
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Clozapine sensing through paper-based microfluidic sensors directly modified via electro-deposition and electro-polymerization†
Mohammad Hossein
Ghanbari
 ab,
Markus
Biesalski
b,
Oliver
Friedrich
c and
Bastian J. M.
Etzold
*ab
ab,
Markus
Biesalski
b,
Oliver
Friedrich
c and
Bastian J. M.
Etzold
*ab
aPower-To-X Technologies, Friedrich-Alexander-Universität Erlangen-Nürnberg, 90762 Fürth, Germany. E-mail: bastian.etzold@fau.de
bTechnische Universität Darmstadt, Ernst-Berl-Institute for Technical Chemistry and Macromolecular Science, Peter-Grünberg-Straße 8, 64287, Darmstadt, Germany
cInstitute of Medical Biotechnology, Department of Chemical and Biological Engineering, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Paul-Gordan-Str. 3, 91052, Erlangen, Germany
First published on 28th August 2024
Abstract
Microfluidic electrochemical sensors (μCS) can be portable, highly sensitive, and low-cost but are less frequently studied nor applied. Additionally, simultaneous electro-deposition of gold nanoparticles (ED (AuNPs)) and electro-polymerization of L-cysteine (EP (L-cys)) are introduced for the first time for modifying the surface of the working electrode through a paper-based microfluidic sensor. This study depicts that by employing such modification, the electrochemically active surface area (ECSA) and the electron transfer rate are increased together and result in improved sensitivity. The modified μCS is depicted to enable sensitive voltametric determination of, e.g., clozapine (CLZ), an anti-psychotic drug to treat schizophrenia. The proposed sensor was characterized by different techniques, and several key parameters were optimized. Under the optimum conditions and using square-wave voltametry (SWV), a linear dose–response for a concentration range from 0.5 to 10.0 μM of CLZ was achieved. The limit of detection and sensitivity resulted in 70.0 nM and 0.045 mA cm−2 μM−1, respectively. Besides, this excellent sensitivity combines with high stability, which was tested for six repetitive measurements with a single device resulting in high reproducibility. Additionally, this procedure was validated with measurements of clozapine in human blood plasma, which demonstrated the excellent applicability of the device, rendering it a promising platform for point-of-care diagnostics and environmental monitoring.
1. Introduction
Schizophrenia is a debilitating mental disorder characterized by disruptions in perceiving reality, often leading to hallucinations and violent behaviors that impede daily activities.1,2 Among the drugs approved by the United States Food and Drug Administration (US-FDA) for treating schizophrenia, clozapine C18H19ClN4 (CLZ), stands out as the most promising drug.3,4 It is unique in that its efficacy can be anticipated by blood testing, with recommended therapeutic levels falling within the range of 1–3 μM in human blood3–5 which is a narrow therapeutic margin. Despite its widespread use, CLZ usage is associated with significant adverse effects, emphasizing the critical need for precise dosage control strategies. Different analytical techniques, such as chromatography and spectrophotometry, are frequently used to monitor CLZ levels in diverse formulations and biological samples.6–9 Yet, despite their established accuracy and dependability, most of these methods come with inherent limitations. Spectroscopic methods, for example, often exhibit lower sensitivity, while high-performance liquid chromatographic (HPLC) methods are time-consuming and relatively costly because of employing expensive reagents, equipment and eluents.1,2On the other hand, electroanalytical techniques are being considered in contrast to other techniques for their distinct advantages. These techniques are favored because of their operational simplicity, rapid analysis, cost-effectiveness, utilization of safe and non-toxic materials, height sensitivity, and the potential for miniaturization, enabling applications, like in vivo biosensors.9–12 Several electrochemical techniques, such as amperometry and voltammetry, have been used to monitor CLZ levels.8–11 However, some of these methods exhibit drawbacks including limited selectivity, high detection limits, low sensitivity and low linear concentration range. As mentioned before, because of the challenge of maintaining CLZ serum levels within a narrow therapeutic range (1–3 μM), the development of a sensitive and reliable measurement method for this drug is important.1–3
The fabrication of a highly sensitive and selective electrochemical sensor hinges on establishing a robust platform to ensure a stable electrochemical response. Utilizing cost-effective and user-friendly screen-printed carbon-based electrodes (SPCE) stands out as a pivotal approach. These SPCEs offer advantages such as demonstrated analytical efficacy, miniature size, ease of handling, minimal sample volumes, high sensitivity and specificity, biodegradability, and the potential for multiplexing capabilities and are thus, intensively studied.13,14 An alternative also offering these advantages and thus, recently attracting attention are microfluidic electrochemical sensors (μCS).15 In a recent study, we compared the performance of SPCE and μCS arrangements for heavy metal sensing and demonstrated that the intrinsic properties of the μCS arrangement lead to superior performance over SPCE in stability tests and real sample analyses, affirming its exceptional sensitivity, selectivity, and stability. This opens the question whether this new platform could not also show advantages in sensing of other analytes.
For all platforms, it is known that modifiers play a crucial role in electrochemical sensing. Using catalytic active modifiers and additives, such as those aimed at enhancing electron or ion transport in electrochemical sensors, typically offers substantial advantages by enhancing sensitivity and selectivity.16,17 An elegant way to deposit metals is electro-deposition, and for polymers, electro-polymerization. These techniques have been successfully applied as external pretreatments of the electrodes and resulted in sensing with enhanced catalytic activity, stability, reproducibility, and active surface area.16,17 Among the metals, nanoparticles, i.e. gold nanoparticles (AuNPs), play a significant role in electrochemical sensors for medical applications as catalytic activity is combined with biocompatibility.18,19 Electro-deposition of AuNPs can occur over a wide pH range from pH 2 to pH 8.16–20
The utilization of organic additives has also garnered considerable attention. L-Cysteine (L-cys), a zwitter-ionic amino acid (depending on pH, it contains both positively charged NH3+ and negatively charged COO− functional groups), is an example which can also be deposited by electro-deposition. The isoelectric point of L-cys is around pH 5.0, which means that at pH values below the isoelectric point, L-cys is positively charged, and at pH values above the isoelectric point, L-cys is negatively charged. Also, it contains a thiol group (SH) which can additionally attract analytes.17,20,21 The electro-polymerization of L-cys is more favored at pH values above the isoelectric point where the negatively charged L-cys can interact more efficiently with the positively charged electrode surface. Hence, electro-polymerization of L-cys is typically performed at a pH range of around 6.0–8.0.18–21 Despite the extensive studies of electro-deposition of AuNPs and electro-polymerization of L-cys in static voltammetry using glassy carbon electrodes or SPCEs, their application to microfluidic systems has been limited,22,23 and to the best of our knowledge, no study reports the electro-deposition and electro-polymerization directly within a paper-based microfluidic sensor.
