
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReal-time detection of TNT analogues in water using fluorescent dendrimer films
Mohammad A.
Ali
,
Shengqiang
Fan
,
Paul L.
Burn
 *,
Ian R.
Gentle
* and
Paul E.
Shaw
*,
Ian R.
Gentle
* and
Paul E.
Shaw
Centre for Organic Photonics & Electronics, School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: p.burn2@uq.edu.au; i.gentle@uq.edu.au
First published on 21st February 2024
Abstract
We have studied the use of fluorescent dendrimer thin films for the detection of nitro-aromatic analogues of and 2,4,6-trinitrotoluene (TNT) in water. The fluorescent sensing material used was a dendrimer composed of first-generation biphenyl dendrons, a 9,9,9′,9′-tetra-n-propyl substituted 2,2′-bifluorene core, and 2-ethylhexyloxy surface groups. The dendrimer had a solid state photoluminescence quantum yield (PLQY) of 49 ± 5% and was insoluble in water. Exposing the film to a nitro-aromatic analyte in water (including seawater) led to a rapid decrease in the fluorescence intensity. The decrease in fluorescence intensity arose from photoinduced electron transfer from the fluorescent dendrimer to the nitroaromatic analyte, with the process being reversible. That is, in the presence of the nitroaromatics the fluorescence was quenched, but on removal of the analyte, the fluorescence was restored. We have also developed a simple paper-based test strip that can be used for detection of the nitro-aromatic analytes in water. Dropping an aqueous solution of 2,4-dinitrotoluene or TNT onto the dendrimer-embedded paper strip led to a rapid visual decrease in the fluorescence intensity. The decrease in intensity could be compared to a standard card to determine the concentration, with the limit of detection using a phone camera being around 0.2 ppm. False positives were not observed when the dendrimer-embedded paper strips were exposed to a range of metal cations or different counteranions at concentrations much greater than are typically found in sea or freshwater.
Introduction
Nitroaromatic compounds, such as 2,4,6-trinitrotoluene (TNT), are inexpensive and are the primary energetic compounds found in land mines and at least 15 standard explosive compositions. These energetic nitrogenous compounds can be released into the environment from munitions, obsolete storage facilities, and unexploded ordnance at shooting ranges, ultimately leading to long-term pollution and contamination of the marine environment or local water supply.1–3 For example, marine ecosystems are still under threat even 50 years after the dumping of unexploded ordnance at sea.4 Humans can develop chronic diseases, such as aplastic anemia, liver disease and cataracts through consumption of water and food contaminated with nitroaromatics.5 Conventional analytical detection techniques, such as high-performance liquid chromatography (HPLC), mass spectroscopy (MS), electrochemical methods, gas chromatography, and ion mobility spectroscopy require sample separation and preconcentration prior to analysis.6–8 Consequently, significant time can elapse between sample collection in the field, analysis of the sample in the laboratory, and the reporting of the result back to the operative in the field. It would therefore be advantageous to have a simple detection method that does not need significant sample preparation and can be used in the field, either for environmental protection or marine security.7Fluorescence-based sensing of explosive and taggant vapours has been widely reported due to its ability to detect such analytes with high sensitivity and that in some cases the sensors are reusable.6,9 Approaches to fluorescence-based detection of explosives and taggants have included depositing the sensing material onto a substrate, which can be of high