
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulating the cathode interface in sodium-beta alumina-based semi-solid-state sodium cells using liquid-organic electrolytes†
Micha P.
Fertig
 *ab,
Christof
Neumann
c,
Matthias
Schulz
b,
Andrey
Turchanin
c and
Michael
Stelter
ab
*ab,
Christof
Neumann
c,
Matthias
Schulz
b,
Andrey
Turchanin
c and
Michael
Stelter
ab
aCenter for Energy and Environmental Chemistry Jena (CEEC Jena), Friedrich Schiller University Jena, Philosophenweg 7a, 07743 Jena, Germany
bFraunhofer Institute for Ceramic Technologies and Systems IKTS, Michael-Faraday-Str. 1, 07629 Hermsdorf, Germany. E-mail: micha.philip.fertig@ikts.fraunhofer.de
cInstitute of Physical Chemistry, Center for Energy and Environmental Chemistry Jena (CEEC Jena), Friedrich Schiller University Jena, Lessingstraße 10, 07743 Jena, Germany
First published on 8th January 2024
Abstract
Solid-state sodium batteries using solid electrolytes have attracted attention as sustainable and powerful electrochemical energy storage systems. Even though the sodium-beta alumina solid electrolyte shows excellent properties, the limited interface contact with both electrodes has limited its wide utilization in room-temperature cell systems. However, interface modifiers can reduce the interfacial resistance, enabling good electrochemical performance. In this paper, we test four different liquid-organic electrolytes (LOE) to modulate the cathode interface. The effect of 1 M NaClO4 in propylene carbonate on the cell performance and the oxidic solid electrolyte was further investigated. We show that the LOE displacement, not the interphases, is the main reason for the impedance growth using a three-electrode cell, a setup rarely employed in solid-state batteries. The realized semi-solid-state sodium cells with modulated cathode interface achieve an area-specific resistance as low as 63 Ω cm2 and a high capacity retention of 99.8% after 100 cycles at 1.0C and 30 °C. Hence, the presented approach is a simple and cost-effective way to enhance the cathode interface in sodium-beta alumina-based cells.
1 Introduction
Solid-state sodium batteries (Na-SSB) are considered “post Li-ion” cell systems. SSBs promise higher specific energy and power due to bipolar stacking options and the usage of a high-capacity sodium metal (qth = 1166 mA h g−1) anode.1 Thanks to the inorganic solid electrolyte with high cation transference number, fast charging capabilities, and higher device safety are expected.2However, modulating the solid-state interface is critical for fabricating all-solid-state cells. Interface modifications enable intimate solid–solid interface contact, reducing interface impedance while facilitating charge transport and, thus, good electrochemical performance.3 Modulation is necessary for all material classes and includes many methods, e.g., compositional tuning,4 artificial solid electrolyte interphases (SEI),5 liquid interlayers,6 carbon coating,7 metal coating,8–10 and alloying.9,11–13 It especially applies to the interface between the polycrystalline Na+ conductor sodium-beta alumina solid electrolyte (BASE) and the negative electrode (NE, hereafter called the anode)14,15 as well as to the positive electrode (PE, hereafter called cathode).16 Several methods exist for modulating the BASE|PE interface, e.g., adding ionic liquids,17–19 polymers,20,21 or additional electrolytes.16
Small amounts of liquid-organic electrolytes (LOE) can also modify the interfaces. In general, LOEs used in Na-ion batteries rely on conducting salts (NaClO4, NaPF6, or NaSO3CF3) dissolved in linear carbonates, cyclic carbonates, or their mixtures. Additives are added to improve the electrolyte's properties because the LOE heavily influences the cell's electrochemical performance.22 For example, adding the film-forming additive fluoroethylene carbonate (FEC) to the organic electrolyte suppresses anomalous side reactions between the metal anode and the organic electrolyte.23–25 Beneficial as well as detrimental effects of FEC were reported for different cathode chemistries and operating temperatures. Nimkar et al. observed a significant improvement in cathode performance because surface films generated on a Na0.44MnO2 cathode blocked further electrolyte decomposition on the electrode surface.26 On the contrary, FEC formed a thick layer on a Na3(VO1−xPO4)2F1+2x cathode, deteriorating the rate performance, as He et al. have shown.27 Similar processes were discovered for a lithium spinel, where FEC dehydrofluorination had been accelerated by an elevated temperature of 55 °C, resulting in a thick surface layer and poorer cathode performance.28 Several groups have reviewed mechanisms and factors influencing surface film build-up, also called cathode electrolyte interphase (CEI), on Na-ion cathodes. The studies show that the influence of CEI depends on factors such as operating temperature, voltage range, electrolyte composition, cathode chemistry, and even anode chemistry.29,30
Hence, exploring LOEs to modify SSB's interfaces might be beneficial. The strategy has been successfully demonstrated in Li-SSBs, wetting either cathodic, anodic, or both interfaces.31–33 The modifications result in hybrid battery concepts, so-called “quasi-solid-state batteries”33 or “semi-solid-state batteries”.34 The LOE achieves low interface impedance by acting as a wetting agent, enabling intimate contact and facile intra-electrode conduction.2 It fills the solid electrolytes and electrode's pores, increasing the active area and lowering the electrode's impedance.34 Meanwhile, high specific energy metal anodes still can be used due to the inorganic electrolyte acting as an ion conductor and separator, shielding the LOE from the strong reductant. The lower flammable organic solvents make the approach safer than liquid electrolytes. It is expected to overcome the poor performance of inorganic solid electrolytes,33 approaching “the most feasible strategy”.34 For long-term operation, the solid electrolyte should be thermodynamically or at least kinetically stable against the LOE to prevent degradation and the growth of resistive interphases (solid–liquid electrolyte interphase, SLEI).2,35
In this study, we modulate the cathode interface in a sodium-beta alumina-based semi-solid-state sodium cell. Therefore, we wet the BASE|cathode interface with four commonly employed liquid-organic electrolyte (LOE) mixtures.22,36 We assessed the cycling performance of the cells using the LOE combinations in a two-electrode setup, choosing the most promising combination for further investigations. Its effect on the solid electrolyte's interface was studied using X-ray photoelectron spectroscopy to investigate the interphases post-mortem. We tracked the resistance contributions to the cell's internal resistance using a three-electrode setup, which is seldom employed in solid-state cells, elucidating that the LOE is displaced from the cathode interface during cycling. Hence, this work clarifies the effect of LOEs on the sodium-beta alumina solid electrolyte and the charge/discharge behavior. We demonstrate that using semi-solid-state electrolytes is a promising approach for sodium-beta alumina-based solid-state sodium batteries, resulting in a stable cycling performance at an operating temperature as low as 30 °C.
2 Methods
2.1. Materials
Na3V2(PO4)3 (NVP) was purchased from Gelon Lib. Poly(vinylidene fluoride) (PVDF) (Mw = 534![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) and carbon black (ENSACO® 350 G, Imerys S.A.) were used as binder and conductive carbon, respectively. N-Methyl-pyrrolidone (NMP) from Merck Schuchardt OHG (≥99.5%) was used as the solvent. Anhydrous NaClO4 (Alfa Aesar, 98–102%) and anhydrous NaPF6 (Thermo Fisher (Kandel) GmbH, ≥99%) were used as conducting salt and dissolved in anhydrous propylene carbonate (PC) (Acros Organics, 99.5%). Both conducting salts were thoroughly pestled and dried at 120 °C under a vacuum prior to usage. Fluoroethylene carbonate (FEC) from Sigma Aldrich Co. acted as an electrolyte additive when applicable. All electrolyte solutions were mixed and stored in a nitrogen-filled glovebox (GS ALPHA X-Line, GS Glovebox) with O2 and H2O < 1.5 ppm. We obtained the disk-shaped sodium-beta alumina solid electrolytes (BASE, d = 20 mm) following a solid-state reaction procedure described in previous work.37 Before application, BASE was thoroughly dried at 650 °C to remove superficially adsorbed residues.
000 g mol−1) and carbon black (ENSACO® 350 G, Imerys S.A.) were used as binder and conductive carbon, respectively. N-Methyl-pyrrolidone (NMP) from Merck Schuchardt OHG (≥99.5%) was used as the solvent. Anhydrous NaClO4 (Alfa Aesar, 98–102%) and anhydrous NaPF6 (Thermo Fisher (Kandel) GmbH, ≥99%) were used as conducting salt and dissolved in anhydrous propylene carbonate (PC) (Acros Organics, 99.5%). Both conducting salts were thoroughly pestled and dried at 120 °C under a vacuum prior to usage. Fluoroethylene carbonate (FEC) from Sigma Aldrich Co. acted as an electrolyte additive when applicable. All electrolyte solutions were mixed and stored in a nitrogen-filled glovebox (GS ALPHA X-Line, GS Glovebox) with O2 and H2O < 1.5 ppm. We obtained the disk-shaped sodium-beta alumina solid electrolytes (BASE, d = 20 mm) following a solid-state reaction procedure described in previous work.37 Before application, BASE was thoroughly dried at 650 °C to remove superficially adsorbed residues.
