
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInjectable liposome-containing click hydrogel microparticles for release of macromolecular cargos†
Luisa L
Palmese
 a,
Paige J.
LeValley
c,
Lina
Pradhan
c,
Amanda L.
Parsons
d,
John S.
Oakey
d,
Mathew
Abraham
e,
Suzanne M.
D’Addio
f,
April M.
Kloxin
*ac,
Yingkai
Liang
*f and
Kristi L.
Kiick
*ab
a,
Paige J.
LeValley
c,
Lina
Pradhan
c,
Amanda L.
Parsons
d,
John S.
Oakey
d,
Mathew
Abraham
e,
Suzanne M.
D’Addio
f,
April M.
Kloxin
*ac,
Yingkai
Liang
*f and
Kristi L.
Kiick
*ab
aMaterials Science and Engineering, University of Delaware, Newark, DE, USA. E-mail: akloxin@udel.edu; kiick@udel.edu
bBiomedical Engineering, University of Delaware, Newark, DE, USA
cChemical and Biomolecular Engineering, University of Delaware, Newark, DE, USA
dChemical and Biomedical Engineering, University of Wyoming, Laramie, WY, USA
eTranslational Imaging, Merck & Co., Inc., West Point, PA, USA
fDiscovery Pharmaceutical Sciences, Merck & Co., Inc., West Point, PA, USA. E-mail: yingkai.liang@merck.com
First published on 5th January 2024
Abstract
Hydrogel microparticles ranging from 0.1–100 μm, referred to as microgels, are attractive for biological applications afforded by their injectability and modularity, which allows facile delivery of mixed populations for tailored combinations of therapeutics. Significant efforts have been made to broaden methods for microgel production including via the materials and chemistries by which they are made. Via droplet-based-microfluidics we have established a method for producing click poly-(ethylene)-glycol (PEG)-based microgels with or without chemically crosslinked liposomes (lipo-microgels) through the Michael-type addition reaction between thiol and either vinyl-sulfone or maleimide groups. Unifom spherical microgels and lipo-microgels were generated with sizes of 74 ± 16 μm and 82 ± 25 μm, respectively, suggesting injectability that was further supported by rheological analyses. Super-resolution confocal microscopy was used to further verify the presence of liposomes within the lipo-microgels and determine their distribution. Atomic force microscopy (AFM) was conducted to compare the mechanical properties and network architecture of bulk hydrogels, microgels, and lipo-microgels. Further, encapsulation and release of model cargo (FITC-Dextran 5 kDa) and protein (equine myoglobin) showed sustained release for up to 3 weeks and retention of protein composition and secondary structure, indicating their ability to both protect and release cargos of interest.
Introduction
Hydrogels are attractive materials for incorporating and releasing functional molecules (e.g. proteins, peptides, growth factors) for targeted biological applications such as cancer treatment,1,2 wound healing,3 cell therapies,4 tissue regeneration,5,6 and vaccines.7 Over the past few decades, hydrogels with high protein/peptide loading capacity have been developed to exhibit tunable release profiles based on their material design. While bulk hydrogels can be tuned via crosslinking density, microstructure, and incorporation of reversible crosslinks, they are still limited in specific applications (e.g. applications where injectability and smaller sizes are required).8 In order to retain the properties of hydrogels that make them advantageous in biological applications while allowing for a more modular approach to the incorporation of therapeutic species, drug carriers formulated into microgels, nanogels, and nanoparticles have emerged as attractive strategies for the delivery of biologics.9–12Microgels are hydrogel particles ranging from 0.1–100 μm that are especially attractive for biological applications because of their injectability coupled with the ability to combine mixed populations of therapeutics for tailored combination delivery. Their size can also be tuned to either allow for retention at the injection site or circulation if desired. Several strategies have been employed to generate microgels, including batch emulsion, bulk fragmentation, and microfluidic methods.8 While each method provides unique benefits, the use of microfluidics to generate microgels is especially attractive for biological applications: preparation occurs under aqueous conditions without the need for organic solvents; size distributions can be tightly controlled; and the loading of sensitive cargo is enabled.13–16 Microfluidic approaches for generating polymeric microgels encompass both different methods of droplet formation, such as flow-focusing, T-junction, terrace-like, and co-flowing stream geometries, as well as different mechanisms of gelation following generation of the droplets, such as radical polymerization, phase transition, ionic cross-linking, and thermal or light based methods.17,18 The fabrication method, material, and chemistry are chosen to best suit the future application of the particles. To develop microgels suitable for drug delivery applications, significant efforts have been made to broaden methods for microgel production and increase the materials and chemistries by which they are made. Alginate, gelatin, hyaluronic acid, and poly(ethylene) glycol (PEG) have been used to synthesize microgels for applications including microcarriers for cell culture,19–22 generation of micro and macroporous annealed hydrogels,23–26 and encapsulation and delivery of proteins and nanoparticles.14,27 Click chemistries in combination with droplet microfluidics has become of interest due to both the high selectivity of biorthogonal reactions and the adaptability of microfluidic systems.28 Microfluidic methods have been successfully employed to yield PEG-microgels not only via radical polymerization, but also via Michael addition (as we have previously reported).29–31 This class of reactions allows for the generation of versatile biocompatible hydrophilic microgels that can be used for drug and protein delivery as well as cell encapsulation. Increasingly specialized hybrid microgels have been more recently explored, with the development of microgels incorporating nanoparticle carriers and nanomaterials.15,32–34 In particular, while liposomes have shown efficacious for delivery of cargoes of a range of sizes (e.g. small molecules, mRNA), there are limited accounts of their integration within injectable microgels.
In this work, we have established a method for producing both PEG-based microgels with and without chemically crosslinked liposomes (lipo-microgels) via droplet-based microfluidics methods that allow for the incorporation and release of model macromolecular cargoes.30 The microgels were formed via Michael-type addition between four-arm PEG-vinyl sulfone and four-arm PEG-thiol in 0.1x PBS upon mixing in a three-channel flow-focusing microfluidic device. After their formation, the microgels were characterized by brightfield and confocal microscopy, scanning electron microscopy, and atomic force microscopy in order to determine their morphology, dimensions, and Young's modulus, respectively. Microgel suspensions were evaluated via steady shear rheology to determine their shear thinning behavior and viscosity of relevance for injectability. The ability of both the microgels and lipo-microgels to carry and release cargo was evaluated by encapsulating and releasing 4K FITC-Dextran as a model drug; dextran release from both the microgels and lipo-microgels was characterized via quantitation of fluorescence over a 3-week period, as well as via confocal imaging of the microgels. Further, to demonstrate that both the microgels and lipo-microgels have the potential to deliver proteins without impacting their structure following release, myoglobin was loaded in microgels both freely and in a liposomal formulation. Following release, the protein was characterized via circular dichroism and electrospray ionization mass spectrometry to confirm maintenance of structure.