optical quality or simply paper. There are now several different classes of fluorescent sensing materials reported for the detection of nitro-based explosives and taggants including conjugated polymers, dendrimers and metal–organic frameworks.1,9,10 Despite the range of sensing materials reported to be capable of vapour detection,11,12 there are fewer examples of solid state sensors for the detection of explosives in water.1,11,13–16 Two key properties required for fluorescent sensing materials to be used in detecting analytes in water are that they should not react appreciably with water upon photoexcitation and they should be insoluble in water. Conjugated polymers can meet these criteria and have been successfully used for fluorescence-based sensing of explosives in water.17,18 Other approaches have included covalently attaching fluorescent sensing molecules onto a substrate,19 fabrication of fluorescent membranes,20 and suspensions of fluorescent nanoshell sensors comprising 100 nm hollow silica nanoparticles coated with a copolymer of silafluorene and fluorene, which were reported to be able to detect TNT and hexogen (RDX) in aqueous media.21
We have previously reported that fluorescent dendrimer films can rapidly detect trace vapours of nitro-based explosives, taggants, and accelerants.22–25 We were therefore interested in determining whether fluorescent dendrimers could also be used to detect explosives dissolved in water. In this study we used a previously synthesised first-generation dendrimer comprising a 9,9,9′,9′-tetra-n-propyl substituted 2,2′-bifluorene core, biphenyl dendrons, and 2-ethylhexyloxy surface groups (see Fig. 1).22 The dendrimer was chosen as it was solution processable from organic solvents and forms good quality amorphous thin films,26 is insoluble in water, has a photoluminescence quantum yield of 49 ± 5% in the solid state, and has been previously shown to reversibly detect nitro-based explosive and taggant vapours in the solid state via photoinduced electron transfer (PET). We report the thin film detection of nitroaromatics in water (including seawater) as well as analyte detection in a paper test strip format.
 | ||
| Fig. 1 The chemical structure of the first generation dendrimer used for the sensing measurements and structures of the TNT analogues [2,4-dinitrotoluene (DNT), p-nitrotoluene (pNT) and 1,4-dinitrobenzene (DNB)] studied. | ||
Methods
Thin film measurements
A custom-built sample chamber was used for the film measurements (Fig. 2). A syringe pump was used to inject the aqueous analyte solution over a 10 s period into the container where the sensing film was mounted. The film was excited using a light emitting diode (LED) with peak emission at 365 nm (note the absorption λmax of the dendrimer was at 352 nm),22 and a lens coupled to an optical fiber was used to collect the emission for detection by an AvaSpec-3648 CCD photodetector. The setup allowed continuous monitoring of the PL intensity. Averaged data used for analysis was collected from three measurements using freshly prepared films. | ||
| Fig. 2 Schematic of the custom-built experimental setup. The analyte solution was injected using a syringe pump. The LED used for excitation of the film was held at 45° to the optical fibre used for collecting the fluorescence from the sensing film to minimize the detection of the reflected excitation. A long-pass filter was used to block scattered excitation. | ||
Sensing films were deposited onto fused silica substrates by spin-coating (4000 rpm for 1 min) from a toluene solution of the dendrimer. The thickness of the sensing films was controlled by varying the concentration of the solution (10 mg mL−1 and 20 mg mL−1). The film thicknesses were measured using a Veeco Dektak 150 surface profilometer.