2.2. Materials characterization
Scanning electron microscope (SEM) images and energy-dispersive X-ray spectroscopy (EDX) scans were obtained from a Carl Zeiss' Ultra 55+ microscope. For microstructure analysis, BASE's fracture surface was polished water-free and thermally etched at 1500 °C for 30 minutes.XRD patterns were recorded using a Bruker D8 Advance diffractometer in the range of 5° to 90° 2θ and Cu Kα radiation of a pristine solid electrolyte. Quantitative XRD evaluation by the Rietveld refinement method was performed with Autoquan 2.8.0.2.
Using ten pristine BASE samples, the ball-on-three-balls method (Zwick 100, Zwick) examined the fracture strength.
X-ray photoelectron spectroscopy (XPS) and SEM were measured on pristine BASE and cycled BASE samples to investigate the surfaces and interphase build-up. The cycled samples were gathered from the cells after disassembling them inside the glovebox. Here, the surface contacting the cathodic side and LOE was investigated. XPS measurements were performed using a K-Alpha X-ray Photoelectron Spectrometer System (Thermo Fisher Scientific) with a monochromatic X-ray source (Al Kα) with a spot diameter of 400 μm and an electron detector with an energy resolution of 0.5 eV. A flood gun was used to compensate for the potential surface charge due to electrically insulating species. The spectra were calibrated using the C 1s peak (284.6 eV) and fitted using Voigt functions after background subtraction. In order to remove the topmost layer, Ar-ion sputtering was performed using the ion gun of the XPS system with an energy of 1000 eV.
2.3. Electrode fabrication, cell configuration, and electrochemical characterization
We prepared NVP cathodes by mixing NVP active material, conductive carbon, and 5% PVDF in NMP solution in a weight ratio 7:2:1 and adding additional NMP solvent to achieve a homogenous slurry. The slurry was doctor-bladed onto Al current collectors. The layer was dried in a vacuum oven at 70 °C. The 200 μm thick cathodes were punched out, resulting in cathodes with a diameter of 18 mm. The positive electrode mass is shown in Table S1.† The cathode was soaked with 1 M electrolyte before cell assembly (Table 1). Table S1† shows the amount added to the cathode. Na metal (99.8%, Acros Organics BVBA, Thermo Fisher Scientific) was used as anode. It was freshly cut from a Na bar, flat-rolled, punched out (d = 14 mm, ≈50 mg), and pressed onto BASE's surface by hand. No liquid electrolyte was added to the Na|BASE interface. Therefore, sodium was available in surplus. No oxidation layer was visible.
| Electrolyte abbreviation | Conducting salt | Solvent, additive |
|---|---|---|
| NaC | NaClO4 | PC |
| NaC–FEC | NaClO4 | PC:FEC 20:1 by volume |
| NaP | NaPF6 | PC |
| NaP–FEC | NaPF6 | PC:FEC 20:1 by volume |
All cells were assembled under a nitrogen atmosphere (O2, H2O < 1.5 ppm). A sodium-beta alumina solid electrolyte paired with two cathodes or two sodium anodes for symmetrical cells. Four probes two-electrode measurements were performed in half-cell configurations using sodium as the anode and NVP cathodes. A ring-shaped sodium reference electrode (EL-CELL GmbH) also contacted the solid electrolyte on the anodic side for the four probe three-electrode measurements. The counter and counter sense probes were connected to the counter electrode, the working and working sense to the working electrode, and the reference to the sodium reference ring. Hence, both half-cells were measured independently in addition to the full cell in a single experiment (Fig. S1†).
All electrochemical tests were performed in gas-tight measuring cells (PAT cell, EL-CELL GmbH), which were inserted in a temperature-controlled docking station and connected to a potentiostat (VMP-3 and SP240, Biologic). Galvanostatic cycling with potential limits (GCPL) was performed from 2.8–3.8 V at different C-rates, as shown in Table S1.† The retentions were calculated using the second cycle, i.e., the first full cycle in the applied voltage limits. The applied currents were calculated using 118 mA h g−1 as the theoretic specific capacity qth of the active material, assuming the V4+/V3+ redox couple and Na3V2(PO4)3 stoichiometry. Potentiostatic impedance spectroscopy (PEIS) was performed in the frequency range from 1 MHz to 100 mHz using 24 measurement points per decade while averaging two measurement points. A sinusoidal amplitude of 20 mV (30 °C) or 10 mV (80 °C) was applied.
3 Results and discussion
3.1. Characterization of the solid electrolyte
The oxidic, polycrystalline, Na+-conducting sodium-beta alumina solid electrolyte (BASE) was chosen to examine the effect of four different liquid-organic electrolytes (LOE) in a semi-solid-state cell. The BASE combines advantageous properties, e.g., good electrochemical performance,15 a wide electrochemical window,38 stability against metallic sodium,39 high mechanical stability, and abundant precursors. The fine-structured solid electrolyte with some grains bigger than 50 μm (Fig. 1a and S2†) exhibits a density ρ of 3.19 g cm−3 (98.2% ± 0.1%) measured with Archimedes' principle in toluene. An ionic conductivity σionic of 2.3 mS cm−1 and 11.1 mS cm−1 was measured in symmetrical Na cells at 30 °C and 80 °C, respectively. According to the Arrhenius-type equation, the coefficient of determination (R2 = 0.99) indicates a thermally-activated conduction process with an activation energy EA of 0.32 eV (Fig. 1a). The disks show a high phase purity of 96% for the well-conducting Na-β′′-alumina crystal phase40 (Fig. 1b), determined with Rietveld refinement. The remaining 4% are correlated to NaAlO2. NaAlO2 is a common impurity in BASE due to the solid-state fabrication process.41 The diffractogram is shown in detail in Fig. S3.