Materials and methods
Materials and general characterization
Thiol end-functionalized four-arm PEG (5 kDa, PEG-SH4) and vinylsulfone end-functionalized four-arm PEG (5 kDa, PEG-VS4) were purchased from JenKem Technology USA Inc. (Allen, TX). Fluorescent FITC-dextran (4 kDa) was purchased from Sigma-Aldrich (St. Louis, MO). All lipids were purchased from Avanti Polar Lipids (Alabaster, AL), including: 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DOPG), 1,2-dioleoylsn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidophenyl)butyramide] (MPB-PE), and 1,2-dioleoyl-sn-glycero-3-phosphocholine-N-(Cyanine 5) (Cy5-PC).Microfluidic device design and fabrication
Microfluidic devices were fabricated according to previously established protocols.30 Briefly, patterning of a silicon wafer (Wafer World, West Palm Beach, FL, USA) with SU-8 negative photoresist (Kayaku, Westborough, MA, USA) and UV contact photolithography was performed to create the flow-focusing device master mold. Microfluidic devices were fabricated with molded polydimethylsiloxane (PDMS) (Ellsworth Adhesives, Centennial, CO, USA). Briefly, PDMS mixed at a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (elastomer to curing agent) ratio, was poured over the wafer and degassed under vacuum for 20 min before being cured at 70 °C for 2.5 h. Devices were then cut and a sharpened 20G blunt syringe tip was used to punch out the inlets and outlet. Devices were then rinsed with ethanol, dried with pressurized air, and oxygen plasma treated for 30 s at ∼750 mtorr (Harrick Scientific, Ithaca, NY, USA) for bonding to flamed glass slides (Fisher Scientific, Waltham, MA, USA). Following bonding, devices were heat treated at 100 °C in an oven for 4 h to accelerate hydrophobicity recovery.
:1 (elastomer to curing agent) ratio, was poured over the wafer and degassed under vacuum for 20 min before being cured at 70 °C for 2.5 h. Devices were then cut and a sharpened 20G blunt syringe tip was used to punch out the inlets and outlet. Devices were then rinsed with ethanol, dried with pressurized air, and oxygen plasma treated for 30 s at ∼750 mtorr (Harrick Scientific, Ithaca, NY, USA) for bonding to flamed glass slides (Fisher Scientific, Waltham, MA, USA). Following bonding, devices were heat treated at 100 °C in an oven for 4 h to accelerate hydrophobicity recovery.
Liposome synthesis and characterization
Liposomes were prepared following the dehydration–rehydration method followed by extrusion through 200 nm and 100 nm polycarbonate membrane filters (Avanti Polar Lipids, Alabaster, AL).35,36 Briefly, DOPC, DOPG, and MPB-PE (maleimide functionalized lipid) were dissolved in chloroform at a molar ratio of 4:1:5. For fluorescently labeled liposomes 0.5% Cy5-PC or PE-TopFluor®AF594 was added, and the amount of DOPC or MPB-PE, respectively, was adjusted to accommodate this addition. A lipid film was formed via evaporation of the chloroform under N2 overnight. The film was rehydrated with 0.1x PBS pH 6 for unloaded liposomes and a 15 mg mL−1 solution of 4K FITC-dextran in 0.1x PBS pH 6 for loaded liposomes, for a final 10 mM lipid concentration, and sonicated for 5 minutes. Following sonication, the suspension was extruded through a 200 nm polycarbonate membrane 10 times and through a 100 nm polycarbonate membrane 15 times. The size distribution of liposomes was determined via dynamic light scattering (DLS) on a Zetasizer (Malvern, Worcestershire, UK). For analysis a scattering angle of 173° was used; the refractive index for the liposomes was assumed to be that of lipid (RI = 0.141); and automatic attenuation was used.
Formation of microgels using microfluidic devices
Microgels were formed using the above-described microfluidic device. The discontinuous, or aqueous, phase consisted of the PEG monomers and a buffer stream flowed in through three separate inlets, and the continuous phase consisted of 2 wt% 008-FluoroSurfactant in HFE7500 (RAN Biotechnologies, Beverly, MA). Three aqueous inlets were used to keep the monomer solutions separate until droplet formation to ensure the microfluidic device channels were not clogged due to premature gelation. The linear velocity of the aqueous streams is estimated to be 1 cm per second, based on a calculated flow rate of 2 μL per minute, which corresponds to a Reynolds number of 1 × 10−11 and Peclet number of ∼40000 during transit of the device. In this regime, both convective and diffusive mixing are negligible while the monomer streams are in contact in the device prior to droplet formation. All components were introduced into the microfluidic device through a 30-cm length of Tygon Microbore tubing (ID = 0.01′′). All aqueous streams were then added to the microfluidic device using Fluigent Flow Ez Pressure Pumps (Fluigent, North Chelmsford, MA) with a pressure set to 30 mbar. The oil stream was controlled using a positive displacement syringe pump (New Era Pump Systems Inc., Farmingdale, NY) at a flow rate of 10 μL min−1. After the flow of all streams was stabilized, the pressures of the three aqueous streams were slightly adjusted such that each component occupied a third of the channel cross section via visual inspection, indicating equal volumetric flow rates prior to droplet formation at the pinch-point where the oil is introduced (Fig. S1, ESI†). Following this point, chaotic advection in the droplets enhances mixing during their path through the device and tubing to the collection vial.37
For 10 wt% PEG microgels, 15 wt% PEG-SH4 was dissolved in 0.1x PBS pH 6 and 15 wt% PEG-VS4 was dissolved in 0.1x PBS pH 6. The center buffer channel was 0.1x PBS pH 6 or 30 mg mL−1 4K FITC-Dextran dissolved in 0.1x PBS pH 6. For lipo-microgels, PEG-SH4 and PEG-VS4 concentrations were adjusted to 15.5 wt% and 14.5 wt%, respectively, to account for the additional maleimide functionality of the liposomes (Fig. S1, ESI†). Both were dissolved in 10 mM liposome suspensions and the center buffer channel was 0.1x PBS at pH 6.
Following collection from the outlet stream, microgels were presumed to be partially-crosslinked based on bulk in situ rheology indicating a cross-over point of approximately 1 hour. To ensure all microgels were crosslinked at some level and prevent crosslinking between microgels, they were left on the bench for one hour in oil once collection was stopped. Excess oil was then removed and the microgels were resuspended in 1x PBS pH 7.4 to allow for complete polymerization overnight at RT. Subsequently, the microgels were isolated via centrifugation for 10 min at 1200 rcf, the PBS was removed and the microgels were washed with water one time to remove salt as well as oil. The washed microgels were then dried overnight under vacuum in a microcentrifuge tube which allowed for further removal of the oil. The microgels were resuspended in PBS at 20000 particles per mL. To determine the diameters of the microgels, an aliquot of the solution of the resuspended microgels was pipetted onto a microscope slide and imaged via brightfield or confocal microscopy (Zeiss LSM 800, Oberkochen, Germany). It should be noted that these parameters were optimized to reduce the potential for generation of aggregated particles. While largely successful in ameliorating these issues, sample aggregation did present in some syntheses. A representative image of aggregated particles is shown in Fig. S1 (ESI†). To estimate particle concentrations, the number of microgels in small volumes of sample was assessed; three 10 μL samples were analyzed using a hemocytometer to count individual microgels. Particle counts and particle diameters were determined using ImageJ.