In this study, we demonstrate the direct and simultaneous electro-deposition of AuNPs and electro-polymerization of L-cys onto the μCS electrodes. This μCS sensor is then subsequently applied for CLZ sensing. As the μCS sensor showed superior performance, also the analysis of human blood serum samples was carried out and proved the excellent sensitivity, selectivity and stability of such device.
2. Experimental
2.1. Chemicals and apparatus
The chemicals and solvents used in this study were analytical-grade reagents and used as received. These included clozapine (Thermo Fisher Scientific, UK), L-cysteine, 97% (Sigma-Aldrich, Louis, USA), gold(III) chloride trihydrate, 99.9% (Merck, Louis, USA), K4Fe(CN)6, 99.98% (Sigma-Aldrich, Taufkirchen, Germany), K3Fe(CN)6, 98.5% (Sigma-Aldrich, Taufkirchen, Germany), methanol (Sigma-Aldrich, France), Graphite foil (Sigraflex, Meitingen, Germany), Filter paper (referred to as microfluidic paper) with a pore size of 12–15 μm (VWR, France), and other reagents were sourced from Sigma-Aldrich, VWR or Carl Roth and used as received. The paper was cut into small stripes of 5 mm × 35 mm and further used to assemble the paper-based microfluidic sensor as outlined below. Acetate buffer solution (ABS) and phosphate buffer solution (PBS) were prepared using relevant salts. All solutions were prepared employing double-distilled water. CLZ was dissolved in methanol to obtain a 1.0 mM CLZ stock solution.Scanning electron microscope (SEM) images were captured using Zeiss EVO 10-SmartSEM touch. Electrochemical measurements were executed utilizing a Potentiostat, (Ivium (Vertex), Eindhoven, Netherlands) controlled with IviumSoft software.
To assess the impact of various modifiers and additives, cyclic voltammograms (CVs) were conducted using 5.0 mM [Fe(CN)6]3−/4− and 0.1 M KCl as a redox probe solution. To evaluate the efficiency of different platforms for the enhancement of the surface area, the active surface area A (cm2) representing the surface roughness value of the immobilized modifier on the WE surface for μCS. This determination was accomplished by recording CV curves in the redox probe solution at different scan rates (v) from 40 mV s−1 to 200 mV s−1 (Fig. S1†) and applying the Randles–Sevcik equation.24
2.2. Human plasma sample preparation
Human blood serum samples, obtained from the Blood Bank (University Hospital Erlangen Transfusion Medicine and Hemostaseology Department) Erlangen, Germany, were used as real samples in the study. The samples were diluted with 0.1 M PBS at pH 8.0 at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio, and some specific concentrations of the CLZ were spiked into each diluted sample (1 mL of plasma diluted with 4 mL of PBS) to avoid interference from other available higher concentration substances in plasma. Subsequently, the SWV signals for each prepared solution were separately and independently measured by the sensor using standard addition method.
:5 ratio, and some specific concentrations of the CLZ were spiked into each diluted sample (1 mL of plasma diluted with 4 mL of PBS) to avoid interference from other available higher concentration substances in plasma. Subsequently, the SWV signals for each prepared solution were separately and independently measured by the sensor using standard addition method.
2.3. Fabrication of the modified μCS device