Aqueous solutions of thin film measurements
For the thin film measurements, 1.0 M stock solutions of 2,4-dinitrotoluene (DNT, an impurity found in TNT), 4-nitrotoluene (pNT), and 1,4-dinitrobenzene (DNB) were prepared in ethanol. Deionised water or seawater was used to prepare aqueous solutions of the desired analyte concentration. It should be noted that the dilution of the stock solution meant that ethanol only made up a small fraction of the analyte solution used in the measurement. The amount of ethanol in the final analyte solution was dependent on the dilution, but the maximum concentration was for the 5 ppm pNT solution, which only contained around 0.004% ethanol.Fabrication of paper test strip and testing
The filter paper (Whatman 1001-125; diameter: 12.5 cm; pore size: 11 μm) was fully immersed into a 0.1 mg mL−1 solution of the dendrimer in toluene for 1 min. The filter paper was removed from the solution and dried in air. The dried filter paper was then cut into test strips of the required dimensions. For the standards strips, 100 mg L−1DNT (note: the DNT solubility in water at 20 °C is approximately 180 mg L−1) or 50 mg L−1TNT stock solutions were prepared in deionised water and then diluted to the desired analyte concentration. The stock solutions (10 μL) were then placed on the test strips. The interferent studies were evaluated using the following cations: Ag+ (AgNO3), Hg2+ [Hg(OAc)2], Cd2+ [Cd(OAc)2], Ni2+ [Ni(OAc)2], Pb2+ [Pb(NO3)2], Zn2+ [Zn(NO3)2], Cu2+ (CuCl2), and Fe3+ (FeCl3), and anions F− (KF), NO3− (KNO3), SO42− (Na2SO4), SCN− (KSCN), AcO− (NaOAc), CO32− (Na2CO3), HCO3− (NaHCO3), Cr2O72− (Na2Cr2O7), and H2PO4− (KH2PO4). Unless specified otherwise, the concentrations of the two light-absorbing cations, Fe3+ and Cu2+, were 10 mg L−1 in deionized water while the other cations and anions were at a concentration 100 mg L−1.Results and discussion
Thin film quenching behaviour
In the first part of the study we investigated how the electron affinity and lipophilicity of aqueous solutions of the analytes affected the sensing ability of the spin-coated dendrimer films. Sensing films of two different thicknesses (35 ± 5 nm and 85 ± 5 nm) were tested with each of the analytes to determine whether the analytes sorbed into the film. DNB has a similar electron affinity to TNT that makes it an ideal analogue to study the effect of energetics on the PL quenching behavior,27DNT is a common impurity in commercial TNT and is more lipophilic than DNB, whilst pNT has a smaller electron affinity and is less polar. Aqueous solutions of the analyte at different concentrations were injected into the container holding the sensing film, with the change in photoluminescence (PL) intensity from the film versus time shown in Fig. 3. An immediate decrease in the PL intensity was observed upon introduction of the analyte solution, with equilibrium achieved after around 2 minutes (the first 20 seconds are shown in Fig. 3). It should be noted that the introduction of pure water also caused an apparent decrease in the PL signal, which is due to the change in the refractive index of the environment surrounding the film causing the amount of light coupled into and out of the film, and from the water into air to change. However, the magnitude of the change in the PL signal from pure water was significantly less than when water containing the analytes was added. In an important result we found that the degree of PL quenching increased with increasing analyte concentration independent of the film thickness. The measured decrease in PL intensity could arise from the analytes simply being sorbed onto the surface or from them diffusing into the film. Given that exciton diffusion lengths are generally of the order of a 5–10 nm, the fact that both the thin and thick films are quenched by similar amounts for each analyte concentration is strongly indicative that the analytes were diffusing/partitioning from the aqueous solution into the sensing film and not simply being sorbed onto its surface. If the analytes were simply sorbed onto the surface, then the thicker film would exhibit significantly less PL quenching than the thin film as fewer excitons would be able to diffuse to the film surface and interact with an analyte. | ||
| Fig. 3 PL intensity changes of the sensing films at different analyte concentrations versus time. The PL intensity was determined from the integrated PL spectra over the emission range of 400–600 nm with a measurement every 0.5 s. The film thicknesses were (a) 35 ± 5 nm and (b) 85 ± 5 nm. The measurement duration was 5 min with the first 20 s shown to focus in on the initial PL changes before the steady state was reached. | ||