Bulk hydrogel rheological characterization
Polymer pre-cursor solutions were prepared and pipette-mixed several times prior to applying 40 μL of the mixed solution to the Peltier plate of the rheometer. The in situ gelation process of 10 wt% total PEG-SH4 + PEG-VS4 hydrogels was monitored via dynamic oscillatory time sweeps using a cone and plate geometry at an angular frequency of 6 rad s−1, an amplitude of 1% strain, and a gap height of 50 μM on a stress-controlled AR-G2 or DHR3 rheometer for 2 hours (TA Instruments, New Castle, DE) to determine the gel point, plateau modulus, and polymerization time.Microgel injectability
To assess the injectability and quantify the shear-thinning properties of the microgel suspensions, steady shear viscosity was measured using a Discovery Hybrid Rheometer (DHR-3; TA Instruments, New Castle, DE) with a 20 mm diameter parallel geometry and Peltier plate. Samples were pre-sheared (10 seconds at 1 s−1) on the rheometer to ensure similar shear history before performing shear sweeps from 3–1000 s−1 at a gap height of 400 μM. The temperature was maintained at 26 °C and mineral oil was used to prevent evaporation. Microgel solutions were also injected through 27 gauge needles and imaged with bright field before and after injection to confirm microgel stability during injection.AFM characterization of microgels
To determine the elastic modulus of individual microgel particles, a Bruker Bioscope Catalyst Atomic Force Microscope (Billerica, Massachusetts) was used to acquire mechanical properties via indentation in accordance with methods we have previously reported.38 A 0.19 micron tall nitride cylindrical tip with a 1 μm probe combined with a low 0.25 N m−1 spring constant (Bruker, Billerica, Massachusetts) was used to assess microgel, lipo-microgel, and bulk hydrogel controls. For microgel and lipo-microgel samples, 10 μL of the respective suspension was pipetted on a positively charged glass slide and allowed to dry. Prior to measurement, 1x PBS was pipetted onto the slide and the microgels were allowed to swell overnight. For bulk hydrogel control samples, polymer precursor solutions were mixed and pipetted onto a positively charged slide fitted with a cylindrical silicone mold (d = 5 mm h = 1 mm). The resulting 10 wt% PEG bulk hydrogel was left to polymerize at room temperature in accordance with shear rheology data indicating the plateau modulus before being swollen in 1x PBS overnight. To visualize the microgels and accurately position the probe, the Bruker Catalyst AFM was mounted onto a Zeiss AxioObserver inverted microscope such that atomic force and brightfield microscopy could be performed simultaneously. Both the microgels and hydrogels were indented to a 5 nN threshold with a 2 μm ramp size and the elastic modulus was determined by fitting the retraction force curve data to the Hertzian Spherical Model using NanoScope Analysis software. Microgels were each indented in a 5 × 5 array for a total of 25 measurements per microgel and five microgels were tested per slide. Bulk hydrogels were treated similarly, by indenting a 5 × 5 array in 5 distinct regions of the gel.In vitro cargo release
Microgels containing FITC-dextran were re-suspended in 1x PBS at a concentration of 20000 microgels per mL at the start of the release experiment (t = 0). The microgel suspension (100 μL) was added to a 10000 MWCO dialysis cup and submerged in a vial containing 900 μL of 1x PBS (in triplicate). Samples were placed in a 37 °C incubator for the duration of the experiment. At each timepoint (1, 3, 6, 24, 48, 72, 120, and 168 hours), 200 μL of the release buffer was removed and replaced with 200 μL of fresh 1x PBS. Cumulative release profiles were generated using the following:where Vm and Vr indicate the amount of buffer taken out at each release timepoint and remaining volume of solution, respectively; C, is the concentration of the released species determined via fluorescence or absorbance; and i is the number of experimental timepoints.

Confocal microscopy
Microgels containing fluorescent species were visualized using a Zeiss LSM 800 confocal microscope with a 10× air objective. Microgels containing 10K FITC-Dextran were imaged using ex. 488 em. 517 nm and microgels containing Cy5 were imaged using ex.561 em. 670 nm. Super resolution confocal microscopy for visualization of AF594 tagged liposomes within microgels was conducted using a Zeiss LSM 880 microscope with a 40× water objective ex. 561 em. 595 nm. Data was collected using Zeiss AiryScan and deconvolved in Hyugens Professional 22.04.Statistical analysis
Data are expressed as the mean ± standard deviation unless otherwise indicated. Statistical significance was analyzed by performing a one-way ANOVA where p < 0.05 was considered statistically significant. If F test revealed statistical significance, pairwise comparisons were made using Tukey-HSD post-hoc. Statistical analysis was performed using JMP Pro 17 (SAS Institute Inc.).Results and discussion
Formation of PEG microgels and lipo-microgels