The configuration of the component for the μCS device is outlined in Scheme 1, employing a working electrode (WE), counter electrode (CE), and pseudo reference electrode (RE), for which 0.5 mm thick graphite foil was cut into specific shapes. Cleaning procedure involved sonication in ethanol and deionized water mixture (1:1) for 15 minutes at 300 W, followed by air-drying at room temperature before use. To maintain cleanliness and prevent contamination, the poly(methyl methacrylate) (PMMA) substrate and sampling sponge, alongside the graphite foil, were cleaned before each μCS device fabrication. A piece of microfluidic paper was sandwiched between the CE and WE to minimize ohmic resistance, with the RE positioned adjacent to the WE. As shown in Scheme 1, the electrodes were placed on a PMMA substrate for user convenience. An analyte reservoir, composed of a piece of sponge at one end of the paper strip, held 2.0 mL of analyte solution, continuously supplying the analyte solution over a 15 minute period. To prevent concentration changes of the analyte due to water evaporation, a glass vial was used to envelop the sampling sponge. An absorbent pad is located at the opposite end of the paper strip facilitated fluid wicking through the paper strip, maintained a constant analyte flow within the paper strip (the average flow rate of an aqueous analyte is estimated to be 130 μL min−1), and collected the analyte passing through the detection sites.14,15 Finally, the HAuCl4 and L-cys solution was electro-deposited simultaneously on the WE of the μCS through cyclic voltammetry (CV) as mentioned below:
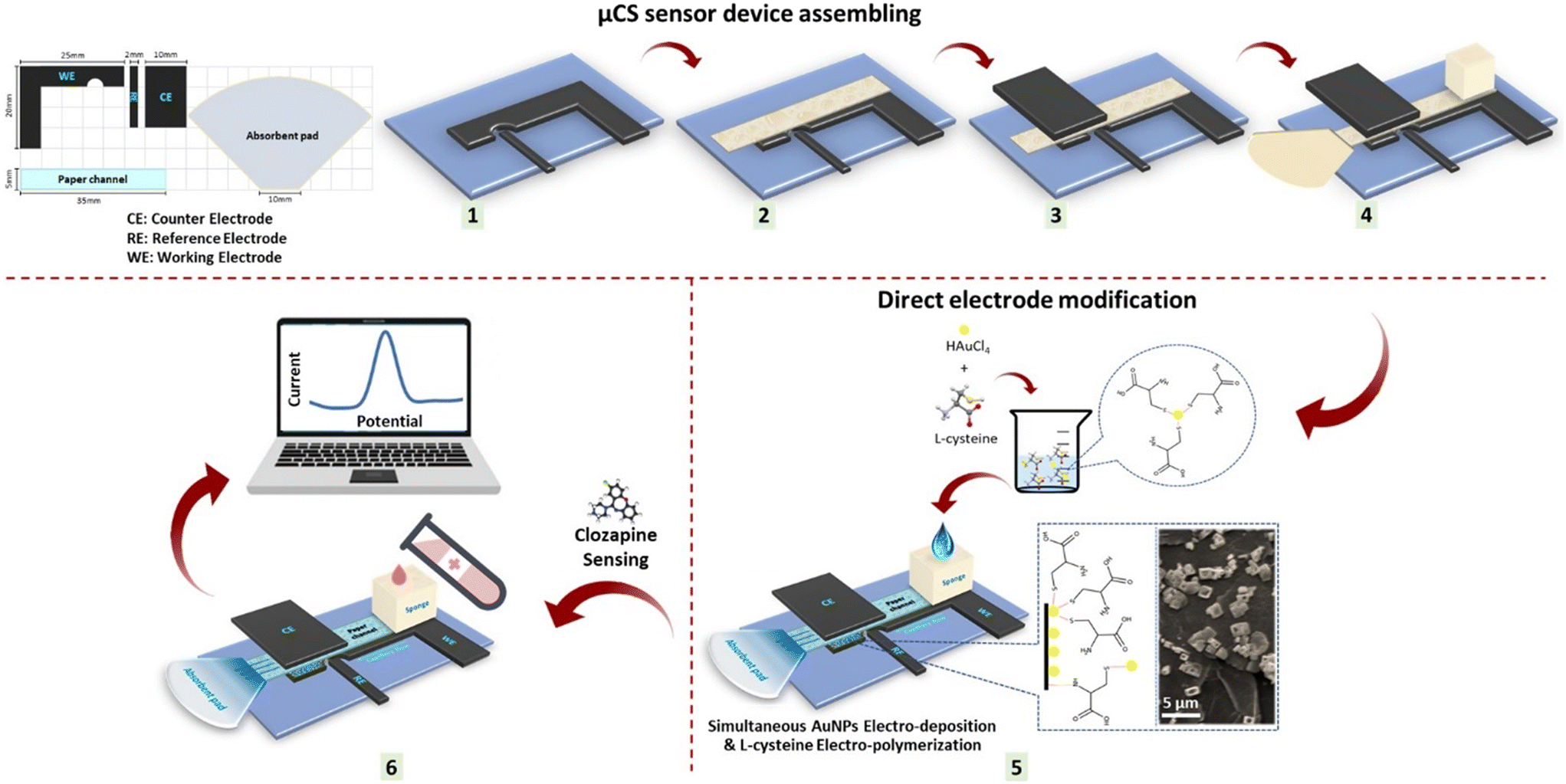 | ||
| Scheme 1 Scheme illustration the ED/EP-sim-μCS sensor device assembling as well as the direct electrode modification and subsequent use in sensing. | ||
(i) AuNPs electro-deposition was carried out for 10 cycles onto the bare WE surface at 0.1 M PBS at pH = 7 comprising 1.0 mM HAuCl4 and 0.1 M KNO3 from 0.0 V to +0.8 V under a scan rate of 50 mV s−1,
(ii) L-Cys electro-polymerization was carried out for 10 cycles onto the bare WE surface at 0.1 M PBS at pH = 6 comprising 1.0 mM L-cys from −1.5 V to +2.2 V under a scan rate of 50 mV s−1.
(iii) AuNPs were electro-deposited, and L-cys were electro-polymerized simultaneously through the μCS by handling a potential window of −1.5 to +2.2 V at a scan rate of 100 mV s−1via CV run for 10 cycles in a solution of 0.1 M PBS at pH = 6.0 consists of 1.0 mM HAuCl4 and 1.0 mM L-cys (Fig. S2†).
As a result, the prepared simultaneous electro-deposition of AuNPs and electro-polymerization of L-cys through a paper-based microfluidic sensor (ED/EP-sim-μCS) was used after three CV runs in 0.1 M PBS at pH = 6.0 and employed as the (bio)sensor.
2.4. Electrochemical sensing
Electrochemical measurements were conducted using an Ivium potentiostat (Vertex) controlled with IviumSoft software. All experiments were conducted at room temperature. Electrochemical impedance spectroscopy (EIS), and cyclic voltammetry (CV), were employed throughout the experiment via [Fe(CN)6]3−/4 as a redox probe solution to compare various unmodified and modified platforms. The EIS experiment was recorded at a frequency range of 0.1–100000 Hz, and the CV experiment was recorded under a scan rate of 100 mV s−1 from −1.0 V to +1.0 V.
CLZ detection was carried out via square wave voltammetry (SWV) with the following parameter settings: pulse height at 25 mV; step height at 10 mV and frequency set to 25 Hz from 0.0 V to +0.8 V. To ensure that the analyte solution reached the surface of the electrodes, electrochemical analyses were started after a 30 s holding time prior to starting the 60 s accumulation time at open circuit potential. Since the baseline is not strictly linear and the peaks tend to show tailing, a baseline is obtained by averaging almost five points to the left and right of the peak and forming a linear equation. After baseline subtraction, the peak is fitted with a Gaussian function.
The limit of detection (LOD) and quantification (LOQ) were calculated by the equation k·s/m (ref. 25) where k was set to 3 for LOD and 10 for LOQ, “s” is the standard deviation of the peak current of the blank (n = 4) and “m” is the slope of the calibration plot for CLZ.
Recovery represents the efficiency with which the sensor can detect and measure the known added amount of analyte within the complex matrix of the human plasma sample. It is typically expressed as a percentage and calculated using the formula:
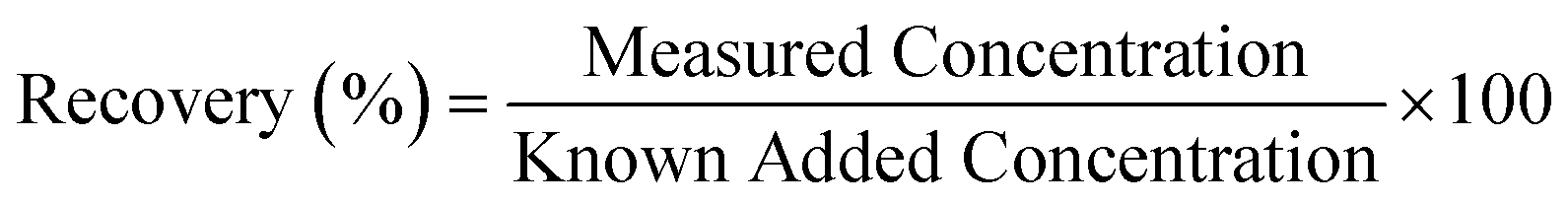
3. Results and discussion
3.1. Direct modification of electrode
As one can see from Fig. 1B showing SEM images to study the morphology of the nanostructures obtained from μCS devices, AuNPs were successfully electro-deposited onto the working electrode surface. Indeed, gold ions (Au3+) in the electrolyte solution are reduced and additionally poly (L-cys) structures electro-polymerized successfully onto the working electrode surface. Thus, a possible interaction of the paper fiber surface with the deposition solution has no negative effect. Interestingly, it is observed that the majority of the polymers are preferentially located at the edges of the graphite flakes, as depicted in Fig. 1B(e–i). These findings suggest that the edge surface positions of graphite flakes, characterized by lower coordination numbers of carbon atoms, may exhibit higher activity in catalyzing the electro-deposition of AuNPs and the electro-polymerization of L-cys. This phenomenon is not entirely unexpected, as it is well-established that electrochemical reactions at graphite edge sites are favored due to the slower kinetics of bond formation at basal plane sites compared to edge/defect sites.15 However, as previously demonstrated, a properly designed sensor configuration is also essential for achieving high sensing performance in these microfluidic electrochemical sensing (μCS) devices. The functional groups present in L-cys enable the modification of electrode surfaces, leading to better analyte detection, selectivity, and overall sensor reliability.26–28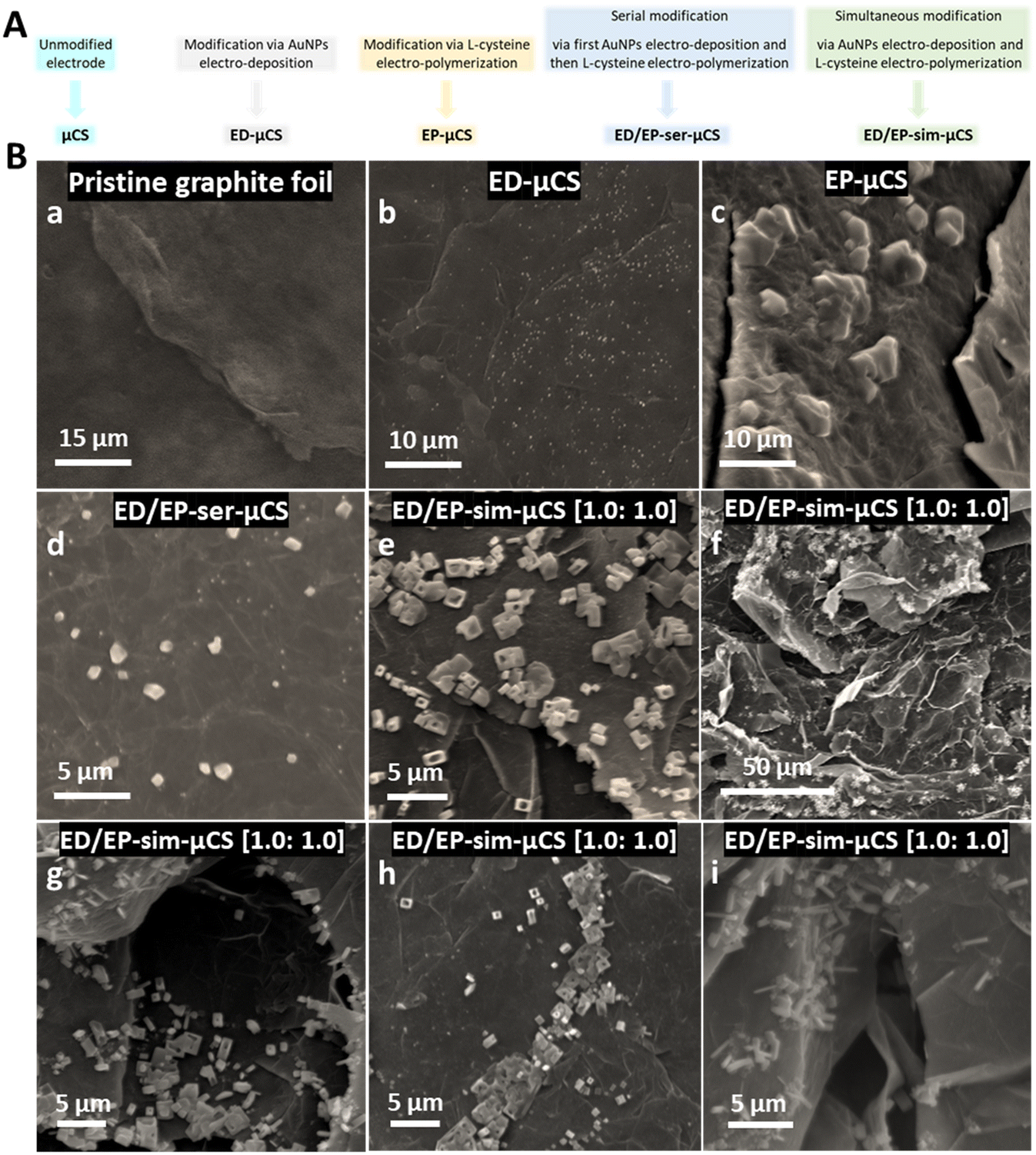 | ||
| Fig. 1 (A) Overview on the materials nomenclature for the different electrodes resulting after the different modification procedures, (B) SEM images of the different electrodes resulting from the different modification procedures. | ||
3.2. Electrochemical characterization of electrodes
To confirm the immobilization of each substrate onto the μCS working electrode surface during the modification process, CVs of the platforms were recorded in the redox probe solution consisting of 5.0 mM of [Fe(CN)6]3−/4−. As can be seen in Fig. 2A, two well-defined redox peaks with a peak-to-peak separation (ΔEp) of around 55 mV is observed for the bare μCS. By modifying the μCS by simultaneous electro-deposition of AuNPs and electro-polymerization of (L-cys) not only did peak currents markedly increase but also ΔEp was decreased to 13 mV for ED/EP-sim-μCS. This decremental behavior indicating the presence of AuNPs and poly (L-cys) enhances the reversible nature of the electrochemical reaction. In total, as one can see from Fig. 2A, particularly the bare electrode shows notable resistive components primarily due to the solution resistance, which results an iR drop. This resistive component is influenced by (i) the electrolyte's resistance, (ii) the concentration of redox species, (iii) the electrode surface area, (iv) the geometry of the electrochemical cell and (v) double layer effects. The iR drop leads to distortions in the voltammogram, such as peak broadening and shifting, which are more pronounced at higher scan rates and currents.24,29–31 Addressing this issue involves a combination of optimizing the electrolyte composition, adjusting the cell design, and electrode configuration (by employing smaller electrodes and minimizing distance between electrodes), and using iR compensation techniques. Each of these strategies can help minimize the impact of the iR drop, leading to more accurate and reliable voltammetric measurements.24,29–31 | ||
| Fig. 2 (A) CVs and (B) EIS of proposed platforms which recorded in a redox probe solution consisting of 5 mM of [Fe(CN)6]3−/4− and 0.1 M KCl under a scan rate of 100 mV s−1 of bare and different modified electrodes. Insets: Equivalent circuits for fitting the plots from the EIS Nyquist plots. a) Bare μCS, b) EP-μCS c) ED-μCS, d) ED/EP-ser-μCS, e) ED/EP-sim-μCS. | ||
In order to study the charge transfer resistance (Rct) parameter (which indicates the kinetics of the probe at the electrode interface), Nyquist plots of the modified and bare platform related to the EIS technique were recorded in the redox probe (Fig. 2B). By comparing the curves related to the bare μCS and ED/EP-sim-μCS, it becomes clear that the Rct value for bare μCS (46 Ω) is higher than ED/EP-sim-μCS (13 Ω). Interestingly, the simultaneous deposition of AuNPs and poly (L-cys) shows the lowest Rct and could show high performance sensor. A summary of the EIS parameters can be found in Table S1.†
To ensure the performance of both simultaneous electro-deoposition of AuNPs and electro-polymerization of L-cys and μCS in the enhancement of the surface area, accordingly, the calculated A of the ED/EP-sim-μCS was estimated to be 1.14 cm2. In comparison with the A value of the bare μCS, (0.31 cm2), this increase is 3.6 times that of the μCS device. Consequently, employing the simultaneous electro-deposition of AuNPs and electro-polymerization of poly (L-cys) onto the WE surface increases the electrochemically active surface area and amplifies the current signal of the modified platforms. All these results confirm that AuNPs and poly (L-cys) were successfully deposited onto the WE surface and show a positive impact. Since the ED/EP-sim-μCS showed the best results, we chose it to investigate its application in CLZ sensing.
Based on these results, the ED/EP-sim modification was chosen for further investigations and further optimization of the electro-deposition and polymerization step carried out. The following parameters were studied in a redox probe solution consisting 5 mM of [Fe(CN)6]3−/4− and 0.1 M KCl: (a) concentration ratio of the gold(III) chloride trihydrate/L-cys; and (b) effect of the potential windows on the electrode modification process; respective text and figures are given in the ESI† (Fig. S3–S5). Briefly, the subsequent experimental parameters were achieved to give best outcomes: (a) concentration ratio of the gold(III) chloride trihydrate/L-cys [1.0 mM: 1.0 mM] in 0.1 M PBS with pH 6.0; and (b) the potential windows on the electrode modification process; from −1.5 V to +2.2 V.
3.3. Comparison of different modified μCS and assessing pH influence
In assessing the performance of the suggested sensors for detecting CLZ (Fig. 3A), the electrocatalytic properties of all modified and unmodified μCS were examined. As shown in Fig. S6,† in the absence of CLZ (in 0.1 M PBS with pH = 8.0), no oxidation peak for CLZ was observed for bare μCS while for the ED/EP-sim-μCS, a just increased capacitive current was observed throughout the CV study. Also, as one can see from Fig. S7,† the oxidation peak for the proposed sensor in a solution containing of 3.0 μM CLZ (in 0.1 M PBS with pH = 8.0) was not clear in the recorded CV which is due to the presence of a high capacitive current. In the presence of 10.0 μM CLZ in pH = 8.0 (0.1 M PBS), the experiments were conducted following 1 min of cathodic accumulation at 0.0 V, followed by sensing via SWV. All platforms distinctly exhibited a stripping (oxidation) peak for CLZ. A comparison of all modified and unmodified μCS, results depict that EP (L-cys), ED (AuNPs), ED/EP serial preparation and ED/EP simultaneous preparation increase the peak current by a factor of 1.9, 2.8, 3.0 and 3.2, respectively, when compared to the unmodified μCS. The previously noted lower charge transfer resistance and increased electrochemical surface area with the ED/EP-sim-μCS consequently results in enhanced capabilities for CLZ sensing. | ||
| Fig. 3 SWVs for 10.0 μM CLZ in pH = 8.0 (0.1 M PBS) and tac = 1 min, (A) for various modified and unmodified μCS, (B) SWVs recorded from pH 4.0 to 9.0, in a solution containing 3.0 μM CLZ. | ||
The pH of the electrolyte solution stands as another critical factor in the catalytic detection of the analyte. Consequently, for the best performing ED/EP-sim-μCS, the impact of electrolyte pH on the peak current of CLZ was studied by altering the pH values from 4.0 to 9.0 via SWV (Fig. 3B). It was depicted that the shape and peak potential of CLZ oxidation depend on electrolyte pH. Accordingly, it is suggested the participation of proton in the CLZ oxidation reaction. As illustrated in Fig. S8,† the Ipa exhibited a rapid rise as the pH values increased from 4.0 to 8.0, however, further elevating the pH to 9.0 resulted in a reduction in peak currents for CLZ detection. The optimal pH for this study was determined to be 8.0. As one can see from Fig. S8,† by plotting Epa against pH, a linear relationship between the oxidation peak potential and pH can be approximated (R2 = 0.96). The calculated slope offers an equivalent contribution of protons and electrons (two electron-two proton) in the oxidation of CLZ at the fabricated electrode within the pH range of 4.0 to 9.0. This alignment supports the proposed mechanism of the electrochemical reaction of CLZ, as depicted in Scheme 2.1–3
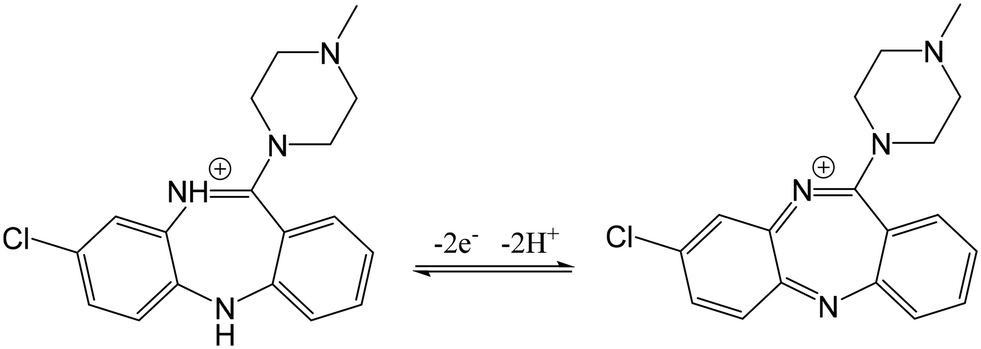 | ||
| Scheme 2 The electrooxidation mechanism of CLZ. | ||
3.4. Analytical performance
 | ||
| Fig. 4 (A) SWVs of the ED/EP-sim-μCS for different concentrations of CLZ in 0.1 M PBS with pH = 8.0, (B) the calibration plot of Ipavs. the different CLZ concentrations. (C) Dependence of Ipavs. replicates. (D) Peak currents for 10.0 μM CLZ in 0.1 M PBS at pH 8.0 detection compared to expected peak currents when various possible interfering biomolecules were present with 30.0 μM as single interfering molecule or a mixture of 30.0 μM of ascorbic acid, glucose and uric acid each (error bar represents the standard deviation for the three different individual experiments (n = 3)). | ||
Our sensor was able to monitor CLZ at clinically relevant concentrations from 10 to 500 ng mL−1, which suggests the ED/EP-sim-μCS device is useful for monitoring CLZ levels in patients.32 Additionally, the outcomes demonstrate that the approach is rapid, cost-effective, and exhibits noteworthy sensitivity. The analytical characteristics of the sensor are compared with various electrochemical methods and devices in CLZ monitoring as presented in Table 1. The results suggest that the proposed strategy is comparable in terms of LOD and/or response linearity ranges and accumulation time.
| Method | Technique | Deposition time (s) | Potential (mV) | pH | Liner range (nM) | LOD (nM) | Real samples | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Titanium oxide nanoparticle modified carbon paste electrode. b Differential pulse voltammetry. c Tungsten trioxide nanoparticles hydride by α-terpineol modified glassy carbon electrode. d Square-wave voltammetry. e Lab-in-a-pencil graphite microfluidic sensing electrode. f Ruthenium doped titanium oxide nanoparticle. g Nafion-modified multi walled carbon nanotube. h Toray paper electrochemical device. i Chronoamperometry. | ||||||||
| TiO2NP/CPEa | DPVb | 420 | 370 | 9.0 | 5 × 102–45 × 103 | 61.0 | Tablet | 5 |
| MWWT/GCEc | SWVd | — | 462 | 7.0 | 1.0 × 102–2.0 × 103 | 30.0 | Urine and blood | 25 |
| μFSEe | Amperometry | — | 320 | 7.4 | 1.0 × 102–1.0 × 104 | 24.0 | Blood | 9 |
| Ru-TiO2f/CPE | SWV | 15 | 210 | 10.4 | 9.0 × 102–4.0 × 104 | 0.43 | Tablet and urine | 13 |
| Nf/MWCNT on CTSg | SWV | 60 | 150 | 7.4 | 1.0 × 102–5.0 × 103 | 83 | Blood | 32 |
| TPEDh | CAi | — | 320 | 7.4 | 5.0 × 102–5.0 × 103 | 10 | Blood | 1 |
| ED/EP-sim-μCS | SWV | 60 | 420 | 8.0 | 5.0 × 10 2 –1.0 × 10 4 | 70 | Blood | This work |
Moreover, the capability for reuse stands out as another important desirable feature for a sensing device, which can considerably cut down on costs and waste during analytical processes. To assess the stability of the ED/EP-sim-μCS, the SWV signal of the sensor was recorded in the presence of 3.0 μM of CLZ in 0.1 M PBS at pH = 8.0 for 6 consecutive SWVs (Fig. S9†), and the peak current response is depicted against the replicate in Fig. 4C. Notably, the anodic stripping current only exhibits a marginal decrease (11%) after 6 repetitions. The sensor maintained 89.0% of its initial peak current with RSD ≤ 10%. This slight reduction is likely attributed to the depletion of the analyte within the sampling sponge or/and the filling or removal of the modifier layer from the WE surface. This investigation also shows the great robustness of ED/EP-sim-μCS for drug sensing applications.
3.5. Selectivity of the sensor
To scrutinize biological samples using the proposed sensor device, an investigation into the impact of common electrochemical species in human blood was studied via SWV. To determine the sensor's ability to CLZ sensing in the presence of possible potentially interfering substances, selectivity studies were carried out. To do so, solutions containing 10.0 μM CLZ at 0.1 M PBS and pH 8.0 were tested alongside other substances, including ascorbic acid, glucose, and uric acid. The resulting peak currents normalized to the peak currents without further substances (I/I0) are outlined in Fig. 4D. I and I0 represent SWV peak current responses of CLZ in the presence and absence of interferences, respectively. Results show that 30.0 μM ascorbic acid or 30.0 μM uric acid did not significantly interfere with 10.0 μM CLZ, and the sensor was able to detect CLZ even in the presence of these common interfering substances. However, in the presence of 30.0 μM glucose, the CLZ signal drops by 20%, while in the presence of 30.0 μM ascorbic acid, 30.0 μM uric acid and 30.0 μM glucose at the same time, they did not significantly interfere with 10.0 μM CLZ. The sensor exhibited a negligible altering current and provided notable selectivity in CLZ sensing in the presence of ascorbic acid, glucose and uric acid as common interfering biomolecules.3.6. Human blood sample analyses
In order to depict the analysis of CLZ in real sample environment, we investigated this ability in the voltametric sensing of CLZ in human blood serum under the optimum conditions. The SWV technique and the standard addition method were used for this purpose. Since the plasma environment is different from the pure buffer solution in terms of the biomolecules present, a calibration plot for the determination of CLZ was prepared under optimal conditions in which the peak currents of CLZ increase linearly with concentration from 1.0 to 9.0 μM (Fig. 5). | ||
| Fig. 5 (A) SWVs of the ED/EP-sim-μCS in human blood serum sample for various concentrations of CLZ in 0.1 M PBS (pH = 8.0). (B) the calibration plot of Ipavs. CLZ concentration and current density vs. the different CLZ concentrations. | ||
Upon the addition of a specific CLZ concentration, the SWV signal was recorded. Subsequently, the acquired concentration values were compared with the spiked concentrations in the serum samples, leading to the determination of RSD values based on three measurements. Notably, the peak intensity exhibited a proportional increase corresponding to the rising CLZ concentration within the range of 4.5 to 8.5 μM. As shown in Table 2 and Fig. S10,† the recovery was ranging from 106% to 99%. Accordingly, a good adaptation is evident between the proposed sensor and the standard method. Moreover, considering the diverse factors influencing CLZ levels in plasma for schizophrenia patients, such as gender, age, and drug dosage, various studies have reported different drug concentrations in the range of 48–1304 ng ml−1 in the literature.25,33,34 Consequently, according to figures of merit of the proposed sensor, sensing of CLZ in admissible concentration of blood plasma (1–3 μM) is possible. These findings also showed that the impact of various biomolecules in blood samples imposes minimal effect on the sensing performance of the device.
| No. | Added CLZ (μM) | Found CLZ (μM) | Recovery (%) |
|---|---|---|---|
| 1 | 0.0 | 0.0 | 100 |
| 2 | 4.5 | 4.8 ± 1.1 | 106 |
| 3 | 8.5 | 8.5 ± 2.4 | 99 |
4. Conclusion
The ED/EP-sim-μCS shows advantages in terms of cost, simplicity, and sensitivity with advantages of microfluidic configuration and 3D electrode layout. This study proofed that also for CLZ sensing, an improved performance was established. SEM analysis and detailed electrochemical characterization proofed that electro-deposition of AuNPs and of L-cys is possible directly within the microfluidic device, and no additional external pretreatment step was needed. Furthermore, the simultaneous electro-deposition and polymerization was also carried out successfully and boosted electrochemical activity and thus, sensitivity of the sensor. The microfluidic properties can be employed to accumulate the analyte prior to the sensing event and thus, further increase the sensitivity. The ED/EP-sim-μCS is capable of detecting CLZ within a wide linear range, low LOD, a larger electrochemical effective surface area, and high sensitivity. It could be further shown that the device is reusable and robust. This promising easy-handling method of μCS devices with 3D electrode arrangement combined with catalytic electrode modifiers could offer some new ideas for other portable electroanalytical/sensing systems intended for pharmacokinetic examinations, and clinical diagnostics to identify different targets.Data availability
All data presented in the main article and in the ESI† are available from an open access repository. Source data are provided at Zenodo. DOI: https://doi.org/10.5281/zenodo.12571068Author contributions
MHG: conceptualization, visualization, investigation, methodology, validation, writing – original draft; MB: funding acquisition, writing – review & editing; OF: writing – review & editing; BE: conceptualization, funding acquisition, supervision, writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge funding for part of the work through the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Projektnummer 465690040 and Projektnummer 405469627. BE acknowledges the support by the Bavarian State Ministry for Science and Arts through the Distinguished Professorship Program. The authors acknowledge the Merck Lab in TU Darmstadt for providing SEMs. The authors also acknowledge Universitätsklinikum Erlangen, Transfusionsmedizinische Abteilung, Blutbank, Ms. Ida-Sophie Löber, and Ms. Maria Leidenberger for facilitating human plasma sample analysis. The cartoon image presented in the graphical abstract was designed by Freepik.References
- S. Kumar, et al., A miniaturized unmodified toray paper-based electrochemical sensing platform for antipsychotic drug analysis, Sens. Actuators, A, 2023, 360, 114520 CrossRef CAS.
- M. H. Ghanbari, et al., Electrochemical determination of the antipsychotic medication clozapine by a carbon paste electrode modified with a nanostructure prepared from titania nanoparticles and copper oxide, Microchim. Acta, 2019, 186, 1–10 CrossRef.
- H. Ben-Yoav, et al., An electrochemical micro-system for clozapine antipsychotic treatment monitoring, Electrochim. Acta, 2015, 163, 260–270 CrossRef CAS.
- D. L. Kelly, et al., Blood draw barriers for treatment with clozapine and development of a point-of-care monitoring device, Clin. Schizophr. Relat. Psychoses, 2018, 12(1), 23–30 CrossRef PubMed.
- M. H. Mashhadizadeh and E. Afshar, Electrochemical investigation of clozapine at TiO2 nanoparticles modified carbon paste electrode and simultaneous adsorptive voltammetric determination of two antipsychotic drugs, Electrochim. Acta, 2013, 87, 816–823 CrossRef CAS.
- S. A. Mohajeri, G. Karimi and M. R. Khansari, Clozapine imprinted polymers: Synthesis, characterization and application for drug assay in human serum, Anal. Chim. Acta, 2010, 683(1), 143–148 CrossRef CAS PubMed.
- C. Frahnert, M. L. Rao and K. Grasmäder, Analysis of eighteen antidepressants, four atypical antipsychotics and active metabolites in serum by liquid chromatography: a simple tool for therapeutic drug monitoring, J. Chromatogr., B, 2003, 794(1), 35–47 CrossRef CAS.
- N. Y. Hasan, et al., Stability indicating methods for the determination of clozapine, J. Pharm. Biomed. Anal., 2002, 30(1), 35–47 CrossRef CAS PubMed.
- M. Senel and A. Alachkar, Lab-in-a-pencil graphite: A 3D-printed microfluidic sensing platform for real-time measurement of antipsychotic clozapine level, Lab Chip, 2021, 21(2), 405–411 RSC.
- M. Kang, et al., Reliable clinical serum analysis with reusable electrochemical sensor: Toward point-of-care measurement of the antipsychotic medication clozapine, Biosens. Bioelectron., 2017, 95, 55–59 CrossRef CAS.
- M. H. Ghanbari, Z. Norouzi and A. Amiri, Application of nickel-doped graphene nanotubes to modified GCE as a sensitive electrochemical sensor for the antipsychotic drug clozapine in spiked human blood serum samples, J. Iran. Chem. Soc., 2023, 1–11 Search PubMed.
- L.-L. Shen, G.-R. Zhang and B. J. M. Etzold, Paper-based microfluidics for electrochemical applications, ChemElectroChem, 2020, 7(1), 10–30 CrossRef CAS PubMed.
- N. P. Shetti, et al., An electrochemical sensor for clozapine at ruthenium doped TiO2 nanoparticles modified electrode, Sens. Actuators, B, 2017, 247, 858–867 CrossRef CAS.
- M. H. Ghanbari, et al., Superior performance of N-doped carbon Nanoonions/Nafion based microfluidic electrochemical Cd2+ sensor when compared to Screen-Printed Carbon-Based electrode devices, Microchem. J., 2024, 110506 CrossRef CAS.
- L.-L. Shen, et al., Modifier-free microfluidic electrochemical sensor for heavy-metal detection, ACS Omega, 2017, 2(8), 4593–4603 CrossRef CAS.
- X. Mei, et al., A novel electrochemical sensor based on gold nanobipyramids and poly-l-cysteine for the sensitive determination of trilobatin, Analyst, 2023, 148(10), 2335–2342 RSC.
- M. H. Ghanbari, et al., Simultaneous electrochemical detection of uric acid and xanthine based on electrodeposited B, N co-doped reduced graphene oxide, gold nanoparticles and electropolymerized poly (L-cysteine) gradually modified electrode platform, Microchem. J., 2022, 175, 107213 CrossRef CAS.
- T. Ndlovu, et al., Electrochemical detection of o-nitrophenol on a poly (propyleneimine)-gold nanocomposite modified glassy carbon electrode, Int. J. Electrochem. Sci., 2010, 5(8), 1179–1186 CrossRef CAS.
- A. Adiraju, et al., Towards Embedded Electrochemical Sensors for On-Site Nitrite Detection by Gold Nanoparticles Modified Screen Printed Carbon Electrodes, Sensors, 2023, 23(6), 2961 CrossRef CAS PubMed.
- M. H. Ghanbari, et al., Utilizing a nanocomposite consisting of zinc ferrite, copper oxide, and gold nanoparticles in the fabrication of a metformin electrochemical sensor supported on a glassy carbon electrode, Microchim. Acta, 2020, 187, 1–11 CrossRef.
- M. H. Ghanbari, Z. Norouzi and B. J. M. Etzold, Increasing sensitivity and selectivity for electrochemical sensing of uric acid and theophylline in real blood serum through multinary nanocomposites, Microchem. J., 2023, 191, 108836 CrossRef CAS.
- C. Vezy, et al., Coupling electropolymerization and microfluidic for the generation of surface anisotropy, Electrochem. Commun., 2010, 12(10), 1266–1269 CrossRef CAS.
- C.-W. Lee, et al., Ultra-sensitive electrochemical detection of bacteremia enabled by redox-active gold nanoparticles (raGNPs) in a nano-sieving microfluidic system (NS-MFS), Biosens. Bioelectron., 2019, 133, 215–222 CrossRef PubMed.
- A. J. Bard, L. R. Faulkner and H. S. White., Electrochemical methods: fundamentals and applications, John Wiley & Sons, 2022 Search PubMed.
- M. R. Fathi and D. Almasifar, Electrochemical sensor for square wave voltammetric determination of clozapine by glassy carbon electrode modified by WO3 nanoparticles, IEEE Sens. J., 2017, 17(18), 6069–6076 CAS.
- W. Liu, et al., Nanopore array derived from l-cysteine oxide/gold hybrids: enhanced sensing platform for hydroquinone and catechol determination, Electrochim. Acta, 2013, 88, 15–23 CrossRef CAS.
- D. He, et al., A novel free-standing CVD graphene platform electrode modified with AuPt hybrid nanoparticles and L-cysteine for the selective determination of epinephrine, J. Electroanal. Chem., 2018, 823, 678–687 CrossRef CAS.
- N. F. Atta, A. Galal and D. M. El-Said, Novel design of a layered electrochemical dopamine sensor in real samples based on gold nanoparticles/β-cyclodextrin/nafion-modified gold electrode, ACS Omega, 2019, 4(19), 17947–17955 CrossRef CAS PubMed.
- L. de Brito Ayres, et al., Rapid detection of Staphylococcus aureus using paper-derived electrochemical biosensors, Anal. Chem., 2022, 94(48), 16847–16854 CrossRef CAS PubMed.
- R. G. Compton and C. E. Banks, Understanding voltammetry, World Scientific, 2018 Search PubMed.
- W. B. Veloso, T. R. L. C. Paixão and G. N. Meloni, 3D printed electrodes design and voltammetric response, Electrochim. Acta, 2023, 449, 142166 CrossRef CAS.
- M. Senel, Electrochemistry Test Strip as Platform for In Situ Detection of Blood Levels of Antipsychotic Clozapine in Finger-Pricked Sample Volume, Biosensors, 2023, 13(3), 346 CrossRef CAS.
- L. V. Rao, M. L. Snyder and G. M. Vallaro, Rapid liquid chromatography/tandem mass spectrometer (LCMS) method for clozapine and its metabolite N-desmethyl Clozapine (Norclozapine) in human serum, J. Clin. Lab. Anal., 2009, 23(6), 394–398 CrossRef CAS.
- E. Spina, et al., Relationship between plasma concentrations of clozapine and norclozapine and therapeutic response in patients with schizophrenia resistant to conventional neuroleptics, Psychopharmacology, 2000, 148, 83–89 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sd00252k |
| This journal is © The Royal Society of Chemistry 2024 |