
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the potential of cobalt hydroxide and its derivatives as a cost-effective and abundant alternative to noble metal electrocatalysts in oxygen evolution reactions: a review
Umair
Shamraiz†
 *a,
Abdul
Majeed†
a,
Bareera
Raza
b,
Noor ul
Ain
c and
Amin
Badshah
d
*a,
Abdul
Majeed†
a,
Bareera
Raza
b,
Noor ul
Ain
c and
Amin
Badshah
d
aDepartment of Chemistry, Technical University of Darmstadt, 64287, Germany. E-mail: umair.shamraiz@tc1.tu-darmstadt.de; umairshamraiz@gmail.com
bCollege of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen 518060, China
cDepartment of Chemistry, Rawalpindi Women University, Rawalpindi 46300, Punjab, Pakistan
dDepartment of Chemistry, Quaid-i-Azam University, 45320 Islamabad, Pakistan
First published on 7th December 2023
Abstract
This review article provides a comprehensive analysis of the use of cobalt hydroxide and its derivatives as catalysts in the oxygen evolution reaction (OER), which is a crucial step in the water-splitting process for hydrogen production. The article covers the various synthetic methods used to prepare cobalt hydroxide and its derivatives, as well as their characterization techniques. The article also discusses the effect of different synthesis parameters on the performance of these materials as OER catalysts. The article goes on to evaluate the performance of cobalt hydroxide and its derivatives under various OER conditions, such as pH, temperature, and applied potential. The article also discusses the advantages and limitations of using these materials as OER catalysts, such as their stability, activity, and cost-effectiveness. The article compares the performance of cobalt hydroxide and its derivatives to other commonly used OER catalysts, such as iridium and ruthenium oxides.
 Umair Shamraiz | Umair Shamraiz, a distinguished researcher in multifunctional electro-catalysts for energy storage and conversion, has spearheaded groundbreaking advancements in the field. Specializing in metal oxide-based palladium/rGO electro-catalysts and α-CoOOH doped with transition metals, Umair has significantly contributed to the development of direct liquid fuel cells and facilitated oxygen evolution through water splitting methodologies. Currently serving as a George Forster Postdoctoral Fellow at the esteemed Technical University of Darmstadt, Umair's pioneering work focuses on investigating novel avenues such as In2S3 and Ag-based thiospinels/tellurospinels for water splitting, pushing the boundaries of sustainable energy technologies. Driven by a vision for the future, Umair's aspirations include pioneering efforts in synthesizing electrolytes tailored for Mg-ion batteries and innovating catalysts that will revolutionize energy storage paradigms. With an unwavering commitment to scientific excellence, Umair Shamraiz continues to lead the charge in advancing the frontiers of electro-catalysis for a more sustainable energy landscape. |
 Abdul Majeed | Abdul Majeed, an esteemed scholar in Materials Science, earned his PhD from the prestigious CAS Institute in China in 2019. His doctoral research focused on the synthesis and exploration of diverse carbon-based materials specifically tailored for efficient water splitting processes, showcasing his expertise in sustainable energy solutions. Continuing his academic journey, Abdul Majeed transitioned to a role as a Postdoctoral Fellow at Shanghai Jiatong University. Here, his research delved into the electrodeposition techniques applied to a range of layered double hydroxide materials, advancing their potential applications in various energy-related fields. Presently serving as an Alexander Von Humboldt Postdoctoral Fellow at TU Darmstadt, Abdul Majeed's dedication to innovative research continues to shine. His significant contributions to the scientific community are evidenced by a portfolio of publications in esteemed journals such as ACS Applied Materials and Interfaces, as well as JMCA, solidifying his position as a distinguished authority in the field of materials science and energy conversion. |
 Bareera Raza | As a dedicated Postdoctoral Fellow at Shenzhen University, Bareera Raza focuses on pioneering advancements in rechargeable energy storage systems, particularly in the realm of non-aqueous electrolyte systems for zinc ion batteries. With a primary goal of developing safe, efficient, and high-current-capable electrolyte solutions, Bareera Raza conducts cutting-edge research aimed at pushing the boundaries of battery technology. Bareera Raza is instrumental in the synthesis of energy-dense, high-voltage cathodes that prioritize compatibility within battery systems. Their work revolves around crafting cathode materials that optimize energy density while ensuring seamless integration with the overall battery architecture. Additionally, Bareera Raza employs a multidisciplinary approach, leveraging a diverse array of battery testing and characterization techniques. Through meticulous design, testing, and analysis, Bareera delves into the intricate chemistry of zinc ion batteries, unraveling insights critical for enhancing battery performance and durability. Driven by a passion for innovation, Bareera Raza stands at the forefront of research, contributing significantly to the development of next-generation rechargeable energy storage systems, shaping the landscape of sustainable energy technologies. |
 Noor ul Ain | Dr Noor ul Ain is an accomplished scholar, holding a distinguished PhD in Chemistry earned from Quaid-i-Azam University in April 2021. Her expertise lies at the intersection of inorganic chemistry, nanotechnology, and energy storage materials, particularly focusing on advancements in lithium-ion batteries. Currently serving as a lecturer (BS 18) at RWU since August 2022, Dr Noor ul Ain has established herself as a reputable researcher with contributions featured in esteemed international journals. Her academic prowess was further acknowledged through her selection for the IRSIP scholarship by HEC, which supported her groundbreaking research during her PhD at University College London. Dr Noor ul Ain's research interests encompass multifaceted environmental applications, specifically targeting the reduction of environmental carcinogens using nanomaterials. Additionally, her work extends to energy storage applications, where she specializes in synthesizing ternary metal oxides tailored as cathode materials for enhancing Li-ion batteries' performance. Moreover, her expertise encompasses the synthesis, characterization, and application of magnetic and non-magnetic copper sulfide nanoparticles, showcasing her versatility in nanomaterial research. Her research endeavors, ranging from environmental applications to advancements in energy storage solutions, are driven by a profound commitment to scientific exploration and innovation. Dr Noor ul Ain's contributions stand as a testament to her dedication to advancing knowledge and addressing critical challenges in the realms of chemistry, nanotechnology, and sustainable energy storage. |
| Amin Badshah Prof. Dr Amin Badshah is a distinguished academician and researcher, having obtained his PhD in 1994 from Quaid-i-Azam University (QAU), Islamabad. With a prolific career in academia, he currently serves as the vice chancellor of the University of Buner and holds the esteemed positions of Ex-Professor Emeritus and Dean of the Faculty of Natural Sciences at QAU, Islamabad. A distinguished fellow of the Pakistan Academy of Sciences (PAS) and the Chemical Society of Pakistan, Dr Badshah's scholarly contributions extend to his role as an associate editor for the Journal of the Chemical Society of Pakistan, showcasing his commitment to advancing scientific discourse and research dissemination. His remarkable achievements in academia and research have been recognized through numerous accolades, including the Research Productivity Award from the Pakistan Council for Science and Technology spanning from 2001 to 2016. Dr Badshah's dedication to excellence has been further underscored by prestigious honors such as the Gold Medal from PAS, Best Teacher and Best Paper Awards from the Higher Education Commission of Pakistan, and the esteemed Tamgha-i-Imtiaz (TI) from the Government of Pakistan. Dr Badshah's illustrious career stands as a testament to his unwavering commitment to scholarly pursuits, academic leadership, and the advancement of scientific knowledge within Pakistan and beyond. |
1 Introduction
Electrocatalytic water splitting is a leading field in producing affordable hydrogen, which is currently the most important requirement for our environment as hydrogen is the greenest fuel.1 Hydrogen has properties such as the highest specific chemical energy density (142 MJ kg−1), and the only byproduct of its use is water, unlike other systems, which release hazardous chemicals upon combustion.2–4 Additionally, we are facing environmental problems due to the excessive consumption of hydrocarbons, and known resources will be depleted by 2080.5,6 Therefore, it is crucial to move towards a hydrogen economy. However, hydrogen production is currently costly and energy-inefficient due to high temperature, pressure, complex technology, corrosive chemicals, and safety measures.7–9 Furthermore, it emits greenhouse gases directly into the air, similar to carbon and hydrocarbons. Steam reforming via hydrocarbons emits dangerous nitrogen, sulfur, carbon, and hydrogen, causing poor cell performance in fuel cells.7–15The problem could be resolved by considering electrocatalytic water splitting, which produces a vast amount of energy from renewables. Additionally, it is the safest method for producing green and pure hydrogen. Water splitting is further divided into three different types, depending on the mode of energy used for catalysis: photocatalytic water splitting,16–21 electrocatalytic water splitting,22–24 and photoelectrochemical water splitting.25 Compared with the electrocatalytic process, the other two are relatively slow in hydrogen production.
In electrocatalytic water splitting, three different types of electrolyzers are used, depending on their working principle. These include proton exchange membrane (PEM) electrolyzers,26–29 anion exchange membrane-based alkaline electrolyzers,30–33 and high-temperature solid-oxide electrolyzers that operate on steam.34 Recently, water splitting in a neutral environment using buffered solutions has gained importance as it is more environmentally friendly and less corrosive.23,34 However, these methods have some disadvantages, such as lower energy efficiency and the need for high cell potentials, and lower current density. On the other hand, many transition metal-based electrocatalysts have shown promising results for oxygen evolution reaction (OER) electrocatalysis under neutral conditions.35–42
Among these electrocatalysts, those that can simultaneously catalyze both the anodic oxygen evolution reaction (OER) and cathodic hydrogen evolution reaction (HER) are considered superior. Many compounds, including metals and their composites, and carbon-based materials, have shown better activity for these reactions. However, the toxic and unstable nature of carbon-based materials hinders their industrial use for hydrogen production.41,42 In contrast, metallic-based electrocatalysts are superior because they offer metallic surfaces enriched with electrons, a variety of oxidation states, and optimized metal–oxygen bond energies, which can facilitate the adsorption and desorption of intermediates.43–46 So far, metals and their compounds such as Pt,25,47,48 Ir,47–50 Ru,51–54 Ti,55 V,56–58 W,59–62 Mo,63–65 Mn,66–68 Fe,69–71 Co,72–78 Ni,79–83 Cu,83,84 and Au85–87 have been successfully used for electrochemical water splitting. Moreover, only Pt is used as a metal, while the others are mostly used in the form of composites. The main purpose of Pt is to catalyze the HER because it can produce hydrogen at 0 V.87 However, it has recently been identified that some phosphides and chalcogenides are capable of carrying out the HER in the same range as Pt.88 On the other hand, Ru and Ir oxides are superior for the OER under acidic conditions.89,90 Unfortunately, these precious metals increase the cost of hydrogen production despite their efficiency. Research is underway to replace Pt with cheaper metals.89,90
Nevertheless, first-row transition metal electrocatalysts including hydroxides and layered double hydroxides of Co have been given preference due to their ease of preparation and better electrocatalytic performance compared to other types.74,75,89,90 Cobalt-based hydroxides coupled with other metals have boosted oxygen evolution reactions. Therefore, a review article is needed to understand the catalysis phenomena of cobalt-based hydroxides and the factors that make them competitive with noble metals. Additionally, the challenges faced by Co-based hydroxides highlight the significance of the OER.91–94
2 Significance of electrocatalytic water splitting
Electrocatalytic water splitting, a non-spontaneous reaction, requires a 1.23 V thermodynamic potential, alongside kinetic hindrances necessitating an additional potential for the anode and cathode.95 The complex oxygen evolution process at the anode contrasts with the simpler hydrogen evolution at the cathode. The detailed mechanism of oxygen evolution involves different pathways and mechanisms like the adsorbate evolution mechanism (AEM) and lattice oxygen participation mechanism (LOM). Co-based hydroxides' catalytic capabilities are showcased, emphasizing their importance in oxygen evolution reactions.22,96AEM
| * + H2O(l) → OH* + H+ + e− |
| OH* → O* + H+ + e− |
| O* + H2O(l) → OOH* + H+ + e− |
| OOH* → O2 + H+ + e− |
LOM
| OH* → (Vo + OO*) + H+ + e− |
| (Vo + OO*) + H2O(l) → O2 + (Vo + OH*) + H+ + e− |


In the context of these equations, ‘Vo’ denotes oxygen vacancies formed due to lattice oxygen involvement in catalysis. The OER mechanism, as depicted in Fig. 1 by Jin et al.,97 elucidates the necessity of four electrons and protons for oxygen evolution. This figure underscores that the OER demands catalysts with variable oxidation states and favorable metal–oxygen bond energies, where cobalt hydroxide/layered double hydroxide stands as a noteworthy catalyst. It highlights the complexity of the OER compared to the HER, necessitating an additional potential for catalysis, typically within 250–500 mV, yet reaching up to 670 mV at an industrial scale. Efforts to diminish overpotential involve creating high surface area composites with varied catalyst loading. Modifying intrinsic activity via doping, particularly with metals like Fe, is another avenue that has been explored.12 Subsequent sections will emphasize the significance of cobalt hydroxide as an OER electrocatalyst under alkaline conditions.
 | ||
| Fig. 1 Generalized mechanism of the OER in acidic (red path) and alkaline (green path) environments. This figure has been reproduced from ref. 97 with permission from American Chemical Society, copyright 2018. | ||
3 Exploring the advantages of cobalt hydroxide in oxygen evolution reactions: electronic and structural properties
3.1 Polymorphs of cobalt hydroxide
Cobalt hydroxide exhibits distinct polymorphs, α-cobalt hydroxide and β-cobalt hydroxide.103,104 The β-form adopts a brucite-like structure with a Co(OH)2 stoichiometry, featuring a closed hexagonal arrangement of OH− ions and Co(II) occupying alternating rows of octahedral sites.98 Conversely, the α-phase showcases a non-stoichiometric composition similar to hydrotalcite-like compounds. This phase includes positively charged Co(OH)2−x layers balanced by interlayer anions (e.g., Cl−, CO32−, NO3−, S2−, etc.), resulting in an extended interlayer spacing exceeding 7 Å, as opposed to the restricted 4.6 Å spacing in the β-form (Fig. 2).98,110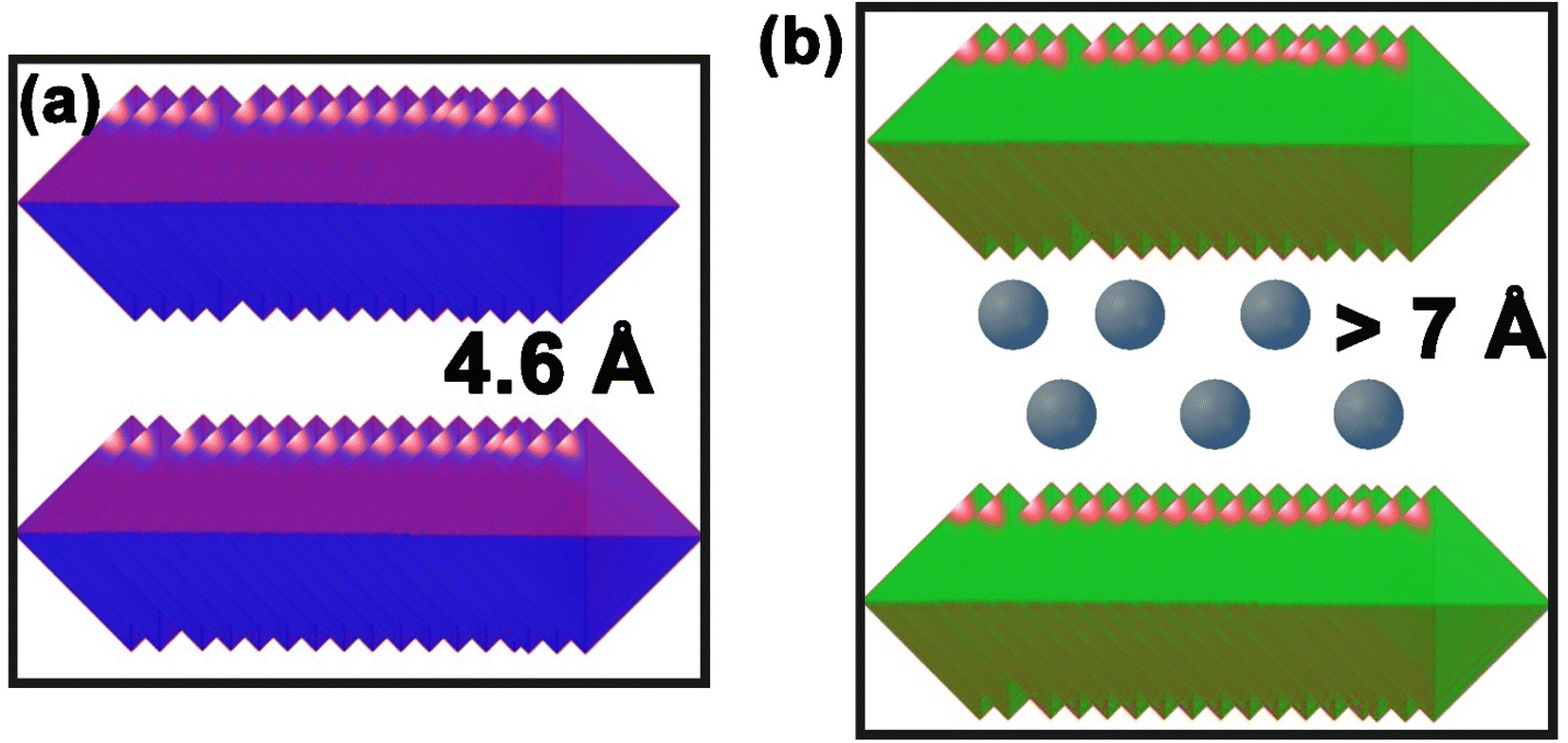 | ||
| Fig. 2 Structure of (a) β-Co(OH)2 and (b) α-Co(OH)2. | ||
Notably, α-Co(OH)2 demonstrates high specific capacity, making it a potential candidate for electrochemical supercapacitors and energy devices.76,77,99,100 On the other hand, β-Co(OH)2's hexagonal layered structure presents an intriguing opportunity as a reactive template for thermoelectric cobaltite ceramics, such as NaCo2O4 and CaxCoO2.101–103 Density functional theory studies reveal the superior electrochemical activity of the α-phase, positioning it as a promising electrode material and a potential candidate for organic magnetic materials through the incorporation of organic anions.104,105
The distinction between the α and β phases extends to their sites; while the β-phase hosts Co2+Oh sites exclusively, the α-phase contains both Co2+Oh and Co2+Td sites.106–108 This variation impacts their electrochemical behavior significantly, with the electroactive Co2+Td sites in the α-phase playing a crucial role. Synthesis of Co2+Td sites is considered vital for electrochemical processes, as indicated by UV-visible spectroscopy displaying distinct absorption bands for both sites in the α-phase, compared to a single absorption band in the β-phase (Fig. 3).109
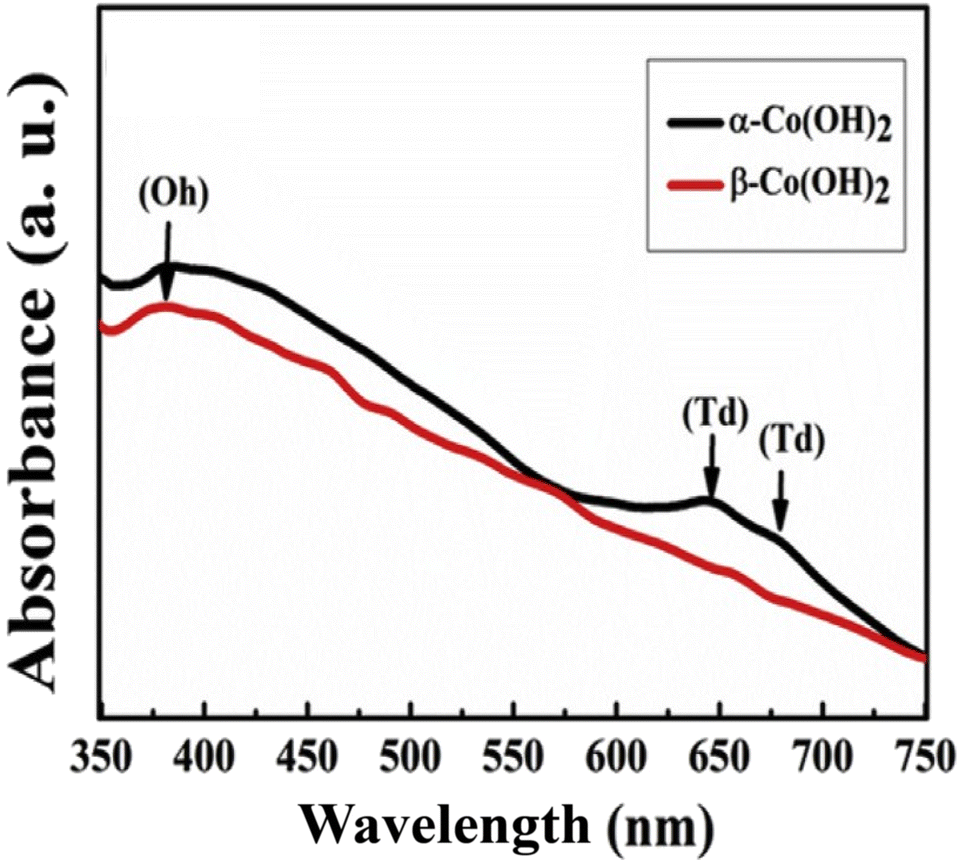 | ||
| Fig. 3 UV-visible absorption spectrum of α- & β-Co(OH)2. This figure has been reproduced from ref. 109 with permission from Elsevier, copyright 2019. | ||
3.2 Cobalt hydroxide and cobalt oxyhydroxide
Cobalt hydroxide manifests in two polymorphs, distinguished by Co2+ ions occupying Co2+Oh and Co2+Td structural sites due to their distinct stoichiometries. Additionally, a structurally akin compound resembling α-Co(OH)2, known as cobalt oxyhydroxide, houses a network of Co2+ and Co3+ cations stabilized by interlayer anions in a hydrotalcite-like configuration.117,118 This cobalt oxyhydroxide, represented by [CoII(1−x)CoIII(x)OH2](Ax)·nH2O (A = NO3−, Cl−, CO32−etc.), accommodates both Co2+ and Co3+ on its layers.117,119,120 The fabrication method and experimental conditions significantly influence the formation of these compounds. The α-Co(OH)2, being a metastable state, gradually transforms into β-Co(OH)2 or CoO(OH), a process heavily dictated by the OH−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co2+ ratio. Fig. 4 illustrates this transformation mechanism. Under specific OH−:Co2+ ratios, the polymorphs' conversion from α-Co(OH)2 to β-Co(OH)2 occurs gradually, while higher ratios prompt immediate transformation. Elevated temperatures expedite the conversion of α-Co(OH)2 to β-Co(OH)2, evident even at lower OH−:Co2+ ratios, indicating a faster transformation at higher temperatures.