Thiol-Michael type addition has been used by ours and other groups for the rapid facile fabrication of bulk hydrogels, and more recently by our group, for the fabrication of microgels.30 Through use of the thiol-Michael type reaction between thiol and maleimide groups, we have previously demonstrated the generation of hybrid-hydrogel materials by incorporation of liposomes chemically crosslinked into bulk hydrogels for the tuned release of multiple therapeutic cargo molecules.35,36 Microgels offer several advantages compared to bulk hydrogels including both their injectability and their modular nature, which can be exploited to develop formulations with facile tuning of molecular payload. For example, the generation of hybrid microgels capable of the incorporation of hydrophobic cargo could serve as a modular delivery system capable of releasing both hydrophobic and hydrophilic cargo in a controlled manner. LeValley et al. demonstrated the ability of flow focusing microfluidics to form thiol-maleimide crosslinked microgels that were degradable in the presence of a reducing agent owing to the presence of degradable thiol-maleimide crosslinks (i.e., those formed with aryl-SH).30 While advantageous in many applications, the rapid gelation that results from the thiol-maleimide reaction has been difficult to employ in microfluidics approaches, as it can cause changes in viscosity and fluid flow that affect the crosslinked properties and uniformity of the microgels, resulting not only in heterogenous network formation but also blockage of the microfluidic channels. Because the rate of thiol-Michael type reactions can be adjusted via alterations in pH, buffer strength, and use of different electron withdrawing groups,39,40 we were able to identify suitable conditions for employing this reaction in flow-focusing microfluidic formation of PEG-based microgels crosslinked via thiol-Michael reaction. Specifically, in situ gelation of 5 kDa four-arm PEG-thiol (PEG-SH4) with 5 kDa four-arm PEG-maleimide (PEG-MI4), 5 kDa four-arm PEG-vinyl sulfone (PEG-VS4), or 5 kDa four-arm PEG-acrylate (PEG-Ac4) was characterized via oscillatory rheology for 10 wt% hydrogels in reduced buffer strength and pH (0.1x PBS pH 6). The gelation time of the hydrogels varied as expected, with PEG-MI4 resulting in the most rapidly achieved crossover point and PEG-Ac4 the slowest. Optimal gelation (crossover point after one hour) was achieved through use of PEG-VS4 (Fig. 1A) and was therefore the selection for all subsequent formulations described (Fig. S2, ESI†). The synthesis of microgels via PEG-SH4 and PEG-VS4 crosslinking is shown in (Fig. 1A–C). While microfluidic chip design closely follows the work done by LeValley et al., use of pressure-controlled pumps to monitor and control the flow of the polymer and buffer streams was implemented instead of positive displacement syringe pumps (Fig. 1B) for more responsive tuning of stream flow during microgel formation.30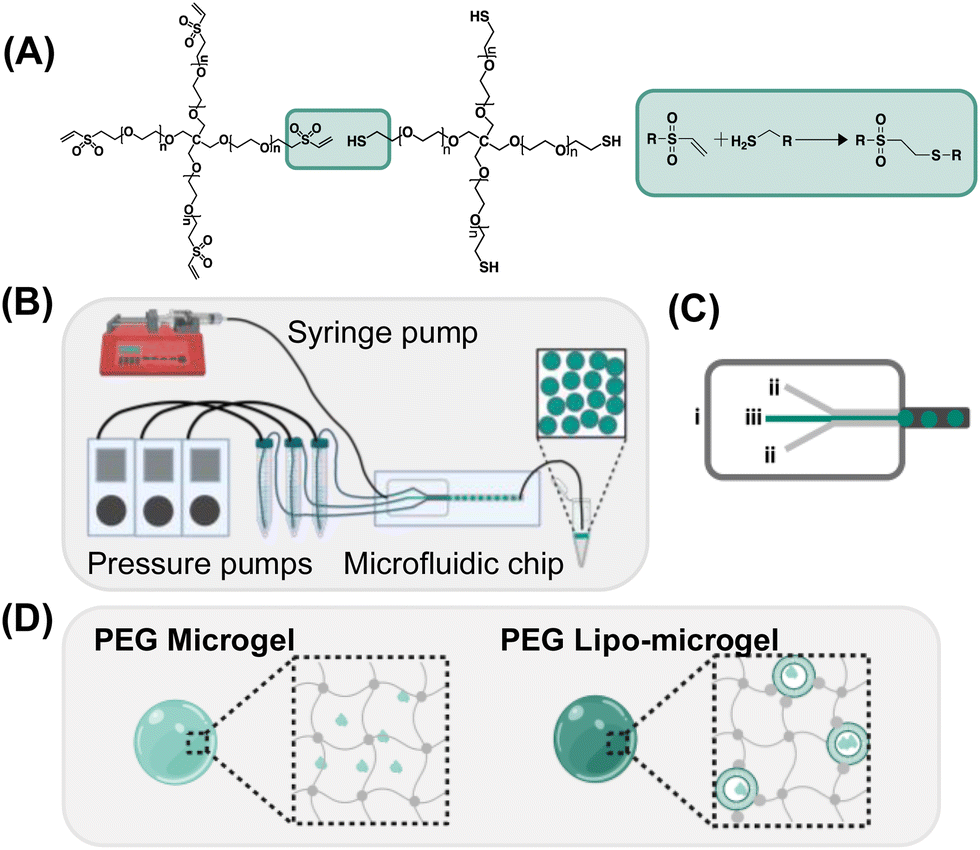 | ||
| Fig. 1 Overview of microgel fabrication (A) Thiol-Michael addition between PEG-4-vinyl-sulfone (5 kDa) (PEG-VS4) and PEG-4-thiol (5 kDa) (PEG-SH4) was used to form hydrogel networks. (B) Flow-focusing water-in-oil microfluidics was utilized to form monodisperse hydrogel microparticles (microgels). Pressure pumps control the flow rate of the hydrogel forming solutions entering the microfluidic device and a syringe pump controls the oil flow rate. (C) Design of microfluidic chip for formation of PEG microgels; (i) oil stream; (ii) PEG-SH4 and PEG-VS4 streams; (iii) buffer stream. (D) Schematic of PEG Microgels and PEG Lipo-microgels where grey depicts polymer and teal depicts FITC-dextran. | ||
Resulting microgels were monodisperse and displayed spherical morphology (Fig. 2A and Fig. S4, ESI†).
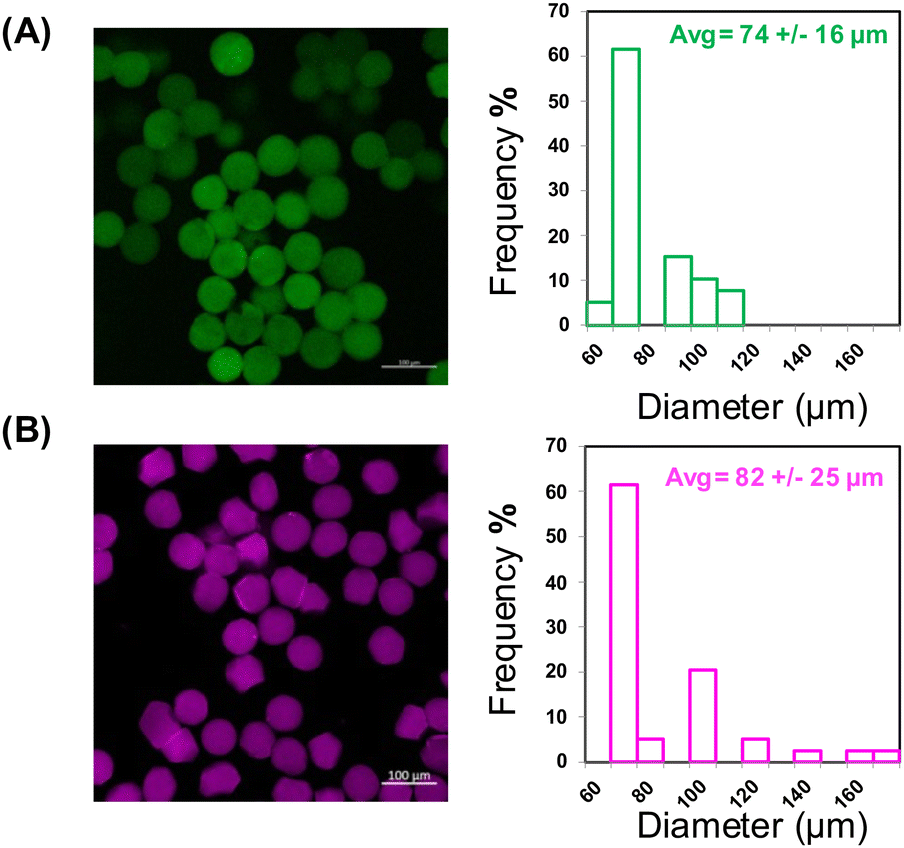 | ||
| Fig. 2 Formation of microgels and lipo-microgels (A) Confocal image of microgels containing FITC-Dextran and histogram of diameters (B) Confocal image of lipo-microgels containing Cy5-labeled liposomes and histogram of diameters. (Scale bar = 100 μm). | ||
Following the successful fabrication of 10 wt% PEG-SH4 PEG-VS4 microgels with diameters of 74 ± 16 μm, maleimide-functionalized liposomes (MI-liposomes) were formulated to permit their chemical crosslinking into the microgels at a (1:0.92:0.08) SH:VS:MI ratio, providing a secondary cargo-loaded carrier as well as a barrier that might slow the release of cargo loaded directly into the PEG network. Bulk oscillatory rheology was performed on the liposome-containing hydrogel which resulted in insignificant differences in gelation, modulus, and mesh size compared to the hydrogel alone (Fig. S2, ESI†). Unloaded and FITC-dextran-loaded liposomes exhibited diameters of 106 ± 1.8 nm (PDI 0.07) and 129 ± 0.86 nm (PDI 0.15) respectively, making them suitable for biological applications and incorporation into the microgels as shown by our previous incorporation of liposomes in bulk hydrogel formulations.35,36 Both PEG-SH4 and PEG-VS4 were dissolved in the MI-liposome suspensions prior to being introduced to the microfluidic device to form liposome-containing microgels (lipo-microgels). MI-liposomes were used in both polymer streams to allow for some pre-reaction to ensure liposome incorporation as well as increased liposome loading in the lipo-microgel without noticeable effects to viscosity due to pre-reaction with PEG-SH4. Liposomes containing a Cy5-labeled lipid were used to enable visualization via confocal microscopy of liposome incorporation in the crosslinked microgels (Fig. 2B). Images indicate incorporation of the liposomes in the microgels as well as a similar size distribution as that observed for microgels lacking liposomes, indicating that the inclusion of liposomes in the polymer streams does not significantly impact microfluidic processing. While the diameters of the microgels and lipo-microgels are similar, the confocal images of the fluorescently tagged lipo-microgels suggest an apparent increase in heterogeneity and change in morphology relative to the microgels alone (Fig. 2), although this difference is not noted in corresponding brightfield images (Fig. S3A and B, ESI†).