† Using a symmetrical Na|BASE|Na setup, we obtain a critical current density of 4.79 mA cm−2 at 25 °C and 0.20 MPa stack pressure with a cycled capacity of 0.25 mA h cm−2 (Fig. 1c), underlining BASE's excellent capability as a solid electrolyte and validating previous results.37Fig. 1c displays an asymmetric voltage hysteresis. The asymmetry can be explained by different point-to-point physical contact between the sodium electrodes and the solid electrolyte. Interface modifications can improve it. For example, Liu's group improved the solid–solid contact using metallic surface coatings11 or interlayers,6 increasing the long-cycle performance and critical current density. The anodic polarization is likely due to the formation of voids and subsequent contact loss at the Na|BASE interface, as reported by Spencer Jolly et al.42 A fracture strength σ0 of 212 MPa is obtained (Fig. 1d).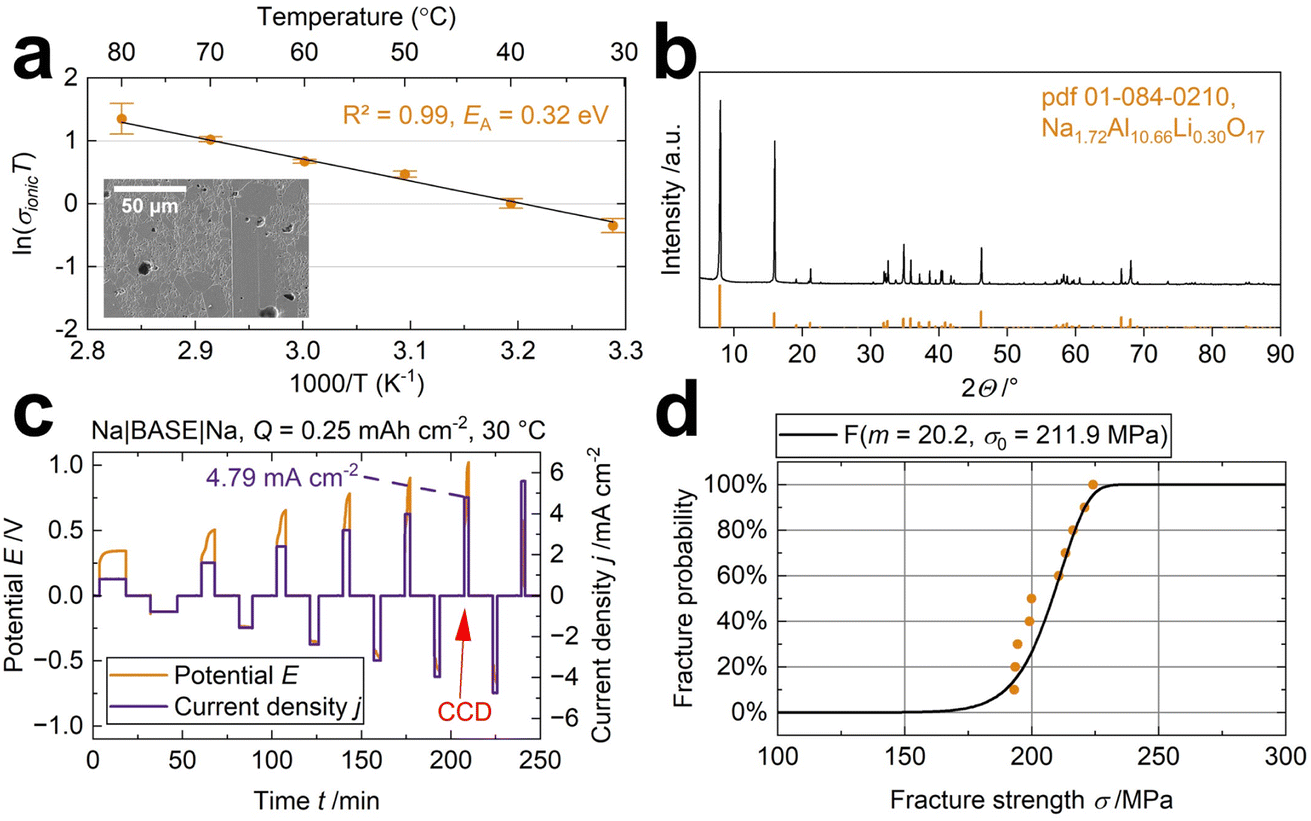 | ||
| Fig. 1 (a) The conductivity measured from 30 °C to 80 °C with Na electrodes show a thermally activated conduction according to the Arrhenius-type equation. The inset shows the specimen's microstructure, detailed in Fig. S2.† (b) The diffractogram of the specimen shows good agreement with literature peaks (pdf 01-084-0210). (c) A critical current density (CCD) of 4.79 mA cm−2 was measured for the specimen. The potential drop indicates the CCD. (d) The Weibull distribution of ten BASE specimens. | ||
3.2. Effect of LOEs on the cycling performance
The cycling performance of two-electrode cells was assessed at 80 °C and 0.3C. Four LOEs were added to modulate the interface between the sodium-beta alumina solid electrolyte (BASE) and the cathode (see Table 1). Fig. 2a shows the specific discharge capacity qsp and the coulombic efficiency of the four cells over 50 cycles. A specific discharge capacity of 101.0 mA h g−1 is achieved for the cell using 1 M NaClO4 without FEC additive (NaC), corresponding to 86% of the active material's theoretical capacity of 118 mA h g−1. After 50 cycles, 97.2% of the initial specific discharge capacity qsp is retained (cell 1, Table S1†). The stable cycling performance of the NaC system is underlined by the voltage profiles with a broad discharge plateau at 3.35 V. The discharge plateau is only 20 mV lower than the charge plateau (Fig. 2b), correlating to the low overvoltage shown in the Nyquist plot in Fig. S4.† However, Fig. S4† reveals that the internal resistance doubles from 40 Ω cm2 in the first cycle to 80 Ω cm2 in the 50th cycle.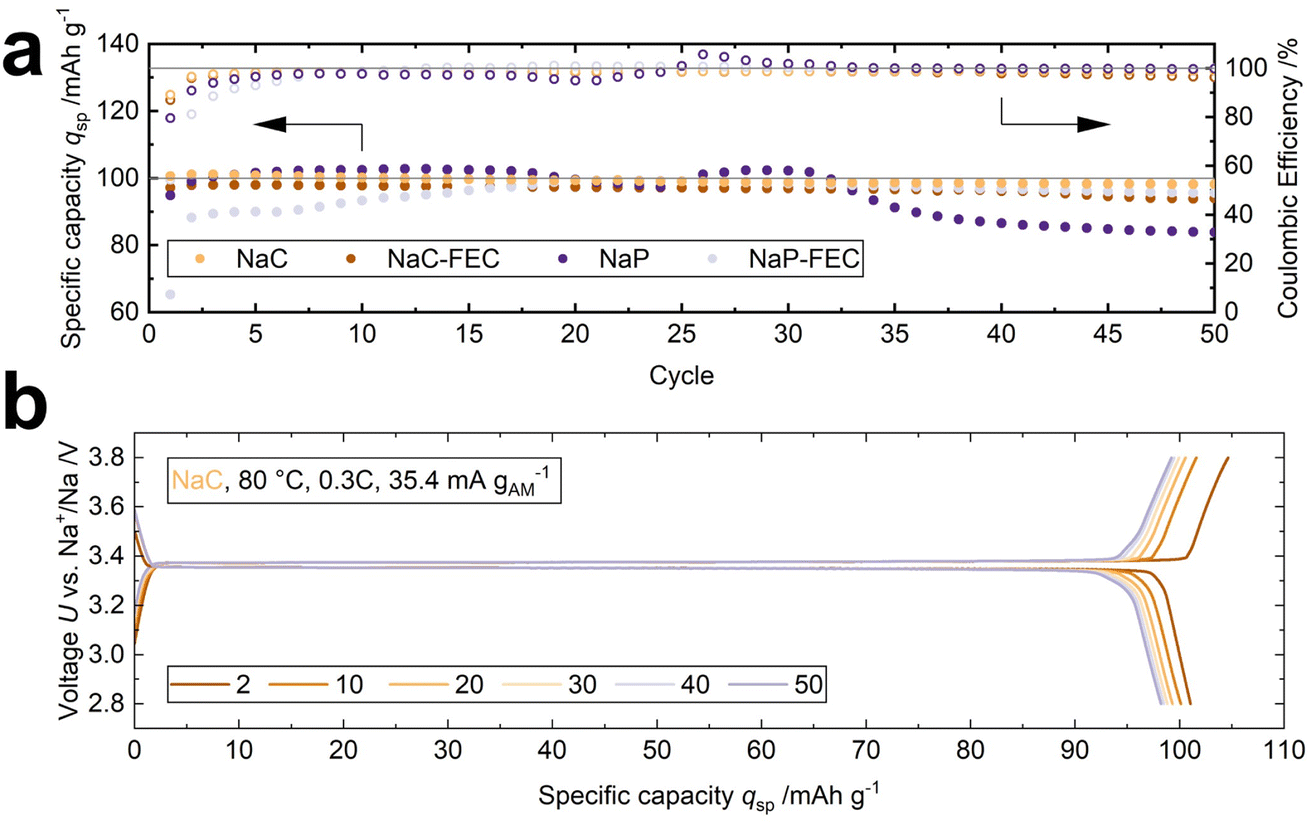 | ||
| Fig. 2 (a) Cycling performance of the cells with four different liquid-organic electrolytes (LOE) at 80 °C and 0.3C. (b) The voltage profile of the NaC cell shows an overvoltage of 20 mV. | ||
Furthermore, the NaC modification shows the best rate capability with a discharge capacity of 96.3 mA h g−1 at 2.0C, while it is 84.8 mA h g−1, 82.0 mA h g−1, and 38.9 mA h g−1 for the NaC–FEC, NaP–FEC, and NaP system, respectively (Fig. S5†).
Hence, while all cells can be charged and discharged, the cells using NaClO4 in the LOE show superior performance, i.e., a more stable cycling behavior, compared to those using NaPF6. This result contradicts findings where NaPF6 was superior to NaClO4.27,43 He et al. attributed the superiority to the “incompatibility and instability of NaClO4” salt with the high potential NVPF cathode,27 which is absent in this work. Furthermore, Ponrouch et al. proved the stability of the NaC system in the given voltage range from 2.2–3.6 V vs. Na+/Na.44 These results suggest that the electrolyte composition must be adapted to the respective system, i.e., solid electrolyte and cathode chemistry, and other parameters, e.g., operating temperature and voltage range.
FEC addition showed no advantage over the pure NaC electrolyte. Although the NaC–FEC cell also shows stable cycling behavior, the discharge capacity is lower than with NaC. The result is in line with observations from He et al. Here, FEC addition formed a cathode surface layer due to its decomposition, enhancing the transfer resistance.27 On the contrary, adding FEC affected the NaP system beneficially. While the specific capacity starts at only 65 mA h g−1 and gradually increases, the cell's capacity retention improves. This observation aligns with findings from Cheng et al., who reported a beneficial effect on the retention by the simultaneous presence of FEC and PF6−.43 However, the corresponding low coulombic efficiency indicates side reactions initially.45 The different effects of FEC on the NaC and NaP system can be explained by a higher presence of fluorinated molecules in the NaP–FEC system, enabling a richer surface chemistry, as described by Aurbach's group.26 Hence, FEC addition affected the NaC and NaP systems in multidirectional ways but showed no advantage over pure NaC in the Na|BASE|LOE|NVP system at 80 °C.
Due to the promising electrochemical performance of the NaC system, we chose to study it in more detail with scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), and three-electrode measurements to clarify the origin of the resistance growth and investigate whether BASE is stable in contact with the LOE.
3.3. Post-cycling analysis of the NaC system
The pristine and cycled cathode's surface was investigated using scanning electron microscopy (SEM). The active material particles of the pristine cathode have a smooth surface, as shown in the center of Fig. 3a. They are embedded in a matrix of smaller active material particles and carbon. Fig. 3b reveals some cracks in the active material's particles after cycling, which is also true for the cycled cathode surface. On the contrary, the pristine cathode in Fig. 3c appears more homogeneous, displaying smaller particles and fewer cracks than the cycled cathode, shown in Fig. 3d.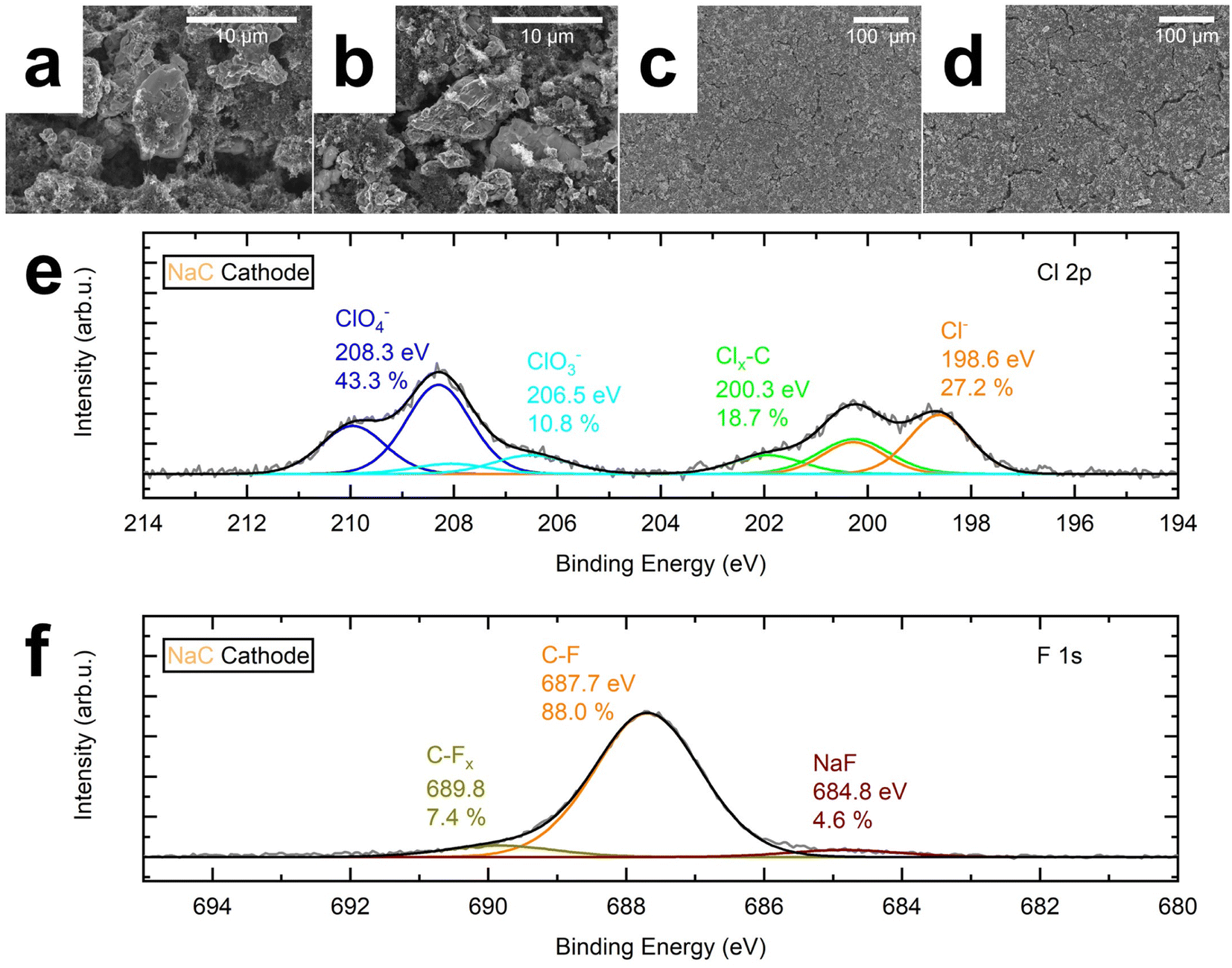 | ||
| Fig. 3 SEM and XPS analysis of the NVP cathodes. (a and c) SEM image of the pristine cathode (b and d) post-mortem SEM image of a cycled NaC cathode. (e) Post-mortem XP spectrum of a cycled NaC cathode's Cl 2p core level. (f) Post-mortem XP spectrum of a cycled NaC cathode's F 1s core level. | ||
We used X-ray photoelectron spectroscopy (XPS) to investigate whether these macroscopic properties translate into a difference in surface composition. The cycled cathode contains higher Na, Cl, and O fractions than the pristine cathode (Table S2†). This result is evident because these elements form the conducting salt NaClO4, added for the cell test. However, the XPS analysis further discloses that the surface species differ mainly due to Cl species on the cycled cathode, as Cl had been absent before cycling (Table S3†). The Cl 2p core level spectrum of the cycled cathode reveals the presence of NaCl at 198.6 eV, C–Clx at 200.3 eV, ClO3− at 206.5 eV, and ClO4− at 208.3 eV on the surface of the cycled cathode (Fig. 3e). ClO4− dominates the spectrum with 43.3%, followed by NaCl (27.2%), C–Clx (18.7%), and ClO3− (10.8%). C–F species related to the PVDF binder dominated the F 1s spectrum at 687.7 eV. Only 4.6% NaF, a common inorganic halide component responsible for CEI formation,29,30,43 was detected at 684.8 eV (Fig. 3f). We assume that the small amounts of film-forming halide species in the NaC system (0.35 at% NaCl, 0.28 at% NaF) result in a thin CEI with little resistance. It induces a low overvoltage and a broad voltage range for cycling and, hence, a higher specific capacity than the fluoride-containing NaC–FEC, NaP, and NaP–FEC mixtures.
SEM imaging was also used to analyze BASE's pristine surface and BASE's surface after cell operation to investigate interphase build-up, which can contribute to resistance increase.2,35 Here, we analyzed the cathode side of the solid electrolyte after removing both electrodes.
While the pristine surface in Fig. 4a appears to be smooth beside some edges, steps, and pores, the surface in Fig. 4b does appear curled and corrugated compared to the original state. Additionally, most pores are no longer visible. Therefore, the solid electrolytes were examined by XPS to investigate the surface composition. The survey spectrum of the cycled BASE, which had been in contact with NaC and the cathode, reveals that the surface composition is dominated by C, O, and Al (Fig. S6a†). Atomic fractions of the components are 12.4%, 56.7%, and 23.2%, respectively (Table S4†). The presence of atomic fractions of 4.4% Na and 1.6% Cl is expected, as Na is part of the solid electrolyte, while Na and Cl form the conducting salt NaClO4. The presence of carbon, illustrated in Fig. S6b,† stems partly from superficially adsorbed carbon species. Adventitious carbon species (C–C/C–H) are detected at 284.6 eV. Oxidized carbon species are detected at higher binding energies of 285.2 eV (C–O), 286.5 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and 289.0 eV (COO−/C–F). Additional species stem from cathodic residues of the conductive carbon and the binder. No CO32− is present, indicated by the absence of a peak at ≈290–291 eV, underlining the approach of reducing superficially adsorbed carbon species by heat treatment.46 Additionally, remains of the cathode, which had been peeled from the BASE, were found on the surface, including C–F species from the PVDF binder found at 687.8 eV and POx from the active material at 133.6 eV. A peak at 685.5 eV hints at the presence of a second fluoride species, likely NaF (Fig. S6c and d†). Compared to the pristine BASE sample, where 1.4% NaF had been detected, it is reduced to 0.7% in the cycled sample.
O), and 289.0 eV (COO−/C–F). Additional species stem from cathodic residues of the conductive carbon and the binder. No CO32− is present, indicated by the absence of a peak at ≈290–291 eV, underlining the approach of reducing superficially adsorbed carbon species by heat treatment.46 Additionally, remains of the cathode, which had been peeled from the BASE, were found on the surface, including C–F species from the PVDF binder found at 687.8 eV and POx from the active material at 133.6 eV. A peak at 685.5 eV hints at the presence of a second fluoride species, likely NaF (Fig. S6c and d†). Compared to the pristine BASE sample, where 1.4% NaF had been detected, it is reduced to 0.7% in the cycled sample.
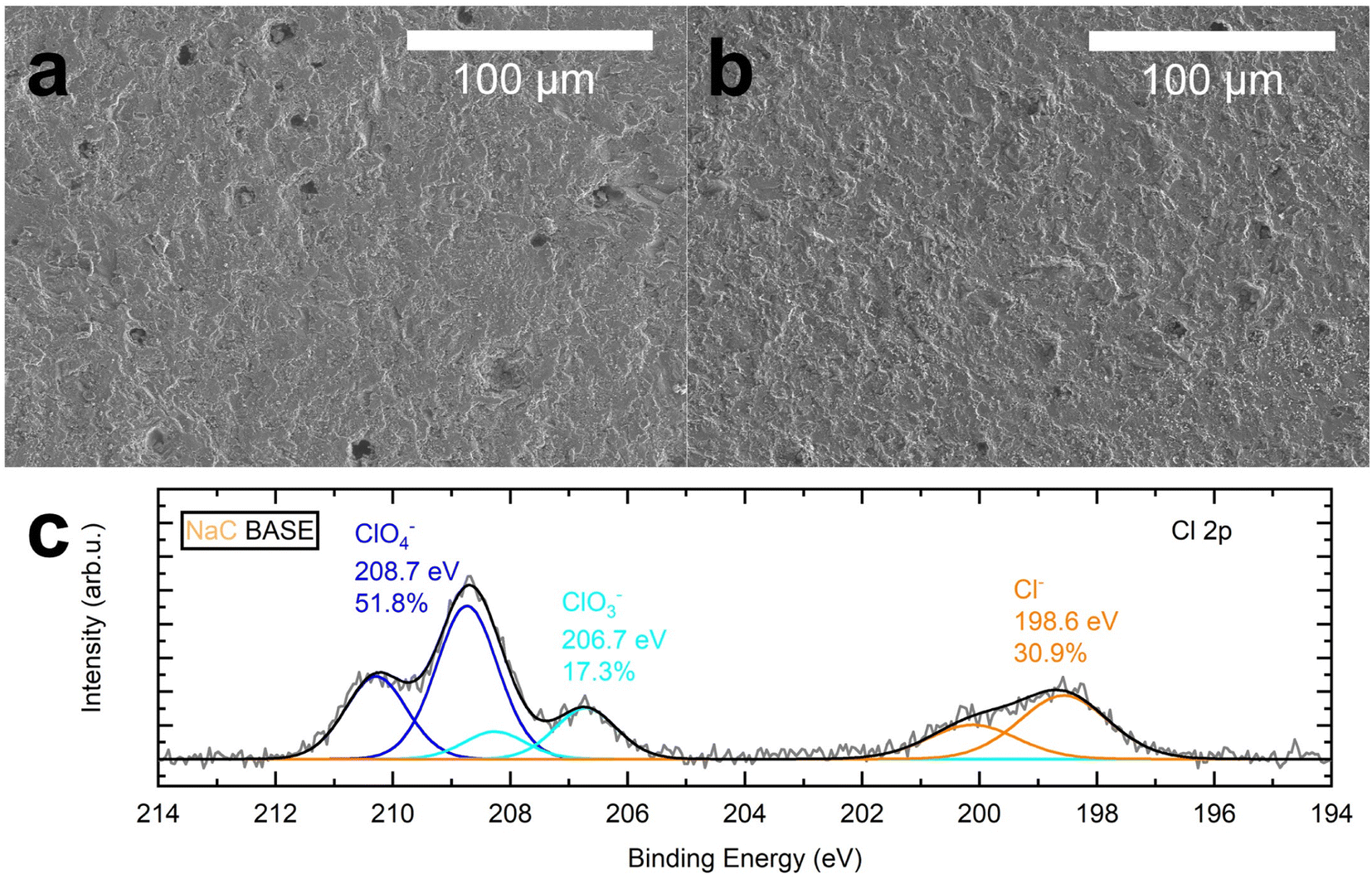 | ||
| Fig. 4 SEM and XPS analysis of the sodium-beta alumina solid electrolyte (BASE). (a) SEM image of a pristine BASE surface. (b) Post-mortem SEM image of a BASE that had been in contact with NaC and the cathode. (c) Post-mortem XP spectrum of BASE's Cl 2p core level. The BASE had been in contact with NaC and the cathode. | ||
The spectrum of the Cl 2p core level in Fig. 3c shows the presence of three Cl species after cycling: chlorides Cl−, likely NaCl or AlCl3, chlorates ClO3−, and perchlorates ClO4− as the main component at 198.6 eV, 206.7 eV, and 208.7 eV, respectively. The spectrum is dominated by the peak attributed to the conducting salt's perchlorate anion, while the chlorate species ClO3− can be seen at a lower binding energy. We determined the proportion of the chlorine species Cl−, ClO4−, and ClO3−, with 30.9%, 51.8%, and 17.3%, respectively. Hence, the atomic fraction of Cl− after cycling is 0.5% compared to 0.2% in the pristine sample, where only the Cl− species had been detected. ClO3− is surprising because NaClO4 in PC is known to be stable in the given voltage range.44 We do not think the species arose due to photoinduced decomposition from the XPS radiation source,47 as no progressive decomposition had been observed in the measurement.
After Ar-ion sputtering, C, Cl, F, and Na levels decrease, and the absence of P is observed, underlining that these elements are present at the surface only. On the contrary, O, Al, and Li increase after sputtering, indicating that these elements had been covered before (Table S4†).
Summarized, SEM imaging and XPS reveal differences between the surfaces of the pristine BASE and the cycled BASE. Positive electrode remains were found on the cycled electrolyte. Additionally, we observed perchlorate and chlorate, which had been absent on the pristine surface. On the contrary, the sodium halogens NaCl and NaF, typical components of interphase layers in sodium cells,48 were found both in the pristine and the cycled BASE samples. While the Cl− levels were elevated on the cycled surface, the F− levels were decreased. In both cases, no CO32− was detected, which occurs often as sodium carbonate in inorganic interphases.48 Therefore, the XPS measurements underline the SEM observation, indicating inorganic interphase build-up to a certain degree. Busche et al. observed the formation of a solid–liquid electrolyte interphase when investigating oxidic Li+ conductors, assuming that any ionic compound will dissolve in the organic electrolyte to a certain extent.35 On the contrary, propylene carbonate contacted BASE in several works.49,50 None of them reported a direct effect of PC to BASE. Will, Mitoff, and Farrington observed a change in electrical resistivity due to an ion exchange. Contrary to interphases, they assumed that the increase in resistivity stems from water impurities in the organic solvent,51,52 an effect we recently confirmed for humid atmospheres.37
3.4. Monitoring resistance evolutions using three-electrode measurements
We used a three-electrode cell with a sodium anode and sodium reference to observe resistance changes at the cathode (working electrode) in situ. Comparing the three-electrode spectrum in Fig. 5a with the two-electrode spectrum in Fig. S4† reveals that both setups yield comparable spectra, validating the employed experimental setup.53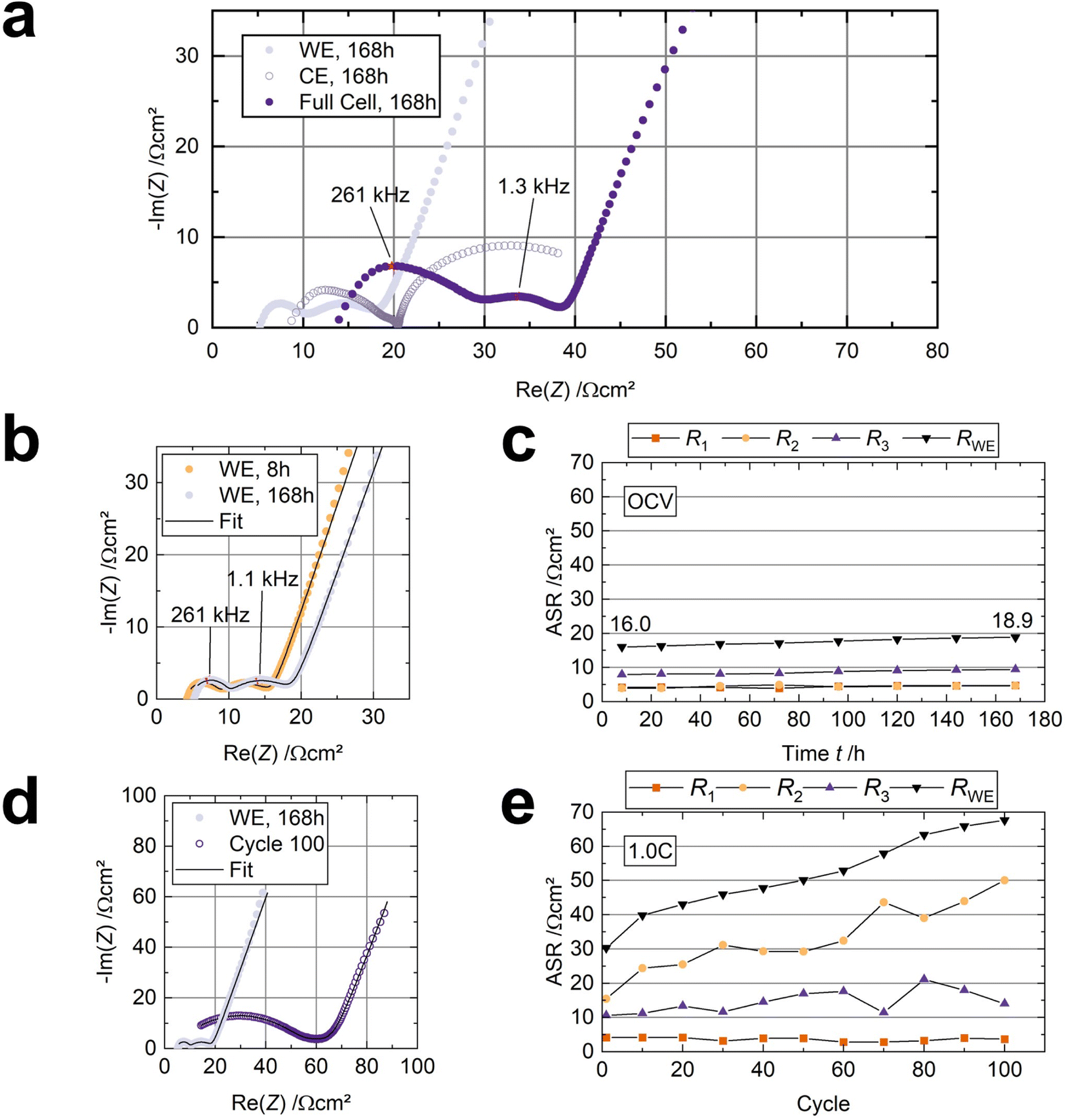 | ||
| Fig. 5 Results of the PEIS measurements in a three-electrode cell (cell 2). The resistance is normalized to the electrode area. (a) Nyquist plot of the cathode, anode, and half-cell after 168 hours at OCV. (b) Nyquist plots were obtained after eight and 168 hours at an OCV of 2.74 V and 2.84 V, respectively. (c) Resistance evolution of the working electrode over 168 hours. (d) Nyquist plot of the cathode after 168 hours at OCV and after 100 cycles. (e) Resistance evolution of the working electrode over 100 cycles. | ||
First, a spectrum was recorded every eight hours at open-circuit voltage (OCV) for 168 h at 80 °C. Surprisingly, the counter electrode's/anode's contribution (RCE) dominates the internal resistance, likely governed by insufficient kinetics (Fig. 5a).34 This observation underlines the need for engineering the Na|BASE interface in future works.3Fig. 5b shows the cathode's Nyquist plots after eight hours and 168 h at OCV and 80 °C. Two semicircles with apex frequencies of 261 kHz and 1 kHz are visible for each spectrum. Thus, three resistance contributions can be deduced. The ohmic resistance (R1), observable at high frequency, stems from the cell components, the solid electrolyte, and the liquid-organic electrolyte. We assign the semicircle in the midfrequency range to the interfacial resistance (R2). The remaining low-frequency semicircle corresponds to the cathode itself, i.e., its charge-transfer resistance (R3).54 We validated the assignment of R2 by adding different amounts of electrolyte to both cathodes in a symmetrical cell (Fig. S7a†). When an additional 10 μl LOE is added to each of the cathodes, R2 decreases significantly (n = 3) from 12.3 ± 1.6 Ω cm2 (3 μl on each cathode) to 5.8 ± 2.6 Ω cm2 (13 μl on each cathode), while R1 stays constant. As the electrodes were peeled from the solid electrolyte to add more NaC to the cathodes, R3 increased slightly but insignificantly (Fig. S7b†). Hence, the midfrequency resistance contribution (R2) correlates to the amount of LOE and the enhancement in interface contact.