To quantify the sensitivity of the sensing material in the solid state to the different analytes and the role of film thickness we carried out a Stern–Volmer like analysis at steady state using
 | (1) |
 | ||
| Fig. 4 Plots of PL quenching versus concentration of the three analytes in water for films with thickness of (a) 35 ± 5 nm and (b) 85 ± 5 nm. The films were exposed to each solution for 2 min before the PL signal was integrated over the emission range of 400 nm to 600 nm with an integration time of 0.5 s. The solid lines show fits to the data with eqn (1). (c) Summary of the values of k obtained from fitting to the steady-state PL quenching data in (a) and (b). The measurement duration was 5 min with the data collected after 2 min at which point it had reached the steady state. | ||
For all three analytes the amount of fluorescence quenching was observed to increase linearly with the analyte concentration in the solution (Fig. 4). Fits to the data using eqn (1) were used to calculate the value of k for the films of different thickness and each analyte (Fig. 4c). Both film thicknesses were found to have similar values for k for each analyte. That is, the efficacy of quenching was not dependent on the thickness of the sensing film. Film thickness independence is useful for manufacturing detection systems as small variances in film formation will not lead to significantly different responses. Furthermore, the lack of thickness dependence in the response is consistent with observations from the analyte concentration experiments discussed earlier whereby the analytes diffuse into the films and are not just sorbed on the surface. DNT shows the largest k value among the three analytes, followed by pNT and DNB. Interestingly, this order is different from that obtained from Stern–Volmer measurements performed with the dendrimer dissolved in an organic solvent. In the latter case the solution Stern-Volmer constant values were in the order of DNB > DNT > pNT, which was attributed to the differences in electron affinities of the analytes.20 The results from the measurements of the analytes dissolved in water interacting with a film of the sensing material suggests that both diffusion from the aqueous solution into the film and electron affinity play an important role in the sensing process. That is, the analytes that contain the lipophilic methyl groups partition into the film more effectively (and thus have a higher concentration) than DNB, with DNT then having superior quenching ability over pNT due to its higher electron affinity.
The dendrimer of this work has been previously shown to detect nitro-based explosive and taggant vapours reversibly. That is, when the analyte was removed from the film under a stream of nitrogen or air the PL intensity increased and returned close to its original value. We were therefore interested to see whether the detection in water was also reversible. To do this we allowed a film to be quenched by DNT until it reached a near equilibrium condition. We then removed the aqueous analyte solution and added ethanol in which DNT is soluble but the dendrimer is not. Upon addition of ethanol the PL intensity was observed to increase as the DNT dissolved from the film into the solution, reaching approximately 90% of the initial value after around 9 minutes (Fig. 5a). The corresponding PL spectra are shown in Fig. 5b and it is important to note that the spectral shape did not change during the experiment indicating that no irreversible chemical reaction between the dendrimer and solvent(s) or analytes had occurred. The fact that the PL intensity did not return to the initial value could arise from the film having partially degraded through photooxidation or some of the DNT not dissolving from the film. It has been shown that the latter case can occur if some of the analyte is irreversibly bound in the dendrimer film.25 To establish whether the former was the reason for the PL intensity not fully recovering we measured the photodegradation of the film in water over the same timescale. As discussed earlier, upon addition of water the PL intensity dropped slightly due to the change in the optical properties of the sample and thereafter there was only a small decrease in the PL intensity due to photooxidation was observed. The 1% decrease in PL due to photooxidation observed after 10 minutes of exposure to UV light was much less than the 10% loss of recovery observed, and hence we assign the latter to residual DNT being trapped in the film.