The suggested morphological differences between the microgels and lipo-microgels are likely thus a result of incorporation of and differences in the distribution of the fluorescently tagged liposome in the microgels. Super-resolution imaging was performed via Airyscan to acquire images of increased resolution of AF594-labled liposomes within the microgels (Fig. 3A and B). Liposome size and polydispersity was evaluated via DLS prior to incorporation in microgels (Fig. S5, ESI†). Images generated via deconvolution of the acquired data show liposomal species throughout the xy plane of the center of a single microgel (Fig. 3A). Similar observations were made for the yz and xz planes, while there is some spherical aberration due to imaging limitations (Fig. S5, ESI†). IMARIS 3D rendering of the liposomes within a microgel indicates an increased concentration of liposomes toward the core of the microgel particle compared to the external surface, consistent with the supposition that the liposome distribution throughout the microgels contribute to the apparent morphological differences indicated by fluorescence microscopy.
 | ||
| Fig. 3 Visualization of liposomes in microgels (A) xy plane obtained on LSM 880 from a z-stack of a microgel containing AF594 tagged liposomes (B) IMARIS 3D rendering of the microgel containing AF594 tagged liposomes. | ||
Mechanical properties of microgels and lipo-microgels
Swollen bulk hydrogel rheological measurements indicate that the storage modulus and mesh size of the 10 wt% hydrogels is 20.5 kPa and approximately 5 nm, respectively (Fig. S6, ESI†). Given our interest in the capability of these microgels to be used for the loading and release of peptides, proteins, and small molecules, the effect on mechanical properties of loading various amounts of relevant cargo was explored. Equine myoglobin was loaded at increasing concentrations in the bulk hydrogels; incorporation of 10, 25, and 50 mg mL−1 equine myoglobin was shown to slightly increase the storage modulus of the swollen bulk hydrogels to ∼25 kPa in all cases (insignificant) however the mesh size remained unchanged (Fig. S6, ESI†). While the mechanical properties of the microgels could be estimated via bulk rheology, the impact of possible differences in crosslinking and network formation during the microgel fabrication process (and the higher surface to volume ratio compared to bulk hydrogels)41 could not be assessed via bulk property measurements.In order to elucidate potential differences in mechanical properties arising between bulk hydrogels and the microgels, AFM was used to determine and compare the Young's moduli of bulk hydrogels, microgels, and lipo-microgels. Microgels were immobilized on positively charged slides and allowed to swell in 1x PBS to conduct quantitative nanomechanical mapping experiments. A bulk hydrogel was also fabricated using a silicone mold on a glass slide to serve as a control. The Young's moduli calculated via AFM for the bulk hydrogel, microgel, and lipo-microgel were 264 kPa, 173 kPa, and 153 kPa, respectively. The distribution of moduli in all cases is high, which has been reported for this method of testing.42,43 While oscillatory shear rheology is capable of assessing the complete mechanical properties of the sample, AFM provides spatial resolution of the microscale properties of the material.44 Due to the differences in material architecture at this scale, it is not surprising that high variability was observed. The Young's modulus determined for the bulk hydrogel is higher than that observed with shear rheology (E ≈ 3G′). Due to confinement by the silicone ring in the case of the bulk hydrogel, it is possible that equilibrium swelling was not reached. The equilibrium swollen modulus estimated via the measured modulus and equilibrium swelling following previous reports in our group was determined to be ∼10 kPa less than experimentally determined.45 While this difference is within the reported error, it is important to note and account for the potential effect of sample preparation on the result. These measurements were reproducible between hydrogels fabricated on different days, and the observation is consistent with other reports for PEG and PNIPAM hydrogels, in which AFM measurements yield higher modulus values compared to bulk shear rheology.44,46 These differences could be due to tip geometry and potential overstressing the material in the sample as well as model limitations for accurately representing adhesion forces.44,47 Due to these observed differences in experimentally calculated values, the Young's moduli of the microgels cannot be definitively reported; however, comparisons can be drawn. Compared to the bulk hydrogel, the Young's modulus of both the microgels and lipo-microgels is lower (Fig. 4A), which could be a result of differences in network formation in the bulk and microgels resulting from either the hydrogel geometry or differences in mixing in the microfluidic system compared to the pipette mixing used when generating bulk hydrogels. It should be noted that some reports indicate a non-negligible effect of local sample slope on modulus measurements obtained via AFM.48 This could partially account for differences in moduli, as the bulk gel control is cylindrical and the microgels are spherical. Regardless of the origins of the observed differences, the desired delivery mechanism as well as the size of the cargo are two parameters that are leveraged to engineer the crosslinking density of hydrogels which will ultimately determine the mesh size. Differences in network architecture between bulk hydrogels and microgels has an effect on the mesh size, which controls both diffusion and release of encapsulated species.49
 | ||
| Fig. 4 Mechanical properties of microgels and microgel suspensions (A) Young's modulus determined via atomic force microscopy for bulk hydrogels, lipo-microgels, and microgels (B) Young's modulus determined via atomic force microscopy of microgels stored dry at room temperature (Dry RT), in 1x PBS at 4 °C (Hydro 4 °C), and in 1x PBS at room temperature (Hydro RT) over 4 weeks. No significant differences observed in all cases. Scale bar represents the mean ± standard deviation n = 5. | ||
Therefore, to tune the release of molecules from microgels it is important to consider that a reduction in mechanical properties is typically an indication of increased mesh size, and thus could result in faster release of molecules incorporated within the network. Additionally, evaluation of the mechanical properties over time is instructive when considering the behavior of the microgels in various storage conditions. To investigate mechanical stability, microgels were stored dry at room temperature (swollen prior to measurement), in suspension at 4 °C, and in suspension at room temperature and tested via AFM after 4 weeks. The moduli of the microgels in each condition was compared to the initial modulus (Fig. 4B). While no statistical significance was determined for any of the storage conditions, it is notable that there is a decrease in the modulus of the microgels stored in aqueous suspension at both 4 °C (Hydro 4 °C) and room temperature (Hydro RT). While these hydrogels are designed to be nondegradable based on their building-block chemistry, over time in these conditions there is the potential for chain rearrangement leading to the observed decrease in modulus. In addition to the mechanical properties of individual microgels, the rheological behavior of the microgel suspension needs to be suitable for injectable delivery through clinically relevant needle gauges. Steady shear viscosity experiments were therefore conducted on suspensions of increasing particle concentration (20000 part per mL, 80000 part per mL, and 200000 part per mL) to establish feasibility of injection. For all concentrations tested, shear-thinning behavior was observed (Fig. 5A), although increasing particle concentration resulted in an increase in initial viscosity. At the highest tested shear rate (1000 s−1), all suspensions exhibited viscosities lower than 0.05 Pa s, which is the reported limit for subcutaneous injection via a 25–27 gauge syringe.50 Because the highest viscosity that can measured by common laboratory rheometers is low in comparison to the higher shear rates commonly associated with injection administration (∼100000 s−1),51 qualitative assessment of injectability was also performed. Suspensions at the highest concentration were injected with little force through a 27 gauge needle with no alterations to microgel size and shape (Fig. 5B). This behavior is in accordance with data indicating that both microgels suspensions and granular hydrogels exhibit desirable shear thinning behavior, making them attractive as translational injectable materials.24,52 Here, a 27 gauge needle was selected to ensure injectability as it has smaller inner diameter than needles typically used clinically (e.g. 22–25 gauge are used for intramuscular injection of vaccines, where higher gauge indicates a narrower needle with smaller opening and inner diameter for injection).53
 | ||
| Fig. 5 Injectability of microgel suspensions (A) Steady shear viscosity of microgel suspensions at increasing concentrations (B) Microgel diameter and brightfield images of trypan blue stained microgels before and after injection through a 27 G syringe. Data represents the mean ± standard deviation. (Scale bar = 100 μm). | ||
Molecule release from microgels and lipo-microgels
To demonstrate that both the microgels and lipo-microgels are capable of encapsulating and releasing cargo on the week-long timescale, a model release molecule, 4K-FITC-Dextran (FITC-Dex), was loaded into microgels and lipo-microgels and its release assessed. In the microgels, the 4K-FITC-Dextran was included in the center stream of the microfluidic device, whereas 4K-FITC-Dextran encapsulated in liposomes (63% encapsulation efficiency) was pre-mixed with the PEG polymer solution to allow for crosslinking between the maleimide groups on the liposomes and thiol groups on the PEG prior to microgel formation as we have previously reported.35 Following the synthesis of these microgels, cumulative release of the FITC-Dex from the microgels at 37 °C was determined (Fig. 6A). Encapsulation efficiencies based on theoretical loading via volumetric analysis were estimated as 65% for microgels and 89% for lipo-microgels. Over 3 weeks, the FITC-Dextran showed sustained release from both the microgels and lipo-microgels with minimal burst release observed. Mathematical fitting of the cumulative release (assuming complete release after 3 weeks in each case) with the Korsmeyer-Peppas model yielded a diffusional exponent (n) less than 0.45 for both microgels and lipo-microgels, indicating diffusion-controlled release (Fig. S7, ESI†). Our observations compare favorably with release in previously reported PEG microgels in which loaded fluoresceine-labeled immunoglobulin (F-IgG) was released over one week, and loaded nerve growth factor (NGF) released over three weeks.18,54 Compared to the microgels in our studies, lipo-microgels released less FITC-Dex over the 3-week period. While this could indicate increased retention in the lipo-microgels, we believe this to be unlikely as FITC-Dex liposomes alone released 100% of their cargo within 1 week (Fig. S8, ESI†). More likely, this is a result of lower cargo loading in the lipo-microgels, indicated by the lower observed fluorescence intensity of the lipo-microgel particles at Day 0 compared to the microgels (Fig. 6B). Differences in loading may be related to how the cargo is introduced in the microfluids (e.g. in the center stream for microgels and in each of the side streams for lipo-microgels, Fig. S1, ESI†), and ongoing work is targeted at determining the reason for these differences in future studies. Confocal microscopy of both microgels and lipo-microgels was performed over 24 h to provide further insight into the release of the labeled species over time. Fig. 6B shows the data collected over 1 hour where it is evident that there is a reduction in fluorescence intensity between both populations of microgels. Due to photobleaching, it was not possible to conduct informative experiments for longer timeframes. To confirm that the microgels exhibit the sustained release suggested by release experiments, we captured confocal images of aliquots of microgels during the duration of the release experiment to show they maintain fluorescence activity over 1 week (Fig. S9, ESI†). At late time points we observed punctate fluorescence in some samples (e.g. FITC-Dex 168 h). Based on similar observations previously made in our group, we believe this to be aggregated release material residing on the surface of the microgels.30 Additionally, preliminary investigation of the impact of injection on release kinetics was conducted to determine the effect, if any, of potential mechanical deformation of microgels when following injection through a 27 gauge needle on the release of FITC-DEX in both microgels and lipo-microgels. In both cases, no differences in release were noted over 1 week (Fig. S10, ESI†). Here, a narrower needle than that employed clinically again was used to ensure relevance for injectability. Note, the experiment as conducted did not investigate large irreversible mechanical deformation that may lead to liposomal dissociation during injection; such studies may be relevant in the future (e.g. if high concentrations of microgels need to be delivered by syringe injection for specific therapeutic applications). Increased particle concentration may increase mechanical deformation during injection, which could ultimately result in higher burst release following injection of more concentrated formulations. Therefore, further experiments are necessary to determine these effects as these new types of lipo-microgels are translated to applications. Overall, the ability to incorporate liposomal carriers within the microgel populations more greatly expands the library of potential delivery modalities. | ||
| Fig. 6 Model release (A) release of FITC-Dextran over 3 weeks from both microgels and lipo-microgels. Each data point represents the mean ± standard deviation for n = 3 (B) confocal microscopy of FITC-dextran microgels and lipo-microgels over 1 h. Scale bars = 100 μm. | ||
To demonstrate the capability of these hydrogels to deliver protein without deleteriously impacting protein structure, microgels loaded with 50 mg mL−1 equine myoglobin were synthesized following the same method previously described. Following their synthesis, microgels were incubated at 37 °C for one week and samples were collected at the specified intervals to generate a release profile (Fig. S11A, ESI†). Circular dichroism of the released myoglobin from the highest concentration sample collected (1 h) and freshly dissolved myoglobin at the same concentration (1 mg mL−1) was performed to compare the secondary structure (Fig. S11B, ESI†). While the spectra obtained from the released myoglobin shows slightly decreased helicity, the characteristic peaks remain, indicating that myoglobin retains secondary structure following release from the microgel.55 Determination of molecular weight of myoglobin before and after release indicates that the myoglobin remains in-tact following the loading and release from the microgels (Fig. S11C, ESI†). The ability to load and release protein from these microgels without impacting structure indicates that these fabrication methods and chemistries could prove helpful in loading compounds that suffer from low stability and require tailored buffer formulations. Our microgel synthesis method allows the molecule of interest to be stored and loaded at its optimal pH and salt concentration, which broadens the range of possible therapeutic targets. Note, myoglobin was loaded at 50 mg mL−1 as a model protein, whereas FITC-Dextran was loaded at 10 mg mL−1 as a model cargo similar in size to peptides; this higher loading concentration in addition to solubility differences between proteins and polysaccharides may account for differences in relative release kinetics, an important consideration as these microgel formulations are translated to therapeutic biomolecules. Additionally, while we were successful in generating lipo-microgels loaded with myoglobin, they were prone to aggregation following drying and resuspension (data not shown). As such, evaluation of the release of myoglobin from the lipo-microgel system would not be comparable to the release from particle suspensions. We speculate this could be related to non-specific interactions of protein not loaded with the liposomes with themselves or reactive species, another design consideration as these systems are translated for the release of specific bioactive cargoes.
Conclusions
In conclusion, we demonstrate the ability to generate microgels and lipo-microgels via the Michael-type addition of thiol and vinyl-sulfone functionalized PEG using a three-channel flow focusing microfluidic device, which also allows for the incorporation of cargo. Microgels and lipo-microgels were spherical and showed monomodal size distribution. Their moduli determined via AFM did not show statistical difference from that of bulk hydrogels, yet, did trend lower, which could indicate differences in network structure and heterogeneity. Microgels in suspension of increasing concentration showed shear-thinning behavior in all cases, and no damage to microgels was observed following injection through a 27 gauge syringe, indicating their injectability. FITC-Dextran was encapsulated and released as a model peptide cargo, and resulted in sustained release for up to 3 weeks. To assess the ability of the microgels to release in-tact protein cargo, myoglobin was loaded and released. The released myoglobin showed no difference in molecular weight or secondary structure following release. While all loading and release experiments detailed in this manuscript were conducted using model cargoes, future work will include the investigation of the loading, distribution, release, and subsequent activity of biologically relevant molecules. Additionally, in the future we aim to load and release proteins of different molecular weight and hydrodynamic radii to be able to mix these microgel populations for the combined and temporally resolved release of multiple protein species. In combination, microgels and lipo-microgels have the potential to improve the delivery of multiple therapeutics with different properties by allowing for the individual tuning of therapeutic encapsulation and release rate.Author contributions
April M. Kloxin, Kristi L. Kiick, Yingkai Liang and Luisa L. Palmese worked together to design and interpret experimental results. Luisa L. Palmese conducted all microgel synthesis and loading and release experiments and completed all figure and manuscript text drafts. Paige J. LeValley and Luisa L. Palmese helped develop microgel synthesis approaches and conduct fluorescence labelling and imaging experiments. Lina Pradhan conducted confocal microscopy experiments and processed corresponding data. Amanda L. Parsons and John S. Oakey constructed the microfluidic devices and consulted on microgel synthesis. Mathew Abraham and Suzanne M. D’Addio provided molecules for loading and release and imaging. April M. Kloxin, Yingkai Liang, and Kristi L. Kiick provided edits to all manuscript and figure drafts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The studies reported in this manuscript were supported by funding from Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA. Microscopy equipment employed in the studies was acquired with shared instrumentation grants (S10 RR027273 and S10 OD016361) and access was supported by the NIH-NIGMS (P20 GM103446), the NIGMS (P20 GM139760), and the State of Delaware. Additionally, the authors acknowledge the use of facilities and instrumentation supported by the National Science Foundation through the University of Delaware Materials Research Science and Engineering Center (DMR-2011824) and the National Institute of General Medical Sciences, part of the National Institutes of Health, through the Delaware COBRE (P20GM104316). The authors also acknowledge technical support from the Delaware Biotechnology Institute Bioimaging Center (Deborah Powell for SEM imaging and Timothy Chaya for super-resolution confocal imaging). Figures created using BioRender. The contents of this manuscript do not necessarily reflect the views of the funding agencies.Notes and references
- M. Sepantafar, R. Maheronnaghsh, H. Mohammadi, F. Radmanesh, M. M. Hasani-sadrabadi, M. Ebrahimi and H. Baharvand, Trends Biotechnol., 2017, 35, 1074–1087 CrossRef CAS PubMed.
- H. T. Ta, C. R. Dass and D. E. Dunstan, J. Controlled Release, 2008, 126, 205–216 CrossRef CAS PubMed.
- Y. Liang, J. He and B. Guo, ACS Nano, 2021, 15, 12687–12722 CrossRef CAS PubMed.
- D. Sivaraj, K. Chen, A. Chattopadhyay, D. Henn, W. Wu, C. Noishiki, N. J. Magbual, S. Mittal, A. M. Mermin-Bunnell, C. A. Bonham, A. A. Trotsyuk, J. A. Barrera, J. Padmanabhan, M. Januszyk and G. C. Gurtner, Front. Bioeng. Biotechnol., 2021, 9, 660145 CrossRef PubMed.
- S. Saravanan, S. Vimalraj, P. Thanikaivelan, S. Banudevi and G. Manivasagam, Int. J. Biol. Macromol., 2019, 121, 38–54 CrossRef CAS PubMed.
- S. Mantha, S. Pillai, P. Khayambashi, A. Upadhyay, Y. Zhang, O. Tao, H. M. Pham and S. D. Tran, Materials, 2019, 12, 3323 CrossRef CAS PubMed.
- G. A. Roth, E. C. Gale, M. Alcántara-Hernández, W. Luo, E. Axpe, R. Verma, Q. Yin, A. C. Yu, H. Lopez Hernandez, C. L. Maikawa, A. A. A. Smith, M. M. Davis, B. Pulendran, J. Idoyaga and E. A. Appel, ACS Cent. Sci., 2020, 6, 1800–1812 CrossRef CAS PubMed.
- A. C. Daly, L. Riley, T. Segura and J. A. Burdick, Nat. Rev. Mater., 2020, 5, 20–43 CrossRef CAS PubMed.
- J. K. Oh, D. I. Lee and J. M. Park, Prog. Polym. Sci., 2009, 34, 1261–1282 CrossRef CAS.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef CAS.
- M. Malmsten, H. Bysell and P. Hansson, Curr. Opin. Colloid Interface Sci., 2010, 15, 435–444 CrossRef CAS.
- H. Bysell, R. Månsson, P. Hansson and M. Malmsten, Adv. Drug Delivery Rev., 2011, 63, 1172–1185 CrossRef CAS PubMed.
- L. Zhang, L.-H. Cai, P. S. Lienemann, T. Rossow, I. Polenz, Q. Vallmajo-Martin, M. Ehrbar, H. Na, D. J. Mooney and D. A. Weitz, Angew. Chem., 2016, 128, 13668–13672 CrossRef.
- J. Pessi, H. A. Santos, I. Miroshnyk, J. Yliruusi, D. A. Weitz and S. Mirza, Int. J. Pharm., 2014, 472, 82–87 CrossRef CAS PubMed.
- J.-W. Kim, A. S. Utada, A. Fernández-Nieves, Z. Hu and D. A. Weitz, Angew. Chem., 2007, 119, 1851–1854 CrossRef.
- B. G. De Geest, J. P. Urbanski, T. Thorsen, J. Demeester and S. C. De Smedt, Langmuir, 2005, 21, 10275–10279 CrossRef CAS PubMed.
- E. Tumarkin and E. Kumacheva, Chem. Soc. Rev., 2009, 38, 2161–2168 RSC.
- C. H. Y. Chung, C. M. L. Lau, D. T. Sin, J. T. Chung, Y. Zhang, Y. Chau and S. Yao, ACS Appl. Bio Mater., 2021, 4, 6186–6194 CrossRef CAS PubMed.
- L. Zhang, K. Chen, H. Zhang, B. Pang, C.-H. Choi, A. S. Mao, H. Liao, S. Utech, D. J. Mooney, H. Wang and D. A. Weitz, Small, 2018, 14, 1702955 CrossRef PubMed.
- A. S. Mao, J.-W. Shin, S. Utech, H. Wang, O. Uzun, W. Li, M. Cooper, Y. Hu, L. Zhang, D. A. Weitz and D. J. Mooney, Nat. Mater., 2017, 16, 236–243 CrossRef CAS PubMed.
- Z. Jiang, B. Xia, R. McBride and J. Oakey, J. Mater. Chem. B, 2017, 5, 173–180 RSC.
- Z. Jiang, R. Shaha, R. McBride, K. Jiang, M. Tang, B. Xu, A. K. Goroncy, C. Frick and J. Oakey, Biofabrication, 2020, 12, 035006 CrossRef CAS PubMed.
- D. R. Griffin, W. M. Weaver, P. O. Scumpia, D. Di Carlo and T. Segura, Nat. Mater., 2015, 14, 737–744 CrossRef CAS PubMed.
- L. R. Nih, E. Sideris, S. T. Carmichael and T. Segura, Adv. Mater., 2017, 29, 1606471 CrossRef PubMed.
- A. C. Suturin, A. J. D. Krüger, K. Neidig, N. Klos, N. Dolfen, M. Bund, T. Gronemann, R. Sebers, A. Manukanc, G. Yazdani, Y. Kittel, D. Rommel, T. Haraszti, J. Köhler and L. De Laporte, Adv. Healthcare Mater., 2022, 11, 2200989 CrossRef CAS PubMed.
- S. Xin, O. M. Wyman and D. L. Alge, Adv. Healthcare Mater., 2018, 7, 1800160 CrossRef PubMed.
- L. Deveza, J. Ashoken, G. Castaneda, X. Tong, M. Keeney, L. H. Han and F. Yang, ACS Biomater. Sci. Eng., 2015, 1, 157–165 CrossRef CAS PubMed.
- T. Heida, O. Otto, D. Biedenweg, N. Hauck and J. Thiele, Polymers, 2020, 12, 1760 CrossRef CAS PubMed.
- C. Sonnet, C. L. Simpson, R. M. Olabisi, K. Sullivan, Z. Lazard, Z. Gugala, J. F. Peroni, J. M. Weh, A. R. Davis, J. L. West and E. A. Olmsted-Davis, J. Orthop. Res., 2013, 31, 1597–1604 CrossRef CAS PubMed.
- P. J. LeValley, A. L. Parsons, B. P. Sutherland, K. L. Kiick, J. S. Oakey and A. M. Kloxin, Pharmaceutics, 2022, 14, 1062 CrossRef CAS PubMed.
- G. A. Foster, D. M. Headen, C. González-García, M. Salmerón-Sánchez, H. Shirwan and A. J. García, Biomaterials, 2017, 113, 170–175 CrossRef CAS PubMed.
- M. van Elk, C. Lorenzato, B. Ozbakir, C. Oerlemans, G. Storm, F. Nijsen, R. Deckers, T. Vermonden and W. E. Hennink, Eur. Polym. J., 2015, 72, 620–631 CrossRef CAS.
- J. Yang, Y. Zhu, F. Wang, L. Deng, X. Xu and W. Cui, Chem. Eng. J., 2020, 400, 126004 CrossRef CAS.
- E. Dharmesh, S. Stealey, M. A. Salazar, D. Elbert and S. P. Zustiak, Front. Biomater. Sci., 2023, 2 Search PubMed.
- Y. Liang and K. L. Kiick, Biomacromolecules, 2016, 17, 601–614 CrossRef CAS PubMed.
- L. L. Palmese, M. Fan, R. A. Scott, H. Tan and K. L. Kiick, J. Biomater. Sci., Polym. Ed., 2021, 32, 635–656 CrossRef CAS PubMed.
- J. M. Ottino, S. R. Wiggins, M. R. Bringer, C. J. Gerdts, H. Song, J. D. Tice and R. F. Ismagilov, Philos. Trans. R. Soc., A, 2004, 362, 1087–1104 CrossRef PubMed.
- H. K. Lau, L. Li, A. K. Jurusik, C. R. Sabanayagam and K. L. Kiick, ACS Biomater. Sci. Eng., 2017, 3, 757–766 CrossRef CAS PubMed.
- N. J. Darling, Y.-S. Hung, S. Sharma and T. Segura, Biomaterials, 2016, 101, 199–206 CrossRef CAS PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- E. Battista, F. Causa and P. A. Netti, Gels, 2017, 3, 20 CrossRef PubMed.
- A. Burmistrova, M. Richter, M. Eisele, C. Üzüm and R. Von Klitzing, Polymers, 2011, 3, 1575–1590 CrossRef CAS.
- K. D. Nyberg, K. H. Hu, S. H. Kleinman, D. B. Khismatullin, M. J. Butte and A. C. Rowat, Biophys. J., 2017, 113, 1574–1584 CrossRef CAS PubMed.
- M. Galluzzi, C. S. Biswas, Y. Wu, Q. Wang, B. Du and F. J. Stadler, NPG Asia Mater., 2016, 8, e327–e327 CrossRef CAS.
- M. S. Rehmann, J. I. Luna, E. Maverakis and A. M. Kloxin, J. Biomed. Mater. Res. A, 2016, 104, 1162–1174 CrossRef CAS PubMed.
- C. J. G. Abrego, L. Dedroog, O. Deschaume, J. Wellens, A. Vananroye, M. P. Lettinga, J. Patterson and C. Bartic, Macromol. Chem. Phys., 2022, 223, 2100366 CrossRef CAS.
- M. E. Dokukin and I. Sokolov, Langmuir, 2012, 28, 16060–16071 CrossRef CAS PubMed.
- K. Heinze, O. Arnould, J. Y. Delenne, V. Lullien-Pellerin, M. Ramonda and M. George, Ultramicroscopy, 2018, 194, 78–88 CrossRef CAS PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS PubMed.
- M. A. Miller, J. D. Engstrom, B. S. Ludher and K. P. Johnston, Langmuir, 2010, 26, 1067–1074 CrossRef CAS PubMed.
- N. Rathore, P. Pranay, J. Bernacki, B. Eu, W. Ji and E. Walls, J. Pharm. Sci., 2012, 101, 4472–4480 CrossRef CAS PubMed.
- D. B. Emiroglu, A. Bekcic, D. Dranseikiene, X. Zhang, T. Zambelli, A. J. deMello and M. W. Tibbitt, Sci. Adv., 2022, 8, eadd8570 CrossRef CAS PubMed.
- H. S. Gill and M. R. Prausnitz, J. Diabetes Sci. Technol., 2007, 1, 725–729 CrossRef PubMed.
- J. Stukel, S. Thompson, L. Simon and R. Willits, J. Biomed. Mater. Res., Part A, 2015, 103, 604–613 CrossRef PubMed.
- N. A. Nicola, E. Minasian, C. A. Appleby and S. J. Leach, Biochemistry, 1975, 14, 5141–5149 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm01009k |
| This journal is © The Royal Society of Chemistry 2024 |