The cell's resistance contributions during OCV, obtained by fitting the impedance data with an R + (RQ) + (RQ) + Q equivalent circuit, are shown in Fig. 5c. Overall, the resistance from the working electrode/cathode (RWE = R1 + R2 + R3), which is in contact with the LOE, increases slightly from 16.0 Ω cm2 to 18.9 Ω cm2. R1 is about 5 Ω cm2, corresponding well to BASE's thickness and ionic conductivity (≈4.5 Ω cm2). R1 and R2 both increase slightly by 0.7 Ω cm2. The charge-transfer resistance R3, increasing from 7.9 Ω cm2 to 9.4 Ω cm2, contributes 50% to the overall resistance of the working electrode RWE. RWE does not reach a constant value after one week (168 h) at OCV. Noteworthy, the internal resistance Rcell of 63 Ω cm2 (RWE + RCE) before cycling is lower compared to other Na-SSB approaches, where polymers21 (Rcell ≈ 500 Ω cm2), hydroborates16 (Rcell ≈ 200 Ω cm2), and even ionic liquids18 (Rcell ≈ 300 Ω cm2) were used as interface modifiers.
After 168 h at OCV, the three-electrode cell was cycled at 80 °C and 1.0C (cell 2, Table S1†) to investigate the resistance evolution under operation. The spectra were recorded from the charged cell. The voltage profiles and electrochemical performance are shown in Fig. S8a and b,† respectively. Fitting the impedance data obtained from the three-electrode setup (Fig. 5d) reveals that the interfacial resistance R2 now governs the working electrode's resistance (Fig. 5e). R1 remains nearly unchanged, and R3 increases slightly from 10 Ω cm2 to 15 Ω cm2 during the subsequent 100 cycles.
After disassembly, we noticed that the cathodes were not wetted by the LOE anymore. Considering only minor changes in the composition of the surface of the BASE disk and the cathode, which were determined by XPS measurement (Section 3.3) and the results of the symmetric cell measurement (Fig. S7†), we conclude that the main reason for the R2 increase, and thus, increase in internal resistance, is the displacement of the liquid electrolyte from the interface. We suspect the liquid electrolyte was displaced due to the electrodes' volume changes during cycling. We rule out solvent evaporation despite the operating temperature of 80 °C since no increase in interfacial resistance was detected during the 168 hours of OCV measurement. As R1 and R3 barely change, the interphases observed with XPS do not considerably affect the cell's resistance.
We confirmed this with another three-electrode cell, cycled at a lower current of 0.3C at 80 °C, where we added 15 μl of LOE, decreasing R2 to nearly zero (cell 3, Table S1 and Fig. S9†). After ten cycles, the semicircle corresponding to the interfacial resistance R2 starts evolving in the medium-frequency range (Fig. S9a†), which subsequently dominates the cathode resistance after 50 cycles and reaches 300 Ω cm2 after 250 cycles (Fig. S9b†). The result underlines that the resistance increase is independent of the applied current. The displacement theory is corroborated by the fact that we decreased R2 to 20 Ω cm2 by adding 3 μl of NaC to the positive electrode after 250 cycles (Fig. S9b†). We assume that the detachment of the active material particles from the positive electrode and the interphase build-up, as shown in the XPS analysis, is responsible for the slight increase in R3 in Fig. S9b.†53 The cell's performance of 250 cycles with 80.1% capacity retention is illustrated in Fig. S9c.†
The effectiveness of using NaC as an interface modifier is additionally demonstrated by operating a cell at 30 °C for 250 cycles (cell 4, Table S1† and Fig. 6a). A BASE with a thickness of 300 μm was employed to minimize internal resistance. An initial discharge capacity qsp of 88.9 mA h g−1 is achieved in the first cycle at 0.5C. 77.7 mA h g−1 are retained after 150 cycles. The cell shows nearly no loss in specific discharge capacity (qret = 99.8%) during the subsequent 100 cycles at 1.0C with qsp = 60.1 mA h g−1 in the 250th cycle. The overlap of the voltage profiles indicates that the capacity retention, voltage retention, and thus, the energy retention approaches 100%, too (Fig. 6b). The performance parameters of the four cells highlighted in this work are compared in Table S1.†
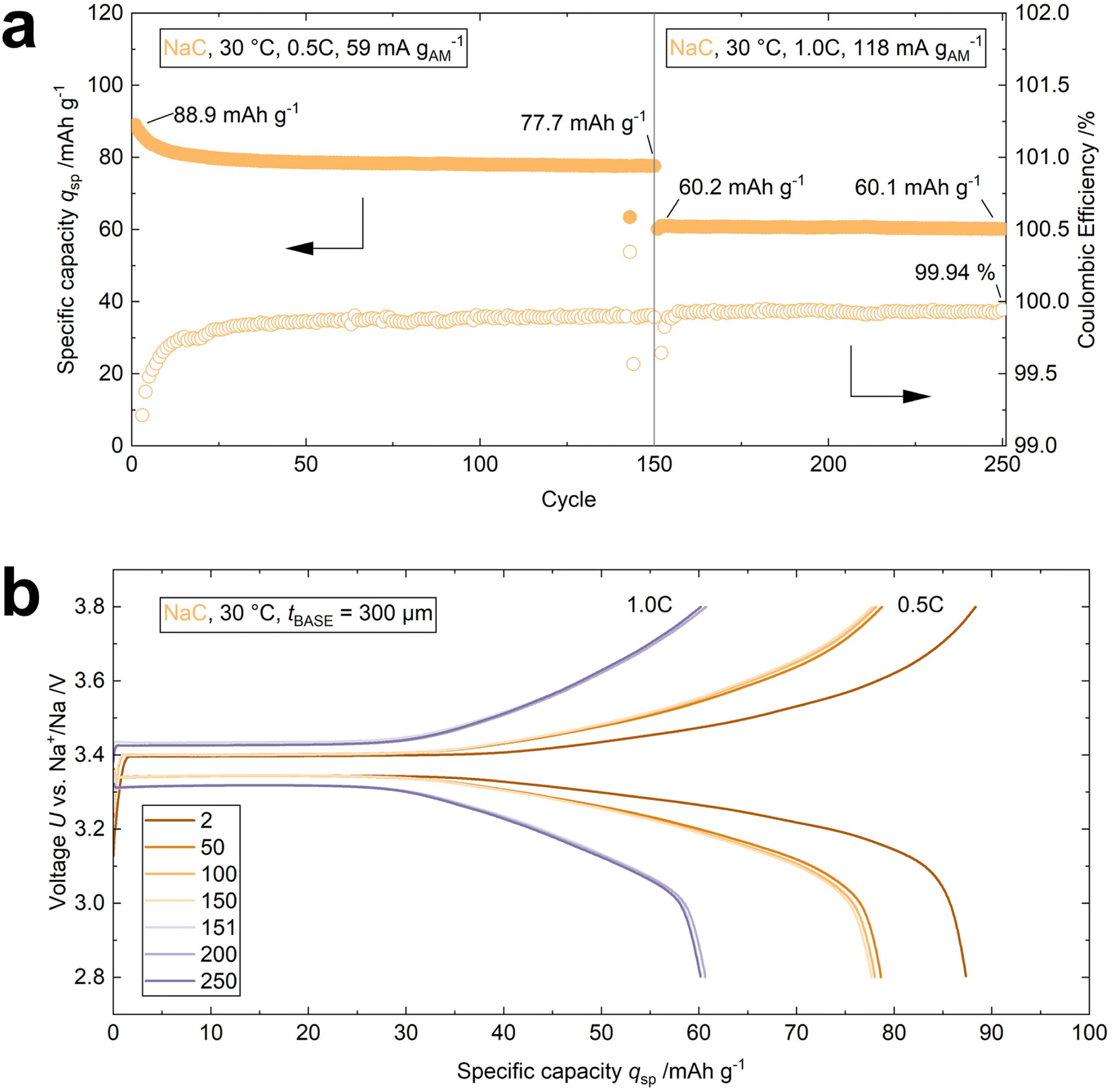 | ||
| Fig. 6 GCPL data of the two-electrode cell cycled at 30 °C with 0.5C for 150 cycles and 1.0C for the subsequent 100 cycles (cell 4). The thickness of the BASE was 300 μm. (a) The cell delivers a specific discharge capacity of 77.7 mA h g−1 after 150 cycles. The cell delivers a specific discharge capacity of 60.1 mA h g−1 after 250 cycles (100 cycles at 1.0C) with a coulombic efficiency of 99.94%. The capacity retention is >99.8%, considering the first discharge at 1.0C. (b) The voltage profiles indicate a loss in specific discharge capacity during the initial 50 cycles. Afterward, the capacity stays constant. The voltage profiles overlap for the cycling at 1.0C, showing high capacity, voltage, and energy retention. | ||
4 Conclusion
Semi-solid-state sodium cells operating at 80 °C and 30 °C were realized, in which the BASE|cathode interface impedance was successfully reduced using a liquid-organic electrolyte (LOE, 1 M NaClO4 in propylene carbonate) as interface modifier. The LOE is capable of infiltrating the positive electrode, effectively establishing a percolation path for facile ion transport and intimate interface contact. X-ray photoelectron spectroscopy discloses only small amounts of film-forming halide species in the NaC system. Using symmetrical cells and three-electrode setups, we were able to correlate the increase in interfacial resistance to a displacement of the LOE during cycling. Hence, future works must aim to retain the LOE, e.g., inside a porous electrode or sealing the cathode to the electrolyte, contributing to additional complexity in the cell production of solid-state cells. Sodium metal can still be used as an anode when the LOE is limited to the cathode interface. Additionally, the cell retains all the advantages of solid-state cells using only a tiny amount of LOE. The LOE approach is a good compromise between high performance and cost and, therefore, a more practical approach than sophisticated interface modifications. Thus, the study demonstrates that – similar to Li- and Na-ion systems – tuning the salt/electrolyte system results in improved electrochemical performance. We suggest tuning the electrolyte system depending on the solid electrolyte and the cathode chemistry.Data availability
The data presented in this study are available on request from the corresponding author.Author contributions
Micha P. Fertig: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing—original draft, writing—review & editing, visualization. Christof Neumann: validation, formal analysis, investigation, data curation, writing—review & editing. Matthias Schulz: conceptualization, resources, writing—review & editing, project administration, funding acquisition. Andrey Turchanin: resources, writing—review & editing, funding acquisition. Michael Stelter: conceptualization, resources, supervision, writing—review & editing, funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge support by the Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, grant number 03XP0404, KeNaB-ART) and by the European Fonds for Regional Development (Europäischer Fonds für Regionale Entwicklung, EFRE-OP 2014–2020, Project No. 2021 FGI 0035, NanoLabXPS) as part of the REACT-EU program. M. P. F. is grateful for financial support from the Landesgraduiertenakademie of the Friedrich Schiller University Jena. All authors thank Cornelius Dirksen for performing Rietveld refinement, Heidi Dohndorf for the powder preparation, Anja Harpf for the SEM/EDX measurements, Thomas Mensinger for polishing the disks, and Benjamin Schüßler for providing the cell scheme. We are grateful to EL-Cell GmbH for providing the instruments and materials used in this study.References
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Fundamentals of inorganic solid-state electrolytes for batteries, Nat. Mater., 2019, 18, 1278–1291, DOI:10.1038/s41563-019-0431-3.
- J. Janek and W. G. Zeier, Challenges in speeding up solid-state battery development, Nat. Energy, 2023, 8, 230–240, DOI:10.1038/s41560-023-01208-9.
- Y. Lu, L. Li, Q. Zhang, Z. Niu and J. Chen, Electrolyte and Interface Engineering for Solid-State Sodium Batteries, Joule, 2018, 2, 1747–1770, DOI:10.1016/j.joule.2018.07.028.
- Q. Ma, et al., Scandium-Substituted Na3Zr2 (SiO4)2(PO4) Prepared by a Solution-Assisted Solid-State Reaction Method as Sodium-Ion Conductors, Chem. Mater., 2016, 28, 4821–4828, DOI:10.1021/acs.chemmater.6b02059.
- W. Zhou, Y. Li, S. Xin and J. B. Goodenough, Rechargeable Sodium All-Solid-State Battery, ACS Cent. Sci., 2017, 3, 52–57, DOI:10.1021/acscentsci.6b00321.
- W. Meng, J. Liu, L. Wang, L. Dai and S. Liu, In Situ Construction of a Liquid Film Interface with Fast Ion Transport for Solid Sodium-Ion Batteries, Nano Lett., 2022, 22, 5214–5220, DOI:10.1021/acs.nanolett.2c00983.
- D. Landmann, G. Graeber, M. Heinz, S. Haussener and C. Battaglia, Sodium plating and stripping from Na-β"-alumina ceramics beyond 1000 mA/cm2, Mater. Today Energy, 2020, 18, 100515, DOI:10.1016/j.mtener.2020.100515.
- Y. Hu, Z. Wen, X. Wu and Y. Lu, Nickel nanowire network coating to alleviate interfacial polarization for Na-beta battery applications, J. Power Sources, 2013, 240, 786–795, DOI:10.1016/j.jpowsour.2013.04.056.
- K. Ahlbrecht, C. Bucharsky, M. Holzapfel, J. Tübke and M. J. Hoffmann, Investigation of the wetting behavior of Na and Na alloys on uncoated and coated Na-β-alumina at temperatures below 150°C, Ionics, 2017, 23, 1319–1327, DOI:10.1007/s11581-017-2017-x.
- D. Reed, et al., Wetting of sodium on β′′-Al2O3/YSZ composites for low temperature planar sodium-metal halide batteries, J. Power Sources, 2013, 227, 94–100, DOI:10.1016/j.jpowsour.2012.11.034.
- S. Cai, et al., Tuning Na3Hf2Si2PO12 electrolyte surfaces by metal coating for high-rate and long cycle life solid-state sodium ion batteries, J. Mater. Chem. A, 2022, 10, 1284–1289, 10.1039/d1ta09693a.
- J. Suo, et al., Designing a Quasi-Liquid Alloy Interface for Solid Na-Ion Battery, ACS Nano, 2023, 17, 10229–10235, DOI:10.1021/acsnano.3c00397.
- M. M. Gross, et al., Tin-based ionic chaperone phases to improve low temperature molten sodium–NaSICON interfaces, J. Mater. Chem. A, 2020, 8, 17012–17018, 10.1039/d0ta03571h.
- B. Tang, P. W. Jaschin, X. Li, S.-H. Bo and Z. Zhou, Critical interface between inorganic solid-state electrolyte and sodium metal, Mater. Today, 2020, 41, 200–218, DOI:10.1016/j.mattod.2020.08.016.
- M. P. Fertig, et al., From High- to Low-Temperature: The Revival of Sodium-Beta Alumina for Sodium Solid-State Batteries, Batteries Supercaps, 2022, e202100131, DOI:10.1002/batt.202100131.
- M.-C. Bay, R. Grissa, K. V. Egorov, R. Asakura and C. Battaglia, Low Na-β′′-alumina electrolyte/cathode interfacial resistance enabled by a hydroborate electrolyte opening up new cell architecture designs for all-solid-state sodium batteries, Materials Futures, 2022, 1, 31001, DOI:10.1088/2752-5724/ac8947.
- L. Liu, et al., Toothpaste-like Electrode: A Novel Approach to Optimize the Interface for Solid-State Sodium-Ion Batteries with Ultralong Cycle Life, ACS Appl. Mater. Interfaces, 2016, 8, 32631–32636, DOI:10.1021/acsami.6b11773.
- T. Deng, et al., Interfacial-engineering-enabled practical low-temperature sodium metal battery, Nat. Nanotechnol., 2022, 17, 269–277, DOI:10.1038/s41565-021-01036-6.
- D. Wang, M. Takiyama, J. Hwang, K. Matsumoto and R. Hagiwara, A Hexafluorophosphate-Based Ionic Liquid as Multifunctional Interfacial Layer between High Voltage Positive Electrode and Solid-State Electrolyte for Sodium Secondary Batteries, Adv. Energy Mater., 2023, 13, 2301020, DOI:10.1002/aenm.202301020.
- T. Wu, et al., Disordered carbon tubes based on cotton cloth for modulating interface impedance in β′′-Al 2 O 3 -based solid-state sodium metal batteries, J. Mater. Chem. A, 2018, 6, 12623–12629, 10.1039/C8TA01883A.
- M. P. Fertig, K. Skadell, K. Wegner, M. Schulz and M. Stelter, A Medium-Temperature All-Solid-State Sodium Battery Utilizing Sodium-Beta Alumina and a Polymeric Composite Positive Electrode, J. Electrochem. Soc., 2023, 170, 50501, DOI:10.1149/1945-7111/accf39.
- G. G. Eshetu, et al., Electrolytes and Interphases in Sodium-Based Rechargeable Batteries: Recent Advances and Perspectives, Adv. Energy Mater., 2020, 10, 2000093, DOI:10.1002/aenm.202000093.
- S. A. Webb, L. Baggetto, C. A. Bridges and G. M. Veith, The electrochemical reactions of pure indium with Li and Na: Anomalous electrolyte decomposition, benefits of FEC additive, phase transitions and electrode performance, J. Power Sources, 2014, 248, 1105–1117, DOI:10.1016/j.jpowsour.2013.10.033.
- K. Pan, et al., Understanding the Electrochemical Compatibility and Reaction Mechanism on Na Metal and Hard Carbon Anodes of PC-Based Electrolytes for Sodium-Ion Batteries, ACS Appl. Mater. Interfaces, 2018, 10, 39651–39660, DOI:10.1021/acsami.8b13236.
- S. Komaba, et al., Fluorinated ethylene carbonate as electrolyte additive for rechargeable Na batteries, ACS Appl. Mater. Interfaces, 2011, 3, 4165–4168, DOI:10.1021/am200973k.
- A. Nimkar, et al., Unraveling the Role of Fluorinated Alkyl Carbonate Additives in Improving Cathode Performance in Sodium-Ion Batteries, ACS Appl. Mater. Interfaces, 2021, 13, 46478–46487, DOI:10.1021/acsami.1c03844.
- J. He, T. Tao, F. Yang and Z. Sun, Optimizing the Electrolyte Systems for Na3(VO1-xPO4)2F1+2x (0≤x≤1) Cathode and Understanding their Interfacial Chemistries Towards High-Rate Sodium-Ion Batteries, ChemSusChem, 2022, 15, e202102522, DOI:10.1002/cssc.202102522.
- H. Shin, J. Park, A. M. Sastry and W. Lu, Effects of Fluoroethylene Carbonate (FEC) on Anode and Cathode Interfaces at Elevated Temperatures, J. Electrochem. Soc., 2015, 162, A1683–A1692, DOI:10.1149/2.0071509jes.
- Y. Huang, et al., Electrolytes and Electrolyte/Electrode Interfaces in Sodium-Ion Batteries: From Scientific Research to Practical Application, Adv. Mater., 2019, 31, e1808393, DOI:10.1002/adma.201808393.
- M. Á. Muñoz-Márquez, M. Zarrabeitia, S. Passerini and T. Rojo, Structure, Composition, Transport Properties, and Electrochemical Performance of the Electrode-Electrolyte Interphase in Non-Aqueous Na-Ion Batteries, Adv. Mater. Interfaces, 2022, 9, 2101773, DOI:10.1002/admi.202101773.
- C. Wang, et al., Boosting the performance of lithium batteries with solid-liquid hybrid electrolytes: Interfacial properties and effects of liquid electrolytes, Nano Energy, 2018, 48, 35–43, DOI:10.1016/j.nanoen.2018.03.020.
- B. Xu, H. Duan, H. Liu, C. Wang and S. Zhong, Stabilization of Garnet/Liquid Electrolyte Interface Using Superbase Additives for Hybrid Li Batteries, ACS Appl. Mater. Interfaces, 2017, 9, 21077–21082, DOI:10.1021/acsami.7b05599.
- L. Mazzapioda, et al., Quasi-solid-state electrolytes - strategy towards stabilising Li|inorganic solid electrolyte interfaces in solid-state Li metal batteries, Energy Mater., 2023, 3, 300019, DOI:10.20517/energymater.2023.03.
- H. Huo and J. Janek, Solid-state batteries: from 'all-solid' to 'almost-solid', Natl. Sci. Rev., 2023, 10, nwad098, DOI:10.1093/nsr/nwad098.
- M. R. Busche, et al., Dynamic formation of a solid-liquid electrolyte interphase and its consequences for hybrid-battery concepts, Nat. Chem., 2016, 8, 426–434, DOI:10.1038/nchem.2470.
- A. Ponrouch, et al., Non-aqueous electrolytes for sodium-ion batteries, J. Mater. Chem. A, 2015, 3, 22–42, 10.1039/C4TA04428B.
- M. P. Fertig, C. Dirksen, M. Schulz and M. Stelter, Humidity-Induced Degradation of Lithium-Stabilized Sodium-Beta Alumina Solid Electrolytes, Batteries, 2022, 8, 103, DOI:10.3390/batteries8090103.
- R. O. Ansell, The Chemical and Electrochemical Stability of Beta-Alumina, J. Mater. Sci., 1986, 365–379 CrossRef CAS.
- S. Wenzel, et al., Interfacial Reactivity Benchmarking of the Sodium Ion Conductors Na3PS4 and Sodium β-Alumina for Protected Sodium Metal Anodes and Sodium All-Solid-State Batteries, ACS Appl. Mater. Interfaces, 2016, 8, 28216–28224, DOI:10.1021/acsami.6b10119.
- R. C. Vries and W. L. Roth, Critical Evaluation of the Literature Data on Beta Alumina and Related Phases: I, Phase Equilibria and Characterization of Beta Alumina Phases, J. Am. Ceram. Soc., 1969, 52, 364, DOI:10.1111/j.1151-2916.1969.tb11956.x.
- X. Lu, J. P. Lemmon, V. L. Sprenkle and Z. Yang, Sodium-beta alumina batteries: Status and challenges, JOM, 2010, 62, 31–36, DOI:10.1007/s11837-010-0132-5.
- D. Spencer Jolly, et al., Sodium/Na β′′ Alumina Interface: Effect of Pressure on Voids, ACS Appl. Mater. Interfaces, 2020, 12, 678–685, DOI:10.1021/acsami.9b17786.
- F. Cheng, et al., Electrolyte Salts for Sodium-Ion Batteries: NaPF6 or NaClO4?, ACS Nano, 2023, 17, 18608–18615, DOI:10.1021/acsnano.3c07474.
- A. Ponrouch, E. Marchante, M. Courty, J.-M. Tarascon and M. R. Palacín, In search of an optimized electrolyte for Na-ion batteries, Energy Environ. Sci., 2012, 5, 8572, 10.1039/c2ee22258b.
- B. Kishore, L. Chen, C. E. J. Dancer and E. Kendrick, Electrochemical formation protocols for maximising the life-time of a sodium ion battery, Chem. Commun., 2020, 56, 12925–12928, 10.1039/d0cc05673a.
- M.-C. Bay, et al., Sodium Plating from Na-β′′-Alumina Ceramics at Room Temperature, Paving the Way for Fast-Charging All-Solid-State Batteries, Adv. Energy Mater., 2020, 10, 1902899, DOI:10.1002/aenm.201902899.
- R. G. Copperthwaite and J. Lloyd, Photoinduced decomposition of sodium perchlorate and sodium chlorate when studied by X-ray photoelectron spectroscopy, J. Chem. Soc., Dalton Trans., 1977, 1117, 10.1039/dt9770001117.
- J. Song, B. Xiao, Y. Lin, K. Xu and X. Li, Interphases in Sodium-Ion Batteries, Adv. Energy Mater., 2018, 8, 1703082, DOI:10.1002/aenm.201703082.
- K. V. Kravchyk, M. Walter and M. V. Kovalenko, A high-voltage concept with sodium-ion conducting β-alumina for magnesium-sodium dual-ion batteries, Commun. Chem., 2019, 2, 84, DOI:10.1038/s42004-019-0186-4.
- F. Sagane, T. Abe and Z. Ogumi, Sodium-ion transfer at the interface between ceramic and organic electrolytes, J. Power Sources, 2010, 195, 7466–7470, DOI:10.1016/j.jpowsour.2010.04.054.
- F. G. Will and S. P. Mitoff, Primary Sodium Batteries with Beta-Alumina Solid Electrolyte, J. Electrochem. Soc., 1975, 122, 457–461, DOI:10.1149/1.2134239.
- G. C. Farrington, Interfacial Na+ Transport in the Beta Alumina/Propylene Carbonate System, J. Electrochem. Soc., 1976, 123, 591–595, DOI:10.1149/1.2132891.
- L. A. Middlemiss, A. J. Rennie, R. Sayers and A. R. West, Characterisation of batteries by electrochemical impedance spectroscopy, Energy Rep., 2020, 6, 232–241, DOI:10.1016/j.egyr.2020.03.029.
- P. Vadhva, et al., Electrochemical Impedance Spectroscopy for All-Solid-State Batteries: Theory, Methods and Future Outlook, ChemElectroChem, 2021, 8, 1930–1947, DOI:10.1002/celc.202100108.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se01258a |
| This journal is © The Royal Society of Chemistry 2024 |