 | ||
| Fig. 5 PL quenching and recovery of the film. (a) The PL signal is integrated over the emission range of 400 nm to 600 nm with an integration time of 0.5 s. (b) Emission spectra of the film. | ||
Sensing analytes in seawater
Having established that the dendrimer film could detect the desired analytes in deionised water we were interested to determine whether the same analytes could be detected in seawater or whether the PL would be strongly affected by the contaminants in the water. We collected seawater from the Pacific Ocean at Noosa Heads, Queensland, Australia for the measurements. In the first part of the study we compared the effect of the untreated seawater and deionised water on the film PL intensity (Fig. 6a). As was observed for deionised water the PL intensity decreased when the film was immersed in the seawater but not to a significantly greater extent. Furthermore, the films were not more susceptible to photooxidation in seawater and the PL intensity was recovered upon removal of the seawater. This is an important result as it indicates that the pollutants and microorganisms in the water do not strongly quench the PL intensity, which would lead to false positive responses. We then prepared solutions of the three analytes in seawater and injected them onto the films. At a concentration of 2.5 ppm the same relative PL quenching of the three analytes was observed indicating that the use of seawater did not change the uptake of the analytes into the film (Fig. 6b). That is, PL quenching by DNT was again greater than pNT, which was greater than DNB. Furthermore, it suggests that seawater and the associated contaminants would not result in false positive or negative responses. | ||
| Fig. 6 (a) PL intensity versus time for the sensing film when immersed in deionised or seawater. (b) PL response to the different analytes in seawater. The concentration was 2.5 ppm for each of the analyte solutions. | ||
Test strip detection of analytes dissolved in water
We next evaluated the use of dendrimer sorbed paper test strips (12.5 mm × 50 mm) as a rapid, potential in-field visual detection method (Fig. 7). We tested both DNT and TNT as well as a range of potential anion and metal cation interferents. In the initial experiments a 10 μL drop of DNT in deionised water was placed on the centre of the black line on the paper strip. By adjusting the concentrations of DNT from 0 to 50 ppm, a standard card, as shown in Fig. 7a, was created. The card was placed under a portable UV lamp, and a photograph of the illuminated card was taken using a phone camera. From the images it can be seen that the PL quenching gradually increased (darker image) with increasing analyte concentration. The standard card image was saved on the phone as a reference and then used to compare the PL quenching of random tests (Fig. 7a). Visually comparing the PL quenching of the random tests of DNT dissolved in water using the same test strips with the standard card enabled the DNT concentration in the aqueous solution to be estimated. Using this simple test strip detection method, we were able to achieve a limit of detection of around 0.2 ppm, which was similar to that measured in the time dependent measurements using the CCD photodetector (Fig. 3). To test whether a range of anions and cations were potential interferents we used a dendrimer-coated filter paper (37.5 mm × 100 mm) partitioned into three strips. Each strip was further divided into smaller sections, each measuring 12.5 mm × 12.5 mm. The central strip was used to create the standard by dropping 10 μL of DNT in water into each square across the concentrations range of 0 to 25 ppm. Each anion and cation were then deposited on the strips for comparison with the DNT standard. On the left strip, each anion was assigned to an individual section, while the the different cations were loaded on the sections of the right strip as per the labelling in Fig. 7b. It should be noted that the concentrations of the anions and cations used are orders of magnitude greater than those typically found in sea and freshwater.28,29 It can be seen in Fig. 7b that none of the potential interferents elicited a change in the luminescence of the test strip at the concentrations tested. We also carried out a similar experiment with TNT as the test analyte (Fig. 7c). We found that the limit of detection for TNT in water was 0.1 ppm. We also note that when iron(III) and copper(II) salts were at a concentration of 100 mg L−1 there was an apparent decease in the PL intensity. The reduction in PL intensity at these very high concentrations was not due to quenching by the salts but rather attenuation of the excitation and/or emission by the weakly absorbing metal cations.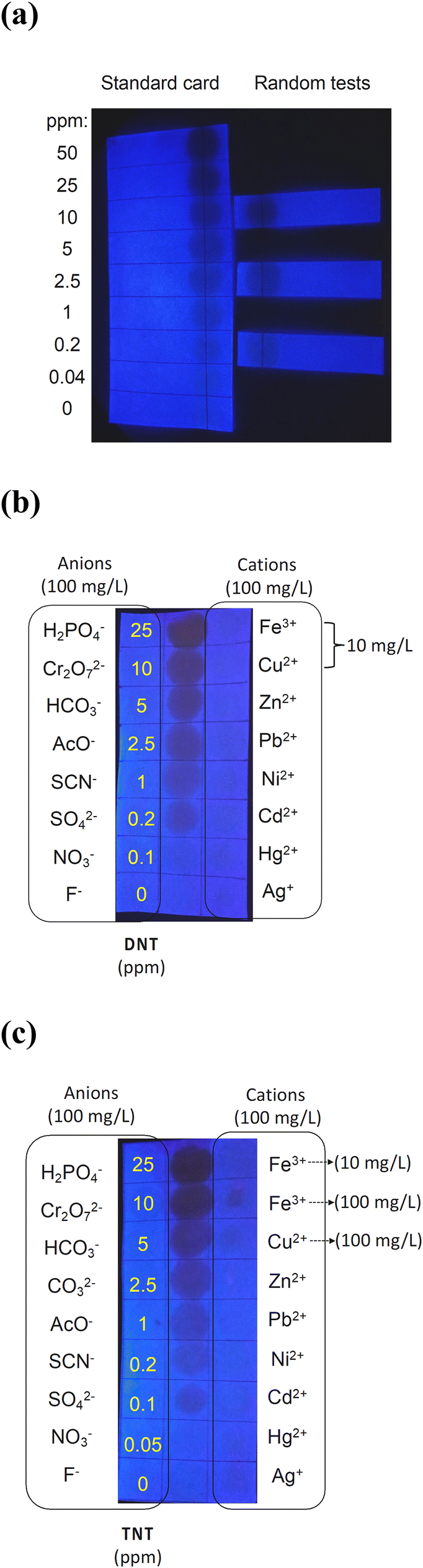 | ||
| Fig. 7 (a) Images of the test strips taken under hand-held UV lamp using a phone camera after placing 10 μL drops of aqueous DNT onto the black line. (b) Test strips showing the lack of response for different cations and anions against the DNT standard. (c) Test strips showing the lack of response for different cations and anions against a TNT standard. | ||
Conclusion
We have demonstrated that a dendrimer composed of a 9,9,9′,9′-tetra-n-propyl substituted 2,2′-bifluorene core, first-generation biphenyl dendrons, and 2-ethylhexyloxy surface groups can effectively detect TNT analogues in freshwater and seawater through quenching of the photoluminescence upon contact with the aqueous analyte solution. Films of the light-emitting dendrimer were insoluble and stable in water. While the dendrimer films could be used to sensitively detect nitro-aromatic analytes, they were not affected by the pollutants and microorganisms found in seawater. Moreover, we show that fluorescent dendrimers can potentially be used as the basis of a simple, effective and low-cost test strip for on-site, real-time detection of TNT in water. In this format, potential anionic and cationic interferents did not elicit a response. Thus, fluorescent dendrimers are promising materials for the detection of explosives in water.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge funding from the Australian Research Council, DP170102072.References
- B. Wang, X.-L. Lv, D. Feng, L.-H. Xie, J. Zhang, M. Li, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 6204–6216 CrossRef CAS PubMed.
- S. M. Tan, C. K. Chua and M. Pumera, Analyst, 2013, 138, 1700 RSC.
- R. W. Bishop, M. A. Hable, C. G. Oliver and R. J. Valis, J. Chromatogr. Sci., 2003, 41, 73–79 CAS.
- M. R. Darrach, A. Chutjian and G. A. Plett, Environ. Sci. Technol., 1998, 32, 1354–1358 CrossRef CAS.
- S. Letzel, Th. Göen, M. Bader, J. Angerer and T. Kraus, Occup. Environ. Med., 2003, 60, 483–488 CrossRef CAS PubMed.
- D. S. Moore, Rev. Sci. Instrum., 2004, 75, 2499–2512 CrossRef CAS.
- J. Yinon, TrAC, Trends Anal. Chem., 2002, 21, 292–301 CrossRef CAS.
- J. S. Caygill, F. Davis and S. P. J. Higson, Talanta, 2012, 88, 14–29 CrossRef CAS PubMed.
- X. Sun, Y. Wang and Y. Lei, Chem. Soc. Rev., 2015, 44, 8019–8061 RSC.
- Y. Salinas, R. Martínez-Máñez, M. D. Marcos, F. Sancenón, A. M. Costero, M. Parra and S. Gil, Chem. Soc. Rev., 2012, 41, 1261–1296 RSC.
- L. Li, X. Lyu, S. Liang and Z. Liu, Dyes Pigm., 2023, 220, 111651 CrossRef CAS.
- L. M. Martelo, L. F. Marques, H. D. Burrows and M. N. Berberan-Santos, Explosives Detection: From Sensing to Response. in Fluorescence in Industry. Springer Series on Fluorescence, ed. B. Pedras, Springer, Cham., 2019, vol. 18, pp. 293–320 Search PubMed.
- T. Langston, Sci. World J., 2010, 10, 546–562 CrossRef CAS PubMed.
- H. Sohn, R. M. Calhoun, M. J. Sailor and W. C. Trogler, Angew. Chem., Int. Ed., 2001, 40, 2104–2105 CrossRef CAS PubMed.
- O. S. Taniya, A. F. Khasanov, L. K. Sadieva, S. Santra, I. L. Nikonov, W. K. A. Al-Ithawi, I. S. Kovalev, D. S. Kopchuk, G. V. Zyryanov and B. C. Ranu, Materials, 2023, 16, 6333 CrossRef CAS PubMed.
- M. Basak and G. Das, ChemPlusChem, 2023, 88, e202300179 CrossRef CAS PubMed.
- A. S. Tanwar, S. Hussain, A. H. Malik, M. A. Afroz and P. K. Iyer, ACS Sens., 2016, 1, 1070–1077 CrossRef CAS.
- R. N. Gillanders, I. A. Campbell, J. M. E. Glackin, I. D. W. Samuel and G. A. Turnbull, Talanta, 2018, 179, 426–429 CrossRef CAS PubMed.
- J. Kang, L. Ding, F. Lü, S. Zhang and Y. Fang, J. Phys. D: Appl. Phys., 2006, 39, 5097–5102 CrossRef CAS.
- C. Jian and W. R. Seitz, Anal. Chim. Acta, 1990, 237, 265–271 CrossRef CAS.
- J. Yang, S. Aschemeyer, H. P. Martinez and W. C. Trogler, Chem. Commun., 2010, 46, 6804 RSC.
- H. Cavaye, P. E. Shaw, X. Wang, P. L. Burn, S.-C. Lo and P. Meredith, Macromolecules, 2010, 43, 10253–10261 CrossRef CAS.
- D. A. Olley, E. J. Wren, G. Vamvounis, M. J. Fernee, X. Wang, P. L. Burn, P. Meredith and P. E. Shaw, Chem. Mater., 2011, 23, 789–794 CrossRef CAS.
- M. A. Ali, S. Shoaee, S. Fan, P. L. Burn, I. R. Gentle, P. Meredith and P. E. Shaw, ChemPhysChem, 2016, 17, 3350–3353 CrossRef CAS PubMed.
- Y. Geng, M. A. Ali, A. J. Clulow, S. Fan, P. L. Burn, I. R. Gentle, P. Meredith and P. E. Shaw, Nat. Commun., 2015, 6, 8240 CrossRef CAS PubMed.
- H. Cavaye, P. E. Shaw, A. R. G. Smith, P. L. Burn, I. R. Gentle, M. James, S.-C. Lo and P. Meredith, J. Phys. Chem. C, 2011, 115, 18366–18371 CrossRef CAS.
- J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 11864–11873 CrossRef CAS.
- S. Sivakumar, Y. C. Song, I. S. Park, S. H. Cho, C. Y. Lee and B. G. Kim, Environ. Monit. Assess., 2010, 165, 449–460 CrossRef CAS PubMed.
- S. C. Apte, G. E. Batley, R. Szymczak, P. S. Rendell, R. Lee and T. D. Waite, Mar. Freshwater Res., 1998, 49, 203–214 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |