fit the data for all our investigated samples in the entire q-range. The model is described in detail in the ESI.†The quality of our model fits was optimised with respect to four fitting parameters, in addition to a scaling parameter and a residual background parameter. The two semi-axes a and b of the individual oligomeric fibrils come from the form factor, whereas the parameters R and D come from the structure factor. The minor semi-axis a of the spheroids appeared to be rather small, i.e. equal or slightly below about 1 nm, but due to lack of data above about 0.25 Å−1 we could not determine its value precisely.
R may be interpreted as an independent measure of the size of the individual fibrils or, alternatively, the size of stiff units making up the fractal network. The parameter D describes the fractal dimension. The values of D correspond to the power law exponent. A more negative value of D may be interpreted as a more compact network.
From the values of a and b, as determined from the SAXS data analysis and assuming a ≈ 1 nm, we have been able to calculate the average number of molecules in an oligomeric fibril by estimating (through theoretical modelling) the volume of a pramlintide molecule to 6.55 nm3.49 Hence, from the results of the model fitting analysis, the oligomers formed by pramlintide in solution are close to dimers (cf.Tables 1 and 2).
| Solvent | Semi-axis b (nm) | Volume (nm3) | Molecules/particle | R (nm) | D |
|---|---|---|---|---|---|
| Water | 3.4 ± 0.2 | 14.2 | 2.2 | 7.1 ± 0.3 | 2.5 ± 0.1 |
| NaSCN | 2.0 ± 0.3 | 8.55 | 1.3 | 1.1 ± 0.1 | 1.7 ± 0.1 |
| NaCl | 3.2 ± 0.2 | 13.4 | 2.0 | 4.5 ± 0.1 | 1.9 ± 0.1 |
| NaF | 3.2 ± 0.2 | 13.4 | 2.0 | 7.6 ± 0.4 | 2.4 ± 0.1 |
| Solvent | Semi-axis b (nm) | Volume (nm3) | Molecules/particle | R (nm) | D |
|---|---|---|---|---|---|
| 1% DMSO | 3.1 ± 0.1 | 12.9 | 2.0 | 4.6 ± 0.1 | 1.9 ± 0.1 |
| 5% DMSO | 3.1 ± 0.2 | 13.1 | 2.0 | 4.8 ± 0.3 | 2.0 ± 0.1 |
| 10% DMSO | 2.3 ± 0.2 | 9.53 | 1.5 | 2.0 ± 0.1 | 1.7 ± 0.1 |
| 20% DMSO | 11.9 ± 0.2 | 49.8 | 7.6 | 1.2 ± 0.1 | 1.4 ± 0.1 |
The values of R are in the same order of magnitude as the size of the oligomers according to the major semi-axis b (cf.Table 1). This is expected since both parameters are measures of the size of the individual oligomeric fibrils. However, the parameters have been determined independently from different parts of the model and the volumes of the fibrils as determined from R and a and b, respectively, do not exactly agree.
The parameter D follows the same pattern as R for the different solvents, i.e. it decreases with increasing size of added ion, or amount of DMSO. This means that the clusters of supramolecular networks become more compact in samples with larger individual fibrils.
Pramlintide in pure water
The scattering curves of pramlintide in water have a pronounced plateau in the mid q-region. The results of the model-fitting analysis show that the size of the oligomeric fibrils is between 2 and 3 pramlintide molecules per fibril, and parameter R is between 6–8 nm (cf.Fig. 2, Table 1 and Fig. S3, Table S1, ESI†). There is an indication that the SLS data levels out at low q, which might indicate smaller supramolecular network clusters as compared with the other samples. However, in absence of added electrolyte strong inter-aggregate interactions are expected to influence the scattering behaviour in the low q-regime, making it difficult to quantitatively determine the size of these clusters from our scattering data.Salts from the Hofmeister series
The scattering curves for the samples with pramlintide in 1 mM salt solutions appear different as compared to the ones with pramlintide dissolved in pure water (cf.Fig. 2). In our studied solutions, pramlintide is positively charged with acetate as counter-ion. Adding small amounts of salt may give rise to an exchange in counter-ions. According to the Hofmeister series, the three counterions SCN−, Cl−, and F− are expected to have different effects on protein aggregation.50 The scattering curve of pramlintide in 1 mM NaSCN has the least pronounced plateau regime, whereas 1 mM NaF appears similar to the scattering curve of pramlintide in pure water (cf.Fig. 2). Even at low concentrations of added salt, the scattering curves were affected, which indicates that the self-assembled structures formed by pramlintide are dependent on the type of added ion.The scattering curves of pramlintide in 1 mM NaSCN did not display the pronounced plateau seen in the case of water (cf.Fig. 2 and Fig. S4, ESI†). The fitted results also show that the parameter R is lower than for water, NaCl, and NaF, indicating that smaller oligomeric fibrils are found in NaSCN (cf.Table 1). According to the Hofmeister series, the SCN− ion increases the salting in-effect of peptides,50 which could explain why the size of the oligomeric fibrils is smaller than in 1 mM NaCl and 1 mM NaF.
The scattering curve of pramlintide in 1 mM NaCl fall between the curves for NaSCN and NaF (cf.Fig. 2), which is also reflected in the results from the model fitting analysis (cf.Table 1). Pramlintide in 1 mM NaF gives a scattering curve with the largest plateau in the intermediate q-region (cf.Fig. 2) and the largest size of the oligomeric fibrils, as compared with NaSCN and NaCl (cf.Table 1). Fluoride is described as a kosmotrope and, according to Hofmeister ion effects, it is expected to enhance peptide aggregation. A stronger ‘salting-out’ effect, could explain why pramlintide, in presence of fluoride as an added ion, forms larger oligomeric fibrils and a more compact structure of the supramolecular networks than the other ions.
The effect of the different salts on the structural behaviour of pramlintide in solution can be attributed to the different properties of the added ions. The Hofmeister effect states that the hydration of the different ions accounts for the different effects on proteins in solution.2 However, as identified in several studies,51–56 the explanation of the specific ion effects is much more complicated, and cannot be described only by the hydration of ions. However, the differences in appearance of the scattering curves can be discussed from the different sizes of the added ions. The molar masses of the added ions range from 19 g mol−1 for fluoride to 58 g mol−1 for thiocyanate. The results indicate that a large added ion promotes the formation of smaller oligomeric fibrils with larger interfacial curvature.
Dimethyl sulfoxide (DMSO)
Small amounts of dimethyl sulfoxide (DMSO) are usually added to sparingly soluble proteins and peptides to enhance the dissolving process and appear to have a significant influence on the structural behaviour of peptides.The plateau in the mid-q regime became less pronounced as an increasing amount of DMSO was added to a pure water pramlintide solution (cf.Fig. 3). The scattering curve of pramlintide in 1 wt% DMSO appears rather similar to that of pramlintide in water and NaF, but with a slightly less pronounced plateau, and slightly lower values of R. Only slight differences could be seen between the scattering curves of pramlintide in 1 wt% and 5 wt% DMSO, which did not yield any significant differences in the fitting results.
According to our results, the size of the oligomeric fibrils decreases with increasing amounts of DMSO up to about 10 wt%. For pramlintide in 20 wt% DMSO, the scattering curve appearance is conspicuously different as compared with the other solvents. The oligomeric fibrils were significantly larger as compared with the other samples (on average 7.6 pramlintide molecules in an oligomeric fibril), although the parameter R was found to be smaller than at lower DMSO concentrations. This might indicate a certain flexibility of the larger fibrils. These samples were also slightly opaque indicating that addition of large amounts of DMSO speeds up the aggregation process, and increases the size of the supramolecular network structures. Interestingly, the conspicuously larger size of the rod-like oligomers in this sample indicates a rapid growth of oligomers in the aggregation process as the supramolecular clusters are becoming bigger.
The reduction in size of the fibrils upon addition of small amounts of DMSO could be due to the DMSO molecules arranging around the peptide molecule, thus interacting with the hydrophobic elements of the peptide chain. With DMSO around the hydrophobic parts of the peptide, instead of water, the interfacial tension between peptide and surrounding water is expected to decrease, and the driving force for peptide-peptide aggregation must be lower.
The values of the parameter D follow a trend similar to R; highest value for water, and lowest for high amounts of DMSO (cf.Tables 1 and 2). This means that the supramolecular networks become less compact when small amounts of salt or DMSO are added to the solvent.
Dynamic light scattering (DLS)
Dynamic light scattering was measured at 90 degrees for 10 mg cm−3 pramlintide in all solvents except 1 mM NaF (cf. Fig. S10, ESI†). The average hydrodynamic radius falls in the range of 100–400 nm. However, the distribution of radii is wide and several micrometre large particles were also recorded. These particles, with wide distribution of particle sizes, correspond to the supramolecular network clusters. The clusters are big, but can likely be broken by shear stress, and form fragments of various sizes.Cryogenic transmission electron microscopy (cryo-TEM)
As a compliment to scattering techniques, we have also studied 10 mg cm−3 pramlintide in water and 1% DMSO with cryogenic transmission electron microscopy. However, it must be noted that in cryo-TEM the sample is prepared in a thin film with thickness of 100–500 nm. According to our DLS results the supramolecular structures are several hundred nanometres in diameter, that is in the same order of magnitude or larger than the thickness of the film. Hence, we do not expect the structures observed with cryo-TEM to be identical to what is observed with scattering techniques. Our studied systems are not equilibrium systems and we cannot exclude that the structures formed have been irreversibly affected as a result of the sample preparation process during the cryo-TEM measurements.A typical cryo-TEM image for the sample pramlintide in water is shown in Fig. S11 in the ESI.† The image indicates that the supramolecular structures consist of rather large and stiff elongated fibrils that have clustered together in a rather randomised fashion. The structure displayed in cryo-TEM is not entirely consistent with our SAXS data, indicating that the sample preparation procedure in cryo-TEM indeed influences the aggregate structures. The cluster formation of fibrils indeed supports the fractal dimensional structure observed with SAXS. However, while the cross-section dimensions of the fibrils agree fairly well with our results for spheroidal oligomers as observed with SAXS, the lengths of the fibrils observed in cryo-TEM are considerably larger than those found from our SAXS results. The discrepancy in fibril length observed with SAXS and cryo-TEM, respectively, may indicate that the aggregation process has accelerated as a result of the sample preparation procedure in the latter case.
Interestingly, some cryo-TEM images reveal a twisted structure of the fibrils indicating the structure of the oligomers where two or a few peptide molecules are twisted, probably to avoid direct contact between hydrophobic parts of the peptide and water.
Conclusions
The amyloid-forming peptide pramlintide aggregates in aqueous solutions to form supramolecular network clusters. The solutions are initially transparent when dissolving the peptide, but after 2–3 months the appearance of the samples changes. Network clusters grow in size with time and, eventually, phase separates into a gel-like dispersion. Adding large amounts of salt or dimethyl sulfoxide (DMSO) to pramlintide solutions speeds up the precipitation process.By employing a relatively simple model, we have been able to analyse SAXS data of transparent pramlintide solutions. In accordance with our model, pramlintide in aqueous solutions aggregates into small oligomeric fibrils that cluster together in loose supramolecular networks with fractal dimensions. As indicated by a combination of SAXS and cryo-TEM, the fibrils consist of two or more peptide molecules twisted around each other to avoid contact between hydrophobic parts of the peptide and water. Since hydrophobic parts of the peptides cannot be completely eliminated in the oligomeric structure, the fibrils become “sticky”, thus generating the supramolecular network structure. The supramolecular network structure built up by the oligomeric species is several hundred nanometres in size, as evidenced by SLS, and DLS.
The specific anion in small amounts of added salt has a significant impact on the size of oligomeric fibrils as well as the fractal dimension. The larger SCN− ion gives smaller fibrils and less compact networks whereas F− gives a more compact network with larger fractal dimension. We can conclude that DMSO significantly affects aggregation behaviour of pramlintide in solution. Low concentrations of DMSO (1–5 wt%) have a similar effect on the scattering profile as the addition of small amounts of NaCl. Adding 10 wt% DMSO reduces the fractal dimension, resulting in less compact network structures. At 20 wt% DMSO, the aggregation process speeds up and a slightly opaque solution with considerably longer fibrils is observed.
Oligomeric species of amyloid assemblies are considered the most toxic form of amyloids and investigating their structure becomes crucial for future research on the immunogenicity of peptide aggregates. This study provides a method to study the geometry of the oligomers and their supramolecular network and demonstrates the effect of different ions and DMSO on the structure of the networks.
Conflicts of interest
K. S. was at the time of work an employee of AstraZeneca.Acknowledgements
Small-angle X-ray scattering was performed on beamline B21 at Diamond Light Source, UK. This study is part of the science program of the Swedish Drug Delivery Center (SweDeliver) and financial support from Vinnova (Dnr 2019-00048) was provided.References
- K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122–128 CrossRef CAS PubMed.
- M. C. Manning, D. K. Chou, B. M. Murphy, R. W. Payne and D. S. Katayama, Pharm. Res., 2010, 27, 544–575 CrossRef PubMed.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- C. J. Roberts, J. Phys. Chem. B, 2003, 107, 1194–1207 CrossRef CAS.
- S. K. Maji, D. Schubert, C. Rivier, S. Lee, J. E. Rivier and R. Riek, PLoS Biol., 2008, 6, e17 CrossRef PubMed.
- N. Zhou, X. Gao, Y. Lv, J. Cheng, W. Zhou and K. Liu, J. Pept. Sci., 2014, 20, 868–875 CrossRef CAS.
- C. A. Gilroy, K. M. Luginbuhl and A. Chilkoti, J. Controlled Release, 2016, 240, 151–164 CrossRef CAS PubMed.
- W. F. Weiss, T. M. Young and C. J. Roberts, J. Pharm. Sci., 2009, 98, 1246–1277 CrossRef CAS.
- K. L. Zapadka, F. J. Becher, A. L. Gomes dos Santos and S. E. Jackson, Interface Focus, 2017, 7, 20170030 CrossRef PubMed.
- E. L. Opie, J. Exp. Med., 1901, 5, 527–540 CrossRef CAS PubMed.
- J. D. Sipe, M. D. Benson, J. N. Buxbaum, S. Ikeda, G. Merlini, M. J. M. Saraiva and P. Westermark, Amyloid, 2014, 21, 221–224 CrossRef.
- C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS PubMed.
- K. Blennow, M. J. de Leon and H. Zetterberg, Lancet, 2006, 368, 387–403 CrossRef CAS PubMed.
- W. Dauer and S. Przedborski, Neuron, 2003, 39, 889–909 CrossRef CAS.
- V. N. Uversky, Cell. Mol. Life Sci. CMLS, 2003, 60, 1852–1871 CrossRef CAS.
- P. Hammarström, X. Jiang, A. R. Hurshman, E. T. Powers and J. W. Kelly, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16427–16432 CrossRef PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS PubMed.
- K. D. Ratanji, J. P. Derrick, R. J. Dearman and I. Kimber, J. Immunotoxicol., 2014, 11, 99–109 CrossRef CAS PubMed.
- E. M. Moussa, J. P. Panchal, B. S. Moorthy, J. S. Blum, M. K. Joubert, L. O. Narhi and E. M. Topp, J. Pharm. Sci., 2016, 105, 417–430 CrossRef CAS PubMed.
- C. Bobo, S. Chaignepain, S. Henry, H. Vignaud, A. Améadan, C. Marchal, E. Prado, J. Doutch, J.-M. Schmitter, C. Nardin, S. Lecomte and C. Cullin, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 1168–1176 CrossRef CAS PubMed.
- S. Henry, H. Vignaud, C. Bobo, M. Decossas, O. Lambert, E. Harte, I. D. Alves, C. Cullin and S. Lecomte, Biomacromolecules, 2015, 16, 944–950 CrossRef CAS PubMed.
- K. Sörgjerd, T. Klingstedt, M. Lindgren, K. Kågedal and P. Hammarström, Biochem. Biophys. Res. Commun., 2008, 377, 1072–1078 CrossRef.
- M.-G. Choi, M. J. Kim, D.-G. Kim, R. Yu, Y.-N. Jang and W.-J. Oh, PLoS One, 2018, 13, e0195339 CrossRef PubMed.
- M. Vivoli Vega, R. Cascella, S. W. Chen, G. Fusco, A. De Simone, C. M. Dobson, C. Cecchi and F. Chiti, ACS Chem. Biol., 2019, 14, 1593–1600 CrossRef CAS PubMed.
- J. T. Jarrett and P. T. Lansbury, Cell, 1993, 73, 1055–1058 CrossRef CAS.
- A. M. Ruschak and A. D. Miranker, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12341–12346 CrossRef CAS PubMed.
- S. I. A. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 CrossRef CAS.
- E. T. Powers and D. L. Powers, Biophys. J., 2008, 94, 379–391 CrossRef CAS PubMed.
- E. Přáda Brichtová, M. Krupová, P. Bouř, V. Lindo, A. Gomes dos Santos and S. E. Jackson, Biophys. J., 2023, 122, 2475–2488 CrossRef PubMed.
- C. Betsholtz, L. Christmansson, U. Engström, F. Rorsman, V. Svensson, K. H. Johnson and P. Westermark, FEBS Lett., 1989, 251, 261–264 CrossRef CAS PubMed.
- P. Westermark, U. Engström, K. H. Johnson, G. T. Westermark and C. Betsholtz, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5036–5040 CrossRef CAS PubMed.
- D. C. da Silva, G. N. Fontes, L. C. S. Erthal and L. M. T. R. Lima, Biophys. Chem., 2016, 219, 1–8 CrossRef CAS PubMed.
- A. N. Roberts, B. Leighton, J. A. Todd, D. Cockburn, P. N. Schofield, R. Sutton, S. Holt, Y. Boyd, A. J. Day and E. A. Foot, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 9662–9666 CrossRef CAS PubMed.
- A. A. Young, W. Vine, B. R. Gedulin, R. Pittner, S. Janes, L. S. L. Gaeta, A. Percy, C. X. Moore, J. E. Koda, T. J. Rink and K. Beaumont, Drug Dev. Res., 1996, 37, 231–248 CrossRef CAS.
- M. M. Rosa and T. Dias, in Handbook of Clinical Neurology, ed. J. Biller and J. M. Ferro, Elsevier, 2014, vol. 120, pp. 809–824 Search PubMed.
- O. Schmitz, B. Brock and J. Rungby, Diabetes, 2004, 53, 233–238 CrossRef PubMed.
- E. Hartter, T. Svoboda, B. Ludvik, M. Schuller, B. Lell, E. Kuenburg, M. Brunnbauer, W. Woloszczuk and R. Prager, Diabetologia, 1991, 34, 52–54 CrossRef CAS PubMed.
- P. Westermark, A. Andersson and G. T. Westermark, Physiol. Rev., 2011, 91, 795–826 CrossRef CAS PubMed.
- P. A. Hollander, P. Levy, M. S. Fineman, D. G. Maggs, L. Z. Shen, S. A. Strobel, C. Weyer and O. G. Kolterman, Diabetes Care, 2003, 26, 784–790 CrossRef CAS PubMed.
- R. E. Ratner, L. L. Want, M. S. Fineman, M. J. Velte, J. A. Ruggles, A. Gottlieb, C. Weyer and O. G. Kolterman, Diabetes Technol. Ther., 2002, 4, 51–61 CrossRef CAS PubMed.
- M. C. Riddle, Diabetes Care, 2020, 43, 518–521 CrossRef PubMed.
- G. J. Ryan, L. J. Jobe and R. Martin, Clin. Ther., 2005, 27, 1500–1512 CrossRef CAS PubMed.
- R. A. Kenley, S. Tracht, A. Stepanenko, M. Townsend and J. L’Italien, AAPS PharmSciTech, 2000, 1, 1–7 Search PubMed.
- A. M. Morris, M. A. Watzky and R. G. Finke, Biochim. Biophys. Acta, Proteins Proteomics, 2009, 1794, 375–397 CrossRef CAS PubMed.
- B. Derjaguin and L. Landau, Acta Physicochim. URSS, 1941, 14, 633 Search PubMed.
- E. J. W. Verwey and J. T. G. Overbeek, Theory of the Stability of Lyophobic Colloids: The Interaction of Sol Particles Having an Electric Double Layer, Elsevier Publishing Company, 1948, pp. 1–205 Search PubMed.
- D. I. Svergun and M. H. J. Koch, Rep. Prog. Phys., 2003, 66, 1735 CrossRef CAS.
- S. Ciccariello, J. Goodisman and H. Brumberger, J. Appl. Crystallogr., 1988, 21, 117–128 CrossRef.
- N. M. Santa Fe, Oetoolkits, Oct version, Cadence Molecular Sciences, 2015.
- K. P. Gregory, G. R. Elliott, H. Robertson, A. Kumar, E. J. Wanless, G. B. Webber, V. S. J. Craig, G. G. Andersson and A. J. Page, Phys. Chem. Chem. Phys., 2022, 24, 12682–12718 RSC.
- S. Funkner, G. Niehues, D. A. Schmidt, M. Heyden, G. Schwaab, K. M. Callahan, D. J. Tobias and M. Havenith, J. Am. Chem. Soc., 2012, 134, 1030–1035 CrossRef CAS PubMed.
- A. W. Omta, M. F. Kropman, S. Woutersen and H. J. Bakker, Science, 2003, 301, 347–349 CrossRef CAS PubMed.
- J. Paterová, K. B. Rembert, J. Heyda, Y. Kurra, H. I. Okur, W. R. Liu, C. Hilty, P. S. Cremer and P. Jungwirth, J. Phys. Chem. B, 2013, 117, 8150–8158 CrossRef PubMed.
- W. Kunz, Curr. Opin. Colloid Interface Sci., 2010, 15, 34–39 CrossRef CAS.
- M. Boström, D. R. M. Williams and B. W. Ninham, Curr. Opin. Colloid Interface Sci., 2004, 9, 48–52 CrossRef.
- Y. Zhang and P. S. Cremer, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15249–15253 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm01172k |
| This journal is © The Royal Society of Chemistry 2024 |