:Co2+ ratio. Fig. 4 illustrates this transformation mechanism. Under specific OH−:Co2+ ratios, the polymorphs' conversion from α-Co(OH)2 to β-Co(OH)2 occurs gradually, while higher ratios prompt immediate transformation. Elevated temperatures expedite the conversion of α-Co(OH)2 to β-Co(OH)2, evident even at lower OH−:Co2+ ratios, indicating a faster transformation at higher temperatures.
 | ||
| Fig. 4 Illustration of the formation of Co3O4 and CoOOH crystals. This figure has been reproduced from ref. 110 with permission from Royal Society of Chemistry, copyright 2011. | ||
4 Investigating the use of cobalt hydroxide as an OER electrocatalyst in water splitting
4.1 β-Cobalt hydroxide (β-Co(OH)2)
In our general discussion, the review starts with β-Co(OH)2, as it is the most stable polymorph and is commonly produced by researchers, either intentionally or unintentionally.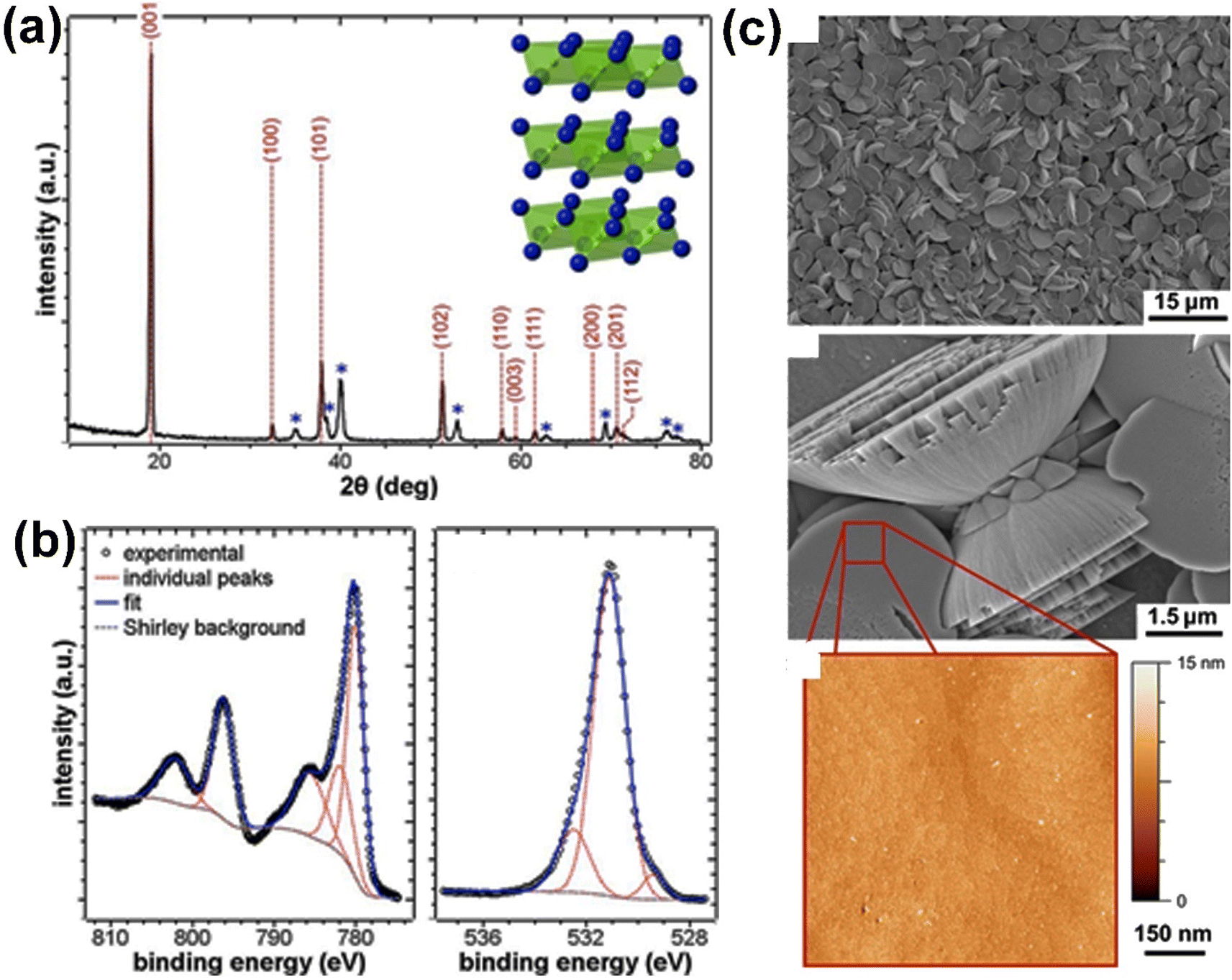 | ||
| Fig. 5 The results of the XRD, XPS, and morphology analysis of electrodeposited Co(OH)2. (a) Shows the XRD pattern of the Co(OH)2, with the reflections of Co(OH)2 indicated in red (JCPDS no. 30-0443) and the blue asterisks representing the Ti substrate. The inset of the panel shows the structure of the Co(OH)2. (b) Shows the XPS spectra of the electrodeposited Co(OH)2, with the Co 2p and O 1s binding energy ranges represented. (c) Shows the morphology of the Co(OH)2 electrodeposited at a charge density of 0.6 C cm−2, including SEM images of the film and an AFM image of a flat disk surface. This figure has been reproduced from ref. 111 with permission from American Chemical Society, copyright 2013. | ||
For electrochemical purposes, they deposited the material on a Ti electrode and LSV curves were measured for both Co3O4 and Co(OH)2 for comparative purposes. Both electrodes showed a similar profile, and Co3O4 shows better activity per geometric surface area (Fig. 6). However, the activity was undoubtedly increased for Co(OH)2 when optimized to the active surface area. This enhanced activity was attributed to the roughness factor possessed by the Co(OH)2 in comparison with the smooth Co3O4. But in our view, it could be mere speculation, as no experimental evidence is provided to prove the conclusion, and the SEM shows very smooth cones of Co(OH)2.
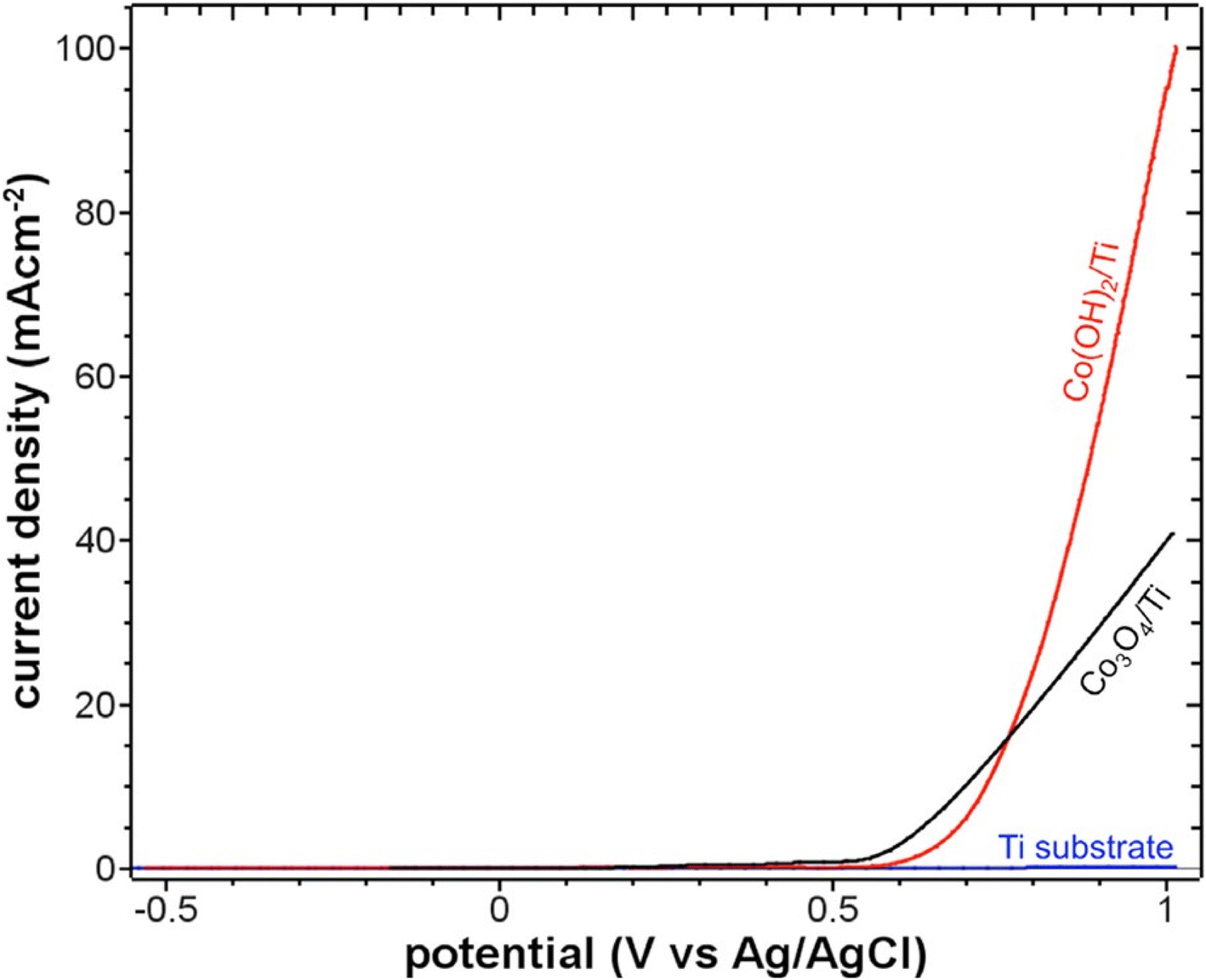 | ||
| Fig. 6 LSVs measured at 1 mV s−1 scan rate in 1 M KOH at the electrodeposited Co(OH)2 (red), crystalline Co3O4 (black), and uncoated Ti substrate (blue). This figure has been reproduced from ref. 111 with permission from American Chemical Society, copyright 2013. | ||
Zhan et al.112 synthesized β-Co(OH)2 hexagonal nanoplates via the well-known hydrothermal method and exploited them for the OER in an alkaline medium. The phase purity was confirmed via PXRD analysis, showing only the presence of peaks for β-Co(OH)2. Furthermore, they applied heat treatment on the synthesized β-Co(OH)2 to make CoO and Co3O4, which was confirmed by PXRD analysis and demonstrated that under the corrosive heat treatment, the OH− molecules leave the surface leaving behind pores to increase the surface area, confirmed by BET analysis. The TEM analysis showed the presence of uniform hexagonal plates having an approximate thickness of 14 nm with an aspect ratio of 5.7. The LSV curves show that the OER activity is much better with Co(OH)2 in comparison with CoO and Co3O4. The Co(OH)2 attained 10 mA cm−2 OER activity at an overpotential of 450 mV, while CoO and Co3O4 required 460 and 510 mV overpotentials, respectively. Moreover, after 400 cycles, the activity was lost, and post-stability characterization confirmed the conversion of Co(OH)2 to Co3O4 under prolonged alkaline conditions. Post-stability analysis revealed Co(OH)2 oxidation to Co3O4 under extended anodic exposure: XRD indicated the presence shift from Co(OH)2 to Co3O4, supported by XPS measurements showing Co 2p1/2 and Co 2p3/2 binding energy shifting from 798.5/783.0 eV to 794.8/779.8 eV, respectively. This oxidation mechanism elucidates Co(OH)2 degradation under prolonged anodic potential, necessitating strategies to enhance its stability for sustained performance. The corresponding Tafel analysis shows faster reaction kinetics of the OER on the surface of Co(OH)2 as compared with the CoO and Co3O4 owing to the Tafel slope values of 60 mV dec−1, 65 mV dec−1 and 75 mV dec−1, respectively. Using hydroxides over oxides simplifies achieving excellent bifunctional activity; Co(OH)2 showcases superior performance due to optimal Co–OH bond strength, facilitating efficient oxygen electrocatalysis by bypassing certain kinetic steps. This study proposes Co(OH)2 as a more cost-effective and potent alternative to complex oxides like MnCoFeO4, offering competitive ORR and OER capabilities, positioning it favorably against precious metal-based bifunctional catalysts.
 | ||
| Fig. 7 (a) XRD patterns of Co(OH)2 nanosheets prepared under different hydrothermal conditions: 120 °C for 12 h, 160 °C for 12 h and 160 °C for 16 h. (b) SEM, TEM and AFM images of Co(OH)2 nanosheets prepared under hydrothermal conditions at 160 °C for 12 h; SEM, TEM and AFM images of Co(OH)2 nanosheets prepared under hydrothermal conditions at 160 °C for 16 h. (c) Polarization curves, (d) Tafel plots and (e) electrochemical impedance spectra. This figure has been reproduced from ref. 113 with permission from Elsevier, copyright 2017. | ||
AFM analysis was carried out to determine the thickness of the 2D Co(OH)2 nanosheets, and Fig. 7b displays tapping-mode images, revealing an average vertical height of approximately 4.0 nm. This direct confirmation underscores the ultrathin attribute of the acquired Co(OH)2 nanosheets. The Co(OH)2, termed as ultrathin nanosheets, produced a current density of 10 mA cm−2 at only 327 mV overpotential, and the kinetics were relatively faster with a Tafel slope of 78 mV dec−1 for the sample with a thickness of 3.9 nm. The authors attributed this enhanced activity to the fine, thin nanosheets.
The ECSA calculations suggested the largest surface area for the sample synthesized at lower temperatures, which is the main contributing factor towards its better activity. The increase in thickness results in an increase in both onset potential and overpotential at 10 mA cm−2, illustrating that morphology and size have a pronounced effect on the intrinsic activity of the electrocatalysts. This effect is also seen in the highest TOF of the sample with the lowest thickness among the assembly of electrocatalysts tested. Moreover, the authors claimed that at elevated temperatures, hexagonal nanoplates are a favored morphology. The nanoplates have a thickness much higher than those of the other two samples in the series and have inferior activity. These results are consistent with the results discussed above reported by Zhan et al.112 Hence, nanosheets with an amorphous nature are better electrocatalysts for the OER than well-defined hexagonal nanoplates.114 The smooth surface does not offer effective participation of the underlying atoms during electrocatalysis, while a rough surface is advantageous as it allows more atoms to contribute to the process, resulting in a better activity. These results are also supported by the synthesis of amorphous Co(OH)2 by the group of P. O'Mullane.115 They reported enhanced OER activity when they subjected the amorphous Co(OH)2 as an electrocatalyst requiring an overpotential of only 360 mV to generate a current density of 10 mA cm−2 with a Tafel slope of 58 mV dec−1. These two results are consistent with the enhancement of electrochemically active surface area due to the rough and amorphous nature of the electrocatalysts. However, Tong et al.113 proposed that no transformation of the parent β-Co(OH)2 occurred during the OER. Moreover, they have suggested that the Co(OH)2 is converted into Co3O4 during the OER. Furthermore, to support their results and to make a better scientific contribution, they have tested their catalysts for the OER at different electrodes. They demonstrated that the Au electrode exhibits superior OER electrocatalysis as compared with the Pd, Cu, and GC electrodes, and the order of activity was found to be Au > Pd > Cu.
Moreover, they supported their results with the role of electronegativity of the electrodes subjected to the OER. Among these electrodes, Au is more electronegative and will definitely facilitate the oxidation of Co(II) to the active Co(III) and then to Co(IV) during the OER as compared to the less electronegative electrodes in the series. These results are also supported by DFT calculations which demonstrate that Au positively shifts the d-band center of Co by 0.74 eV compared to pure Co. The elevated d-band center favors the oxidation of Co.116 Similarly, Babar et al.117 fabricated Co(OH)2 nanosheets directly on Ni foam by electrodeposition and subjected it to heat treatment up to 400 °C in air. The better activity of Co(OH)2/NF over Co3O4/NF was attributed to the enhanced surface area, availability of more active sites, and better charge transport ability. These results are in line with the results discussed so far in this review. The annealing of the electrodeposited Co(OH)2/NF catalysts has two pronounced effects: there is a transformation of Co(OH)2 to Co3O4 and also the crystallinity of the sample is increased. This is the main reason why Co(OH)2 is a potent candidate for the OER; the amorphous nature is the driving force behind this superiority. Along with the amorphous nature, it was also identified that annealing has reduced the interlayer spacing between the layers owing to the aggregation, which results in the reduction of the active surface area. These results suggested that the activity enhancement was due to the amorphous and porous nature of Co(OH)2, both of which were changed upon annealing and resulted in the loss of activity. The OER activity reported here is much better than the reports so far dealing with the nanosheets, but the author did not draw a conclusive statement for this enhancement. In conclusion, the better activity could be due to the NF electrode used, having high surface and porosity results which have a pronounced effect not only on the activity but also on the stability of the material as well. Ghanem et al.118 used a liquid crystal template to fabricate mesoporous cobalt hydroxide. Due to the oxidation peak of cobalt, the authors reported 25 mA cm−2 at 1.52 V, which is 130 mV lower in comparison with bulk cobalt hydroxide. Nevertheless, it shows that the overall overpotential should be lower, between 250 and 270 mV at a current density of 10 mA cm−2. Moreover, they performed the OER in 2.0 M KOH as well, which displayed even better activity, and a current density of 20 mA cm−2 was achieved at 1.50 V. It is obvious that the enhanced activity was due to the increased electrochemically active surface area because of the porous nature of the material.
Yang et al.119 explored Co–OOH bond elongation in Co(OH)2 nanosheets via crystal distortion for enhanced OER. By incorporating Ga (gallium) into the Co(OH)2, there's a unique alteration in the crystal lattice that stretches out the Co–OOH bonds specifically on the 120 facets, generating active sites. Despite Ga's inclusion showing no distortion in the parent structure, PXRD revealed peak shifts, enhancing the OER due to longer metal–oxygen bond lengths. Catalysts exhibited exceptional OER at 265 mV overpotential for 10 mA cm−2 with improved conductivity. Zhang et al.120 designed self-supported hexagonal β-cobalt hydroxide via a painting–alloying–dealloying method, attaining a low 332 mV overpotential for 10 mA cm−2 in the OER without a binder. The ultrathin structure and in situ growth on Ga-coated Co plates enhanced electronic conductivity and electrolyte movement. Dileep et al.121 exfoliated β-Co(OH)2 to smaller sheets, achieving 390 mV overpotential for 10 mA cm−2. The group pf Coleman122 used liquid exfoliated Co(OH)2, obtaining 440 mV overpotential. Nanosheet optimizations in length, edges, and loading concentrations improved activity. Anchoring on carbon nanotubes reduced overpotential to 295 mV, demonstrating superior OER.
Zhang et al.125 synthesized needle-like 3D carbonate-incorporating Co(OH)2 superstructures for the OER. The synthesis was a simple hydrothermal treatment using CoCl2·6H2O as a source of cobalt, urea as a precipitating agent, and sodium glycolate as a surfactant. The reaction was carried out at 150 °C. The 3D structure is formed by the nucleation of the 1D needle-like structures. The authors did not emphasize the factors responsible for the formation of these unique 3D structures. In conclusion, the most probable factor could be the use of the sodium glycolate surfactant, which could be responsible for the formation of needle-like 3D structures. The 3D superstructures delivered a current density of 10 mA cm−2 at the expense of merely 240 mV overpotential. The authors drew a few conclusive points depending on the reaction conditions. The apparent size of the CCHS appeared to be in micrometers, but they are constructed from the nanoneedles having a fine nanometer size range. Nevertheless, the CCHS has an influence on the transport of the electrolyte onto the active sites owing to good electrolyte–surface compatibility. Furthermore, the samples synthesized at 150 °C for 15 h and 120 °C for 12 h agglomerated quickly, and the structures were too thick to transport electrolytes effectively. Moreover, the samples synthesized at 150 °C for 6 h and 180 °C for 12 h appeared to be too loose for ion and electron transport.
Darbandi et al.126 reported hexagonal mesoporous rings of β-Co(OH)2 by a chemical precipitation method. The ring-like structure is formed with the H2O2-assisted method, while the rest of the procedure is similar to the reports discussed above. The rings appeared as aggregates of small fine nanoparticles.
Dhawale et al.127 adopted a microwave-assisted synthesis for the fabrication of Co(OH)2 nanorods and later on, their thermal conversion into porous Co3O4 nanorods. They used CTAB as a surfactant during the synthesis of Co(OH)2. CTAB is a very powerful surfactant for making the nanorod-like morphology of nanomaterials. They tested the OER in 0.1 M KOH solution. The sample calcined at 600 °C showed better performance as compared to the other samples in the list. The authors claimed that the enhanced performance was due to the increase in active sites at a higher temperature. Liu et al.128 fabricated Co(OH)2 nanocages and obtained a current density of 10 mA cm−2 at an overpotential of 280 mV.
4.2 α-Cobalt hydroxide (α-Co(OH)2)
In the subsequent sections the discussion will be focused on the impact of different morphologies of α-Co(OH)2 on the oxygen evolution reactions.Bu et al.130 studied the OER activity of Co3(PO4)2 (CP), Co(OH)2 (CH), Co5(PO4)4OH2 (CPH), and commercial RuO2. The study uses linear sweep voltammetry and Tafel plots to evaluate the performance of the catalysts. The LSV results show that CP and CH have relatively high overpotentials (370 and 360 mV, respectively) at a current density of 10 mA cm−2, whereas CPH and RuO2 have significantly lower overpotentials (254 and 256 mV, respectively) at the same current density illustrated in Fig. 8a. The Tafel plots give further insight into the reaction kinetics. The Tafel plots show that CPH has a smaller Tafel slope (57 mV dec−1) than CH (210 mV dec−1) and CP (246 mV dec−1) in the range of high overpotentials, indicating that CPH is more active for the OER as illustrated in Fig. 8b. This can be attributed to the presence of phosphorus and hydrogen in CPH, which helps to create more active sites and improve the catalytic activity. Analysis describes the reaction mechanism for the OER in Co-based catalysts such as CP, CH, and CPH, which involves bonding interactions within intermediates (M–OH, M–O, and M–OOH) followed by O–O formation. The relatively large Tafel slopes of CH and CP suggest that the rate-limiting step of the OER process is in the first step of this mechanism, where one electron is transferred, which is consistent with previous reports. The small Tafel slope of CPH, on the other hand, suggests that the rate-limiting step is different, indicating that CPH is highly active for the OER. The electrochemically active surface area (ECSA) plays an important role in electrocatalytic reactions. CPH has Cdl of 14 mF cm−2, which is nearly 3.5 times and 2.3 times larger than those of CP (4 mF cm−2) and CH (6 mF cm−2), respectively shown in Fig. 8c. This suggests that CPH has more electrochemical active sites and a larger surface area than CP and CH, which improves its electrocatalytic performance. Additionally, the EIS plots revealed that the ionic and ohmic resistance of CPH is smaller than that of CP and CH, indicating that CPH has better electrical conductivity, which contributes to its excellent OER performance demonstrated in Fig. 8d. Moreover, the ECSA normalized current densities also revealed that the CPH has a lower onset potential as compared with the other samples in the series shown in Fig. 8e. The CPH was further evaluated for a long-term stability test at different current densities such as 10 mA cm−2 and 30 mA cm−2, and it showed excellent stability under alkaline conditions illustrated in Fig. 8f.
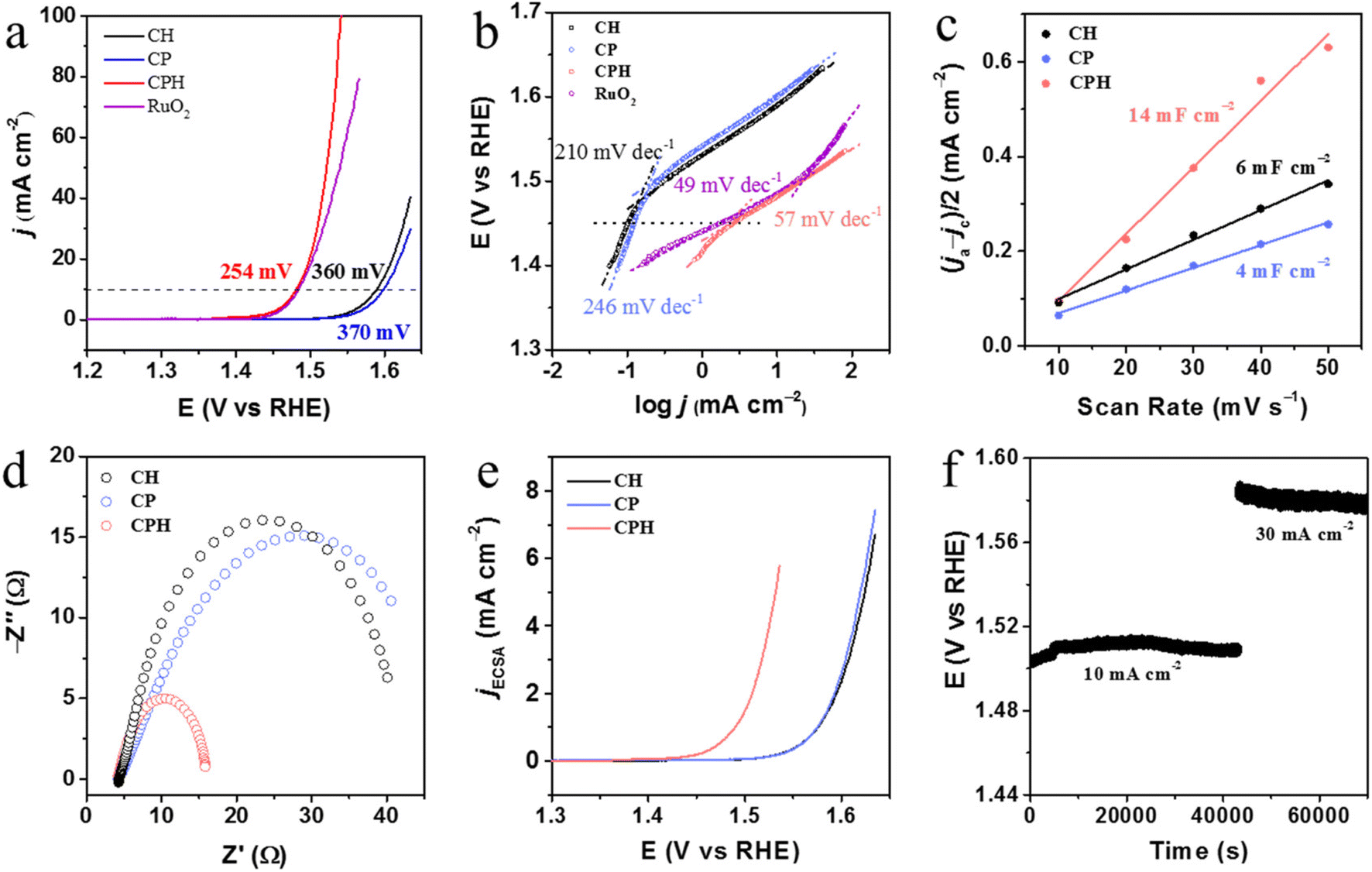 | ||
| Fig. 8 (a) OER polarization curves of CP, CH, and CPH deposited onto glassy carbon electrodes. The sweep rate is 5 mV s−1 in 1 M KOH. (b) Corresponding Tafel slope plots. (c) ECSA evaluation from the plots of current density at 0.15 V vs. RHE. (d) EIS spectra of all samples at an overpotential of 350 mV. (e) Specific current densities of all samples obtained via normalizing the geometric current densities to the ECSA. (f) Stability test of CPH at the current density of 10 mA cm−2 and then 30 mA cm−2. This figure has been reproduced from ref. 130 with permission from American Chemical Society, copyright 2019. | ||
Cui et al.131 investigated the electrocatalytic activity of α-Co(OH)2 nanosheets towards the OER in a 0.1 M KOH solution. The nanosheets were prepared using a MOF-mediated method and were characterized in terms of surface area, electrocatalytic activity, and stability. For comparison, they also investigated samples prepared with urea (Co(OH)2-urea) and with a 4 hour solvothermal treatment (Co(OH)2-4 h) using the same mass loading (0.25 mg cm−2). The results showed that the prepared α-Co(OH)2 nanosheets (Co(OH)2-12 h) had the lowest overpotential of 340 mV at a current density of 10 mA cm−2, while the Co(OH)2-urea had the highest overpotential of 480 mV. The study also found that the Tafel slope of Co(OH)2-12 h was significantly lower than that of Co(OH)2-urea, indicating more favorable reaction kinetics. Furthermore, it was found that the surface area of the samples correlated with their OER activity, with higher surface area samples exhibiting better activity. The study suggests that the well-dispersed structure and highly exposed surface area of the prepared α-Co(OH)2 nanosheets were responsible for their superior OER activity compared to the other samples. The results were supported by measurements of ECSA and EIS. Moreover, the prepared α-Co(OH)2 nanosheets had high stability under OER conditions.
Jash et al.132 investigated the electrocatalytic activity of different materials for the OER in a 1 M NaOH electrolyte solution at pH 13.9. They used CV to measure the activity of the materials and compared them. The exfoliated single layer (SL-Co(OH)2) required the lowest overpotential (350 mV) to achieve a current density of 10 mA cm−2, which is significantly lower than those of α-o(OH)2 (380 mV) and β-Co(OH)2 (437 mV). The Tafel slope also decreased from 85 to 57 mV dec−1 from β-Co(OH)2 to SL-Co(OH)2, further indicating that SL-Co(OH)2 is the best catalyst among this series of catalysts. SL-Co(OH)2 had a remarkable mass activity of 153.8 A g−1 and an intrinsic activity parameter TOF of 0.146 s−1, which were 3.8 and 16.2 times higher for SL-Co(OH)2 than those of α-Co(OH)2 and β-Co(OH)2 respectively. These results suggest that SL-Co(OH)2 is a more active catalyst for the OER. The improved conductivity and exposed metal ions of SL-Co(OH)2 are the reason behind this high catalytic activity, as they provide more active sites to tune the band gap states and support the electron transfer during the OER process.
In an interesting study Huang et al.133 used XAFS to investigate the reaction mechanism of the OER on α-Co(OH)2. In this study they used LSV to measure the OER activity of an α-Co(OH)2 nanosheet in 1 M KOH electrolyte. The α-Co(OH)2 nanosheet had a notably small overpotential of 320 mV at a current density of 10 mA cm−2 for the OER, which is lower than that of the state-of-the-art IrO2 electrocatalyst (overpotential of 340 mV at 10 mA cm−2). The α-Co(OH)2 nanosheet had excellent durability under alkaline conditions and it exhibited good electrochemical stability. To understand the relationship between the structure and activity of the α-Co(OH)2 nanosheet, the researchers used X-ray absorption fine structure (XAFS) measurement to characterize its oxidation state and structure before and after the OER. The results of XAFS measurement suggested that the α-Co(OH)2 nanosheet undergoes a phase change from α-Co(OH)2 to γ-CoOOH during the OER procedure, with an increased oxidation state and proportion of Co sites that contain a formal oxidation state of Co(IV). The results of the XRD pattern and EXAFS data also confirm this phase change. This suggests that the excellent catalytic activity of the α-Co(OH)2 nanosheet originated from the formation of γ-CoOOH, which is facilitated by the presence of O vacancies in the surface Co octahedron during the OER, thus leading to the formation of hydroperoxy (OOH) species.
The detailed analysis of the study done by Jiang et al.134 showed that the electrocatalytic activity of PI/CNT-Co(OH)2 films was investigated in 1 M KOH using a standard three-electrode system. The Co(OH)2 2 min film showed an lower onset potential at around 1.50 V vs. RHE with a rapid increase of OER current. In comparison, the Co(OH)2 products at 0.5, 1, 3, 4, and 5 min had higher onset potentials, and their OER current dropped below that of the Co(OH)2 2 min film. The Co(OH)2 2 min film generated a current density of 10 mA cm−2 at an overpotential of 317 mV, whereas the Co(OH)2 products at other deposition times exhibited more positive overpotentials. The prior peak of Co(OH)2 around 1.05 V was assigned to the Co(II)/Co(III or IV) redox process. In the electrochemical process, Co(OH)2 was first oxidized to CoOOH, and then the CoOOH was further oxidized to CoO2. Both CoOOH and CoO2 can catalyze the OER, with CoO2 being more efficient for the OER. The electrochemical process of Co(OH)2 at the PI/CNT film can be described by the following reactions:
| Co(OH)2 + OH → CoOOH + H2O + e |
| CoOOH + OH → CoO + H2O + e |
The Co(OH)2 2 min film showed the lowest overpotential and the fastest increase of OER current among the products, indicating that the PI/CNT-Co(OH)2 2 min film exhibited better electrocatalytic activity for the OER, mainly determined by the catalytically active sites of Co(OH)2 nanosheets. With the deposition time increasing from 0.5 to 2 min, an increasing number of Co(OH)2 nanosheets were formed on the PI/CNT film, providing a large amount of active sites for the OER. However, further extension of the reaction time (3–5 min) resulted in the aggregation of nanosheets, decreasing the active site density of Co(OH)2 and weakening the OER electrocatalytic activity of Co(OH)2. To obtain further insights into the OER activity, Tafel plots of PI/CNT-Co(OH)2 films at different deposition times were studied. The Co(OH)2 2 min film showed a small Tafel slope of 49 mV dec−1, which is close to the Tafel slopes of well-established OER catalysts Ir/C (40 mV dec−1) and IrO2 (49 mV dec−1). The Co(OH)2 products at 0.5, 1, 3, 4, and 5 min displayed relatively higher Tafel slopes of 87, 70, 57, 65, and 79 mV dec−1.
Liang et al.135 revealed that the ultrathin α-Co(OH)2 nanosheets have a high electrocatalytic activity for the OER due to their unique structure and composition. The ultrathin α-Co(OH)2 nanosheets have a thickness of around 50 nm and a hexagonal plate-like structure, which allows for a large surface area and efficient charge transfer. Additionally, the Tafel slope of the α-Co(OH)2 nanosheets was 64.9 mV dec−1, which is much smaller than that of hexagonal α-Co(OH)2 plates (81.2 mV dec−1) and commercial RuO2 (78.7 mV dec−1), indicating superior reaction kinetics in terms of OER. Furthermore, pH-dependent studies of the α-Co(OH)2 nanosheets revealed that the current density increases with increasing pH values, which indicates that the Co species in the nanosheets are oxidized easily at high concentrations of OH−. This suggests that the ultrathin α-Co(OH)2 nanosheets may have potential applications in pH-sensitive electrocatalytic systems. α-Co(OH)2 nanosheets had good durability, as demonstrated by the CPE test. After galvanostatic conditioning at 1.65 V for about 25 h, the current density was still maintained at around 11 mA cm−2, indicating good stability of the α-Co(OH)2 nanosheets. Overall, the study suggests that the ultrathin α-Co(OH)2 nanosheets have a high electrocatalytic activity for the OER, which is attributed to their unique structure, composition, and pH-dependent behavior. Ultrathin α-Co(OH)2 nanosheets have good durability and stability. These findings may have potential implications for the development of efficient and durable electrocatalysts for water splitting and other electrochemical applications.
Mao et al.136 investigated the electrocatalytic activity of a modified electrode composed of α-Co(OH)2, polypyrrole (PPy), and graphene oxide (GO) for the OER in an alkaline solution. LSV was used to measure the OER activity of the modified electrode and compare it to other modified electrodes. The results showed that the PPy modified α-Co(OH)2 electrode had a much lower onset potential for the OER than other modified electrodes (262 mV compared to 523 mV for the GO-modified GCE), indicating that the addition of PPy significantly improves the activity of the electrocatalyst. Moreover, the electrical conductivity of the as-synthesized pristine PPy powders is 0.6 S cm−1, while the conductivity of pure GO and PPy/GO is 0.5 × 10−4 and 1.2 × 10−4 S cm−1, respectively. This information helps to explain why the addition of PPy improves the conductivity of the modified electrode, allowing for more efficient electron transfer and promoting the OER in the positive direction. Furthermore, they determined that the double-layer capacitance of the α-Co(OH)2/PPy/GO modified GCE is 15.6 mF cm−2, which is higher than that of the α-Co(OH)2 modified GCE (3.53 mF cm−2) and α-Co(OH)2/GO modified GCE (11.0 mF cm−2). This further supports the conclusion that the modified electrode has a higher electrochemically active surface area due to the addition of PPy. Additionally, the Tafel slope of the α-Co(OH)2/PPy/GO modified GCE is 74.5 mV dec−1, much lower than those of GO (387 mV dec−1), PPy/GO (168 mV dec−1), α-Co(OH)2 (94.8 mV dec−1), and α-Co(OH)2/GO (84.3 mV dec−1).
Wang et al.137 reported the OER activity of a composite material made of graphene nanoribbons (GNRs) and α-Co(OH)2. The study evaluated the OER activity of the composite material using a three-electrode system in a 1 M KOH solution. The results show that the GNRs/Co(OH)2 composite had the lowest onset potential of 1.46 V (versus RHE) and an overpotential of 280 mV for reaching a current density of 10 mA cm−2. This is in contrast to the control samples, with a-Co(OH)2 alone showing a larger overpotential of 350 mV at 10 mA cm−2, and GNRs exhibiting a sluggish polarization due to their inertness for the OER process. A combination of GNRs and Co(OH)2 leads to a synergetic effect, with the GNRs providing good electron conductivity and the Co(OH)2 being an active OER catalyst. The study also suggests that the morphology and state of the Co(OH)2 nanoparticles formed on the GNRs play an important role in their OER activity. Specifically, the thin and well-dispersed nanoparticles on the GNRs lead to more exposed surface active sites and better contact with the substrate. This is supported by TEM analysis, which showed that thin and entangled Co(OH)2 nanoparticles are formed on the GNR substrate, while irregular and large Co(OH)2 particles are produced when CNTs are used as the substrate. Furthermore, GNRs/Co(OH)2 displayed a lower slope of 66 mV dec−1, corresponding to the fastest kinetics, compared to CNT/Co(OH)2 (81 mV dec−1), α-Co(OH)2 (109 mV dec−1) and GNRs (287 mV dec−1). EIS measurement was also used to determine the charge transfer resistance (Rct) of the samples, with the GNRs/Co(OH)2 showing the lowest resistance of 24 X, while the resistance of α-Co(OH)2 alone was 150 X. The long-term durability of the GNRs/Co(OH)2 catalyst was also tested by the chronoamperometry method under a constant potential and found to be very stable for the OER process. In conclusion, the results of the study suggested that the GNRs/Co(OH)2 composite material is a highly active and stable catalyst for the OER process, with the synergetic effect between GNRs and Co(OH)2 leading to the enhancement of OER activity. It also highlights the importance of the morphology and state of Co(OH)2 nanoparticles in the OER activity, with thin and well-dispersed nanoparticles on the GNRs leading to more exposed surface active sites and better contact with the substrate. Similar results have been reported by Pan et al.138 that the a-Co(OH)2-NS electrode has a faster electron transfer process during the OER compared to α-Co(OH)2-NF and bare NF, as indicated by the smaller charge transfer resistance (Rct) value. The ECSA of α-Co(OH)2-NS is also higher than those of α-Co(OH)2-NF and bare NF. Additionally, α-Co(OH)2-NS showed good long-term stability and negligible changes in current density at high potentials and high current densities.
Ge et al.92 synthesized benzoate anion containing Co(OH)2 and demonstrated an overpotential of only 291 mV at a current density of 50 mA cm−2 which is far better than that of Co(OH)2 with CO32− as interlayer anions. The enhanced activity was due to the increase in the interlayer spacing because of the large size of the benzoate anion as compared with that of the CO32− anion. We concluded that the increase in the interlayer spacing added two attributes to the Co(OH)2; one is the availability of more exposed active sites and other is the increase in the ECSA evident from the increase in the Cdl of 1.43 mF cm−2 for carbonate intercalated Co(OH)2 to the 1.61 mF cm−2 for benzoate intercalated Co(OH)2. Both of these factors have a significant contribution to the enhanced OER activity. The α-Co(OH)2 nanosheets exhibit promising potential as oxygen evolution reaction (OER) electrocatalysts, holding considerable practical implications across various industries. Their unique structural properties, characterized by ultrathin, single-crystalline features, suggest high efficiency in catalyzing OER processes. These nanosheets could find applications in energy storage systems, particularly in the development of more efficient and sustainable electrochemical devices such as water-splitting technologies. Additionally, their uniformity and well-defined structures pave the way for advancements in catalyst design, enhancing the performance and durability of electrocatalytic systems in industrial settings, driving forward innovations in renewable energy production and storage.
000 s of chronopotentiometric electrolysis testing. The improved performance of α-Co(OH)2 nanomeshes is attributed to their abundant mesopores, which expose more active sites. XANES analysis of the α-Co(OH)2 nanomeshes showed an increase in the Co valence state from Co2+ to Co3+ and the XAFS showed a decrease in intensity for the Co–O peak, indicating more oxygen vacancies in the exposed Co octahedrons.
An interesting study done by Zha et al.140 discussed the use of hollow nano-dodecahedrons of α-Co(OH)2 for OER activity. To synthesize α-Co(OH)2 and Cl-doped α-Co(OH)2 hollow nano-dodecahedrons, the procedure was initiated by obtaining ZIF-67 dodecahedrons in advance, following the methodology outlined in ref. 17 with specific modifications. Initially, a solution comprising 1.43 g (∼4.91 mmol) of Co(NO3)2·6H2O and 3.24 g (∼39.5 mmol) of 2-methylimidazole in 100 mL methanol underwent magnetic stirring at room temperature for 2 hours. The resultant solid product, obtained via centrifugation and subsequent methanol washes, was vacuum dried at 60 °C. Subsequent steps involved the addition of 20 mg ZIF-67 and 150 mg hexamethylenetetramine (HMT) into a 30 mL water–methanol mixture (with a volume ratio of 9:1) in the presence or absence of 50 mg NaCl. Following a 5 minutes stirring period, the system underwent thermal treatment at 40 °C for 4 hours within a Teflon-lined stainless-steel autoclave. After natural cooling to room temperature, a green precipitate was collected, washed thoroughly with deionized water and ethanol, and dried under vacuum conditions at 60 °C. The catalysts were activated by 10 CV cycles at 100 mV s−1. The results showed that the Cl-doped α-Co(OH)2/GCE had the most favorable OER kinetics and the highest electrocatalytic activity among the various catalysts tested, with a Tafel slope of 57 mV dec−1 and a current density of 110.7 mA cm−2. This is due to the transformation from Co(OH)2 to CoOOH that occurs during the activation process. The hollow nano-dodecahedrons were synthesized through a hydrothermal method and were used as a substrate for the catalysts. The hollow structure of the nano-dodecahedrons is composed of a thin wall of the material and a large internal void. This structure provides a large surface area for the catalysts to adsorb and increases the number of active sites available for the electrocatalytic reactions, which in turn enhances the activity. Additionally, the hollow structure allows for a higher mass transport rate for the electrolyte which is important for the OER, since it involves the transfer of oxygen ions from the electrolyte to the electrode. This allows for a more efficient transfer of oxygen ions, leading to a higher current density. Furthermore, the hollow structure also provides a suitable environment for the catalysts to grow on, which can affect the stability and durability of the catalysts. The large internal void can also provide space for the formation of intermediate products and reduce the possibility of poisoning the catalysts, which can improve the stability and durability of the catalysts. Therefore, the use of the hollow nano-dodecahedrons as a substrate for the catalysts not only enhances the electrocatalytic activity but also improves the stability and durability of the catalysts which is important for long-term use. A similar study was performed by Liu et al.141 discussing the use of different types of Co(OH)2 as electrocatalysts for the OER. Different types of Co(OH)2 were synthesized by homogeneous precipitation of dilute CoCl2 solutions with HMT in the presence and absence of NaCl. α-Co(OH)2-Cl, which contains chloride anions, had superior OER performance compared to α-Co(OH)2-DS and β-Co(OH)2, with a small onset overpotential of ∼240 mV and a high current density of 10 mA cm−2 at an overpotential of ∼320 mV. α-Co(OH)2-Cl had a more efficient kinetics for the OER and had the largest active surface area. The enhanced activity of the electrochemically etched α-Co(OH)2-Cl is increased not just by the ECSA, but also by the intrinsic activity after electrochemical etching which plays a more important role. Overall, the study suggests that α-Co(OH)2-Cl is a promising electrocatalyst for the OER in water splitting.
Bose et al.142 described a study on the electrocatalytic OER of a catalyst, Y-CoII(H2O)6, by coating it with carbon on an electrode surface and conducting experiments in 0.1 M KOH (pH 13). The α-Co(OH)2 film, which forms on the zeolite surface in the presence of KOH solution, plays a crucial role in boosting the OER. The structure of the electrode coating was observed using FESEM, which showed that the zeolite particles were well separated from each other, but the surface texture of the zeolite became corrugated with the appearance of α-Co(OH)2 film/sheet. The average size of the particles is approximately 1 mm. The α-Co(OH)2 film plays a crucial role in promoting the OER by providing surface-bound redox-active species. Overall, the α-Co(OH)2 film, which forms on the zeolite surface in the presence of KOH solution, plays a crucial role in boosting the OER by providing a smooth and uniform surface texture of the zeolite, promoting surface-bound redox-active species, and promoting higher-valence Co-species.
Co(OH)(C6H5COO)·H2O nanobelts are a new type of catalyst for the OER and have excellent performance compared to other Co-based catalysts, as well as benchmark catalysts such as RuO2.143 The nanobelts are formed by the incorporation of benzoate anions (C6H5COO−) into the Co(OH)2 lattice, creating a unique catalyst structure. One key factor in the improved OER activity of these nanobelts is the large ECSA. The ECSA of the Co(OH)(C6H5COO)·H2O nanobelts was calculated to be 19.5 μF cm−2, which is significantly larger than that of the Co(OH)2 (13.0 μF cm−2) and Co3O4 (14.0 μF cm−2) counterparts. This large surface area allows for more active sites for the OER to occur which is reflected in the higher OER current observed for the nanobelts. Another important factor is the relatively low overpotential required for the nanobelts to reach a current density of 10 mA cm−2. The overpotential of the nanobelts was 360 mV, which is significantly lower than that of the Co(OH)2 (440 mV) and Co3O4 (387 mV) counterparts. This means that less energy is required to drive the OER, making the nanobelts a more efficient catalyst. The Tafel slope was also found to be lower for the nanobelts (76 mV dec−1) than for the Co(OH)2 (118 mV dec−1), Co3O4 (123 mV dec−1) and Pt/C (128 mV dec−1) catalysts, but slightly higher than that of the benchmark catalyst RuO2 (58 mV dec−1). This indicates that the rate of the OER on the nanobelts is more favorable compared to that on other Co-based catalysts and Pt/C, but slightly slower than that on the benchmark RuO2. Overall, the incorporation of benzoate anions into the Co(OH)2 lattice to form the Co(OH)(C6H5COO)·H2O nanobelts results in a unique catalyst structure with large electrochemical surface area, low overpotential, and favorable kinetics for the OER, making them a promising catalyst for the OER. Similarly, Liu et al.144 described a new method for the synthesis of α-Co(OH)2 nanoplates using Al3+ metal ions and ammonium solution, and the electrochemical performance of these nanoplates as electrode materials for supercapacitors and as OER electrocatalysts was measured. The OER of α-Co(OH)2 nanoplates was found to be significantly better than that of β-Co(OH)2 particles, with a smaller onset potential and faster current increase. The mass activity and TOF of α-Co(OH)2 nanoplates were also found to be much higher than those of β-Co(OH)2 particles. The mass activity of α-Co(OH)2 nanoplates was found to be 28.2 A g−1 at a quite small overpotential of 348 mV, while it was only 0.2 A g−1 for β-Co(OH)2 particles. The overpotential of 449 mV at 10 mA cm−2 for β-Co(OH)2 particles was evidently higher than that of α-Co(OH)2 nanoplates. The α-Co(OH)2 nanoplates also exhibit a linear increase in TOF with overpotential, affording much higher TOFs than β-Co(OH)2 particles. EIS characterization was used to determine that the electronic properties of α-Co(OH)2 nanoplates are related to the improved OER activity. The transport resistance of α-Co(OH)2 nanoplates was found to be decreased.
The following conclusions could be drawn depending on the properties of intercalated anions in α-Co(OH)2.
• Chlorides: chloride ions are known to increase the conductivity of the electrolyte and lower the overpotential for the OER. This is because chloride ions increase the electron transfer rate by facilitating the formation of a stable and conductive film on the surface of the electrode. However, at high concentrations, chloride ions can also lead to corrosion and dissolution of the Co(OH)2, which can negatively affect the performance of the OER.
• Carbonates: carbonate ions are known to improve the OER performance by providing a basic environment that stabilizes the Co(OH)2 and increases the pH of the electrolyte. This can increase the activity of the Co(OH)2 and lower the overpotential for the OER. However, at high concentrations, carbonates can also lead to carbonate-induced dissolution of the Co(OH)2, which can negatively affect the performance of the OER.
• Nitrates: nitrate ions are known to increase the conductivity of the electrolyte and lower the overpotential for the OER. This is because nitrate ions increase the electron transfer rate by facilitating the formation of a stable and conductive film on the surface of the electrode. However, at high concentrations, nitrate ions can also lead to corrosion and dissolution of the Co(OH)2, which can negatively affect the performance of the OER.
• Phosphates: phosphate ions are known to improve the OER performance by increasing the stability of the cobalt hydroxide in aqueous environments and providing a basic environment that stabilizes the Co(OH)2 and increases the pH of the electrolyte. This can increase the activity of the Co(OH)2 and lower the overpotential for the OER.
• Benzoates: benzoate ions are known to improve the OER performance by increasing the stability of the cobalt hydroxide in aqueous environments and providing a basic environment that stabilizes the Co(OH)2 and increases the pH of the electrolyte. This can increase the activity of the Co(OH)2 and lower the overpotential for the OER.
It is important to note that the specific effects of these interlayer anions on the OER in α-Co(OH)2 will depend on the concentration of the anions and the specific conditions of the reaction. Additionally, the effect of interlayer anions on the OER can be different depending on the type of Co(OH)2, the surface area, particle size and morphology of the Co(OH)2, and the potential and temperature of the reaction. Therefore, it is important to conduct a detailed study of the OER in the presence of these interlayer anions to understand their specific effects and optimize the performance of the reaction. It is also important to note that this information is based on the current research available, and new discoveries and developments may occur in the future which may change the current understanding of the specific properties and trends.
4.3 Cobalt oxyhydroxide (CoOOH)
In the subsequent sections we will discuss the impact of different morphologies of α-Co(OH)2 on the oxygen evolution reactions. | ||
| Fig. 9 (a) Polarization curves of CoOxHy/CC, CoOxHy-NO/CC, RuO2/CC and CC in 0.1 M PBS (iR-corrected, 2 mV s−1). (b) The corresponding Tafel plots. (c) Capacitive current densities at 1.25 V versus RHE as a function of scan rate for CoOxHy/CC, CoOxHy-NO/CC and RuO2/CC. (d) CVs of CoOxHy/CC and CoOxHy-NO/CC in 0.1 M PBS (iR-corrected, 5 mV s−1). This figure has been reproduced from ref. 145 with permission from Royal Society of Chemistry, copyright 2019. | ||
Chen et al.146 showed that the Co/PANI HNSs are a hierarchical porous hybrid material composed of cross-linked PANI and ultrathin CoOOH nanosheets. They were fabricated by combining a wetness-impregnation method and a subsequent electrochemical process. LSV was performed to evaluate the OER activities of PANI, CoOOH, and Co/PANI HNSs. The results showed that the Co/PANI HNS catalyst has the lowest onset potential and highest anodic current density among the three samples, even better than those of the reference catalyst of IrO2. The Co/PANI HNSs only require 291 mV overpotential to reach a current density of 10 mA cm−2, significantly lower than those of PANI (474 mV), CoOOH (407 mV), and even IrO2 (344 mV). The catalytic performance of Co/PANI HNSs is comparable or even superior to that of some recently reported state-of-the-art OER electrocatalysts based on CoOOH-type active species under alkaline conditions. The exceptional performance is attributed to the hierarchical porous hybrid material composed of cross-linked PANI and ultrathin CoOOH nanosheets, which is conducive to the adsorption, activation, and conversion of the reactants and also the large ECSA of the material. The CoOOH nanosheets are strongly bound to PANI through the formation of Co–N coordination bonds, which act as a structural bridge between the two components. This not only improves the stability of the structure but also reduces the resistance to charge transfer at the interface. The interaction between Co d-orbitals and PANI's π-conjugated ligands in Co/PANI HNSs allows for efficient electron transfer from CoOOH to PANI, which improves the adsorption energy of reactants and products during the OER process. Additionally, the presence of mesopores and macropores in Co/PANI HNSs allows for easy access of electrolytes to active sites, leading to faster reaction kinetics. These factors contribute to the exceptional performance of Co/PANI HNSs as an OER electrocatalyst.
Meng et al.147 fabricated laser induced oxygen vacant CoOOH for performing the OER. LSV curves were obtained at a scan rate of 5 mV s−1, and bulk CoOOH (B-CoOOH) and commercial RuO2 were used as references. The results showed that L-CoOOH required a lower overpotential of 330 mV to reach a current density of 10 mA cm−2, which is better than those of RuO2 (340 mV) and BCoOOH (410 mV). Additionally, the Tafel slope of L-CoOOH (63.2 mV dec−1) is lower than that of B-CoOOH (75.4 mV dec−1) and close to that of RuO2 (64.9 mV dec−1), indicating favorable OER kinetics for L-CoOOH. After 10 hours of testing, RuO2 only maintained 69% of its initial current density, while L-CoOOH retained 95% of its initial current density. The morphology and composition of L-CoOOH remained unchanged after the OER durability test. The Cdl value of L-CoOOH was found to be 7.4 times higher than that of B-CoOOH, indicating the existence of more active sites in L-CoOOH. The electrical conductivity of L-CoOOH was also higher than that of B-CoOOH due to a smaller Rct. Finally, the study used X-ray photoelectron spectroscopy (XPS) to explore the surface valence state of L-CoOOH and B-CoOOH. The XPS results showed that L-CoOOH had a higher ratio of Co2+ to Co3+ than B-CoOOH, which was attributed to the increased active sites and faster electron transfer in L-CoOOH. Overall, it is concluded that L-CoOOH is a promising OER catalyst with excellent performance and durability and that its superior performance is due to the increased active sites and faster electron transfer.
The research conducted by Wang et al.148 described a new method for synthesizing hierarchical ultrathin sheet-based CoOOH hollow nanospheres using a room-temperature self-template strategy. The researchers used Pi-doped Co-glycerate hollow nanospheres as the self-sacrificing templates, which were then converted to CoOOH hollow nanospheres through an outside-in pathway involving ionization, anion-exchange reaction, and oxidation in an alkaline environment. To maintain the spherical structure, the precursors were coated with a Nafion membrane, which was easily removed by washing with ethanol. The resulting CoOOH hierarchical hollow nanospheres have several desirable properties, including ultrathin nanosheets and abundant oxygen vacancies, which provide abundant catalytic active sites, shorten charge/ion diffusion paths, reduce charge-transport resistance, and enhance the stability for the OER. Therefore, the CoOOH hierarchical hollow nanospheres obtained by this method have extraordinary electrocatalytic activity and good stability for the OER. The hierarchical CoOOH hollow nanospheres have been shown to have high catalytic activity in the OER, with an overpotential as low as 275 mV at a current density of 10 mA cm−2. It is superior to the γ-CoOOH nanosheets (overpotential of 304 mV) and the bare Ni foam (overpotential of 362 mV) at the same current density. The hierarchical CoOOH hollow nanospheres also have lower charge-transfer resistance and faster charge transfer kinetics than the γ-CoOOH nanosheets. Additionally, the hierarchical CoOOH hollow nanospheres have a higher electrochemical double-layer capacitance (22.6 mF cm−2) than the γ-CoOOH nanosheets (4.6 mF cm−2), indicating more active sites on the surface of the hierarchical CoOOH hollow nanospheres. The hierarchical CoOOH hollow nanospheres also showed long-term stability, with no change in polarization curves even after 1000 cycles. However, the crystalline structure of the hierarchical CoOOH hollow nanospheres was not preserved after the OER test.
Ye et al.149 investigated the electrocatalytic activity of a material termed Fe0.33Co0.67OOH PNSAs/CFC as an oxygen evolution reaction (OER) catalyst. The material exhibited the highest OER electrocatalytic activity among the materials tested, which included commercial IrO2 coated on Ni foam (IrO2/NF) and CoOOH PNSAs/CFC. The Fe/Co molar ratio in the Fe0.33Co0.67OOH PNSAs/CFC material was controlled by adjusting the time of in situ anodic oxidation of α-Co(OH)2 in a solution of 0.01 M (NH4)2Fe(SO4)2. The study found that the Fe/Co molar ratio in the material can be increased to about 1:2 and remains constant after 30 minutes, and the catalysts composed of Fe/Co = 0.493 (Fe0.33Co0.67OOH PNSAs/CFC) exhibit the highest OER electrocatalytic activity. The Fe0.33Co0.67OOH PNSAs/CFC material required a much lower overpotential than the other materials tested, specifically, 266 mV at 10 mA cm−2, whereas CoOOH PNSAs/CFC and IrO2/NF catalysts required overpotentials of about 331 and 290 mV, respectively. The Fe0.33Co0.67OOH PNSAs/CFC also showed much higher mass electrocatalytic activity for the OER than CoOOH PNSAs/CFC and IrO2/NF. The Tafel slope of Fe0.33Co0.67OOH is only about 30 mV dec−1, which is much lower than those of CoOOH PNSAs/CFC (56.4 mV dec−1) and IrO2/NF (77.2 mV dec−1). The EIS of Fe0.33Co0.67OOH PNSAs/CFC and CoOOH PNSAs/CFC, and the corresponding circuit model fitting analysis were also performed. The EIS results showed that the Rct of Fe0.33Co0.67OOH PNSAs/CFC was smaller than that of CoOOH PNSAs/CFC, suggesting a fast electron transfer is realized in Fe0.33Co0.67OOH PNSAs/CFC, which would expedite the OER and reduce the overpotential due to its high conductivity. The study also measured the TOF of the electrocatalysts, which showed that the TOF of Fe0.33Co0.67OOH PNSAs/CFC was much higher than that of CoOOH PNSAs/CFC, further illustrating the improved activity of the material.
Bergmann et al.150 used DFT calculations to explain the change in site occupancy of Co2+ Td ions to empty Oh sites with the μ-O vacancy formation in the reaction zone during the OER, as calculated for the Co3O4(100) surface. Oxygen vacancies also probably migrate into the subsurface; thus, the authors consider this process to induce the restructuring of the near-surface at elevated O2 evolution rates. Oxygen vacancy formation on the Co3O4(110) surface has less impact on the local atomic structure. XANES was used to determine the change in the cobalt K XANES spectra, which suggests oxidation of the metal ion to Co4+. The lower charge transfer energy in Co3+/4+Ox(OH)y also allows the formation of hole density on the oxygen ligand, as identified for IrOx(OH)y. Tafel slopes decreased as the mean electrochemical reducibility of the Co3+–O sites increased, providing evidence that this reducibility is closely correlated with favorable electrocatalytic properties. Better electrocatalytic properties with a more pronounced Co2+/Co3+ redox transition was also identified under phosphate-free, alkaline reaction conditions. Additionally, the redox charge of Co2+/Co3+ is a better descriptor of the OER active site density than the overall number of redox sites. In summary, the study found that the structural transformation of Co2+ oxides into Co3+ Oh-containing CoOx(OH)y during OER conditioning occurred in the near-surface region, leading to a structurally unified catalytically active state with a higher mean cobalt oxidation state and a contracted CoO6 bonding environment. DFT calculations showed that this transformation was caused by the formation of oxygen vacancies in the reaction zone. The electrocatalytic properties of CoOx(OH)y were found to be correlated with the near-surface electronic structure and degree of defects. The Tafel slopes were found to decrease as the mean electrochemical reducibility of the Co3+–O sites increased, indicating that this reducibility is closely correlated with favorable electrocatalytic properties.
Cui et al.151 studied the promotion of the OER via the synergistic effect of coupling CoOOH with MnOOH. The MnOOH is a well-known OER catalyst because of its high activity and stability. It has been shown to have a low overpotential and small Tafel slope, indicating fast OER kinetics. However, the activity of MnOOH can be further improved by synergistically coupling it with other catalysts, such as cobalt oxide (CoOOH). B-MCO−0.1 catalyst was found to have a lower overpotential (η10 = 313 mV) and smaller Tafel slope (87 mV dec−1) compared to control samples such as γ-MnOOH (494 mV and 122 mV dec−1) and CoOOH (420 mV and 140 mV dec−1). This suggests that the interfacial interaction between the γ-MnOOH and CoOOH particles in the B-MCO−0.1 catalyst results in faster OER kinetics. The study also found that the B-MCO−0.1 had the highest mass activity (4.3 A mg−1) and TOF (50.3 s−1) among the control samples. These results indicate that the interfacial interaction between γ-MnOOH and CoOOH in the B-MCO−0.1 catalyst not only improves the activity, but also the stability of the surface species. One possible mechanism for the synergy between MnOOH and CoOOH is that the CoOOH particles act as a conductive matrix, facilitating electron transfer during the OER process. The γ-MnOOH particles, on the other hand, provide a large number of active sites for the OER to occur. The strong interfacial interaction between these two catalysts results in a higher number of active sites per unit area and faster electron transfer, leading to improved OER activity. Additionally, as seen in the study, the B-MCO−0.1 also showed the biggest Cdl value (11.5 mF cm−2) and the largest ECSA among the control samples, indicating more accessible active sites in the OER; both of these factors also contribute to the improved activity of the B-MCO−0.1 catalyst.
In a similar study, Xu et al.152 evaluated the OER electrochemical performance of a NiSe@CoOOH core–shell structure as a 3D integrated electrode in a 1 M KOH aqueous electrolyte. The NiSe@CoOOH core–shell structure was found to have superior OER activity compared to bare Ni foam, NiSe NWs/NF, and RuO2/NF electrodes. The NiSe@CoOOH core–shell structure had a sharp onset potential at 217 mV with greatly enhanced OER current, requiring an overpotential of only 292 mV to reach 100 mA cm−2. The Tafel slope of the NiSe@CoOOH core–shell structure electrode was approximately 65 mV dec−1, which is lower than that of the other electrodes tested, indicating that the core–shell structure accelerates the catalytic kinetics for the OER. The authors suggest that the as-prepared NiSe@CoOOH/NF provides the real catalytic mediator CoOOH nanosheets with more surface-active sites for the OER and the core–shell structure accelerates the charge transfer during the OER. The unique structure of the NiSe core and the coating with 200 nm CoOOH nanosheets may improve the structural stability and accelerate fast electron transfer which is beneficial for fast transportation of electrons from the NiSe core to the CoOOH shell. Additionally, the stability of the electrocatalyst was found to be good, showing consistent results even after 12 hours of electrolysis.
Moreover, Liu et al.153 put in some effort to unveil the role of Co3+ in the OER with CoOOH based materials. The OER was evaluated in an O2-saturated 1 M KOH (pH ∼ 14). The results of LSV measurements with a sweep rate of 5 mV s−1 showed that the Co3+-33 h sample exhibited prominently enhanced catalytic activity for the OER compared to other Co3+ samples and β-Co(OH)2. The low overpotential of 395 mV at 10 mA cm−2 indicated its good catalytic activity towards the OER and is superior to many recently developed catalysts. The enhanced activity was attributed to the unique Co3+-rich surface composition (Co(OH)2/CoOOH) of Co3+-33 h, in which the Co3+ ions served as the active sites for the OER. The varied charge distribution, as evidenced by the negative shift of Co 2p3/2 in Co3+-33 h compared with Co(OH)2, might result in a lower energy barrier for the adsorption of H2O during water oxidation. Furthermore, the varied catalytic activity between Co3+-24 h, Co3+-33 h, and Co3+-42 h can be related to the content of Co3+ on the surface, as revealed by the XPS analysis. The constant current activation time increased, more active Co3+ ions were generated and the content of Co3+ reached a maximum after activation of 33 h. In summary, the enhanced OER activity of the Co3+-33 h sample was due to the unique Co3+-rich surface composition (Co(OH)2/CoOOH) of Co3+-33 h, in which the Co3+ ions served as the active sites for the OER, and the varied charge distribution, which resulted in a lower energy barrier for the adsorption of H2O during the OER, leading to improved OER performance.
Reikowski et al.154 discussed the preparation of pure phase epitaxial Co3O4(111) and CoOOH(001) thin layers on Au(111) through electrodeposition. The stability of the CoOOH(001) layer and the restructuring of the Co3O4(111) spinel oxide near the surface region are discussed, as well as the catalytic properties of both layers. The stability of the different cobalt oxides is defined by the following redox reactions:
| 3Co(OH)2 + 2OH− → Co3O4 + 4H2O + 2e− |
| Co3O4 + OH− + H2O → 3CoOOH + e− |
| CoOOH + OH− → CoO2 + H2O + e− |
Conclusively, this study highlighted the stability and structural changes of the CoOOH(001) thin layer on Au(111) when subjected to different potentials. According to thermodynamic, the CoOOH film exhibits a wide stability range and does not exhibit structural changes down to 0.4 V. The stability of the CoOOH layer is attributed to its smooth surface, which is composed of close-packed (001)-terminated terraces. The absence of structural changes in the OER range suggested that the CoOOH lattice remains unchanged in this range and the natural (001) cleavage plane is stable and active during the OER. DFT calculations support this observation, showing that the CoOOH(001) surface termination gradually changes from OH to O in this potential regime, and reaches a fully O-terminated surface at 1.5 V vs. RHE. Additionally, DFT calculations also exclude the formation of bulk crystalline CoO2, even at high potentials where its formation should be thermodynamically favorable. These findings provide a deeper understanding of the stability and activity of the CoOOH(001) thin layer as a potential electrocatalyst in OER applications. Potential-dependent restructuring of Co3O4 film surfaces refers to the changes in the surface structure of a Co3O4 film that occur at different applied potentials. The article discussed the formation of a “skin layer” on the surface of the Co3O4 film that occurs at potentials between 1.2 and 1.67 V. This skin layer is a reversible, ≈1 nm thick layer that forms on the top and side walls of the Co3O4(111) grains. The formation of this skin layer is not directly related to the catalytic reaction (OER) as previously assumed, but rather reflects the thermodynamics of the Co3O4/CoOOH phase transition. It is proposed that the formation of this skin layer is caused by the displacement of Co2+ cations from tetrahedral to octahedral symmetry, as the Co oxidation state increases from Co2+/Co3+ to Co3+ for potentials above 1.2 V, which is well below the thermodynamic limit of the OER required to initiate the reaction. Absolutely, the formation of this “reversible skin layer” seems to be linked more to the chemical changes within the oxide itself rather than being directly tied to the OER. This means that the transformation of the material from, say, Co3O4 to CoOOH and back, which forms this skin layer, is happening independently or prior to the onset of the OER process.
The charge transfer that occurs during this specific chemical transformation contributes significantly to what's known as pseudocapacitive current. This current, observed before the OER, stems from this transformation process within the material rather than the electrochemical reactions involved in oxygen evolution. Understanding and harnessing this reversible skin layer formation could potentially lead to advancements in pseudocapacitive-based energy storage or electrochemical devices. This restructuring requires significant transport of oxygen and Co ions through the oxide lattice, which can occur rapidly due to the low thickness of the skin layer. The skin layer does not form on a perfectly smooth Co3O4(111) surface but rather on nanoscale 3D islands that expose other facets to the electrolyte, which may facilitate the layer formation by providing channels for cation migration within the oxygen framework. The catalytic properties of epitaxial samples of Co3O4(111) and CoOOH(001) films indicate that a smooth epitaxial CoOOH(001) layer is nearly as good a catalyst as a Co3O4(111) layer, which is in contrast with general assumptions in the literature. The study also suggests that the OER active sites in the films are di-μ-oxo-bridged ion clusters in amorphous CoCat catalyst films and μ3-O/OH sites on the (001) plane of CoOOH. The findings of the study may have significant implications for the understanding of the nature of OER active sites on Co oxide catalysts and suggest that well-defined epitaxial films may be useful in future studies. In conclusion, the electrochemical charge (Q0) measured in the pre-OER region for CoOOH(001) is attributed to adsorption/desorption on the oxide surface, specifically protonation/deprotonation of the outermost oxygen atoms on the top surface plane and within the grain boundaries. In the case of Co3O4, the charge Q0 corresponds to a charge transfer that involves the bulk of the Co3O4 epitaxial film. This is supported by optical reflectivity measurements and previous observations for CoCat. The changes in the bulk charge state are linked to reversible changes in unit cell volume. However, the nature of the charges associated with the substantial pseudocapacitive charge/discharge in Co3O4 is still not clear, and more work is necessary to check the hypothesis of intercalation of solution species. It is important to note that the current in the pre-OER range cannot be easily employed to estimate the electrochemical surface area or redox-active surface sites, as dividing the pseudocapacitance of the oxide by a standard double layer capacitance leads to an erroneous ECSA that is several orders of magnitude larger than derived from independent structural characterization.
The mechanism proposed by the group of Xile Hu155 in Fig. 10 for the OER on Co-based catalysts is consistent with spectroscopic and electrokinetic data. The mechanism suggests that the oxidation of Co(III) to Co(IV) is reconciled by considering a di-Co active site, where the oxidation of the first Co(III) ion is accompanied by a loss of 2 protons, and the oxidation of the second Co(III) ion is accompanied by a loss of 1 proton. This results in the formation of a Co(IV)–O˙ intermediate, which is proposed to be in equilibrium with a bridging Co-superoxide-Co species. The mechanism proposed in Fig. 10 predicts that the concentration of the superoxide species would increase with increasing potentials, which is supported by the observed data. The potential-determining step is proposed to be the oxidation of the second Co(III) to Co(IV) ion. It is different from previous mechanisms proposed for Co-based catalysts, which mostly invoke Co(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or Co(III)–O˙ as the highest valence Co species responsible for O–O bond formation.
O or Co(III)–O˙ as the highest valence Co species responsible for O–O bond formation.
 | ||
| Fig. 10 Proposed OER mechanism. This figure has been reproduced from ref. 155 with permission from American Chemical Society, copyright 2020. | ||
• The first step in the proposed mechanism is the oxidation of the first Co(III) ion, which is accompanied by a loss of 2 protons. This results in the formation of a Co(III)-(μ-O)2-Co(IV) species. The oxidation of the second Co(III) ion is accompanied by a loss of 1 proton and results in the formation of a Co(III)-(μ-O)2Co(IV)-OH species. These two steps represent the proton-coupled electron transfer (PCET) steps in the mechanism.
• The next step in the mechanism is the oxidation of the resting state CoO2 by 1 electron. This occurs at the O anion to give a Co(IV)–O˙ species, which is proposed to be in equilibrium with a bridging Co-superoxide-Co species. This species is the active oxygen species observed in the Raman spectra. The oxidation of the resting state CoO2 to the Co(IV)–O˙ species is considered a pre-equilibrium.
The rate-determining step (RDS) in the mechanism is the release of O2 from the Co-superoxide-Co species with a concomitant H2O binding. This step is followed by the binding of another H2O and further 1-electron oxidation, which regenerates the starting Co(III) form of the catalyst (CoOOH). This sequence would give a Tafel slope, a first order in [OH−], as well as D-EIE and -KIE that agree with experimental values. The proposed mechanism is consistent with previous experimental and theoretical studies, which show that the formation of Co(IV)–O˙ is necessary for O–O bond formation catalyzed by various small Co-oxide fragments. A theoretical study suggested that assuming 4 consecutive PCETs as the mechanism of OER, the most active site of crystalline β-CoOOH has a resting state of Co(III) instead of Co(IV). However, such a mechanism would not apply to this system as the electrokinetic data cannot be fit by 4 PCETs. It is worth noting that dioxygen release as a rate-determining step in the OER has not been proposed for Co-based catalysts, but it does have several precedents in other types of catalysts.
4.4 Doped-cobalt hydroxides as OER electrocatalysts
In recent years, researchers have been exploring different doping strategies to optimize the performance of doped cobalt hydroxide catalysts for the OER. For example, alloying cobalt hydroxide with other metals has been shown to improve the catalytic activity and stability, and using different synthetic methodologies can result in different morphologies which can affect the catalytic properties. Furthermore, doped cobalt hydroxide can be used under both acidic and basic conditions, which makes it versatile for different types of applications. Also, it has a good performance at both low and high current density, this feature makes it suitable for different types of devices.The study performed by Zhang et al.156 discusses the evaluation of the catalytic performance of CCHH-based electrocatalysts for the oxygen evolution reaction (OER) in 1 M KOH using a three-electrode system. One of the main findings of the study is that the catalytic activities were greatly boosted after Fe doping. The optimal Fe-CCHH/NF-30 exhibited the best OER performance, requiring a much smaller overpotential of 200 mV to deliver a current density of 10 mA cm−2. This means that it requires less energy to drive the reaction to the same current density compared to other catalysts, making it more efficient. The OER activity of Fe-CCHH/NF-30 was also found to be better than that of the commercial RuO2 with the same loading, and this is quite competitive when compared to other CCHH-based or Co-based catalysts. The study suggests that the superior OER performance of Fe-CCHH/NF-30 is due to the tailored electronic structure that favors the reaction kinetics. The XPS spectrum of Co 2p demonstrates a noticeable negative shift upon doping with Fe, indicating a lower oxidation state of Co compared to its undoped counterpart. This phenomenon contributes significantly to the in situ conversion of Co to higher oxidation states during electrochemical investigations, thereby augmenting its overall activity. The tailored electronic structure is likely a result of the atomically doped Fe, which can greatly enhance the intrinsic activity of CCHH. It is an important finding, as it suggests that single atom catalysts may be more effective than traditional catalysts that rely on multiple atoms to drive the reaction. The study also suggests that the approach of in situ electrochemical transformation may be a promising strategy to design single atom catalysts with high activity and stability. In situ electrochemical transformation involves introducing the dopant atoms (in this case Fe) during the electrocatalyst synthesis process, rather than adding them afterward. This allows for better control over the electronic structure of the catalyst, which in turn leads to better performance. Finally, the Fe-CCHH/NF-30 had excellent durability under long-time stability and large current density conditions. This is an important factor for practical applications, as catalysts that degrade quickly or lose activity over time would not be useful in real-world settings. However, further research is required to understand the potential of single atom catalysts in real-world applications and how to design these catalysts for better performance and stability.
Jin et al.157 suggested that the incorporation of Fe into Co(OH)2 plays a crucial role in promoting the OER. The catalytic ability of Fe-incorporating Co(OH)2 (α-Co4Fe(OH)x) is notably higher than that of pure Co(OH)2 (α-Co(OH)2). This indicated that Fe promotes the OER by increasing the catalytic activity of Co(OH)2. Additionally, the polarization curve of α-Co4Fe(OH)x revealed a small onset potential of 0.26 V for the OER, indicating that the Fe-incorporating Co(OH)2 catalyst is able to initiate the OER at a lower potential compared to pure Co(OH)2. This suggests that Fe may be facilitating the OER by making it easier for electrons to transfer to the catalytic sites, leading to a lower activation energy for the reaction. Furthermore, the α-Co4Fe(OH)x catalyst also achieved a high TOF of 0.027 s−1 at an overpotential of 0.3 V, which is 11 and 5 times higher than those of α-Co(OH)2 and IrO2, respectively. This suggests that the Fe incorporation increases the rate of OER by increasing the number of active sites available for the reaction to occur. In summary, the study suggested that Fe plays a significant role in promoting the OER by increasing the catalytic activity, decreasing the onset potential, and increasing the turnover frequency of the Co(OH)2 catalyst.
The LSV curves of the Co(OH)2 and NixCoy(OH)2 materials were measured at a scan rate of 10 mV s−1 by Wang et al.158 The enlarged LSV curve demonstrates the oxidation peaks of CoII/CoIII (∼1.15 V vs. RHE) and CoIII/CoIV (∼1.43 V vs. RHE). When doping the Ni in this system (15%), the OER activity increases (overpotential = 383 mV). The Ni0.25Co0.75(OH)2 exhibits the best OER performance with an overpotential of 352 mV. This is because the Ni0.25Co0.75(OH)2 has the highest intrinsic activity for the OER, which is likely due to the specific electronic structures that are beneficial for the OH− adsorption. The Ni doping leads to a positive shift of the oxidation peak of Co species, and the Ni0.25Co0.75(OH)2 has the largest CoII/CoIII oxidation peaks compared to other samples. This suggests that Ni doping causes a change in the electronic structure of Co atoms, leading to the difference in OER behavior. Furthermore, the voltammetry of NixCoy(OH)2 shows a strong dependence of the CoII/III potential on the Ni content, suggesting a strong electronic coupling between Ni and Co in the solid. The Ni0.25Co0.75(OH)2 has the largest number of tri- and tetra-valent metal cations on the surface of the nanosheet material before the occurrence of the OER. Therefore, the Ni0.25Co0.75(OH)2 has the smallest overpotential since these high-valence Co cations are regarded as active sites of the OER. This indicates that Ni0.25Co0.75(OH)2 has a much larger active surface area compared to other samples. In summary, the doping of Ni into Co-based hydroxide materials enhances the OER activity by changing the electronic structures of Co atoms, leading to a positive shift of the oxidation peak of Co species and a stronger electronic coupling between Ni and Co. Similar results on the role of Ni in boosting the OER of Co(OH)2 could be found in the study by Guo et al.159
In a similar study Zeng et al.160 suggested that the addition of iron and nickel to cobalt hydroxide (Co(OH)2) improved the performance of the material as an electrocatalyst for the OER. The LSV data from the study suggests that the Co–Ni–Fe511 compound nanosheets have a higher polarization current and lower overpotential compared to Co(OH)2 and also show better performance than Co–Ni511 and Co–Fe511. The Tafel plots also indicate that the incorporation of Fe and Ni results in a more rapid OER rate and a smaller Tafel slope. Additionally, the electrochemical impedance spectroscopy results suggest that the Co–Ni–Fe511 catalyst has faster electrode kinetics and higher faradaic efficiency. Overall, it is suggested that the synergistic effect of Fe and Ni dopants enhances the OER activity of cobalt hydroxide.
Cao et al.161 synthesized α-Co(OH)2 using different solvents and also doped it with different transition metal ions (Ni2+, Cu2+, and Fe2+) to study their effect on the electrochemical performance of the material as an OER electrocatalyst. They used CV and LSV to measure the OER performance of the different samples. The results show that α-Co(OH)2 synthesized in pure water exhibited poor OER performance, while the addition of Cu2+ or Fe2+ significantly reduced the overpotential, with the best performance being obtained when the samples contained 30% Cu2+ or 20% Fe2+ in a mole. In contrast, doping with Ni2+ did not improve the performance. However, it also contrasts with the initial studies where doping with Ni enhances the OER activity. They also found that Fe–Co(OH)2 not only had the lowest overpotential but also had excellent stability and the highest TOF value which was 3 times higher than that of α-Co(OH)2. Additionally, Fe–Co(OH)2 had the lowest Tafel slope, indicating its quicker electrocatalytic kinetics and higher OER electrochemical catalytic activity.162 Furthermore, EIS was conducted to evaluate the electron transport ability and the results show that Fe–Co(OH)2 and Cu–Co(OH)2 had smaller Rct values, indicating more efficient charge transport during the electrochemical OER process than α-Co(OH)2. A similar study on the role of Cu doping to increase the OER activity of Co(OH)2 is reported by Chen et al.163
In an interesting study, electrochemical tests of pure and modified CoOOH nanowires were performed by Chen et al.164 The results showed that the maximum current density of NiCoOOH-A (99% greater than pure CoOOH) and the potential required to attain 10 mA cm−2 (decreased by 20 mV) were improved when compared to those of pure CoOOH. When the surface Ni concentration was increased, the OER activity also increased. Specifically, at a concentration of 9.7% Ni, the OER activity increased to 400% at 1.55 V when compared to pure CoOOH. However, when the surface Ni concentration was increased to 15%, the OER activity decreased from the optimal value. It was also observed that Ni–CoOOH–C with 15% surface Ni has lower activity than 5.5% Ni but exhibits higher activity than pure CoOOH. A comparison of the OER activity of Ni–CoOOH samples to NiOxHy nanoparticles illustrates that Ni-modification of CoOOH creates more active OER sites than does CoOOH or NiOxHy alone. On the other hand, Mn modification showed no improvement or decrease in the OER activity of pure CoOOH at all levels of Mn incorporation. The apparent lack of influence of Mn incorporation on the OER activity can be explained by the formation of both inactive Mn4+ and active Mn3+ ions from Mn incorporation. It was recently suggested that accidental Fe doping may have been the reason for the improved OER activity of certain Co/Ni oxide or oxyhydroxide catalysts. However, the authors of this study believe that the trends in OER activity reported here are due to Ni and Mn incorporation as opposed to accidental Fe doping. Inspection of the Tafel regions in LSV of Ni– and Mn–CoOOH samples revealed a decrease in the Tafel slope for Ni–CoOOH compared to pure CoOOH, specifically from 53 mV dec−1 to 36 mV dec−1. However, no change was observed upon Mn modification.
Nevertheless, a contradictory study on Mn doping in CoOOH for boosting the OER was done by Huang et al.165 The research presents the water oxidation performance of electrocatalysts made of Mn-confined CoOOH nanosheets. The electrocatalysts were evaluated using steady-state electrochemical measurements in 1 M KOH solution, specifically cyclic voltammetry (CV) and polarization curves. Mn-doped CoOOH nanosheets showed improved OER performance compared to pure CoOOH nanosheets. The onset potential for the OER was 195 mV for the Mn-doped CoOOH nanosheets, while it was around 300 mV for pure CoOOH nanosheets. This means that the Mn-doped CoOOH nanosheets required less energy to initiate the OER. The anodic current of the Mn-doped CoOOH nanosheets was also higher than that of pure CoOOH nanosheets at a low overpotential of 255 mV, where it reached 10 mA cm−2. This is in contrast to pure CoOOH nanosheets, which required a larger overpotential of around 300 mV to reach the same current density. This data suggests that the Mn-doping effectively boosts the catalytic activity of CoOOH nanosheets for the OER. Furthermore, the Tafel slopes of the Mn-doped CoOOH nanosheets were lower than those of pure CoOOH nanosheets and IrO2, which means that the rate of OER is faster for Mn-doped CoOOH nanosheets. The Tafel slope for 2% Mn-doped CoOOH nanosheets is 38 mV dec−1 and 33 mV dec−1 for 5% Mn-doped CoOOH nanosheets, whereas it is 40 mV dec−1 for pure CoOOH nanosheets and 52 mV dec−1 for IrO2. Electrochemical double-layer capacitance (Cdl) results also showed that the Mn-confined CoOOH nanosheets possess larger Cdl than pure CoOOH nanosheets. The largest Cdl of 5% Mn-doped CoOOH nanosheets (8.52 mF cm−2) is twice that of pure CoOOH nanosheets (3.38 mF cm−2) as obtained from the Cdl results, confirming more effective surface area and better charge storage capacity for Mn-doped CoOOH nanosheets. Overall, the data suggest that Mn-doping effectively boosts the OER performance of CoOOH nanosheets by lowering the onset potential, increasing the anodic current at low overpotentials, reducing the Tafel slope, and increasing the electrochemical double-layer capacitance. The exact mechanism of how Mn-doping improves OER performance is not clear, but it is proposed that Mn ions may act as active sites for the OER by participating in the adsorption and dissociation of water molecules, or by changing the electronic properties of CoOOH.
Chen et al.166 used operando techniques, specifically in situ SERS to observe the structural changes of the catalysts under anodic polarization during the OER. This allows for the real-time observation of the catalysts' structural changes while they are actively participating in the OER. The study found that the Co-RT and Ni4.1Co-RT catalysts had similar OER activities, with both catalysts having similar electrochemical properties such as the Tafel slope and exchange current density. However, Raman spectroscopy revealed that the structural changes in the catalysts under anodic polarization were different. For the Co-RT sample, the Raman spectra showed a monotonic decrease in the A1g Raman band intensity at potentials greater than that for the resting state. This decrease in intensity was more pronounced at high oxygen evolution rates (around 4 mA cm−2). This structural change continues at relatively high oxygen evolution rates, indicating that the structural transformation of Co3O4 to CoO(OH) is closely related to the OER activity. On the other hand, for the Ni4.1Co-RT catalyst, the Raman spectra were similar to those for the Co-RT samples and showed no significant changes under anodic polarization. This suggests that Ni incorporation did not alter the catalyst structure when the Ni4.1Co-RT catalyst was prepared by the long-time annealing procedure. For the Ni5.3Co-100 sample, the Raman spectra showed a significant structural transformation at 0.2 V. At this potential, the A1g band intensity was attenuated by about 45%, and the E g (487 cm−1) and F 2g (531 cm−1) modes completely disappeared. Simultaneously, two broad peaks at 542 and 600 cm−1, as well as a small shoulder at 490 cm−1, appeared. These new peaks suggest a different local structure for this catalyst at the 0.2 V potential, which may be related to improved OER activity. In summary, the study found that the incorporation of Ni in the Co3O4 spinel lattice leads to a structural transformation that may improve OER activity in the Ni5.3Co-100 sample, but this is not confirmed by the study. The Ni4.1Co-RT catalyst showed no significant change in the structure under anodic polarization.167
In a study, Mn doping was found to play an important role in improving the performance of OER catalysts.168 The authors of that study synthesized three samples with different ratios of Mn ions in LDH structure and evaluated their OER performance through a three-electrode configuration in 1 M KOH. The results indicate that the sample with the optimal amount of Mn (Co1Mn1 LDH/NF) has the lowest overpotential of 285 mV at 10 mA cm−2, which is lower than those of other samples and some reported excellent Co-based materials. Additionally, this sample has a current density that is nearly 3 times higher than that of the CoCo LDH/NF sample at 1.8 V vs. RHE, which further illustrates the significant synergistic effect of Mn doping on the improvement of OER performance. Additionally, Mn doping leads to faster reaction kinetics as observed through the Tafel slope which is smaller in the sample with optimal Mn doping. It also increases the stability of the catalyst as observed through the chronoamperometry (i–t) test, where the retention rate of current density for the sample with optimal Mn doping is almost 98% even after 20 hours of testing, and the morphology could also be retained well. Furthermore, the faradaic efficiency was also tested to evaluate the oxygen conversion rates, and the sample with optimal Mn doping is found to have 94% faradaic efficiency, thus exhibiting more stability and efficient energy-conversion rate in the material. In summary, Mn doping plays a crucial role in the OER by significantly improving the performance of the catalyst by lowering the overpotential, increasing the current density, and leading to faster reaction kinetics, greater stability, and higher efficiency.
Karmakar and Srivastava studied the electrocatalytic activity of Co1.9Ni0.1(CO3)(OH)2/GP and Co0.95Mn0.05CO3/GP measured for the OER at a scan rate of 5 mV s−1 with iR-compensation and the corresponding polarization curves.169 It was found that NiCCH exhibited the lowest overpotential (η) values at all current densities and even outperformed the commercial state-of-the-art RuO2 electrocatalyst. The improvement in OER performance was attributed to the presence of multiple valence oxidation states and increased active metal centers due to the substitution of Co with heterometal atoms (Ni or Mn). Additionally, the larger specific surface area of NiCCH and MnCCH compared to CCH for their superior electrochemical activity was also considered. The 2D rectangular nanoplates of NiCCH and 3D octahedral MnCCH particles could also facilitate electrolyte penetration for better contact with available active sites for the OER. Furthermore, the findings showed that the lowest Tafel slope for NiCCH indicated faster OER kinetics in comparison to RuO2, CCH, and MnCCH. The calculated Rct values suggested a faster electron transfer rate in NiCCH and MnCCH compared to CCH. Overall, the study suggests that transition-metal substitution effectively modified the electronic structure of NiCCH and MnCCH, enhancing their electrical conductivity and OER performance.
The study performed by Zhang et al.93 has shown that the CoIr-0.2 catalyst, which is composed of atomic iridium and Co(OH)2, exhibited excellent OER performance in a neutral electrolyte (1.0 M PBS, pH = 7) and alkaline solution (1.0 M KOH, pH = 14). This is evidenced by its low overpotential (η) required to achieve a current density of 10 mA cm−2, which is lower than that of commercial IrO2 in both solutions. The Tafel slope for CoIr-0.2 is also much smaller than those of other CoIr-x samples and IrO2, indicating that it requires less energy for electrochemical processes. Additionally, the stability of CoIr-0.2 in both neutral and alkaline solutions is excellent. X-ray absorption spectra were used to confirm the presence of atomic iridium in the CoIr-0.2 catalyst and to understand the effect of iridium incorporation on the electrocatalytic performance. Overall, the study suggests that the CoIr-0.2 catalyst, composed of atomic iridium and cobalt hydroxide, has great potential for practical applications in the OER.
The study reported by Li et al.170 demonstrated that the Co(OH)2/BP nanosheets with a mass ratio of 5:1 showed the best OER activity. The overpotential (η) of Co(OH)2/BP 5:1 nanosheets at a current density of 10 mA cm−2 was 276 mV, which is significantly lower than that of pure Co(OH)2 (370 mV). Furthermore, the current density (at 320 mV) of Co(OH)2/BP 5:1 was 39.6 mA cm−2, which is about 20.8 times that of Co(OH)2. The study also demonstrated that the Tafel slope of Co(OH)2/BP 5:1 is reduced to 57 mV dec−1, compared to 88 mV dec−1 for Co(OH)2 nanosheets, indicating a sharp increase of current density with increasing the overpotential and facile charge transfer in the nanosheets. Additionally, the study also reported that the Co(OH)2/BP 5:1 nanosheets possess a much larger ECSA than Co(OH)2, indicating a significantly improved active surface. The study suggests that the addition of black phosphorus (BP) to Co(OH)2 greatly improves the OER activity and stability of the catalyst. It is not specified in the study how exactly BP boosts the OER in this specific case; however, in general, it is believed that BP has high conductivity, good thermal stability, and high surface area which makes it a good candidate for electrocatalysis. BP has a layered structure which makes it a good support for catalytic active materials; it also has a high surface area which allows for more active sites for catalytic reactions. Also, it has high electrical conductivity which can facilitate charge transfer. In addition, its unique properties such as high stability, high surface area, and high conductivity may enhance the electronic interaction between the Co(OH)2 and BP, which could lead to a significant enhancement in the OER activity. It's worth noting that this is only a speculation, and the actual mechanism of how BP boosts the OER of Co(OH)2 in the specific case is not reported in the study.
Dong et al.171 found that the catalytic effect of Ag-decorated cobalt hydroxide nanosheets is much better than that of pure Co(OH)2 nanosheets. The amount of Ag in the Co(OH)2 nanosheets also had a significant impact on the performance, where a certain range of Ag can decrease the overpotential. However, excessive Ag can reduce the catalytic performance of the material by occupying the active sites of Co(OH)2. The optimum catalytic performance was found to be at 10% Ag-decorated Co(OH)2 nanosheets, which had the highest current density of 37.6 mA cm−2 and the lowest overpotential of 270 mV. It also had a lower Tafel slope of 67 mV dec−1 compared to other Ag-decorated Co(OH)2 nanosheets, indicating a remarkable OER kinetic increment. Overall, the 10% Ag-decorated Co(OH)2 nanosheets showed better catalytic performance than other materials in the study and previous research results.172–175 Kim et al.176 discussed the use of Ag NW@Co NS2 and Ag later@Co NS3 catalysts in the conversion of solar energy to fuels with 10% efficiency. Fig. 11a–d depict the SEM, TEM, HRTEM, and EDX mapping analyses of the sample. These analyses confirm the sheet-like morphology of the samples enveloped by clearly defined Ag nanowires. The EDX mapping demonstrates substantial concentrations of Co, Ag, and O within the sample, validating their presence in significant quantities. The AgO layer on the Ag NW provides a high concentration of active sites for the OER, which increases the catalytic activity of the catalyst. Ag NW also has a well-assembled core–shell structure, which provides more stability to the catalyst layer, and allows for the facile release of O2 bubbles without damage to the catalyst. The study quantifies and compares the OER performance of different catalysts. To explore the surface redox characteristics of the pure Co NS, Ag NW, and Ag NW@Co NS3 catalysts, CV was conducted in a nitrogen-saturated 1 M KOH solution shown in Fig. 11e. The efficiency of the OER was assessed through LSVs depicted in Fig. 11f. Notably, the Ag NWs displayed minimal reactivity within the tested potential range, aside from an anodic peak at approximately 1.6 V (vs. RHE), which was attributed to the oxidation of Ag2O. TOFs at overpotentials of 300 (1.53 V vs. RHE) and 350 mV (1.58 V vs. RHE), respectively, are illustrated in Fig. 11g. The Ag NW@Co NS2 and NS3 catalysts exhibited TOFs of 0.011 and 0.016 s−1, which are 6.9 and 10 times that of the pure Co NS catalyst (0.0016 s−1), respectively, further confirming the higher activity of these catalysts. The stability values evaluated via chronoamperometry of the Ag NW@Co NS2 (1.56 V) and Ag NW@Co NS3 (1.55 V) catalysts were found to be lower than that of the pure Co NS (1.63 V) catalyst, indicating their higher efficiency in the OER illustrated in Fig. 11h. The Tafel slope of the Ag NW@Co NS2 and NS3 catalysts (75.8 mV dec−1, 75.4 mV dec−1) was significantly lower than that of the physically mixed catalyst (82.5 mV dec−1).
 | ||
| Fig. 11 (a) SEM, (b) TEM and (c) HRTEM focused on the Co NS part and (d) EDX mapping images of the Ag NW@Co NS3. (e) CVs of the Ag NW@Co NS3, pure Co NS and Ag NW catalysts in an N2-saturated 1 M KOH electrolyte at a scan rate of 50 mV s−1. (f) LSVs in an N2-saturated 1 M KOH electrolyte at a rotation rate of 1600 rpm and a scan rate of 10 mV s−1, (g) TOFs at overpotentials of 300 and 350 mV and (h) CP tests of the Ag NW@Co NS (2 and 3) and pure Co NS at a current density of 10 mA cm−2 for 10 h. This figure has been reproduced from ref. 176 with permission from Royal Society of Chemistry, copyright 2016. | ||
Liu et al.177 studied the effect of boron (B) and iron (Fe) on the catalysts' OER performance. B is used as a precursor to produce B-α-CoxFe LDH catalysts. B is found to have an effect on the catalytic activity of B-α-CoxFe LDH catalysts, with the best activity obtained when using 1 mmol NaTPB. This is likely due to the fact that B can act as a support for the catalytic metal ions and can also affect the electronic and crystal structures of the catalysts, which can improve their electrocatalytic activity. Fe, on the other hand, is one of the metal ions used in the B-α-CoxFe LDH catalysts. The study found that the B-α-Co5.8Fe LDH had outstanding electrocatalytic activity among the catalysts studied, which suggests that the Fe content plays a crucial role in the catalytic activity. Fe ions may act as active sites or may have a synergistic effect with other metal ions to enhance the catalytic activity. Additionally, Fe can also affect the electronic and crystal structures of the catalysts, which can also improve their electrocatalytic activity.
Wang et al.178 synthesized P-doped NiMoO4 parallel arrays anchored on cobalt carbonate hydroxide (NiMoP@CoCH). In this study, the OER activity of electrodes was investigated using corrected LSV curves. The NiMoP@CoCH/CC-2 electrode was found to have a lower overpotential of 267 mV at 40 mA cm−2, compared to the other electrodes tested, and also had the highest ECSA. The superior performance of the NiMoP@CoCH/CC-2 electrode is attributed to its hierarchical heterostructures, which create a “highway” for electrolyte and gas bubble mass transfer, and the incorporation of phosphate dopant and oxygen vacancies, which improves electrical conductivity and increases the number of active defects. Additionally, the electrode's structural and electrochemical stability is enhanced by the growth of NiMoP nanosheets on CoCH nanoarrays. Nevertheless, this electrode has the potential for use in various other fields.179,180
The study performed by He et al.181 is focused on the electrochemical performance of as-fabricated catalysts towards the OER in 0.1 M KOH electrolyte. The catalysts studied include sol–gel-derived SFM, as-spun SFM nanowires (SFM-NF), bare SFM hollow nanofibers, and Co(OH)2/SFM-NF. The OER activity of these catalysts was initially assessed through LSV polarization curves in a three-electrode system. The sol–gel-derived SFM catalyst was used as a benchmark for the study, and its performance was compared to that of the other catalysts. The SFM-NF catalyst was found to have a smaller onset potential of 1.58 V and a larger current density compared to the sol–gel-derived SFM catalyst. This improved OER activity is likely attributed to the larger surface area of the nanofibers. The Co(OH)2/SFM-NF catalyst was found to have the best OER performance among the catalysts studied, with an onset potential of 1.54 V and the highest current density. This improved performance was attributed to the optimized 10 nm Co(OH)2 decoration on the SFM-NF. The OER current of Co(OH)2/SFM-NF largely exceeded that of the studied IrO2 sample beyond 1.60 V, even though the latter had a relatively lower onset potential (1.51 V). The Co(OH)2/SFM-NF sample required a low overpotential of 387 mV to generate 10 mA cm−2, which is significantly lower than the overpotentials required by IrO2 (447 mV), SFM-NF (468 mV), SFM (548 mV), and Co(OH)2 (529 mV). The Co(OH)2/SFM-NF catalyst also exhibited a superior OER activity compared to other reported Fe-based perovskite oxides such as 3D ordered microporous LaFe0.8Co0.2O3 (460 mV), phosphorus-doped LaFeO3−δ (465 mV), PrBa0.85Ca0.15Fe2O5−δ (410 mV), and SrTi0.1Fe0.85Ni0.05O3−δ/CNT (480 mV). Additionally, Co(OH)2/SFM-NF had a smaller Tafel slope of 71 mV dec−1 than IrO2 (88 mV dec−1), SFM-NF (99 mV dec−1), and commercial IrO2 catalysts (79 mV dec−1). In conclusion, the study found that the Co(OH)2/SFM-NF catalyst has the best electrochemical performance towards the OER among the catalysts studied, with a smaller onset potential of 1.54 V and the highest current density. The improved performance is attributed to the larger surface area of the nanofibers and the optimized 10 nm Co(OH)2 decoration on the SFM-NF. The Co(OH)2/SFM-NF catalyst also exhibited a superior OER activity compared to other reported Fe-based perovskite oxides and required a low overpotential of 387 mV to generate 10 mA cm−2.
4.5 Cobalt based bimetallic hydroxides/layered double hydroxides
:Co 3:1 deposit showed the lowest η10 mA of ∼262 mV, and after 1000 accelerated stability test cycles and 6 hours of chronopotentiometry at j = 10 mA cm−2, the same samples were immediately subjected to stepped chronopotentiometry from 10 to 100 mA cm−2 over a 5 h period. Current densities were stepped up by 10 mA cm−2 increments every 30 minutes. Ni:Co 3:1 outperforms plain Ni(OH)2 and Co(OH)2 with η10 mA of 262 mV versus 278 and 304 mV for the pure samples respectively. This difference in performance is further amplified at higher current densities with an η100 mA value of 417 mV versus 475 and 508 mV respectively (Fig. 12c). The study also found that despite the prolonged and harsh testing conditions, the deposit showed consistently low overpotentials of ∼255 mV (Fig. 12d) at j = 10 mA cm−2 and achieved higher current densities (∼15% @ η370 mV) in the OER region compared to the 1 min deposit. However, the performance of the Co(OH)2 sample appears to have deteriorated in the CV curve in Fig. 12b. But the chronopotentiometry carried out at 10 mA cm−2 after the 1000 cycles of accelerated stability testing shows that the Co(OH)2 sample returns to the η10 mA values exhibited before the harsh cycling process (Fig. 12e). The apparent loss of performance seen in the CV after 1000 cycles may be a function of phase change caused by overcharging during the accelerated stability testing between 1.3 and 1.8 V vs. RHE. Overall, the study provides evidence that the use of nanodendrites as catalysts for the OER can lead to improved performance compared to traditional catalysts, with lower overpotentials and higher current densities. However, further research is needed to fully understand the underlying mechanisms of these nanodendrites and how to optimize their performance.
 | ||
| Fig. 12 (a) HRTEM image of a Ni(OH)2 nanodendrite. Inset shows the selected-area diffraction pattern indicating the amorphous nature of the structure. (b) Tilted cross-sectional view of electrodeposited hierarchical 3-D Ni(OH)2 nanodendrite forests. Inset shows a near spherical air bubble making contact with the deposited surface under water due to ultralow bubble adhesion. (c) Comparison of cyclic voltammograms of nanodendrite (1 min) deposits with varying Ni:Co ratios after 10 stabilizing cycles and (e) after 1000 cycles of accelerated stability testing. All CV curves were recorded at 5 mV s−1. (d) Chronopotentiometry curves for various Ni:Co deposits recorded at j = 10 mA cm−2 showcase their excellent stability. (f) Chronopotentiometry curves of Ni:Co 3:1 compared to Ni(OH)2 and Co(OH)2 deposits recorded at current densities ranging from 10 to 100 mA cm−2. Current density values were stepped up by 10 mA cm−2 every 0.5 h. This figure has been reproduced from ref. 182 with permission of American Chemical Society, copyright 2017. | ||
Prataap et al.183 designed the porous structure of the NiCo alloy electrode by a single-step electrodeposition method, where hydrogen evolution during the deposition process acted as a dynamic template. This method resulted in the formation of a porous structure with a large surface area, which exposed more electrochemically active sites to the electrolyte. This increased surface area was one of the main reasons for the improved electrocatalytic activity toward oxygen evolution. The porous NiCo alloy electrode exhibited the highest electrocatalytic activity with the lowest onset potential of 1.47 V and the current density of 10 mA cm−2 was obtained at 1.53 V. The porous structure of the electrode also enabled more H2O adsorption and accelerated O2 desorption, which contributed to the low OER onset potential and high current density achieved. The porous structure of the electrode also improved the electrode's electrochemical properties. The calculated ECSA value for the porous NiCo alloy electrode was 1.73 cm2 and the TOF was 0.0025 s−1, indicating higher catalytic activity. The low solution resistance and charge transfer resistance across the electrode–electrolyte interface, as indicated by low Rs and Rct values, also contributed to the improved electrocatalytic activity of the porous NiCo alloy electrode. In summary, the porous structure of the NiCo alloy electrode played a significant role in improving its electrocatalytic activity towards oxygen evolution in alkaline solutions. The porous structure increased the electrode's surface area, exposed more electrochemically active sites to the electrolyte, enabled more H2O adsorption, accelerated O2 desorption, and improved the electrode's stability, electrochemical properties, and catalytic activity. Similarly, Wang et al.184 synthesized a porous Co0.75Ni0.25(OH)2 nanosheet catalyst using laser ablation in the liquid method. The catalyst was found to have low overpotentials for the HER and OER and achieved a current density of 10 mA cm−2 at an external voltage of 1.56 V, which is much lower than those of commercial Ir/C–Pt/C catalysts. The porous structure and the presence of Co3+ ions on the pore wall were found to be responsible for the catalyst's high activity. The researchers believe that this laser ablation in the liquid method can be used to create other novel catalysts for energy conversion.185
Wang et al.186 studied the electrocatalytic OER performance of CoxNi1−x(OH)(BzO)$H2O catalysts with varying Ni/Co ratios. The introduction of a certain amount of Co in Ni(OH)(BzO)$H2O greatly enhances the OER activity. The Co0.8Ni0.2(OH)(BzO)$H2O catalyst shows the most superior performance among all the samples, with an onset potential of 1.50 V vs. RHE and the smallest overpotential of 319 mV at the current density of 10 mA cm−2. The introduction of inorganic–organic species and the use of nanobelts have a positive impact on OER activity. It is possible that the inorganic–organic species and nanobelts were used as a way to synthesize the catalysts and their effect on the OER activity was observed as a result. The inorganic–organic species could have played a role in creating catalytically active sites or increasing the active surface area of the catalysts, which would in turn enhance the OER activity. The use of nanobelts may have increased the surface area and improved the electrical conductivity of the catalysts, which would also enhance the OER activity.
CoNi-LDH NSs (nanosheets) were electrodeposited onto a Ni foam by Yoon et al.,187 but the catalytic stability was low (18% degradation) after 10 hours of water electrolysis due to separation from the Ni foam. To improve adhesion, Ni nanocrystals (NCs) were electrodeposited onto the foam to increase the surface area and enhance adhesion. The 0.3/0.2 CoNi-LDH NSs/Ni NCs/Ni foam exhibited the best catalytic activity and the highest current density (111.5 mA cm−2 at 1 V). The Ni NCs also had a positive effect on overpotential and stability, with a current decrease of only 7.4% after 10 hours. Additionally, the Ni NCs did not affect the morphology of the CoNi-LDH NSs. TOF for the CoNi-LDH NSs with the NCs increases from 0.035 to 0.060 s−1 in the range from 0.05/0.2 to 0.3/0.2. However, when the Co/Ni ratio is increased further to 0.4/0.2, the TOF decreases to 0.046 s−1.
Keisuke Awaya and Shintaro Ida188 discussed the electrochemical activity of ultrathin hexagonal nickel–cobalt hydroxide nanosheet films for the OER and ORR. The authors found that a Ni1Co4–OH nanosheet film had the lowest overpotential of all the Ni/Co ratios tested and that increasing the layer number of the film also lowered the overpotential. However, the overpotential decreases per layer were larger for 1–4 layers compared to 4–12 layers. The authors also mention that ultrathin films created by stacking nanosheets are ideal for electrochemical reactions and that controlling the layer number of the nanosheets can be useful for investigating the most effective thickness for these reactions.
Wang et al.189 described the synthesis of a potato chip-like cobalt nickel-layered double hydroxide@polypyrrole-cotton pad (CoNi-LDH@PCPs) by in situ polymerization. This process involves the simultaneous formation of the material and its polymerization. The composite material has a unique potato chip-like structure, which helps to improve the kinetics of the electrode reactions. This material was used as a positive electrode for wearable supercapacitors and it was found to have a high specific capacitance of 387.2 C g−1 at 1 A g−1 and excellent flexibility. As an OER catalyst, the PCP-based electrode had a low overpotential of 350 mV at a current density of 10 mA cm−2 and a Tafel slope of 58 mV dec−1. The theoretical calculations suggested that the synthesis strategy used in this study can be used to control the formation of hollow CoNi-LDH arrays, which could lead to the development of high-performance electrodes for energy storage and electrochemical catalysis.
Chen et al.190 presented a new approach for fabricating efficient and low-cost electrocatalysts for hydrogen energy production by fusing NiCo LHS nanowire arrays with ZIF-67 to create three-dimensional flower-like structures on a Ni–Fe foam support. The electrocatalytic performance was improved through simple oxidation, carbonization, sulfurization, and selenization. The optimized S-doped catalyst had the highest electrocatalytic performance, with low overpotentials of 170 and 100 mV for the OER and HER at 10 mA cm−2 in 1 M KOH, respectively. This was attributed to the strong interfacial coupling between the NiCo-LHSs and ZIF-67 derivatives, as well as the rational electronic structures, dense catalytic active sites, and large specific surface area. The results are promising for the development of next-generation catalytic materials for electrocatalysis and other renewable energy applications.191
Chodankar et al.192 reported non-noble, precious nano–micro structured Ni2P2O7 microsheets decorated with nickel–cobalt hydroxide (Ni–Co-hydroxide) nanoflakes. The designed material exhibited superior catalytic activity towards the OER with a relatively low overpotential of 197 mV at a current density of 10 mA cm−2 in 1.0 M KOH, coupled with excellent long-term stability (12 h). Outstanding OER activity resulted from the nano–microstructure of the prepared electrode with excellent redox activity in an alkaline electrolyte. The material is stable after 2000 CV cycles at a scan rate of 100 mV s−1 with almost no degradation and shows strong long-term electrochemical stability. It demonstrates high performance with non-noble metal catalysts and a low-cost electrode for oxygen generation. The Ni2P2O7 microsheets in the material serve as the support structure for the Ni–Co-hydroxide nanoflakes. The vertically aligned microsheets provide numerous sites for the electrochemical reactions to take place, as well as a surface to which the Ni–Co-hydroxide nanosheets adhere. The amorphous nature of the Ni–Co-hydroxide with a number of unsaturated metal atoms provides more reactive sites on the catalyst surface, facilitating improved OER. Additionally, the microsheets provide stability to the overall structure, allowing for the long-term stability of the material.
Sun et al.193 deposited an amorphous boron oxide layer in situ on NiCo-LDH nanosheet arrays through a simple chemical vapor deposition process. This results in a NiCo-LDHs@B2O3/carbon paper (NiCo-LDHs@B2O3/CP) electrode with a low overpotential of 213 mV at a current density of 10 mA cm−2 in 1 M KOH, which is 123 mV lower than the overpotential of NiCo-LDHs/CP and slightly better than that of the noble metal catalyst RuO2/CP (230 mV). Additionally, at 50 mA cm−2 in 1.0 M KOH, the overpotential of NiCo-LDHs@B2O3/CP is still low at 320 mV, which is much better than that of NiCo-LDHs/CP (441 mV) and RuO2/CP (373 mV). The introduction of amorphous B2O3 layers promotes the formation of borate interfaces, which effectively enhances the reactivity of active species and improves the cycling stability of the electrode. This research could inspire the development of low-cost, efficient OER electrocatalysts for energy conversion using NiCo-LDHs@B2O3/CP. The B2O3 is a known catalyst for the OER. The addition of B2O3 to an electrode can increase the rate of the OER and improve the overall efficiency of the process. This is because B2O3 can facilitate the formation of active sites on the electrode surface, where the OER can take place. Additionally, B2O3 can also stabilize the intermediate products of the OER and lower the overpotential required for the reaction to occur, which further enhances the efficiency of the process.
A facile cation-exchange process was developed by Zhou et al.91 to synthesize Fe-doped Ni(OH)2 and Co(OH)2 nanosheets with enriched active sites for enhanced OER. Compared to typical NiFe LDH nanosheets, the Fe-doped Ni(OH)2 nanosheets exhibit higher ECSA and improved surface wettability resulting in significantly enhanced catalytic activity. Specifically, the Fe-doped Ni(OH)2 nanosheets require a low overpotential of only 245 mV to reach a current density of 10 mA cm−2 with a low Tafel slope of 61 mV dec−1, which is greatly decreased as compared with that of NiFe LDH (310 mV and 78 mV dec−1). Additionally, this cation-exchange process can be extended to prepare Fe-doped Co(OH)2 nanosheets with improved catalytic activity for oxygen evolution. We believe that this novel strategy would shed light on developing high-performance heteroatom-doped catalysts with abundant active sites. Fe is a commonly used dopant in catalysts for the OER because it can significantly increase the activity of the catalyst. This is due to the fact that Fe can form strong chemical bonds with oxygen, which makes it an effective oxidizing agent. Additionally, Fe has multiple oxidation states, which allows for a greater range of reaction pathways for the OER to occur. Fe dopants are commonly used in a variety of catalysts such as transition metal oxides and hydroxides and have been found to be particularly effective in boosting the OER in non-noble metal catalysts like perovskite oxides and spinel oxides.194
Mixed cobalt and nickel-based LDHs and mixed metal oxides (MMOs) doped with gallium were synthesized by Chakrapani et al.195 in various compositions by constant pH co-precipitation and calcination. The materials were found to exhibit electrocatalytic activity for the oxygen evolution reaction in an alkaline solution. The highest activity was found for catalysts containing both transition metal cations Co and Ni. However, the effect of calcination was found to depend on the composition of the LDHs. The optimal cation composition of the LDHs was Co1.5Ni0.5Ga with an overpotential of 382 mV. The highest performance among the MMOs was encountered for the Co0.5Ni1.5Ga composition, reaching a similar overpotential. In particular, gallium has been found to enhance the activity and stability of electrocatalysts used in the OER process. It does this by forming a protective layer on the surface of the catalyst, which helps to prevent the catalyst from degrading or becoming inactive. Additionally, gallium is thought to increase the number of active sites on the catalyst surface, which can further improve the efficiency of the OER process.
The proposed method by Sakita et al.198 is a one-step, facile, and fast method for obtaining an effective catalyst for the OER. It involves the direct electrodeposition of CoFe alloy thin films covered with LDH in a free-nitrate bath using potentiostatic mode in a continuous or pulsed regime. The catalyst is formed directly on glassy carbon substrates and exhibits a mixed morphology of granular and dendritic CoFe alloy covered with a sponge-like structure of CoFe-LDH containing a chloride (Cl) interlayer. The best-prepared catalyst material showed an overpotential of 286 mV at 10 mA cm−2, with a Tafel slope of 48 mV dec−1 for the OER, displaying enhanced catalytic activity. The presence of Co and Fe oxidized species at the surface of the composite material and the LDH structure intercalating chloride directly formed on the glassy carbon substrate make it an efficient and cost-effective electrocatalyst for water splitting. The adjustment of the electrosynthesis potential and pulses is crucial to avoid the formation of pure alloy or excessive hydroxides. Additionally, a new approach to preparing a binder-free electrocatalyst has been developed.
A cobalt iron hydroxide (CoFe/NF) electrode was synthesized by Babar et al.199 through a simple electrodeposition method and showed high catalytic activity and excellent durability in a 1 M KOH electrolyte. The CoFe/NF electrode required overpotentials of 220 mV for the OER and 110 mV for the HER to reach a current density of 10 mA cm−2. The higher catalytic activity was attributed to synergistic effects between Co and Fe as active sites. The CoFe/NF films were used as bifunctional catalysts for overall water splitting and achieved a current density of 10 mA cm−2 at 1.64 V and excellent stability for 50 hours. This transition metal hydroxide can be effectively applied for mass production as a highly efficient bifunctional catalyst for overall water splitting. The specific effects may depend on the loading method, loading amount, and other factors. Additionally, other advantages such as cost-effectiveness, high conductivity, and high surface area make Ni foam an attractive substrate for electrocatalysts.200,201
Guo et al.202 used a simple and efficient electrospinning method to fabricate amorphous cobalt (Co)–iron (Fe) hydroxide on a foamed nickel substrate for the first time. This material showed excellent oxygen-evolution advantages over a typical reference catalyst (RuO2) when tested in an untreated alkaline medium. Additionally, when tested in a three-electrode system in 1 M KOH, the amorphous PVP/CoFe1.3 nanofibers had high catalytic activity and stability for the OER, with a low overpotential of 0.267 V at 100 mA cm−2 and a low Tafel slope of 47.43 mV dec−1. The electrospinning method is considered a feasible and effective synthetic technique for the fabrication of mixed metal hydroxide-based nanofibers with a large surface area, small and uniform grain size, and high morphological uniformity. The study demonstrates the potential of amorphous CoFe hydroxide as a high-efficiency OER catalyst and opens possibilities for large-scale and environmentally friendly production of water splitting. The amorphous nature of the CoFe hydroxide is thought to enhance the OER in several ways. One possible explanation is that the amorphous structure has a higher surface area and a larger number of active sites for the OER to occur, leading to higher catalytic activity. Additionally, the amorphous structure may have a more homogeneous distribution of the Co and Fe atoms, which can promote the synergistic effect between the two elements, further enhancing the OER activity. Furthermore, amorphous materials are often more stable than crystalline materials, which can lead to increased durability of the catalyst over time.203
Defect rich CoFe-based LDHs have been successfully synthesized by Liu et al.204 as a bifunctional, non-noble-metal electrocatalyst for overall water splitting. The delamination and exfoliation process in the DMF–ethanol solvent (CoFe LDH-F) has resulted in an enhancement of both OER and HER activity. The exfoliation process creates more coordinatively unsaturated metals and improves the intrinsic electronic conductivity, which are important factors in water electrolyzer reactions. The CoFe LDH-F catalyst outperforms the commercial IrO2 electrocatalyst in activity and stability for the OER and approaches the performance of Pt for the HER, with a current density of ∼10 mA cm−2 at the applied voltage of 1.63 V for the long-term electrolysis test. The performance and cost-effectiveness of CoFe LDH-F make it a promising material to replace noble-metal-based catalysts for large-scale water splitting. The defects in the CoFe LDH-F materials play an important role in boosting the OER, which can be further studied to improve the catalytic activity. The defects in the CoFe-LDH structure are believed to enhance its catalytic activity by providing active sites for the OER. In addition, the ultrathin thickness of the CoFe-LDH allows for a high surface area-to-volume ratio, which further increases its catalytic activity.
The investigation found that the Fe sites in the catalyst films were well integrated into the CoOOH structure and that the Fe sites underwent potential-dependent structural changes. Oxidized Fe species play a key role in the OER mechanism on Co(Fe)OxHy and the reaction mechanism proceeds through Fe-based active sites with an intermediate that has Fe in an oxidation state higher than 3+.205
Dai et al.206 described the synthesis of ultrathin carbon-coated, nitrogen and iron-modified cobalt carbonate hydroxide needle arrays on carbon cloth (C@NFeCoCH/CC) using a hydrothermal reaction and low-temperature calcination process. The combination of Fe and N modification results in high OER activity of C@NFeCoCH/CC due to the formation of highly active Co/Fe–N–C species on the interface of the carbon layer and needles. The thin carbon layer, N and Fe modification, and continuous conductive carbon cloth allow for fast and stable charge transfer. The optimized C@NFe1Co1CH/CC has a low overpotential of 235 mV at 10 mA cm−2, a mass activity of 681.4 A g−1, and superior stability over 30 hours at 10 mA cm−2. The introduction of active sites is a promising approach for the application of metal carbonate hydroxides in electrocatalysis. Carbon protection of CoFe LDHs can increase their OER activity by providing a more stable and efficient catalytic surface for the reaction to occur on. Carbon-protected CoFe LDHs have been shown to have higher OER activity and better stability compared to unmodified CoFe LDHs. This is due to the carbon layer protecting the CoFe LDHs from degradation and providing a more efficient catalytic surface for the OER to occur on. Additionally, carbon protection can also increase the electrical conductivity of the CoFe LDHs, which also improves the OER activity.
A stable single atomic ruthenium catalyst was developed by Li et al.207 through anchoring it on the surface of CoFe LDH. The catalyst was found to have a strong electronic coupling between the Ru and LDH. With a low Ru loading of 0.45 wt%, the catalyst exhibited high activity and stability in the OER, requiring only 198 mV overpotential to drive a current density of 10 mA cm−2 in an alkaline solution. The activity and stability of the catalyst were attributed to the synergetic electron coupling between the single-atom noble metal and the LDH. These findings have potential implications for the development of cost-effective and high-performance electrocatalysts for energy conversion-related applications.
The OER activity of CoMn LDH was found to be significantly improved by anodic conditioning (AC).212 AC is a process where the material is exposed to an anodic current density, in this case, J = 10 mA cm−2, for a certain period. After 3 hours of galvanostatic conditioning at this current density, the overpotential (@ J = 10 mA cm−2) decreased by about 20 mV for CoMn LDH, but only negligibly (3 mV) for Co(OH)2 + Mn2O3. This suggests that the AC process specifically enhances the OER activity of CoMn LDH. Further, it was found that the activity enhancement was durable, with the current density remaining at 16.5 mA cm−2 at 300 mV for more than 10 hours. The faradaic efficiency, or the efficiency of the catalytic process, was also found to be quantitative during a 5 hour electrolysis experiment. When comparing the activity of CoMn LDH to other OER catalysts, the researchers used TOF which is a measure of the intrinsic activity of a catalyst. The TOF of CoMn LDH at 350 mV (1.05 s−1) was more than 20 times higher than those of Co–Pi, CoOx, MnOx, and their composites (such as mixed Co/Mn spinel), and 9 times higher than that of IrO2. This makes CoMn LDH the most active Co or Mn-based OER catalyst to date. The presence of Mn likely enhances the activity through synergistic effects between the Co and Mn ions, or by providing active sites for the OER process. The researchers mention that the high activity of CoMn LDH is unique and not observed in other Co or Mn-based OER catalysts, which suggests that the combination of Co and Mn ions in this specific CoMn LDH structure plays a crucial role in boosting OER activity.
CoGeO2(OH)2 is a cobalt–germanium hydroxide material that has been shown to have potential for use as an electrocatalyst in the OER.213 The results of the LSV measurements showed that the onset of CoGeO2(OH)2 occurs before 1.50 V vs. RHE, which means that the electrode begins to produce oxygen at a lower voltage than other cobalt oxides. Additionally, the 10 mA cm−2 current density occurs at an overpotential of ∼340 mV, which is lower than the overpotential of 400 mV for most typical bulk cobalt oxides, indicating good OER activity. The mass activation of CoGeO2(OH)2 at 350 mV overpotential is about 49.5 A g−1, which is larger than those of other cobalt compounds, such as Co(OH)2, Co3O4, and CoO. A higher mass activation value indicates that the material is able to produce more current per unit mass and is therefore considered to be more active. This research suggests that increasing the specific surface area of CoGeO2(OH)2 may be an efficient route to optimize the OER activity of the material. This is because increasing the surface area would increase the number of active sites available for the OER to occur, leading to a higher overall reaction rate. This has been demonstrated in previous studies on nanostructured Co-based catalysts, where the OER activity was improved by increasing the surface area.
Yang et al.214 reported on the use of freestanding CoV-hydroxide nanoneedles as OER catalysts. The best performance was observed with the Co0.75V0.25-HNN catalyst, which had overpotentials of only 266 and 350 mV at current densities of 10 and 100 mA cm−2, respectively. This is the first report of such low overpotentials for freestanding CoV-hydroxide nanoneedles as OER catalysts and is comparable to most of the reported Co-based and V-based catalysts. The incorporation of V into Co-based hydroxide nanoneedles (NF@Co1−xVx-HNNs) was found to greatly enhance their catalytic activity as OER catalysts. The V incorporation modified the morphology, composition, and electronic structure of the samples, leading to improved OER performance. The V incorporation was found to change the electronic properties of the Co-based hydroxide nanoneedles, resulting in multivalent oxidation reactions, including the V3+ to V5+ and Co2+ to Co4+ oxidation. This change in electronic properties is thought to be the main reason for the improved OER performance, as it leads to lower overpotentials and higher corresponding currents in the voltage range of 1.25–1.65 V (vs. RHE). The incorporation of V also resulted in an increased active surface area and active catalytic sites, as well as faster reaction kinetics, as indicated by the lower Tafel slopes and higher TOF of the V-incorporating samples. In conclusion, the study shows that the incorporation of V into Co-based hydroxide nanoneedles is an effective strategy to improve their OER performance, by modifying their electronic properties and increasing the active surface area and active catalytic sites.
Ping et al.215 aimed to investigate the use of a novel electrocatalyst, 3DGN/CoAl-NS, for the OER in an aqueous solution of 1.0 M KOH. The 3DGN/CoAl-NS electrocatalyst was prepared by coating CoAl-LDH nanosheets onto 3D graphene. The loading amount of CoAl-NSs on 3DGN was measured to be about 0.05 ± 0.01 mg cm−2, using inductively coupled plasma atomic emission spectroscopy. The Tafel slope was found to be 36 mV dec−1 for the 3DGN/CoAl-NS electrocatalyst. This value is lower than the Tafel slope of other reported catalysts, indicating that the 3DGN/CoAl-NS electrocatalyst has a higher reaction rate. The overpotential, which is a measure of the energy required to drive the OER, was found to be 252 mV at a geometrical current density of 10 mA cm−2. The geometrical current density, which is a measure of the amount of current produced by the catalyst, was found to be 45.37 and 91.74 mA cm−2 at overpotentials of 300 and 350 mV, respectively. The TOF, which is a measure of the number of OERs that can occur per second, was found to be 0.56 and 1.14 s−1 at overpotentials of 300 and 350 mV, respectively. When compared to state-of-the-art LDH-related OER electrocatalysts, the 3DGN/CoAl-NS electrocatalyst was found to have comparable or even better catalytic activity. Additionally, control experiments were carried out to compare the performance of 3DGN/CoAl-NS with other Al-based LDH nanosheet-coated 3DGN, with 3DGN/CoAl-NS showing the best performance. Overall, the results of this research suggested that the 3DGN/CoAl-NS electrocatalyst has promising potential as a catalyst for the OER in water splitting and other applications. The role of Al in this research was to provide structural support for the electrocatalyst. The Al in the CoAl-LDH nanosheets helped to create a layered structure with a high surface area, which is beneficial for the OER. Additionally, the Al in the CoAl-LDH nanosheets may also contribute to the catalytic activity of the electrocatalyst, by promoting the adsorption of reactant molecules and by facilitating the transfer of electrons during the OER.
4.6 Cobalt based ternary hydroxides as OER electrocatalysts
Wang et al.216 evaluated the OER performance of NiCoFe-LDH/CFC in comparison to bare CFC and NiCo-LDH/CFC. The bare CFC substrate had a very low OER current density and a large onset potential, indicating that the CFC substrate had negligible catalytic activity for the OER process. However, when coated with NiCo-LDH, the material displayed a much-reduced onset potential and an increased current density for the OER. The introduction of Fe to the NiCo-LDH material further reduced the onset potential and enhanced the current density for the OER. The best NiCoFe-LDH/CFC material with 1 hour co-precipitation (CER) time showed a low overpotential of 280 mV, which was 50 mV less than that of the commercial Ir/C based electrode. This demonstrates the superior OER activity of the NiCoFe-LDH/CFC material compared to current commercial OER catalysts. It was found that increasing the CER duration to 3 and 6 hours resulted in a steady or reduced OER activity. This is attributed to the loss of active materials during the CER process, as one mole of Fe(III) replaced 1.5 moles of Co(II) or Ni(II). The overall activity of a material is determined by the intrinsic activity and the amount of active material. In the 3 hour CER sample, the increased intrinsic activity of the electrocatalyst and the loss of active material resulted in a trade-off, while in the 6 hour CER sample, the loss of active material outweighed the benefit of the enhanced intrinsic activity of the material, leading to reduced OER activity. They used Tafel plots to demonstrate the improved kinetics of the Fe-modified material for the OER. The linear range of the Tafel plots of the materials with different Fe content was fitted by the Tafel equation to extract the slope (overpotential). The researchers found that the NiCoFe-LDH/CFC samples with 1 and 3 hours of CER time had a low Tafel slope, indicating improved kinetics for the OER. The lowest Tafel slope was obtained by the 6 hour NiCoFe-LDH/CFC material, indicating an evolution of rate-determining steps. The Tafel slope of NiCoFe-LDH/CFC was lower than that of NiCo-LDH/CFC, indicating a four-electron transfer determining step, which is a sign of a good electrocatalyst. The excellent OER properties of the as-prepared NiCoFe-LDH/CFC are caused by several factors. The in situ CER method preserves the advantages of the NiCoLDH/CFC material, such as an intimate contact between the catalyst and substrate for efficient charge transport, a large surface area, and multiple channels for the diffusion of electrolyte ions. The incorporation of Fe also helps to enhance the material's performance in the OER process by inducing crystal structure disorder and reducing the thickness of the nanosheets, increasing the number of active sites, changing the charge transport determining step, and increasing the conductivity of the electrode.217–228Guo et al.229 studied the effect of catalyst morphology on the performance of the OER using a specific material, Co4Mn1Fey LTH/NF. XRD patterns of Co4Mn1Fey LTH/NF products with different Fe levels display similar peaks, matching carbonate-containing LDH reflections, indicating successful synthesis on the Ni foam substrate illustrated in Fig. 13a. SEM images reveal distinct morphological changes: without Fe, uniform NWs grow (∼2–3 μm long, ∼53 nm in diameter) on the Ni foam (Fig. 13b). With increasing Fe (up to 3 mmol), a transition occurs, leading to a coexistence of nanosheets and NWs forming oriented hybrid arrays, enhancing electron transport (Fig. 13c–e). At 4 mmol Fe, nanowires vanish, leaving only nanosheets, reflecting a significant morphological transformation (Fig. 13f). A schematic diagram illustrating the impact of Fe addition on morphology is provided in Fig. 13g. Various Co4Mn1Fey LTH/NF samples featuring different levels of Fe addition were evaluated for their respective OER performances. Notably, these obtained materials demonstrate suitability for use as self-supporting electrodes without the need for additional coating treatment. Fig. 13h illustrates oxidation peaks in the range of 1.3–1.45 V vs. RHE, typically associated with the transformation of Ni species from Ni foam into Ni(OH)2 and further into NiOOH via Ni2+/Ni3+ oxidation. Among the series of electrodes examined, the Co4Mn1Fe3 LTH/NF stands out with the most exceptional OER performance. It achieves overpotentials of 226 mV and 251 mV at current densities of 100 mA cm−2 and 200 mA cm−2, respectively. These overpotentials notably outperform those observed for Co4Mn1 LDH/NF (356 mV and 383 mV), Co4Mn1Fe1 LTH/NF (274 mV and 307 mV), Co4Mn1Fe2 LTH/NF (265 mV and 282 mV), and Co4Mn1Fe4 LTH/NF (241 mV and 262 mV) in reaching the corresponding current densities of 100 mA cm−2 and 200 mA cm−2 (Fig. 13j). As depicted in Fig. 13j, Co4Mn1Fe3 LTH/NF demonstrates a notably lower Tafel slope of 55 mV dec−1 compared to other electrodes (Fig. 13i). This lower slope signifies highly satisfactory kinetics, wherein a slight increase in overpotential results in a significant rise in current density.229
 | ||
| Fig. 13 (a) XRD patterns of a: Co4Mn1 LDH/NF, b: Co4Mn1Fe1 LTH/NF, c: Co4Mn1Fe2 LTH/NF, d: Co4Mn1Fe3 LTH/NF, and e: Co4Mn1Fe4 LTH/NF samples; SEM images of (b) Co4Mn1 LDH/NF, (c) Co4Mn1Fe1 LTH/NF, (d) Co4Mn1Fe2 LTH/NF, (e) Co4Mn1Fe3 LTH/NF and (f) Co4Mn1Fe4 LTH/NF samples; (g) proposed mechanism for the effect of Fe addition on morphology construction; (h) LSV curves, (i) Tafel plots and (j) corresponding contrast of Co4Mn1 LDH/NF, Co4Mn1Fe1 LTH/NF, Co4Mn1Fe2 LTH/NF, Co4Mn1Fe3 LTH/NF and Co4Mn1Fe4 LTH/NF. This figure has been reproduced from ref. 229 with permission from Royal Society of Chemistry, copyright 2019. | ||
Hu et al.230 discussed the use of CoFeV LDH/NF catalysts in 1 M KOH for the OER. The CoFeV LDH/NF catalysts are made by doping cobalt ferrite (CoFe) LDH with V ions. The results show that the CoFeV-0.25/NF catalyst has a lower overpotential and Tafel slope compared to the pristine CoFe LDH/NF, commercial RuO2/NF, and other CoFeV LDH/NF samples with different amounts of V dopant. The overpotential of the CoFeV-0.25/NF catalyst at a current density of 10 mA cm−2 is 0.242 V, which is lower than those of CoFe LDH/NF (0.318 V) and commercial RuO2/NF (0.282 V). This means that the CoFeV-0.25/NF catalyst requires less energy to drive the OER compared to the other catalysts. The Tafel slope of the CoFeV-0.25/NF catalyst is 57 mV dec−1, which is lower than those of CoFeV-0.125/NF (66 mV dec−1), CoFeV-0.5/NF (64 mV dec−1) and CoFe LDH/NF (72 mV dec−1). This confirms the faster OER kinetics and superior catalytic activity of the CoFeV-0.25/NF catalyst. The results showed a slight degradation of the current density over time, indicating the durability of the catalyst. The Cdl of the CoFeV-0.25/NF sample is similar to that of the other CoFeV LDH/NF samples, while it is larger than that of the CoFe LDH/NF sample. The intrinsic OER activity of the CoFeV-0.25/NF sample is significantly better than that of the bimetallic CoFe LDH/NF, which demonstrates that the addition of V dopant in CoFe LDH could improve the intrinsic OER activity.
Chen et al.231 presented a comprehensive study on the catalytic performance of CoFeCr (oxy)hydroxides for the OER in alkaline electrolytes. The researchers found that the optimal elemental composition for the Co5Fe3Cr2 (oxy)hydroxides is Co:Fe:Cr = 5:3:2, which exhibited the smallest overpotential (η10) of 232 mV and the highest mass activity of 477.0 A g−1 compared to other CoFe-based catalysts. The amorphous crystal structure of Co5Fe3Cr2 (oxy)hydroxides is also essential for its excellent OER activity as thermally annealing the catalyst at 500 °C resulted in degraded performance. The stability of the catalyst was also evaluated through continuous discharging at 50 mA cm−2 in 1 M KOH electrolyte and was found to be excellent. Comparison of the catalytic performance of Co5Fe3Cr2 (oxy)hydroxides to commercial IrO2 showed that Co5Fe3Cr2 (oxy)hydroxides delivered one of the best catalytic activities among recently reported CoFe-based OER catalysts. The study also compared the catalytic performance normalized by the ECSA of the catalysts, which is important for practical applications. Overall, the research provides a valuable contribution to the field of OER catalysis, and the results suggest that Co5Fe3Cr2 (oxy)hydroxides have the potential to be used as an efficient and stable OER catalyst in alkaline electrolyte.
Wang et al.232 described a study in which a dendritic CoFeCu ternary alloy was grown on a nickel foam electrode using an electrodeposition method. The dendritic morphology was observed to have formed as a result of the introduction of Cu into the solution, and it was found that the CoFeCu/NF electrode had a large ECSA and fast charge transfer rate. The electrocatalytic activity of the CoFeCu/NF electrode for the OER was found to be excellent, with an overpotential of 202 mV at a current density of 10 mA cm−2 in 1.0 M KOH. This overpotential is considered low, which is desirable for the OER process. The introduction of Cu significantly improved the OER performance of the CoFeCu alloy compared to the CoFe alloy alone. This improvement was attributed to the enhanced charge transfer and increased exposure of active sites due to the formation of the dendritic morphology. Some transition metals in the CoFeCu alloy had been converted into their hydrogen oxide or oxyhydroxide after electrolysis, which may have contributed to the improved performance. Overall, the results suggested that the dendritic CoFeCu/NF electrode is a promising electrocatalyst for the OER in water splitting, with great potential for practical application.233,234
5 Shortcomings and limitations of using cobalt-based layered hydroxides as OER electrocatalysts
Cobalt-layered double hydroxides (Co-LDHs) have been studied widely as a potential catalyst for the oxygen evolution reaction (OER), but there are several shortcomings associated with their use.• Stability: Co-LDHs tend to degrade quickly under OER conditions, which limits their long-term effectiveness as a catalyst. Additionally, cobalt ions in the Co-LDH can leach out into the electrolyte, which further reduces the stability of the catalyst.
• Low activity: finally, Co-LDHs are not as active as other catalysts, such as IrO2 and RuO2, which are widely used in the OER. This can lead to slower reaction kinetics and lower overall performance.
• Limited selectivity: Co-LDHs may not be selective for the OER and may also catalyze the reduction of other species in the electrolyte, such as water to hydrogen.
• Insensitivity to pH: the activity of Co-LDHs for the OER is not strongly affected by pH, which can limit their ability to optimize the reaction conditions for maximum efficiency.
• Low durability: Co-LDHs can be easily damaged by exposure to high temperatures, humidity, and other environmental factors, which can further limit their stability and longevity as catalysts.
• Cost: Co-LDHs are relatively expensive to produce, which can make them less cost-effective than other catalysts, such as NiO and Ni(OH)2.
• Low conductivity: Co-LDHs have low electronic conductivity which reduces their performance as catalysts.
Overall, while cobalt-layered double hydroxides have been studied as a potential catalyst for the oxygen evolution reaction, there are several limitations that need to be addressed before they can be considered as a viable alternative to more established catalysts.
6 Strategies to improve OER activity on cobalt-based layered hydroxides
Enhancing the oxygen evolution reaction (OER) activity of cobalt-based layered hydroxides encompasses diverse strategic approaches:(1) Defect engineering: introducing defects within the crystal structure of cobalt-based layered hydroxides stands as an effective approach. These defects create active sites on the material's surface, enhancing its catalytic activity for the OER.
(2) Surface modification with synergistic materials: modifying the surface of cobalt-based layered hydroxides using materials like transition metal oxides or conductive polymers generates a synergistic effect. This modification enhances the OER activity of the cobalt-based catalyst.
(3) Bifunctional catalyst design: employing cobalt-based layered hydroxides in tandem with catalysts such as IrO2 or RuO2 produces bifunctional catalysts. This synergistic combination enhances overall OER activity.
(4) Tailored synthesis methods: utilizing varied synthesis methods to fabricate cobalt-based layered hydroxides with distinct structures (e.g., nanorods, nanosheets) exploits unique properties for improved OER activity.
(5) Optimization of composition: adjusting the cobalt-to-transition metal ion ratio within the layered hydroxide composition optimizes the catalyst's OER activity.
(6) Enhanced support design: designing suitable supports for cobalt-based hydroxides, like employing carbon-based materials, enhances electronic conductivity and stability, augmenting catalyst performance.
(7) Stability enhancement in acidic media: addressing the stability of cobalt-based hydroxides in acidic environments involves functionalizing the surface with appropriate groups or applying protective coatings.
Each of these strategies offers promising pathways for bolstering the OER activity of cobalt-based layered hydroxides, providing avenues for further advancement and application in electrocatalysis for various industrial and environmental applications.
The performance of cobalt-based catalysts, in comparison to commonly used OER catalysts like iridium oxide (IrO2) or ruthenium oxide (RuO2), showcases distinct characteristics. Cobalt-based materials often offer competitive performance with an OER overpotential in the range of approximately 300–500 mV, while IrO2 and RuO2 tend to exhibit lower overpotentials of around 200–300 mV. However, cobalt-based catalysts present advantages in terms of cost-effectiveness and earth abundance, in contrast to the higher cost and scarcity associated with IrO2 and RuO2. While cobalt-based materials might have slightly higher overpotentials, their comparative affordability and greater availability make them promising alternatives in practical applications despite their slightly inferior performance under specific OER conditions.
7 Conclusion
In conclusion, the use of cobalt hydroxide and its derivatives as catalysts for the OER in water splitting devices is a promising area of research. Studies have shown that these materials have high OER activity and stability, making them potentially useful in the development of more efficient and cost-effective water splitting systems. Additionally, cobalt is an earth-abundant and relatively inexpensive material, which is another advantage over other catalysts. However, there is still much to be understood about the mechanisms of action of cobalt hydroxide and its derivatives in the OER. Further research is needed to optimize their performance, such as exploring the effect of different synthesis methods, modifying the surface properties of the materials, and studying their stability under different conditions. It is also important to consider the potential environmental and health effects of using cobalt-based materials in large-scale applications. Cobalt is a toxic material and therefore, safety measures should be taken in the handling and disposal of cobalt-containing materials. Cobalt hydroxide and its derivatives have shown promising results as catalysts for the OER, and they have the potential to be a viable alternative to currently used catalysts in water splitting systems. However, more research is needed to fully understand their mechanisms of action and to optimize their performance. Additionally, safety and environmental concerns should be taken into account when considering their use on a large scale.Conflicts of interest
There are no conflicts to declare.References
- L. Barreto, A. Makihira and K. Riahi, Int. J. Hydrogen Energy, 2003, 28, 267–284 CrossRef CAS.
- K.-H. Liu, H.-X. Zhong, S.-J. Li, Y.-X. Duan, M.-M. Shi, X.-B. Zhang, J.-M. Yan and Q. Jiang, Prog. Mater. Sci., 2018, 92, 64–111 CrossRef CAS.
- Q. Ding, B. Song, P. Xu and S. Jin, Chem, 2016, 1, 699–726 CAS.
- N. Abas, A. Kalair and N. Khan, Futures, 2015, 69, 31–49 CrossRef.
- S. Anantharaj, K. Karthick and S. Kundu, Mater. Today Energy, 2017, 6, 1–26 CrossRef.
- X. Long, Z. Wang, S. Xiao, Y. An and S. Yang, Mater. Today, 2016, 19, 213–226 CrossRef CAS.
- L. Barelli, G. Bidini, F. Gallorini and S. Servili, Energy, 2008, 33, 554–570 CrossRef CAS.
- A. P. Simpson and A. E. Lutz, Int. J. Hydrogen Energy, 2007, 32, 4811–4820 CrossRef CAS.
- D. K. Liguras, D. I. Kondarides and X. E. Verykios, Appl. Catal., B, 2003, 43, 345–354 CrossRef CAS.
- W. Zhu, R. Zhang, F. Qu, A. M. Asiri and X. Sun, ChemCatChem, 2017, 9, 1721–1743 CrossRef CAS.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- B. Steel and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef PubMed.
- Y. Wang, K. S. Chen, J. Mishler, S. C. Cho and X. C. Adroher, Appl. Energy, 2011, 88, 981–1007 CrossRef CAS.
- S. Guo and E. Wang, Nano Today, 2011, 6, 240–264 CrossRef CAS.
- G. P. Gardner, Y. B. Go, D. M. Robinson, P. F. Smith, J. Hadermann, A. Abakumov, M. Greenblatt and G. C. Dismukes, Angew. Chem., Int. Ed., 2012, 51, 1616–1619 CrossRef CAS PubMed.
- M. Barroso, A. J. Cowan, S. R. Pendlebury, M. Grätzel, D. R. Klug and J. R. Durrant, J. Am. Chem. Soc., 2011, 133, 14868–14871 CrossRef CAS PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- C. G. Silva, Y. Bouizi, V. Fornes and H. Garcia, J. Am. Chem. Soc., 2009, 131, 13833–13839 CrossRef PubMed.
- Z. Chen, P. Sun, B. Fan, Z. Zhang and X. Fang, J. Phys. Chem. C, 2014, 118, 7801–7807 CrossRef CAS.
- M. Tayyab, U. E. Kulsoom, Y. Liu, S. Mansoor, M. Khan, Z. Akmal, A. Mushtaq, M. Arif, U. Shamriaz and L. Zhou, Int. J. Hydrogen Energy, 2023, 1400–1413 Search PubMed.
- S. Anantharaj and S. Noda, Small, 2020, 16, 1905779 CrossRef CAS PubMed.
- S. Anantharaj and V. Aravindan, Adv. Energy Mater., 2020, 10, 1902666 CrossRef CAS.
- U. Shamraiz, A. Badshah, A. Alfantazi, H. Hussain, B. Raza, S. Ullah and I. R. Green, J. Electrochem. Soc., 2021, 168, 066510 CrossRef CAS.
- Z. Wu, B. Fang, Z. Wang, C. Wang, Z. Liu, F. Liu, W. Wang, A. Alfantazi, D. Wang and D. P. Wilkinson, ACS Catal., 2013, 3, 2101–2107 CrossRef CAS.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- S. Grigoriev, P. Millet and V. Fateev, J. Power Sources, 2008, 177, 281–285 CrossRef CAS.
- S. Song, H. Zhang, X. Ma, Z. Shao, R. T. Baker and B. Yi, Int. J. Hydrogen Energy, 2008, 33, 4955–4961 CrossRef CAS.
- A. T. Marshall, S. Sunde, M. Tsypkin and R. Tunold, Int. J. Hydrogen Energy, 2007, 32, 2320–2324 CrossRef CAS.
- H. Wendt and G. Imarisio, J. Appl. Electrochem., 1988, 18, 1–14 CrossRef CAS.
- D. Santos, C. Sequeira, D. Macciò, A. Saccone and J. Figueiredo, Int. J. Hydrogen Energy, 2013, 38, 3137–3145 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- E. Fabbri, A. Habereder, K. Waltar, R. Kötz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 RSC.
- A. Brisse, J. Schefold and M. Zahid, Int. J. Hydrogen Energy, 2008, 33, 5375–5382 CrossRef CAS.
- M. Ma, F. Qu, X. Ji, D. Liu, S. Hao, G. Du, A. M. Asiri, Y. Yao, L. Chen and X. Sun, Small, 2017, 13, 1700394 CrossRef PubMed.
- L. Yang, D. Liu, S. Hao, R. Kong, A. M. Asiri, C. Zhang and X. Sun, J. Mater. Chem. A, 2017, 5, 7305–7308 RSC.
- R. Ge, M. Ma, X. Ren, F. Qu, Z. Liu, G. Du, A. M. Asiri, L. Chen, B. Zheng and X. Sun, Chem. Commun., 2017, 53, 7812–7815 RSC.
- M. Ma, Y. Liu, X. Ma, R. Ge, F. Qu, Z. Liu, G. Du, A. M. Asiri, Y. Yao and X. Sun, Sustainable Energy Fuels, 2017, 1, 1287–1291 RSC.
- M. Xie, L. Yang, Y. Ji, Z. Wang, X. Ren, Z. Liu, A. M. Asiri, X. Xiong and X. Sun, Nanoscale, 2017, 9, 16612–16615 RSC.
- T. Wang, X. Zhang, X. Zhu, Q. Liu, S. Lu, A. M. Asiri, Y. Luo and X. Sun, Nanoscale, 2020, 12, 5359–5362 RSC.
- M. Ma, D. Liu, S. Hao, R. Kong, G. Du, A. M. Asiri, Y. Yao and X. Sun, Inorg. Chem. Front., 2017, 4, 840–844 RSC.
- X. Ji, L. Cui, D. Liu, S. Hao, J. Liu, F. Qu, Y. Ma, G. Du, A. M. Asiri and X. Sun, Chem. Commun., 2017, 53, 3070–3073 RSC.
- S. Anantharaj, M. Venkatesh, A. S. Salunke, T. V. Simha, V. Prabu and S. Kundu, ACS Sustain. Chem. Eng., 2017, 5, 10072–10083 CrossRef CAS.
- S. Anantharaj, K. Karthick and S. Kundu, lnorg. Chem., 2018, 57, 3082–3096 CrossRef CAS PubMed.
- K. Karthick, S. Anantharaj and S. Kundu, ACS Sustain. Chem. Eng., 2018, 6, 6802–6810 CrossRef CAS.
- S. Anantharaj, T. S. Amarnath, E. Subhashini, S. Chatterjee, K. C. Swaathini, K. Karthick and S. Kundu, ACS Catal., 2018, 8, 5686–5697 CrossRef CAS.
- S. Anantharaj, P. Karthik and S. Kundu, J. Mater. Chem. A, 2015, 3, 24463–24478 RSC.
- K. A. Stoerzinger, L. Qiao, M. D. Biegalski and Y. Shao-Horn, J. Phys. Chem. Lett., 2014, 5, 1636–1641 CrossRef CAS PubMed.
- M. Yagi, E. Tomita, S. Sakita, T. Kuwabara and K. Nagai, J. Phys. Chem. B, 2005, 109, 21489–21491 CrossRef CAS PubMed.
- R. Kötz and S. Stucki, Electrochim. Acta, 1986, 31, 1311–1316 CrossRef.
- Z. S. Wu, D. W. Wang, W. Ren, J. Zhao, G. Zhou, F. Li and H. M. Cheng, Adv. Funct. Mater., 2010, 20, 3595–3602 CrossRef CAS.
- W. Yong-gang and Z. Xiao-gang, Electrochim. Acta, 2004, 49, 1957–1962 CrossRef.
- L. Burke and M. McCarthy, Electrochim. Acta, 1984, 29, 211–216 CrossRef CAS.
- D. Susanti, D.-S. Tsai, Y.-S. Huang, A. Korotcov and W.-H. Chung, J. Phys. Chem. C, 2007, 111, 9530–9537 CrossRef CAS.
- J. Swaminathan, R. Subbiah and V. Singaram, ACS Catal., 2016, 6, 2222–2229 CrossRef CAS.
- H. Shi, H. Liang, F. Ming and Z. Wang, Angew. Chem., 2017, 129, 588–592 CrossRef.
- J. Jiang, F. Sun, S. Zhou, W. Hu, H. Zhang, J. Dong, Z. Jiang, J. Zhao, J. Li and W. Yan, Nat. Commun., 2018, 9, 1–12 CrossRef PubMed.
- J. Liu, Y. Ji, J. Nai, X. Niu, Y. Luo, L. Guo and S. Yang, Energy Environ. Sci., 2018, 11, 1736–1741 RSC.
- F. Wang, J. Li, F. Wang, T. A. Shifa, Z. Cheng, Z. Wang, K. Xu, X. Zhan, Q. Wang and Y. Huang, Adv. Funct. Mater., 2015, 25, 6077–6083 CrossRef CAS.
- X. Zong, J. Han, G. Ma, H. Yan, G. Wu and C. Li, J. Phys. Chem. C, 2011, 115, 12202–12208 CrossRef CAS.
- H. Zhou, F. Yu, J. Sun, H. Zhu, I. K. Mishra, S. Chen and Z. Ren, Nano Lett., 2016, 16, 7604–7609 CrossRef CAS PubMed.
- L. Cheng, W. Huang, Q. Gong, C. Liu, Z. Liu, Y. Li and H. Dai, Angew. Chem., Int. Ed., 2014, 53, 7860–7863 CrossRef CAS PubMed.
- Y. Zhao, Y. Zhang, Z. Yang, Y. Yan and K. Sun, Sci. Technol. Adv. Mater., 2013, 14, 043501 CrossRef CAS PubMed.
- J. Theerthagiri, R. Senthil, B. Senthilkumar, A. R. Polu, J. Madhavan and M. Ashokkumar, J. Solid State Chem., 2017, 252, 43–71 CrossRef CAS.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957–3971 CrossRef CAS.
- T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 18153–18156 CrossRef CAS PubMed.
- D. Jeong, K. Jin, S. E. Jerng, H. Seo, D. Kim, S. H. Nahm, S. H. Kim and K. T. Nam, ACS Catal., 2015, 5, 4624–4628 CrossRef CAS.
- M. Morimitsu and N. Oshiumi, Chem. Lett., 2009, 38, 822–823 CrossRef CAS.
- J. Chen, F. Zheng, S.-J. Zhang, A. Fisher, Y. Zhou, Z. Wang, Y. Li, B.-B. Xu, J.-T. Li and S.-G. Sun, ACS Catal., 2018, 8, 11342–11351 CrossRef CAS.
- W. D. Chemelewski, H.-C. Lee, J.-F. Lin, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2014, 136, 2843–2850 CrossRef CAS PubMed.
- S. Zou, M. S. Burke, M. G. Kast, J. Fan, N. Danilovic and S. W. Boettcher, Chem. Mater., 2015, 27, 8011–8020 CrossRef CAS.
- M. Mathankumar, S. Anantharaj, A. Nandakumar, S. Kundu and B. Subramanian, J. Mater. Chem. A, 2017, 5, 23053–23066 RSC.
- X. Duan, Y. Yang, C. Liu, M. Zhou, L. Yang, H. He, Y. Zhang and P. Xiao, Appl. Surf. Sci., 2017, 407, 177–184 CrossRef CAS.
- S. Anantharaj, K. Karthick and S. Kundu, lnorg. Chem., 2019, 58, 8570–8576 CrossRef CAS PubMed.
- B. Malik, S. Anantharaj, K. Karthick, D. K. Pattanayak and S. Kundu, Catal. Sci. Technol., 2017, 7, 2486–2497 RSC.
- U. Shamraiz, A. Badshah and B. Raza, Langmuir, 2020, 36, 2223–2230 CrossRef CAS PubMed.
- U. Shamraiz, A. Badshah, B. Raza and I. R. Green, ACS Appl. Energy Mater., 2020, 6486–6491 CrossRef CAS.
- C. Gervas, M. D. Khan, S. Mlowe, C. Zhang, C. Zhao, R. K. Gupta, M. P. Akerman, P. Mashazi, T. Nyokong and N. Revaprasadu, ChemElectroChem, 2019, 6, 2560–2569 CrossRef CAS.
- G. B. Shombe, M. D. Khan, A. M. Alenad, J. Choi, T. Ingsel, R. K. Gupta and N. Revaprasadu, Sustainable Energy Fuels, 2020, 4, 5132–5143 RSC.
- G. E. Ayom, M. D. Khan, T. Ingsel, W. Lin, R. K. Gupta, S. J. Zamisa, W. E. van Zyl and N. Revaprasadu, Eur. J. Chem., 2020, 26, 2693–2704 CrossRef CAS PubMed.
- R. A. Hussain and I. Hussain, J. Solid State Chem., 2019, 277, 316–328 CrossRef CAS.
- J. Skowronski and A. Wazny, J. New Mater. Electrochem. Syst., 2006, 9, 345 CAS.
- J. Deng, M. R. Nellist, M. B. Stevens, C. Dette, Y. Wang and S. W. Boettcher, Nano Lett., 2017, 17, 6922–6926 CrossRef CAS PubMed.
- K. Karthick, S. Anantharaj, S. Patchaiammal, S. N. Jagadeesan, P. Kumar, S. R. Ede, D. K. Pattanayak and S. Kundu, lnorg. Chem., 2019, 58, 9490–9499 CrossRef CAS PubMed.
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, J. Am. Chem. Soc., 2018, 140, 3876–3879 CrossRef CAS PubMed.
- L. Yuan, Z. Yan, L. Jiang, E. Wang, S. Wang and G. Sun, J. Energy Chem., 2016, 25, 805–810 CrossRef.
- U. Shamraiz, B. Raza, H. Hussain, A. Badshah, I. R. Green, F. A. Kiani and A. Al-Harrasi, Int. Mater. Rev., 2019, 64, 478–512 CrossRef CAS.
- J. Masud, P. C. Ioannou, N. Levesanos, P. Kyritsis and M. Nath, ChemSusChem, 2016, 9, 3128–3132 CrossRef CAS PubMed.
- J. Xu, G. Liu, J. Li and X. Wang, Electrochim. Acta, 2012, 59, 105–112 CrossRef CAS.
- D.-Y. Kuo, J. K. Kawasaki, J. N. Nelson, J. Kloppenburg, G. Hautier, K. M. Shen, D. G. Schlom and J. Suntivich, J. Am. Chem. Soc., 2017, 139, 3473–3479 CrossRef CAS PubMed.
- Q. Zhou, Y. Chen, G. Zhao, Y. Lin, Z. Yu, X. Xu, X. Wang, H. K. Liu, W. Sun and S. X. Dou, ACS Catal., 2018, 8, 5382–5390 CrossRef CAS.
- R. Ge, X. Ren, X. Ji, Z. Liu, G. Du, A. M. Asiri, X. Sun and L. Chen, ChemSusChem, 2017, 10, 4004–4008 CrossRef CAS PubMed.
- Y. Zhang, C. Wu, H. Jiang, Y. Lin, H. Liu, Q. He, S. Chen, T. Duan and L. Song, Adv. Mater., 2018, 30, 1707522 CrossRef PubMed.
- J. Jiang, A. Zhang, L. Li and L. Ai, J. Power Sources, 2015, 278, 445–451 CrossRef CAS.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- J. S. Yoo, X. Rong, Y. Liu and A. M. Kolpak, ACS Catal., 2018, 8, 4628–4636 CrossRef CAS.
- H. Jin, C. Guo, X. Liu, J. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem. Rev., 2018, 118, 6337–6408 CrossRef CAS PubMed.
- P. Benson, G. Briggs and W. Wynne-Jones, Electrochim. Acta, 1964, 9, 275–280 CrossRef CAS.
- L. Cao, F. Xu, Y. Y. Liang and H. L. Li, Adv. Mater., 2004, 16, 1853–1857 CrossRef CAS.
- U. Shamraiz, A. Badshah and N. u. Ain, ACS Appl. Energy Mater., 2023, 6, 2489–2496 CrossRef CAS.
- I. Terasaki, Y. Sasago and K. Uchinokura, Phys. Rev. B, 1997, 56, R12685 CrossRef CAS.
- R. R. Salunkhe, K. Jang, H. Yu, S. Yu, T. Ganesh, S.-H. Han and H. Ahn, J. Alloys Compd., 2011, 509, 6677–6682 CrossRef CAS.
- C.-C. Tseng, J.-L. Lee, Y.-M. Liu, M.-D. Ger and Y.-Y. Shu, J. Taiwan Inst. Chem. Eng., 2013, 44, 415–419 CrossRef CAS.
- P. Oliva, J. Leonardi, J. Laurent, C. Delmas, J. Braconnier, M. Figlarz, F. Fievet and A. d. Guibert, J. Power Sources, 1982, 8, 229–255 CrossRef CAS.
- F. Lyu, Y. Bai, Q. Wang, L. Wang, X. Zhang and Y. Yin, Dalton Trans., 2017, 46, 10545–10548 RSC.
- M. Kurmoo, Chem. Mater., 1999, 11, 3370–3378 CrossRef CAS.
- A. Rujiwatra, C. J. Kepert, J. B. Claridge, M. J. Rosseinsky, H. Kumagai and M. Kurmoo, J. Am. Chem. Soc., 2001, 123, 10584–10594 CrossRef CAS PubMed.
- Y. Hou, H. Kondoh, M. Shimojo, T. Kogure and T. Ohta, J. Phys. Chem. B, 2005, 109, 19094–19098 CrossRef CAS PubMed.
- M. Jana, P. Sivakumar, M. Kota, M. G. Jung and H. S. Park, J. Power Sources, 2019, 422, 9–17 CrossRef CAS.
- Z. Huang, Y. Zhao, Y. Song, Y. Li, G. Wu, H. Tang and J. Zhao, RSC Adv., 2016, 6, 80059–80064 RSC.
- J. A. Koza, C. M. Hull, Y.-C. Liu and J. A. Switzer, Chem. Mater., 2013, 25, 1922–1926 CrossRef CAS.
- Y. Zhan, G. Du, S. Yang, C. Xu, M. Lu, Z. Liu and J. Y. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 12930–12936 CrossRef CAS PubMed.
- Y. Tong, J. Xu, H. Jiang, F. Gao and Q. Lu, Chem. Eng. J., 2017, 316, 225–231 CrossRef CAS.
- O. De Silva, M. Singh, S. Mahasivam, N. Mahmood, B. J. Murdoch, R. Ramanathan and V. Bansal, ACS Appl. Nano Mater., 2022, 5, 12209–12216 CrossRef CAS.
- M. A. Sayeed, T. Herd and A. P. O'Mullane, J. Mater. Chem. A, 2016, 4, 991–999 RSC.
- Z. Zhuang, W. Sheng and Y. Yan, Adv. Mater., 2014, 26, 3950–3955 CrossRef CAS PubMed.
- P. Babar, A. Lokhande, B. Pawar, M. Gang, E. Jo, C. Go, M. Suryawanshi, S. Pawar and J. H. Kim, Appl. Surf. Sci., 2018, 427, 253–259 CrossRef CAS.
- M. A. Ghanem, A. M. Al-Mayouf, P. Arunachalam and T. Abiti, Electrochim. Acta, 2016, 207, 177–186 CrossRef CAS.
- H. Yang, Y. Long, Y. Zhu, Z. Zhao, P. Ma, J. Jin and J. Ma, Green Chem., 2017, 19, 5809–5817 RSC.
- J. Zhang, C. Dong, Z. Wang, C. Zhang, H. Gao, J. Niu and Z. Zhang, Electrochim. Acta, 2018, 284, 495–503 CrossRef CAS.
- N. P. Dileep, T. V. Vineesh, P. V. Sarma, M. V. Chalil, C. S. Prasad and M. Shaijumon, ACS Appl. Energy Mater., 2020, 3, 1461–1467 CrossRef CAS.
- D. McAteer, I. J. Godwin, Z. Ling, A. Harvey, L. He, C. S. Boland, V. Vega-Mayoral, B. Szydłowska, A. A. Rovetta and C. Backes, Adv. Energy Mater., 2018, 8, 1702965 CrossRef.
- H. Zhang, B. Chen, H. Jiang, X. Duan, Y. Zhu and C. Li, Nanoscale, 2018, 10, 12991–12996 RSC.
- S. Ullah, U. Shamraiz, A. Badshah, B. Raza, A. S. Farooqi, H. M. Tofil, M. A. Zeb and A. Alfantazi, Int. J. Hydrogen Energy, 2022, 47, 30799–30809 CrossRef CAS.
- S. Zhang, B. Ni, H. Li, H. Lin, H. Zhu, H. Wang and X. Wang, Chem. Commun., 2017, 53, 8010–8013 RSC.
- M. Darbandi, B. Shaabani, A. Alizadeh, P. Yardani, E. Shahryari and M. G. Hosseini, Microporous Mesoporous Mater., 2019, 284, 421–426 CrossRef CAS.
- D. S. Dhawale, P. Bodhankar, N. Sonawane and P. B. Sarawade, Sustainable Energy Fuels, 2019, 3, 1713–1719 RSC.
- J. Liu, J. Nai, T. You, P. An, J. Zhang, G. Ma, X. Niu, C. Liang, S. Yang and L. Guo, Small, 2018, 14, 1703514 CrossRef PubMed.
- M. Huang, W. Liu, L. Wang, J. Liu, G. Chen, W. You, J. Zhang, L. Yuan, X. Zhang and R. Che, Nano Res., 2020, 13, 810–817 CrossRef CAS.
- X. Bu, C. Chiang, R. Wei, Z. Li, Y. Meng, C. Peng, Y. Lin, Y. Li, Y. Lin and K. S. Chan, ACS Appl. Mater. Interfaces, 2019, 11, 38633–38640 CrossRef CAS PubMed.
- C. Cui, J. Wang, Z. Luo, J. Wang, C. Li and Z. Li, Electrochim. Acta, 2018, 273, 327–334 CrossRef CAS.
- P. Jash, P. Srivastava and A. Paul, Chem. Commun., 2019, 55, 2230–2233 RSC.
- J. Huang, Q. Liu, T. Yao, Z. Pan and S. Wei, J. Phys.: Conf. Ser., 2016, 712, 012128 CrossRef.
- Y. Jiang, X. Li, T. Wang and C. Wang, Nanoscale, 2016, 8, 9667–9675 RSC.
- Z. Liang, C. Zhang, Y. Xu, W. Zhang, H. Zheng and R. Cao, ACS Sustain. Chem. Eng., 2018, 7, 3527–3535 CrossRef.
- H. Mao, X. Guo, Y. Fu, Z. Cao, D. Sun, B. Wang, Y. Zhang, Q. Fan and X.-M. Song, Appl. Surf. Sci., 2019, 485, 554–563 CrossRef CAS.
- J. Wang, Y. Bao, C. Cui, Z. Zhang, S. Li, J. Pan, Y. Zhang, G. Tu, J. Wang and Z. Li, J. Mater. Sci., 2019, 54, 7692–7701 CrossRef CAS.
- S. Pan, J. Yu, Y. Zhang and B. Li, Mater. Lett., 2020, 278, 128414 CrossRef CAS.
- B. Zhang, J. Zhang, X. Tan, D. Tan, J. Shi, F. Zhang, L. Liu, Z. Su, B. Han and L. Zheng, Chem. Commun., 2018, 54, 4045–4048 RSC.
- Q. Zha, W. Xu, X. Li and Y. Ni, Dalton Trans., 2019, 48, 12127–12136 RSC.
- P. F. Liu, S. Yang, L. R. Zheng, B. Zhang and H. G. Yang, J. Mater. Chem. A, 2016, 4, 9578–9584 RSC.
- S. Bose, J. Debgupta, R. M. Ramsundar and S. K. Das, Eur. J. Chem., 2017, 23, 8051–8057 CrossRef CAS PubMed.
- L. Huang, J. Jiang and L. Ai, ACS Appl. Mater. Interfaces, 2017, 9, 7059–7067 CrossRef CAS PubMed.
- R. Liu, Z. Jiang, J. Ma, L. Ni, X. Sun, Y. Liu, H. Chen and Q. Liu, RSC Adv., 2017, 7, 3783–3789 RSC.
- M. Chen, Y. Xie, J.-X. Wu, H. Huang, J. Teng, D. Wang, Y. Fan, J.-J. Jiang, H.-P. Wang and C.-Y. Su, J. Mater. Chem. A, 2019, 7, 10217–10224 RSC.
- X. Chen, Y. Chen, Z. Shen, C. Song, P. Ji, N. Wang, D. Su, Y. Wang, G. Wang and L. Cui, Appl. Surf. Sci., 2020, 529, 147173 CrossRef CAS.
- C. Meng, M. Lin, X. Sun, X. Chen, X. Chen, X. Du and Y. Zhou, Chem. Commun., 2019, 55, 2904–2907 RSC.
- H. Wang, E.-M. Feng, Y.-M. Liu and C.-Y. Zhang, J. Mater. Chem. A, 2019, 7, 7777–7783 RSC.
- S. H. Ye, Z. X. Shi, J. X. Feng, Y. X. Tong and G. R. Li, Angew. Chem., Int. Ed., 2018, 57, 2672–2676 CrossRef CAS PubMed.
- A. Bergmann, T. E. Jones, E. Martinez Moreno, D. Teschner, P. Chernev, M. Gliech, T. Reier, H. Dau and P. Strasser, Nat. Catal., 2018, 1, 711–719 CrossRef CAS.
- M. Cui, H. Zhao, X. Dai, Y. Yang, X. Zhang, X. Luan, F. Nie, Z. Ren, Y. Dong and Y. Wang, ACS Sustain. Chem. Eng., 2019, 7, 13015–13022 CrossRef CAS.
- Y.-Z. Xu, C.-Z. Yuan and X.-P. Chen, Appl. Surf. Sci., 2017, 426, 688–693 CrossRef CAS.
- L. Liu, Y. Ou, D. Gao, L. Yang, H. Dong, P. Xiao and Y. Zhang, J. Power Sources, 2018, 396, 395–403 CrossRef CAS.
- F. Reikowski, F. Maroun, I. Pacheco, T. Wiegmann, P. Allongue, J. Stettner and O. M. Magnussen, ACS Catal., 2019, 9, 3811–3821 CrossRef CAS.
- A. Moysiadou, S. Lee, C.-S. Hsu, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2020, 142, 11901–11914 CrossRef CAS PubMed.
- S. Zhang, B. Huang, L. Wang, X. Zhang, H. Zhu, X. Zhu, J. Li, S. Guo and E. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 40220–40228 CrossRef CAS PubMed.
- H. Jin, S. Mao, G. Zhan, F. Xu, X. Bao and Y. Wang, J. Mater. Chem. A, 2017, 5, 1078–1084 RSC.
- Y. Wang, C. Yang, Y. Huang, Z. Li, Z. Liang and G. Cao, J. Mater. Chem. A, 2020, 8, 6699–6708 RSC.
- X. Guo, G. Liang and A. Gu, J. Colloid Interface Sci., 2019, 553, 713–719 CrossRef CAS PubMed.
- G. Zeng, M. Liao, C. Zhou, X. Chen, Y. Wang and D. Xiao, RSC Adv., 2016, 6, 42255–42262 RSC.
- B. Cao, C. Luo, J. Lao, H. Chen, R. Qi, H. Lin and H. Peng, ACS Omega, 2019, 4, 16612–16618 CrossRef CAS PubMed.
- X. Bai and J. Guan, Top. Catal., 2021, 1–10 Search PubMed.
- L. Chen, H. Zhang, L. Chen, X. Wei, J. Shi and M. He, J. Mater. Chem. A, 2017, 5, 22568–22575 RSC.
- Z. Chen, C. X. Kronawitter, Y.-W. Yeh, X. Yang, P. Zhao, N. Yao and B. E. Koel, J. Mater. Chem. A, 2017, 5, 842–850 RSC.
- Y. Huang, X. Zhao, F. Tang, X. Zheng, W. Cheng, W. Che, F. Hu, Y. Jiang, Q. Liu and S. Wei, J. Mater. Chem. A, 2018, 6, 3202–3210 RSC.
- Z. Chen, L. Cai, X. Yang, C. Kronawitter, L. Guo, S. Shen and B. E. Koel, ACS Catal., 2018, 8, 1238–1247 CrossRef CAS.
- N. Ma, C. Gong, H. Xie, C. Shi, J. Sha, C. He, F. He, N. Zhao and E. Liu, Int. J. Hydrogen Energy, 2022, 47, 29762–29770 CrossRef CAS.
- Z. Wang, Y. Hu, W. Liu, L. Xu, M. Guan, Y. Zhao, J. Bao and H. Li, Eur. J. Chem., 2020, 26, 9382–9388 CrossRef CAS PubMed.
- A. Karmakar and S. K. Srivastava, ACS Appl. Energy Mater., 2020, 3, 7335–7344 CrossRef CAS.
- Y. Li, C. Liao, K. Tang, N. Zhang, W. Qi, H. Xie, J. He, K. Yin, Y. Gao and C. Wang, Electrochim. Acta, 2019, 297, 40–45 CrossRef CAS.
- R. Dong, H. Du, Y. Sun, K. Huang, W. Li and B. Geng, ACS Sustain. Chem. Eng., 2018, 6, 13420–13426 CrossRef CAS.
- X.-Z. Song, N. Zhang, F. Liu, Z.-H. Wang, W.-Y. Zhu, G.-Z. Zhang, Z.-Y. Niu, X.-F. Wang and Z. Tan, J. Alloys Compd., 2021, 873, 159766 CrossRef CAS.
- R. Zhang, Z. Hu, W. Ke, M. He, T. Ning, J. Bao, Z. Shang, W. Zhu and G. Zhu, Adv. Powder Technol., 2022, 33, 103728 CrossRef CAS.
- Y.-L. Meng, Y. Li, Z. Tan, X. Chen, L.-L. Wu, L.-W. Ji, Q.-S. Wang, X.-Z. Song and S. Song, Energy Fuels, 2021, 35, 2775–2784 CrossRef CAS.
- M. Duraivel, S. Nagappan, K. H. Park and K. Prabakar, Electrochim. Acta, 2022, 411, 140071 CrossRef CAS.
- H. Kim, Y. Kim, Y. Noh and W. B. Kim, Dalton Trans., 2016, 45, 13686–13690 RSC.
- Y. Liu, Z. Jin, P. Li, X. Tian, X. Chen and D. Xiao, ChemElectroChem, 2018, 5, 593–597 CrossRef CAS.
- F. Wang, K. Ma, W. Tian, J. Dong, H. Han, H. Wang, K. Deng, H. Yue, Y. X. Zhang and W. Jiang, J. Mater. Chem. A, 2019, 7, 19589–19596 RSC.
- Z. Xu, M. Hao, X. Liu, J. Ma, L. Wang, C. Li and W. Wang, Catalysts, 2022, 12, 1417 CrossRef CAS.
- Y.-L. Meng, Y. Li, Z. Tan, X. Chen, L.-L. Wu, L.-W. Ji, Q.-S. Wang, X.-Z. Song and S. Song, Energy Fuels, 2021, 35, 2775–2784 CrossRef CAS.
- B. He, K. Tan, Y. Gong, R. Wang, H. Wang and L. Zhao, Nanoscale, 2020, 12, 9048–9057 RSC.
- A. Balram, H. Zhang and S. Santhanagopalan, ACS Appl. Mater. Interfaces, 2017, 9, 28355–28365 CrossRef CAS PubMed.
- R. V. Prataap and S. Mohan, Chem. Commun., 2017, 53, 3365–3368 RSC.
- X. Wang, Z. Li, D. Y. Wu, G. R. Shen, C. Zou, Y. Feng, H. Liu, C. K. Dong and X. W. Du, Small, 2019, 15, 1804832 CrossRef PubMed.
- Z. Cai, J. Shen, M. Zhang, L. Cui, W. Liu and J. Liu, J. Colloid Interface Sci., 2023, 630, 57–65 CrossRef CAS PubMed.
- Y. Wang, L. Huang, L. Ai, M. Wang, Z. Fan, J. Jiang, H. Sun and S. Wang, Electrochim. Acta, 2019, 318, 966–976 CrossRef CAS.
- S. Yoon, J.-Y. Yun, J.-H. Lim and B. Yoo, J. Alloys Compd., 2017, 693, 964–969 CrossRef CAS.
- K. Awaya and S. Ida, Chem. Lett., 2020, 49, 888–891 CrossRef CAS.
- W. Wang, Y. Lu, M. Zhao, R. Luo, Y. Yang, T. Peng, H. Yan, X. Liu and Y. Luo, ACS Nano, 2019, 13, 12206–12218 CrossRef CAS PubMed.
- W. Chen, Y. Zhang, G. Chen, Y. Zhou, X. Xiang and K. K. Ostrikov, ACS Sustain. Chem. Eng., 2019, 7, 8255–8264 CrossRef CAS.
- X.-Z. Song, N. Zhang, Z.-Y. Niu, Y. Pan, X.-F. Wang and Z. Tan, New J. Chem., 2021, 45, 10199–10203 RSC.
- N. R. Chodankar, I. V. Bagal, S. W. Ryu, Y. K. Han and D. H. Kim, ChemCatChem, 2019, 11, 4256–4261 CrossRef CAS.
- Z. Sun, L. Lin, C. Nan, H. Li, G. Sun and X. Yang, ACS Sustain. Chem. Eng., 2018, 6, 14257–14263 CrossRef CAS.
- Y.-N. Zhou, R.-Y. Fan, Y.-N. Cao, H.-Y. Wang, B. Dong, H.-Y. Zhao, F.-L. Wang, J.-F. Yu and Y.-M. Chai, Nanoscale, 2021, 13, 13463–13472 RSC.
- K. Chakrapani, F. Özcan, K. F. Ortega, T. Machowski and M. Behrens, ChemElectroChem, 2018, 5, 93–100 CrossRef CAS.
- F. Yang, K. Sliozberg, I. Sinev, H. Antoni, A. Bähr, K. Ollegott, W. Xia, J. Masa, W. Grünert and B. R. Cuenya, ChemSusChem, 2017, 10, 156–165 CrossRef CAS PubMed.
- Y. Zheng, R. Gao, Y. Qiu, L. Zheng, Z. Hu and X. Liu, lnorg. Chem., 2021, 60, 5252–5263 CrossRef CAS PubMed.
- A. M. Sakita, R. Della Noce, E. Vallés and A. V. Benedetti, Appl. Surf. Sci., 2018, 434, 1153–1160 CrossRef CAS.
- P. Babar, A. Lokhande, H. H. Shin, B. Pawar, M. G. Gang, S. Pawar and J. H. Kim, Small, 2018, 14, 1702568 CrossRef PubMed.
- J. Cheng, X. Yue, C. Chen, X. Shen, S. Zeng, Z. Ji, A. Yuan and G. Zhu, J. Electroanal. Chem., 2022, 905, 115957 CrossRef CAS.
- X. Bai, Z. Duan, B. Nan, L. Wang, T. Tang and J. Guan, Chin. J. Catal., 2022, 43, 2240–2248 CrossRef CAS.
- Z. Guo, W. Ye, X. Fang, J. Wan, Y. Ye, Y. Dong, D. Cao and D. Yan, Inorg. Chem. Front., 2019, 6, 687–693 RSC.
- W. Liu, H. Liu, L. Dang, H. Zhang, X. Wu, B. Yang, Z. Li, X. Zhang, L. Lei and S. Jin, Adv. Funct. Mater., 2017, 27, 1603904 CrossRef.
- P. F. Liu, S. Yang, B. Zhang and H. G. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 34474–34481 CrossRef CAS PubMed.
- L. J. Enman, M. B. Stevens, M. H. Dahan, M. R. Nellist, M. C. Toroker and S. W. Boettcher, Angew. Chem., 2018, 130, 13022–13026 CrossRef.
- M. Dai, H. Fan, G. Xu, M. Wang, S. Zhang, L. Lu and Y. Zhang, J. Power Sources, 2020, 450, 227639 CrossRef CAS.
- P. Li, M. Wang, X. Duan, L. Zheng, X. Cheng, Y. Zhang, Y. Kuang, Y. Li, Q. Ma and Z. Feng, Nat. Commun., 2019, 10, 1–11 CrossRef PubMed.
- C. Qiao, Y. Zhang, Y. Zhu, C. Cao, X. Bao and J. Xu, J. Mater. Chem. A, 2015, 3, 6878–6883 RSC.
- J. Wang, C. F. Tan, T. Zhu and G. W. Ho, Angew. Chem., 2016, 128, 10482–10486 CrossRef.
- C. Qiu, F. Cai, Y. Wang, Y. Liu, Q. Wang and C. Zhao, J. Colloid Interface Sci., 2020, 565, 351–359 CrossRef CAS PubMed.
- S. Dutta, C. Ray, Y. Negishi and T. Pal, ACS Appl. Mater. Interfaces, 2017, 9, 8134–8141 CrossRef CAS PubMed.
- F. Song and X. Hu, J. Am. Chem. Soc., 2014, 136, 16481–16484 CrossRef CAS PubMed.
- Z. Xu, W. Li, X. Wang, B. Wang, Z. Shi, C. Dong, S. Yan and Z. Zou, ACS Appl. Mater. Interfaces, 2018, 10, 30357–30366 CrossRef CAS PubMed.
- M. Yang, X. Fu, M. Shao, Z. Wang, L. Cao, S. Gu, M. Li, H. Cheng, Y. Li and H. Pan, ChemElectroChem, 2019, 6, 2050–2055 CrossRef CAS.
- J. Ping, Y. Wang, Q. Lu, B. Chen, J. Chen, Y. Huang, Q. Ma, C. Tan, J. Yang and X. Cao, Adv. Mater., 2016, 28, 7640–7645 CrossRef CAS PubMed.
- T. Wang, W. Xu and H. Wang, Electrochim. Acta, 2017, 257, 118–127 CrossRef CAS.
- A. Guzmán-Vargas, J. Vazquez-Samperio, M. A. Oliver-Tolentino, N. Nava, N. Castillo, M. J. Macías-Hernández and E. Reguera, J. Mater. Sci., 2018, 53, 4515–4526 CrossRef.
- L.-M. Cao, J.-W. Wang, D.-C. Zhong and T.-B. Lu, J. Mater. Chem. A, 2018, 6, 3224–3230 RSC.
- C. Dong, L. Han, C. Zhang and Z. Zhang, ACS Sustain. Chem. Eng., 2018, 6, 16096–16104 CrossRef CAS.
- R. Yu, D. Liu, M. Yuan, Y. Wang, C. Ye, J. Li and Y. Du, J. Colloid Interface Sci., 2021, 602, 612–618 CrossRef CAS PubMed.
- C. Cheng, F. Liu, D. Zhong, G. Hao, G. Liu, J. Li and Q. Zhao, J. Colloid Interface Sci., 2022, 606, 873–883 CrossRef CAS PubMed.
- R. Rohit, A. D. Jagadale, S. K. Shinde, D.-Y. Kim, V. S. Kumbhar and M. Nakayama, J. Alloys Compd., 2021, 863, 158081 CrossRef CAS.
- J. Wang, X. Sun, H. Hu, T. Shen, G. Liu, Z. Li, D. Cao, L. He and Y. F. Song, Eur. J. Chem., 2022, 28, e202103601 CrossRef CAS PubMed.
- J. Xia, K. Huang, Z. Yao, B. Zhang, S. Li, Z. Chen, F. Wu, J. Wu and Y. Huang, J. Alloys Compd., 2022, 891, 161934 CrossRef CAS.
- T. Wang, Q. Pang, B. Li, Y. Chen and J. Z. Zhang, Appl. Phys. Lett., 2021, 118, 233903 CrossRef CAS.
- A. H. Imani, R. Ojani and J.-B. Raoof, Int. J. Hydrogen Energy, 2021, 46, 449–457 CrossRef CAS.
- X. Wang, Y. He, Y. Zhou, R. Li, W. Lu, K. Wang and W. Liu, Int. J. Hydrogen Energy, 2022, 47, 23644–23652 CrossRef CAS.
- A. Y. Faid, A. O. Barnett, F. Seland and S. Sunde, Int. J. Hydrogen Energy, 2022, 47, 23483–23497 CrossRef CAS.
- M. Guo, L. Zhou, Y. Li, Q. Zheng, F. Xie and D. Lin, J. Mater. Chem. A, 2019, 7, 13130–13141 RSC.
- Y. Hu, Z. Wang, W. Liu, L. Xu, M. Guan, Y. Huang, Y. Zhao, J. Bao and H.-m. Li, ACS Sustain. Chem. Eng., 2019, 7, 16828–16834 CrossRef CAS.
- J. Chen, H. Li, S. Chen, J. Fei, C. Liu, Z. Yu, K. Shin, Z. Liu, L. Song and G. Henkelman, Adv. Energy Mater., 2021, 11, 2003412 CrossRef CAS.
- Y. Wang, R. Zhu, Z. Wang, Y. Huang and Z. Li, J. Alloys Compd., 2021, 880, 160523 CrossRef CAS.
- L. Zhang, W. Cai and N. Bao, Adv. Mater., 2021, 33, 2100745 CrossRef CAS PubMed.
- H. Singh, R. Biswas, I. Ahmed, P. Thakur, A. Kundu, A. R. Panigrahi, B. Banerjee, K. K. Halder, J. Lahtinen and K. Mondal, ACS Appl. Mater. Interfaces, 2022, 14, 6570–6581 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |