
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShedding new light on quadrupolar 1,4-dihydropyrrolo[3,2-b]pyrroles: impact of electron-deficient scaffolds over emission†
Bartosz
Szymański
ab,
Smruti Ranjan
Sahoo
 c,
Olena
Vakuliuk
a,
Rashid
Valiev
d,
Ruslan
Ramazanov
d,
Piotr
Łaski
b,
Katarzyna N.
Jarzembska
b,
Radosław
Kamiński
b,
Mohammad B.
Teimouri
*ae,
Glib
Baryshnikov
*c and
Daniel T.
Gryko
*a
c,
Olena
Vakuliuk
a,
Rashid
Valiev
d,
Ruslan
Ramazanov
d,
Piotr
Łaski
b,
Katarzyna N.
Jarzembska
b,
Radosław
Kamiński
b,
Mohammad B.
Teimouri
*ae,
Glib
Baryshnikov
*c and
Daniel T.
Gryko
*a
aInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44–52, 01-224 Warsaw, Poland. E-mail: daniel.gryko@icho.edu.pl
bDepartment of Chemistry, University of Warsaw, Żwirki i Wigury 101, 02-089 Warsaw, Poland
cLaboratory of Organic Electronics, Department of Science and Technology, Linköping University, SE-60174 Norrköping, Sweden
dDepartment of Chemistry, University of Helsinki, FI-00014 Helsinki, Finland
eFaculty of Chemistry, Kharazmi University, 15719-14911 Tehran, Iran
First published on 29th November 2024
Abstract
In this work, we disclose a series of seven quadrupolar centrosymmetric 1,4-dihydropyrrolo[3,2-b]pyrroles (DHPPs) linked to the two peripheral, strongly electron-accepting heterocycles such as benzoxadiazole, benzothiadiazole and benzoselenadiazole. This represents the first study probing the influence of electron-deficient heterocycles, rather that small electron-withdrawing substituents, on photophysics of DHPPs. These new acceptor–donor–acceptor hybrid dyes exhibit an appreciable combination of photophysical properties including absorption maxima in the range of 470–620 nm, and emission in the range of 500–720 nm with fluorescence quantum yields reaching 0.88. We discovered that the presence of two 7-nitro-benzoxadiazolyl substituents at positions 2 and 5 of DHPP core, evokes a strong fluorescence in non-polar solvents shifted to 639 nm. This is the most bathochromically shifted emission for quadrupolar, centrosymmetric chromophore bearing exclusively biaryl linkages. Interestingly, 1,2,4,5-tetraaryl-1,4-dihydropyrrolo[3,2-b]pyrrole (TAPP) possessing 4-benzothiadiazolyl groups is strongly emitting in the crystalline state (fluorescence quantum yield = 0.43). The combined photophysical and crystallographic studies point towards existence of intermolecular hydrogen bonds which modify the dihedral angles between the donor and acceptor moieties as a primary reason for this strong emission. Small structural alteration via the replacement of two 2,1,3-benzoxadiazole scaffolds with 2,1,3-benzoxadiazole-2-oxide moieties causes >103 decrease in the fluorescence intensity. Computational studies point out to strong charge transfer originating from exceptionally large dihedral angles as the pivotal reason of this phenomenon. Although internal conversion originating from the charge-transfer state is the prevailing non-radiative deactivation mechanism, intersystem crossing also plays a role. The rational design of DHPPs that enables modulation of emission will advance their applicability.
Introduction
Due to their extraordinary photophysical properties, the quadrupolar, centrosymmetric dyes continue to attract significant interest.1–9 Among many features, excited-state symmetry-breaking (ES-SB) is of particular interest both from theoretical and experimental point of view.10,11 The creative summary published by Terenziani and Painelli12 provided the impetus for the intense studies11,13–15 which culminated with a breakthrough observation of ES-SB in real time via time-resolved IR, by Vauthey and co-workers.16 A major chemical difficulty in enhancing our understanding of ES-SB is the lack of suitable centrosymmetric models. During the last decade, it became clear that one of the best models among quadrupolar centrosymmetric dyes are the 1,2,4,5-tetraaryl-1,4-dihydropyrrolo[3,2-b]pyrroles (TAPPs)17–22 possessing two strongly electron-withdrawing groups at positions 2 and 5.23 The investigation focused so far chiefly on TAPPs possessing 4-cyanophenyl substituents8,24–27 and nitroaryl substituents.28–30 These studies enabled to formulate theories of ES-SB,31,32 study ground-state versus excited state symmetry breaking33 and evaluate the influence of the position of the NO2 group on the fluorescence intensity.34The reported TAPPs feature biaryl linkages which in principle offer weak coupling limiting the possibility to bathochromically shift the absorption and emission. The motivation for our study originated from the desire to shift the emission beyond 600 nm while staying within formally weakly coupled acceptor–donor–acceptor (A–D–A) architectures, i.e. possessing exclusively biaryl linkages. To address these limitations we sought to incorporate electron-deficient heterocycles as substituents, which can effectively enlarge the π-system of resulting dyes. We hypothesised that the combination of the π-expanded electron-withdrawing heterocyclic scaffold with the presence of an additional acceptor such as NO2 group is the key to achieving an extraordinary bathochromic shift of both absorption and emission.
Results and discussion
Design and synthesis
Our design was based on the following logic: benzoxadiazole,35–38 benzothiadiazole39–45 and benzoselenadiazole46–48 are heterocycles possessing strongly electron-deficient character and their direct linking to the exceptionally electron-rich 1,4-dihydropyrrolo[3,2-b]pyrrole (DHPP) core should lead to quadrupolar centrosymmetric dyes with markedly red-shifted absorption and emission. The fluorescence of derivatives of benzoxadiazole and its analogues is very sensitive to changes in the polarity of the solvent. We envisioned that depending on the bridging position and the character of the central heteroatom the photophysics of the resulting dyes can be modulated. Another attractive modification was to replace benzoxadiazolyl substituent with corresponding N-oxide. Consequently, the designed TAPPs 1–7 were prepared via a multicomponent reaction of aldehydes, aromatic primary amines and diacetyl49 (Fig. 1, see ESI† for synthetic details). Most of the required aldehydes (i.e. 4-formyl-2,1,3-benzoxadiazole, 5-formyl-2,1,3-benzoxadiazole, 4-formyl-2,1,3-benzothiadiazole and 5-formyl-2,1,3-benzothiadiazole) were commercially available except for 4-formyl-2,1,3-benzoselenadiazole, 6-formyl-2,1,3-benzoxadiazole 2-oxide and 4-formyl-7-nitro-2,1,3-benzoxadiazole which were synthesized for this project. For example, 6-formyl-2,1,3-benzoxadiazole 2-oxide was prepared from the corresponding 4-chloro-3-nitrobenzaldehyde and NaN3 in DMSO as the solvent via a general method involving the two-step process of SNAr/ring-closing reaction (Scheme S5, see ESI, Section 2† for details).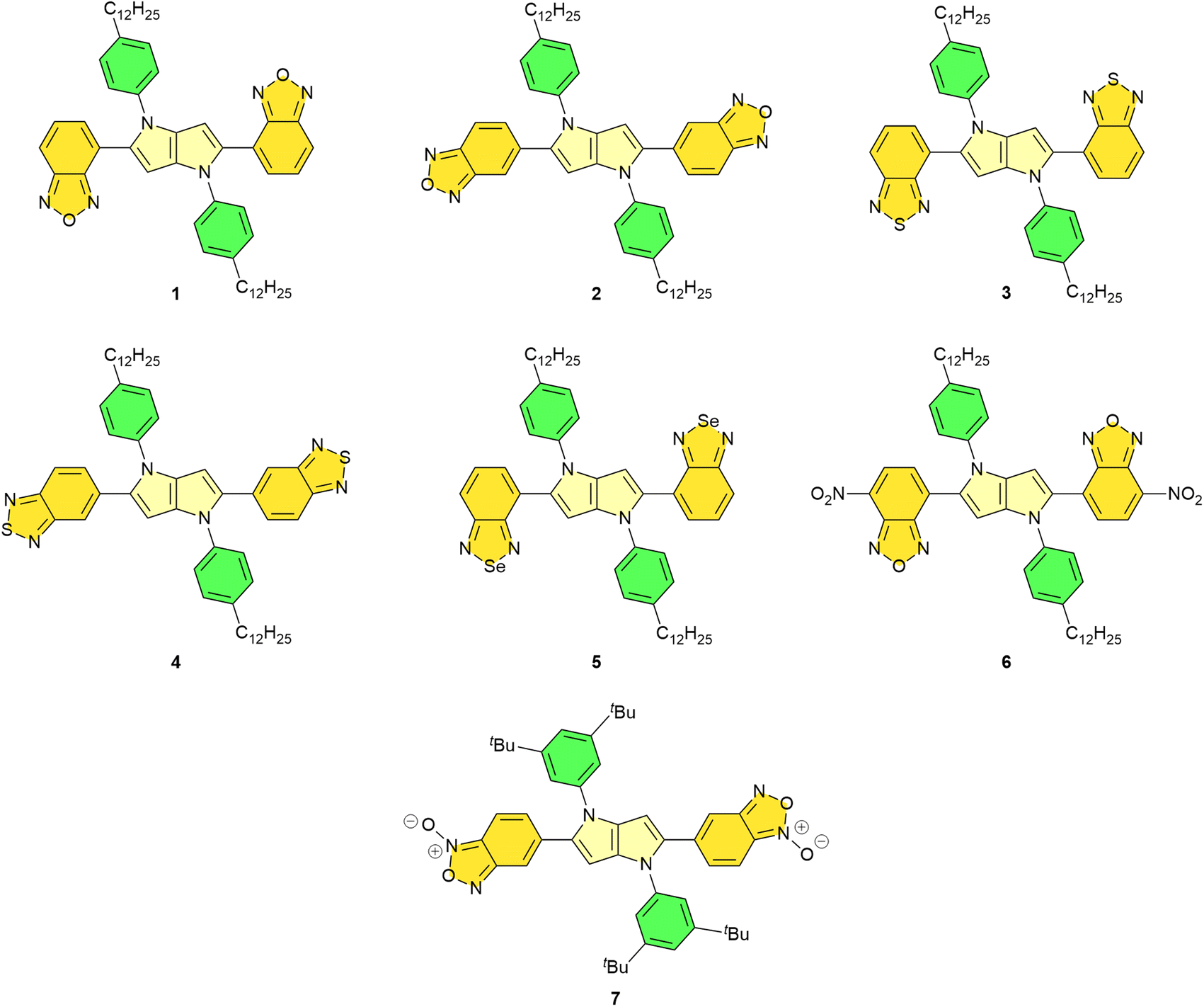 | ||
| Fig. 1 Structures of quadrupolar tetraarylpyrrolo[3,2-b]pyrroles 1–7. | ||
Photophysical properties in solution
Absorption and emission spectra of all obtained TAPPs were measured in solutions with dichloromethane (DCM), tetrahydrofuran (THF), toluene, 1,4-dioxane and cyclohexane used as solvents of different polarity (Fig. 2, Table 1; Section 4, Fig. S1–S16 in ESI†). For compounds 1, 2 and 6 the spectra in dimethyl sulfoxide (DMSO) were also measured, however, remaining TAPPs are insoluble in this solvent. The measured molar absorption coefficients (ε) are between 104 and 105 M−1 cm−1. | ||
| Fig. 2 Normalised absorption (solid lines) and emission (dashed lines) spectra of compounds 1–7 in cyclohexane. | ||
| Dye | Solvent | λ maxabs/nm [ε × 10−4/M−1 cm−1] | λ maxem/nm | Φ fl | Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) /cm−1 /cm−1 |
|---|---|---|---|---|---|
|
a Data from ref. 50.
b Reference: fluorescein in 0.1 M NaOH (Φfl = 0.9, λexc = 490 nm).
c Reference: rhodamine 6G in EtOH (Φfl = 0.95, λexc = 510 nm).
d Reference: cresyl violet in EtOH (Φfl = 0.56, λexc = 610 nm). λmaxabs – absorption maximum, λmaxem – emission maximum, ε – molar absorption coefficient, Φfl – fluorescence quantum yield, Δ – Stokes' shift. Emission peak for dye 7 in THF was fitted with the Lorentz function, due to its very low intensity.
|
|||||
| 1 | Cyclohexane | 499 [2.9], 511 [2.9] | 553, 581 | 0.77b | 1490 |
| THF | 501 [2.5] | 620 | 0.44b | 3910 | |
| DCM | 504 [2.3] | 673 | 0.075b | 4980 | |
| DMSO | 500 [2.2] | 708 | — | 5880 | |
| 2 | Cyclohexane | 448 [3.0], 471 [2.8] | 499, 518 | 0.88b | 1190 |
| THF | 469 [2.9] | 609 | 0.35b | 4900 | |
| DCM | 470 [2.8] | 646 | 0.043b | 5800 | |
| DMSO | 478 [2.8] | 698 | — | 6590 | |
| 3 | Cyclohexane | 506 [1.8] | 572 | 0.52c | 2910 |
| THF | 507 [1.4] | 657 | 0.25c | 4500 | |
| DCM | 498 [1.7] | 716 | 0.030c | 6110 | |
| 4 | Cyclohexane | 464 [2.9], 482 [2.9] | 519 | 0.78c | 1480 |
| THF | 479 [3.0] | 620 | 0.45c | 4750 | |
| DCM | 476 [3.0] | 661 | 0.12c | 5880 | |
| 5 | Cyclohexane | 544 [1.3] | 622 | 0.081d | 2310 |
| THF | 536 [1.4] | 726 | 0.034d | 4880 | |
| DCM | 532 [1.9] | 879 | 0.018d | 7420 | |
| 6 | Cyclohexane | 575 [3.6], 618 [5.6] | 639, 691 | 0.79d | 530 |
| THF | 624 [4.6] | 722 | 0.007d | 2180 | |
| DCM | 648 [4.1] | 737 | 0.004d | 1860 | |
| DMSO | 627 [6.0] | 777 | — | 3080 | |
| 7 | Cyclohexane | 494 [1.6] | 542 | 0.002b | 1790 |
| THF | 497 [2.7] | 586 | 0.002b | 3060 | |
| DCM | 495 [2.6] | — | — | — | |
Bathochromic shift of emission often reaching edge of NIR region (e.g. from 572 nm to 716 nm for TAPP 3) is always accompanied with gradual decrease in fluorescence quantum yield. In non-polar solvents such as cyclohexane, the TAPPs exhibit Φfl as high as 0.88, while with the increase of the polarity (in DCM, THF) decrease to 0.20–0.40 or almost entirely vanishes (in DMSO). For all synthesised TAPPs, the Stokes shifts (Δ) in cyclohexane are below 3000 cm−1 and increase to above 3900 cm−1 in polar solvents (except for 6 and 7). There is a negative correlation between Δ and Φfl within a series probably due to an increased number of relaxation pathways in the case of large Stokes shifts. In the case of TAPP 6 one of the sharpest, for quadrupolar TAPPs, decreases of Φfl with increasing solvent polarity is observed (0.007 in THF compared to 0.79 in cyclohexane). Interestingly, compound 6 exhibits not only solvatofluorochromism but also solvatochromism, with a significant difference in absorption maxima in DCM, compared to cyclohexane (749 cm−1, 30 nm). This may suggest symmetry breaking in the ground state in polar solvents observed earlier for TAPPs bearing simple nitroaryl substituents.33
Solid-state spectroscopy
The obtained TAPPs also exhibit notably different optical properties in the solid state, reflected among the other features in their colour. Four of the obtained TAPPs, namely 1, 2, 4 and 6, show bathochromic shift (i.e. red-shift) of absorption maxima in the solid state when compared to cyclohexane solutions. This is a typical effect for aromatic compounds, which results from the formation of short interatomic contacts in solids. Solely the compound 3 behaves oppositely and exhibits a hypsochromic shift (i.e. blue-shift) of absorption maxima in the solid state (Fig. 3). Plausible reason for it, is much larger dihedral angle between benzothiadiazole and DHPP core compared to the situation in solution (vide infra). Not surprisingly, given its photophysical properties in the solution, the absorption of 6 is the most red-shifted one among the entire series. | ||
| Fig. 3 Absorption spectra of dyes 1–4 and 6 in the solid state (powder samples, red solid lines) and in cyclohexane (CHX; shown for comparison; blue dot-dashed lines). Emission spectra are shown as green dot-dashed lines. All spectra were normalized. | ||
Furthermore, as far as the solid-state emission spectra of compounds 1–4 and 6 are concerned (for dye 5 we could not obtain X-ray quality crystal; TAPP 7 was excluded due to the lack of measurable emission), compounds 2, 4 and 6 show rather low Φfl values, i.e., below 0.07. In turn, TAPPs 1 and 3 exhibit significantly higher Φfl values, which reached 0.20 and 0.43, respectively. Intriguingly, the emission spectrum of dye 1 has an additional peak at ca. 460 nm (Fig. 3). Finally, it is worth noting that 6 exhibits solid-state fluorescence above 700 nm, i.e. in the near-infrared region.
According to the experimental data, compounds 2, 4 and 6 exhibit fluorescence with rather short lifetimes, (i.e. below 1.5 ns, Table 2). Again compound 1 and 3 can be distinguished. 1 shows a longer lifetime (ca. 4 ns), whereas its sulphur analogue 3 stands out, possessing bi-exponential fluorescence decay (lifetimes 1.63(3) and 5.1(3) ns).
| Dye | λ maxabs/nm | Δλmaxabs/nm [cm−1] | λ maxem/nm | Φ fl | τ/ns |
|---|---|---|---|---|---|
| a Bi-exponential decay; two lifetimes are given. | |||||
| 1 | 518 | +19 [+735] | 577 | 0.20 | 3.80(1) |
| 2 | 478 | +7 [+356] | 582 | 0.06 | 1.11(1) |
| 3 | 463 | −43 [−1835] | 564 | 0.43 | 1.63(3)/5.1(3)a |
| 4 | 489 | +7 [+297] | 597 | 0.05 | 0.77(1) |
| 6 | 681 | +63 [+1497] | 737 | 0.02 | 0.91(2) |
| 7 | 539 | +45 [+1690] | 789 | <0.01 | — |
Structural analysis
Different properties of the studied TAPPs in the solid state result not only from molecular differences between the dyes, but also from the way molecules are arranged in the crystal structure. Therefore, to investigate the effect that the molecular structure of the studied systems (e.g. substitution of oxygen atoms by sulphur atoms, longer or shorter alkyl chains) has on the crystal architecture and how it is then reflected in spectroscopic properties, single-crystal X-ray diffraction measurements were conducted. It should, however, be noted that we encountered severe difficulties in the crystallization of compound 2, and therefore its analogue 2a possessing methyl groups instead of long alkyl chains was synthesized (see ESI† for synthetic details) and crystallized (Fig. 5). The same approach, for comparative purposes, was applied to compound 3 (which crystallized readily), resulting in synthesis and crystal structure determination of 3a (Fig. 5). In this case, we wanted to briefly elucidate the effect of the chain length on molecular structure and crystal packing. Overall, the studied compounds crystallize in triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (1, 3) and monoclinic P21/c (4, 3a), P21/n (7), or C2/c (2a, 6) space groups. In all cases, molecules are located at the centres of symmetry, thus only half (or halves) of a molecule is present in the asymmetric unit (ASU). It should be noted that in the case of 1 ASU contains also one DCM molecule (Fig. 4).
(1, 3) and monoclinic P21/c (4, 3a), P21/n (7), or C2/c (2a, 6) space groups. In all cases, molecules are located at the centres of symmetry, thus only half (or halves) of a molecule is present in the asymmetric unit (ASU). It should be noted that in the case of 1 ASU contains also one DCM molecule (Fig. 4).
 | ||
| Fig. 4 Molecular structures of compounds 1, 3, 4, 6 and 7. Thermal motion is shown as ellipsoids with a 50% probability level; selected hydrogen atoms are removed for clarity; only ASU is labelled; large black spheres represent long alkyl chains starting from the labelled carbon atom. For compound 1 only one symmetry-independent molecule is shown and solvent is removed for clarity. For 7 only one symmetry-independent molecule is shown, disorder is removed and some labels of tBu groups are not shown. For details see ESI, Section 6.† | ||
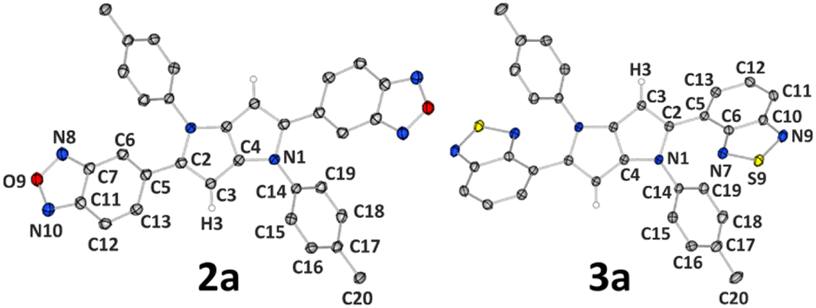 | ||
| Fig. 5 Molecular structures of compounds 2a and 3a. Thermal motion is shown as ellipsoids with a 50% probability level; selected hydrogen atoms are removed for clarity; only ASU is labelled. | ||
The XRD studies serve as a basis for the correlation between the structure and the spectroscopic properties in the solid state. Two main factors are discussed here – molecular planarity and packing in the crystalline state. In our previous studies50 we have shown the degree of planarity of the molecular A–D–A core is well correlated with the fluorescence quantum yield decrease. However, in the crystal the planarity of the molecule can also be significantly influenced by the weak, yet cooperative, intermolecular forces. In this respect, quite significant differences between interactions of long- and short-chained analogues seem interesting. Nevertheless, packing in the crystal structures of dyes 3 and 6, which exhibit 0.43 and 0.02 fluorescence quantum yield in the solid state, respectively, is significantly different. For compound 6, there is a visible head-to-tail arrangement, while for compound 3 the packing is additionally driven by intramolecular and intermolecular interactions, lowering the symmetry (Fig. 6). We observed that the crystals of a lower symmetry exhibit higher fluorescence quantum yields (0.20 for compound 1, 0.43 for compound 3). The key variation in packing of examined structures are caused by the presence of intermolecular hydrogen bonds between one of nitrogen atoms belonging to benzothiadiazole scaffold and hydrogen located at position 3 of DHPP core. Different polarization of diazole ring in benzoxadiazole versus benzothiadiazole results in slightly higher electrostatic potential around nitrogen atoms in the latter one, which in turn enables the formation of intermolecular hydrogen bond. This interaction causes moderate (for TAPP 4) or large (for TAPP 3) increase of dihedral angle versus the optimized geometry for single molecule. The different geometries of studied TAPPs have to be reflected in different excitonic coupling in the solid state. It seems to be the reason for both large emission intensity and hypsochromic shift of emission in the case of TAPP 3. More elaborate discussion of the intermolecular interactions and dependence of absorption maxima on the molecular planarity is reported in ESI, Section 7.†
 | ||
| Fig. 6 Packing diagrams of the crystal structures for compounds 3 (view along X axis) and 6 (view along Z axis). Perspective projection to emphasise layered structure. | ||
As the compound 7 is almost non-emitting both in solution and in solid state, the cause is probably in its molecular structure rather than its arrangement in the crystal. However, the latter is intriguing, due to two symmetry-inequivalent halves of 7 in ASU. One is fully ordered, whereas the another one exhibits disorder in the benzoxadiazole moieties (Fig. 7). The O20 oxygen atom of this part of the molecule can be located either on one or on the other side of the benzoxadiazole moiety with approximately a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 probability ratio (O20A and O20B, respectively), almost exactly as shown for CDCl3 at 25 °C.61 This is due to the equilibrium in solution between two regioisomers (i.e., N1,ring–O1–N2,ring–O2 ⇌ O1–N1,ring–O2–N2,ring) as a result of Boulton–Katritzky rearrangement shown in detail in Scheme S1a (ESI).†62,63 Both isomeric forms co-crystallize together. In consequence, the benzoxadiazole fragments in this molecule occupy two alternative positions, which enable their comparable stabilisation in the crystal structure. Both are involved in weak and cooperative C–H⋯O or C–H⋯N hydrogen bonds (Fig. 7), and to geometrical restrictions. In order to satisfy the energetic requirements, the whole fragment moves slightly away (X = O), or towards (X = N) the methyl group. Note the interactions are present in both sides of the benzoxadiazole moiety.
:1 probability ratio (O20A and O20B, respectively), almost exactly as shown for CDCl3 at 25 °C.61 This is due to the equilibrium in solution between two regioisomers (i.e., N1,ring–O1–N2,ring–O2 ⇌ O1–N1,ring–O2–N2,ring) as a result of Boulton–Katritzky rearrangement shown in detail in Scheme S1a (ESI).†62,63 Both isomeric forms co-crystallize together. In consequence, the benzoxadiazole fragments in this molecule occupy two alternative positions, which enable their comparable stabilisation in the crystal structure. Both are involved in weak and cooperative C–H⋯O or C–H⋯N hydrogen bonds (Fig. 7), and to geometrical restrictions. In order to satisfy the energetic requirements, the whole fragment moves slightly away (X = O), or towards (X = N) the methyl group. Note the interactions are present in both sides of the benzoxadiazole moiety.
 | ||
| Fig. 7 Weak hydrogen bonds present in the vicinity of the disordered benzofuroxane fragment in dye 7 crystal structure. Minor component of disorder is shown in semi-transparent gold colour. | ||
Theoretical studies
To better understand the correlation between the peripheral substituents and optical properties of the centrosymmetric dyes (measured in the solution) we performed quantum chemical calculations within DFT framework using the B3LYP-37/6-31G(d,p) level of theory. Given that computational studies for TAPP 1 have been already performed and published,50 (for details, see ESI, Section 8†). We focused on the most intriguing cases of i.e. TAPPs 5, 6 and 7. HOMOs for all these dyes (calculated at optimized S0 geometries, Fig. 8 and S8.1, ESI†) although mainly located on the central DHPP core, are also present to some extent on peripheral scaffolds. Dye 5 demonstrates a simultaneous shift of frontier molecular orbital levels to higher energies, but the energy gap remains same as for dye 6. Interestingly, the presence of two N-oxide functionalities in the structure of dye 7 has a significant effect on its molecular structure. For instance, we observe larger dihedral angles between two acceptors and the central DHPP core in dye 7 (in the range from 30 to 35°) as compared to dye 6 (in the range from 21 to 27°). This leads to significant destabilization of LUMO and a larger HOMO–LUMO gap for dye 7.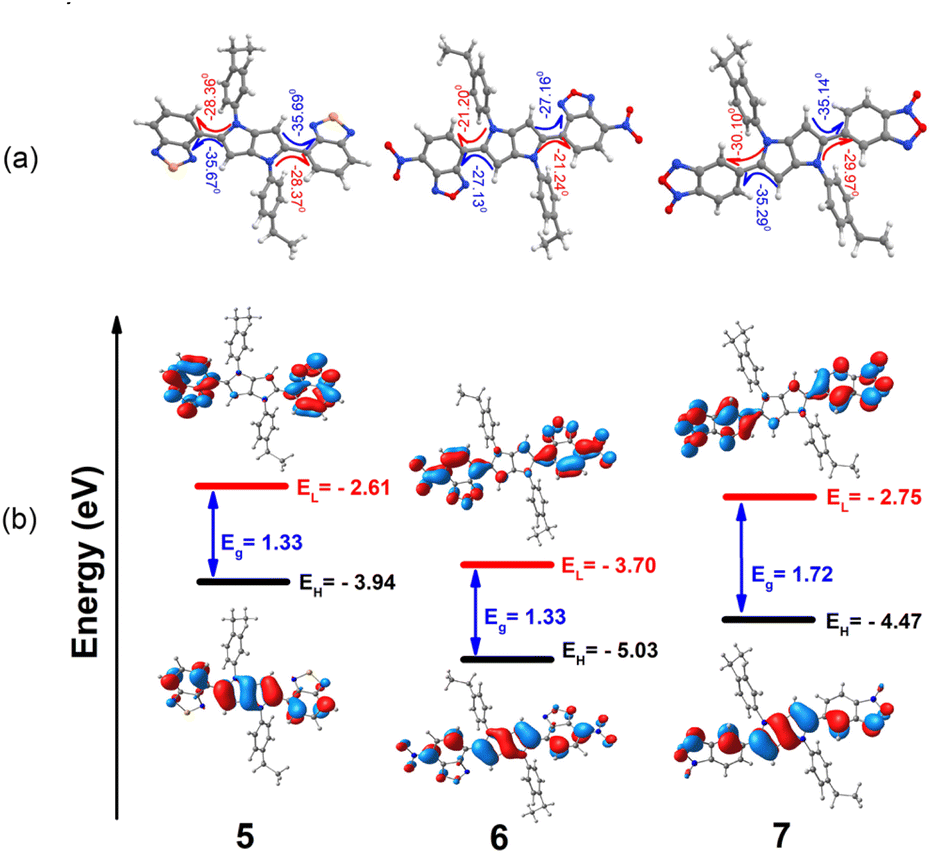 | ||
| Fig. 8 B3LYP-37/6-31G(d,p) theory level computed highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbital (LUMO) plots, HOMO/LUMO electronic energies and HOMO–LUMO (Eg) gaps of investigated dyes 5–7. | ||
Calculated absorption and emission maxima have been summarized in Table 3. In some cases, for example, emission of dye 6 and absorption of dye 5, we observed perfect agreement between the theory and experiment. For other cases, the computed results followed quantitatively the experimental results. The absorption and emission wavelengths of studied dyes are found to be dependent on solvent polarity. The maxima were redshifted with the increase of solvent polarity from cyclohexane to DCM.
| Dye | Solvent | E(S1)/eV | E(T1)/eV | E(T2)/eV | E(T3)/eV | E(T4)/eV |
|
|
|
|
k r × 108/s−1 | k ISC × 107/s−1 | k IC × 108/s−1 | Φ theorfl |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | Cyclohexane | 1.91 | 0.66 | 1.15 | 2.17 | 2.38 | 0.163 | 0.656 | — | — | 1.16 | 0.0021 | 3.70 | 0.24 |
| Dioxane | 1.91 | 0.65 | 1.14 | 2.17 | 2.38 | 0.065 | 0.680 | — | — | 0.93 | 0.082 | 14.0 | 0.06 | |
| Toluene | 1.90 | 0.65 | 1.14 | 2.18 | 2.38 | 0.061 | 0.690 | — | — | 1.04 | 0.026 | 9.10 | 0.10 | |
| THF | 1.82 | 0.60 | 1.14 | 2.18 | 2.39 | 0.139 | 0.824 | — | — | 1.08 | 0.22 | 17.0 | 0.06 | |
| DCM | 1.81 | 0.59 | 1.13 | 2.18 | 2.39 | 0.149 | 0.838 | — | — | 0.75 | 9.40 | 7.50 | 0.08 | |
| 6 | Cyclohexane | 1.95 | 0.69 | 1.48 | 2.18 | 2.27 | 0.009 | 0.286 | — | — | 2.57 | 0.11 | 2.80 | 0.48 |
| Dioxane | 1.92 | 0.68 | 1.49 | 2.17 | 2.28 | 0.009 | 0.285 | — | — | 2.57 | 0.13 | 3.10 | 0.45 | |
| Toluene | 1.91 | 0.68 | 1.48 | 2.17 | 2.28 | 0.008 | 0.285 | — | — | 2.55 | 0.17 | 3.30 | 0.43 | |
| THF | 1.71 | 0.61 | 1.48 | 2.13 | 2.24 | 0.009 | 0.282 | — | — | 2.30 | 1.60 | 8.30 | 0.21 | |
| DCM | 1.69 | 0.60 | 1.48 | 2.13 | 2.24 | 0.009 | 0.283 | — | — | 2.27 | 2.00 | 9.00 | 0.20 | |
| 7 | Cyclohexane | 2.03 | <0.5 | 0.64 | 1.80 | 2.02 | 0.447 | 0.104 | 0.429 | 0.256 | 0.104 | 3.80 | 0.49 | 0.11 |
| Dioxane | 2.03 | <0.5 | 0.65 | 1.79 | 2.02 | 0.441 | 0.102 | 0.422 | 0.255 | 0.121 | 4.10 | 0.51 | 0.12 | |
| Toluene | 2.04 | <0.5 | 0.65 | 1.79 | 2.02 | 0.436 | 0.100 | 0.414 | 0.255 | 0.103 | 15.0 | 0.39 | 0.06 | |
| THF | 2.02 | <0.5 | 0.67 | 1.76 | 2.01 | 0.106 | 0.099 | 0.105 | 0.139 | 0.320 | 0.15 | 0.66 | 0.32 | |
| DCM | 2.02 | <0.5 | 0.67 | 1.75 | 2.00 | 0.107 | 0.098 | 0.104 | 0.141 | 0.182 | 0.16 | 4.70 | 0.04 |


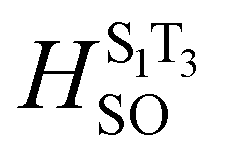

At S1 geometry, we noticed substantial changes in molecular structure. For instance, one acceptor unit of dye 7 has been found nearly orthogonal (twisting angles range from 65 to 74°) to the central DHPP core. This gives rise to significant charge transfer from donor to acceptor in S1 state and as a result weak emission by S1 state in non-polar as well as polar solvents. Concurrently, for dye 7 we computed the very small oscillator strength (f) values 0.05–0.16 for S1 → S0 transition regardless of solvent polarity (ESI†). At the same time, we do not observe dramatic changes in molecular geometry in S1 state of dye 6 in non-polar as well as polar solvents. For this dye, we calculated S1 state donor–acceptor twisting angles, which range from 20 to 23° and are similar to those at their S0 geometry. Consequently, we calculated bright S1 emission in the region 637–732 nm (f value ranges from 1.56 to 1.83) for dye 6 in all studied solvents. The S1 emission sustains a typical redshift with respect to solvent polarity (Table 1). In analogy to dye 6, we also do not see any significant structural changes in the S1 state of dye 5. We observed that donor–acceptor dihedral angles are from 28 to 36° and from 25 to 29° at S0 and S1 geometry of TAPP 5, respectively. For this TAPP, S1 computed emission in the range 648–683 nm also showed red-shift with an increase of solvent polarity.
To verify the singlet excited state properties, we further calculated the fluorescence quantum yields (Φtheorfl) of studied dyes. To numerically analyse the Φtheorfl, we computed triplet excitations and spin–orbit-coupling matrix elements (HSO) at S1 state geometry. Additionally, the radiative fluorescence decay rate (kr) and non-radiative relaxation rate constants such as intersystem crossing, ISC (kISC) and internal conversion, IC (kIC) were also calculated (see detailed theory in ESI†). All calculated results have been summarized in Table 3. As we can see, in solution we found two triplets T1 and T2 below the S1 state for dye 5 and 6, whereas investigated dye 7 sustains four triplet states T1, T2, T3 and T4 below S1. Triplets that are below the S1 were found to contribute to the ISC channel. For dye 6, we calculated large Φtheorfl 0.43–0.48 in non-polar solvents, which are in agreement with experimentally observed 0.79 for cyclohexane. Here, both kr and kIC are comparable and about 102 times higher than the kISC, and hence leading to large fluorescence quantum yields in non-polar solvents. For polar solvents (THF and DCM) however, we predicted larger kIC than kr, that can result in smaller Φtheorfl (Table 3). Therefore, strong fluorescence observed for dye 6 in non-polar solvents originates from the high radiative rate constant kr and weak contribution of IC and ISC rates. The weak fluorescence of dye 7 in all tested solvents (<0.01) is in line with calculated fluorescence quantum yields (0.04–0.11). For this compound, we found that both non-radiative decay rates kISC and kIC are larger than the radiative fluorescence decay rate, consequently small Φtheorfl are obtained (Table 3). In the case of dye 5, kr and kIC largely compete with kISC rates (excluding DCM solvent).
In general, kIC always dominates over kr so the resulting fluorescence quantum yield is moderate in non-polar cyclohexane solvent and decreases substantially with increasing the solvent polarity. However, in DCM kISC additionally contributes to the fluorescence quenching. To conclude, the kIC represents the main channel of fluorescence quenching similar to our previous studies,50 and the solvent-dependent fluorescence efficiency, thus satisfies the energy gap rule between the S1 and S0 states – smaller gap results in larger kIC and smaller Φtheorfl.
Conclusions
We have explored how the chemical structure of the electron-deficient heterocycle and its linkage position affects the photophysics of the quadrupolar, centrosymmetric chromophores by bridging benzoxadiazole, benzothiadiazole and benzoselenadiazole scaffolds with 1,4-dihydropyrrolo[3,2-b]pyrrole core. The electron-poor nature of peripheral heterocycles combined with known exceptionally strong electronic coupling at positions 2 and 5 of DHPP core results in the fact that these both heterocycles effectively form one π-expanded chromophore. Our findings revealed that it is possible to modulate both the emission maxima and the fluorescence intensity by changing the heteroatom and the bridging position. Our study demonstrates that the combination of strongly electron-withdrawing heterocyclic substituent with a nitro group leads to the bathochromic shift of emission beyond 600 nm, previously unachievable for weakly coupled architectures. The presence of 2,1,3-benzoxadiazole-2-oxide in the structure of these quadrupolar dyes results in very low fluorescence quantum yields. Quantum-chemical calculations complement the spectroscopic data and provide rationalization of low emission intensity which is caused by: (a) significant charge-transfer originating from an increase of the already large dihedral angles in the excited state, (b) the dominating role of internal conversion as the main deactivation channel of S1 state and (c) complementing intersystem crossing originating from the number of excited triplet states located below S1. Strong emission of centrosymmetric TAPP possessing NO2 substituents originates from balanced charge transfer which suppresses intersystem crossing and the formation of dark CT states. We discovered that if symmetry of the crystal is lower, which is the case of TAPP possessing benzothiadiazol-4-yl substituents, the fluorescence quantum yield in the solid state is larger. The underlying reason is the formation of intermolecular hydrogen bonds which occurs only if benzothiadiazolyl substituents are present, which originates from different character of nitrogen atoms in benzothiadiazole versus benzoxadiazole, which in turn reflects different electrostatic potential in these two heterocyclic scaffolds. These discoveries provide tools for designing novel quadrupolar dyes with desired photophysical properties and help to translate properties from solution to solid state answering many questions in solid state.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for compounds 1, 2a, 3, 3a, 4, 6 and 7 have been deposited at the CCDC under 2288872 and 2359982–2359987.Author contributions
Conceptualization: D. T. G. and M. B. T.; investigation: B. S., O. V., M. B. T., P. Ł., S. R. S., R. V., R. R.; supervision: D. T. G., G. B., R. K., K. N. J.; visualization: D. T. G., B. S., P. Ł.; writing – original draft: B. S., D. T. G., O. V., S. R. S., G. B., P. Ł., R. K., K. N. J.; writing – review and editing: D. T. G., O. V., R. K., K. N. J., G. B. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre in Poland (OPUS 2020/37/B/ST4/00017 and HARMONIA 2018/30/M/ST5/00460 grants). This project has received funding from the European Union's Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreement no. 101007804. The computations were enabled by resources provided by the National Academic Infrastructure for Super Computing in Sweden (NAISS, 2023/5-77) at the National Supercomputer Centre (NSC) at Linköping University partially funded by the Swedish Research Council through grant agreement no. 2022-06725. G. B. thanks the support by the Swedish Research Council (no. 2020-04600), Swedish Government Strategic Research Area in Materials Science on Advanced Functional Materials at Linköping University (Faculty grant SFO-Mat-LiU no. 2009-00971), and European Union (ERC, LUMOR, 101077649). Views and opinions expressed are, however, those of the authors only and do not necessarily reflect those of the European Union or the European Research Council Executive Agency. Neither the European Union nor the granting authority can be held responsible for them. G. B. and S. R. S. also thanks the support by the Olle Engkvist Byggmastare Foundation (contract no. 212-0136). The in-house X-ray diffraction experiments were carried out at the Department of Physics, University of Warsaw, on Rigaku Oxford Diffraction SuperNova diffractometer, which was co-financed by the European Union within the European Regional Development Fund (program no. POIG.02.01.00-14.122/09). We thank K. Durka (Warsaw, Poland) for possibility to use the integrating sphere.Notes and references
- E. Vauthey, J. Phys. Chem. Lett., 2022, 13, 2064–2071 CrossRef CAS PubMed.
- J. S. Beckwith, C. A. Rumble and E. Vauthey, Int. Rev. Phys. Chem., 2020, 39, 135–216 Search PubMed.
- T. Kim, W. Kim, H. Mori, A. Osuka and D. Kim, J. Phys. Chem. C, 2018, 122, 19409–19415 CrossRef CAS.
- Y. Feng, P. J. Das, R. M. Young, P. J. Brown, J. E. Hornick, J. A. Weber, J. S. W. Seale, C. L. Stern, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2022, 144, 16841–16854 CrossRef CAS PubMed.
- R. Feng, N. Sato, T. Yasuda, H. Furuta and S. Shimizu, Chem. Commun., 2020, 56, 2975–2978 RSC.
- S. Shimizu, A. Murayama, T. Haruyama, T. Iino, S. Mori, H. Furuta and N. Kobayashi, Chem.–Eur. J., 2015, 21, 12996–13003 CrossRef CAS PubMed.
- B. Bardi, M. Krzeszewski, D. T. Gryko, A. Painelli and F. Terenziani, Chem.–Eur. J., 2019, 25, 13930–13938 CrossRef CAS PubMed.
- Z. Szakács, M. Tasior, D. T. Gryko and E. Vauthey, ChemPhysChem, 2020, 21, 1718–1730 CrossRef PubMed.
- P. Verma, M. Tasior, P. Roy, S. R. Meech, D. T. Gryko and E. Vauthey, Phys. Chem. Chem. Phys., 2023, 25, 22689–22699 RSC.
- W. Kim, T. Kim, S. Kang, Y. Hong, F. Würthner and D. Kim, Angew. Chem., Int. Ed., 2020, 59, 8571–8578 CrossRef CAS PubMed.
- T. M. Cooper, J. E. Haley, D. M. Krein, A. R. Burke, J. E. Slagle, A. Mikhailov and A. Rebane, J. Phys. Chem. A, 2017, 121, 5442–5449 CrossRef CAS PubMed.
- F. Terenziani, A. Painelli, C. Katan, M. Charlot and M. Blanchard-Desce, J. Am. Chem. Soc., 2006, 128, 15742–15755 CrossRef CAS PubMed.
- T. Kim, J. Kim, H. Mori, S. Park, M. Lim, A. Osuka and D. Kim, Phys. Chem. Chem. Phys., 2017, 19, 13970–13977 RSC.
- T. Beppu, K. Tomiguchi, A. Masuhara, Y.-J. Pu and H. Katagiri, Angew. Chem., Int. Ed., 2015, 54, 7332–7335 CrossRef CAS PubMed.
- C. L. Anderson, T. Zhang, M. Qi, Z. Chen, C. Yang, S. J. Teat, N. S. Settineri, E. A. Dailing, A. Garzón-Ruiz, A. Navarro, Y. Lv and Y. Liu, J. Am. Chem. Soc., 2023, 145, 5474–5485 CrossRef CAS PubMed.
- B. Dereka, A. Rosspeintner, Z. Li, R. Liska and E. Vauthey, J. Am. Chem. Soc., 2016, 138, 4643–4649 CrossRef CAS PubMed.
- M. Krzeszewski, D. Gryko and D. T. Gryko, Acc. Chem. Res., 2017, 50, 2334–2345 CrossRef CAS PubMed.
- S. Stecko and D. T. Gryko, JACS Au, 2022, 2, 1290–1305 CrossRef CAS PubMed.
- G. Sanil, B. Koszarna, Y. M. Poronik, O. Vakuliuk, B. Szymański, D. Kusy and D. T. Gryko, The Chemistry of 1,4-Dihydropyrrolo[3,2-b]pyrroles, in Advances in Heterocyclic Chemistry, ed. E. F. V. Scriven and C. A. Ramsden, Academic Press, 2022, vol. 138, pp. 335–409 Search PubMed.
- J. A. Clark, D. Kusy, O. Vakuliuk, M. Krzeszewski, K. J. Kochanowski, B. Koszarna, O. O'Mari, D. Jacquemin, D. T. Gryko and V. I. Vullev, Chem. Sci., 2023, 14, 13537–13550 RSC.
- Y. Qin, C. Schnedermann, M. Tasior, D. T. Gryko and D. G. Nocera, J. Phys. Chem. Lett., 2020, 11, 4866–4872 CrossRef CAS PubMed.
- M. B. Teimouri, I. Deperasińska, M. Rammo, M. Banasiewicz, C. W. Stark, Ł. Dobrzycki, M. K. Cyrański, A. Rebane and D. T. Gryko, J. Org. Chem., 2024, 89, 4657–4672 CrossRef CAS PubMed.
- K. Górski, I. Deperasińska, G. V. Baryshnikov, S. Ozaki, K. Kamada, H. Ågren and D. T. Gryko, Org. Lett., 2021, 23, 6770–6774 CrossRef.
- B. Dereka, A. Rosspeintner, M. Krzeszewski, D. T. Gryko and E. Vauthey, Angew. Chem., Int. Ed., 2016, 55, 15624–15628 CrossRef CAS.
- B. Dereka, A. Rosspeintner, R. Stężycki, C. Ruckebusch, D. T. Gryko and E. Vauthey, J. Phys. Chem. Lett., 2017, 8, 6029–6034 CrossRef CAS PubMed.
- B. Dereka and E. Vauthey, J. Phys. Chem. Lett., 2017, 8, 3927–3932 CrossRef CAS PubMed.
- A. I. Ivanov, B. Dereka and E. Vauthey, J. Chem. Phys., 2017, 146, 164306 CrossRef PubMed.
- D. H. Friese, A. Mikhaylov, M. Krzeszewski, Y. M. Poronik, A. Rebane, K. Ruud and D. T. Gryko, Chem.–Eur. J., 2015, 21, 18364–18374 CrossRef CAS PubMed.
- Ł. G. Łukasiewicz, H. G. Ryu, A. Mikhaylov, C. Azarias, M. Banasiewicz, B. Kozankiewicz, K. H. Ahn, D. Jacquemin, A. Rebane and D. T. Gryko, Chem.–Asian J., 2017, 12, 1736–1748 CrossRef PubMed.
- Y. M. Poronik, G. V. Baryshnikov, I. Deperasińska, E. M. Espinoza, J. A. Clark, H. Ågren, D. T. Gryko and V. I. Vullev, Commun. Chem., 2020, 3, 190 CrossRef CAS PubMed.
- A. I. Ivanov, J. Phys. Chem. C, 2018, 122, 29165–29172 CrossRef CAS.
- A. E. Nazarov and A. I. Ivanov, J. Phys. Chem. B, 2020, 124, 10787–10801 CrossRef CAS PubMed.
- Ł. G. Łukasiewicz, M. Rammo, C. Stark, M. Krzeszewski, D. Jacquemin, A. Rebane and D. T. Gryko, ChemPhotoChem, 2020, 4, 508–519 CrossRef.
- K. Górski, D. Kusy, S. Ozaki, M. Banasiewicz, R. Valiev, S. R. Sahoo, K. Kamada, G. Baryshnikov and D. T. Gryko, J. Mater. Chem. C, 2024, 12, 1980–1987 RSC.
- T. Oyama, L. Mendive-Tapia, V. Cowell, A. Kopp, M. Vendrell and L. Ackermann, Chem. Sci., 2023, 14, 5728–5733 RSC.
- F. de Moliner, I. Biazruchka, K. Konsewicz, S. Benson, S. Singh, J.-S. Lee and M. Vendrell, Front. Chem. Sci. Eng., 2022, 16, 128–135 CrossRef CAS.
- P. K. Mehta, K. Ryu, C. K. Kim and K.-H. Lee, New J. Chem., 2022, 46, 7003–7013 RSC.
- S. Benson, F. de Moliner, A. Fernandez, E. Kuru, N. L. Asiimwe, J.-S. Lee, L. Hamilton, D. Sieger, I. R. Bravo, A. M. Elliot, Y. Feng and M. Vendrell, Nat. Commun., 2021, 12, 2369 CrossRef CAS PubMed.
- Y. Wang and T. Michinobu, J. Mater. Chem. C, 2016, 4, 6200–6214 RSC.
- B. A. D. Neto, J. R. Correa and J. Spencer, Chem.–Eur. J., 2022, 28, e202103262 CrossRef CAS PubMed.
- K. Veys, F. de Jong, A. Adriaens, G. Coenen, K. Coenen, Y. De Ligt, Q. Meysman, T. Paredis, J. Stalmans, W. Dehaen, M. Van der Auweraer and D. Escudero, ChemPhotoChem, 2023, 7, e202200262 CrossRef CAS.
- T. Ishi-i, R. Kichise, I. S. Park, T. Yasuda and T. Matsumoto, J. Mater. Chem. C, 2023, 11, 3003–3009 RSC.
- M. Posada Urrutia and C. Dyrager, Chem.–Eur. J., 2024, 30, e202400644 CrossRef CAS PubMed.
- B. A. D. Neto, A. A. M. Lapis, E. N. da Silva Júnior and J. Dupont, Eur. J. Org. Chem., 2013, 2013, 228–255 CrossRef CAS.
- M. R. Anthony Raj, C. Yao, M. Frémont and W. G. Skene, Chem.–Eur. J., 2024, 30, e202401417 CrossRef CAS.
- S. Benson, A. Fernandez, N. D. Barth, F. de Moliner, M. H. Horrocks, C. S. Herrington, J. L. Abad, A. Delgado, L. Kelly, Z. Chang, Y. Feng, M. Nishiura, Y. Hori, K. Kikuchi and M. Vendrell, Angew. Chem., Int. Ed., 2019, 58, 6911–6915 CrossRef CAS.
- R. A. Balaguez, R. Krüger, B. Iepsen, R. F. Schumacher, R. S. Oliboni, T. Barcellos, H. C. Junqueira, M. S. Baptista, B. A. Iglesias and D. Alves, Eur. J. Org Chem., 2018, 2018, 6507–6514 CrossRef CAS.
- S. Uchiyama, K. Kimura, C. Gota, K. Okabe, K. Kawamoto, N. Inada, T. Yoshihara and S. Tobita, Chem.–Eur. J., 2012, 18, 9552–9563 CrossRef CAS PubMed.
- M. Tasior, O. Vakuliuk, D. Koga, B. Koszarna, K. Górski, M. Grzybowski, Ł. Kielesiński, M. Krzeszewski and D. T. Gryko, J. Org. Chem., 2020, 85, 13529–13543 CrossRef CAS PubMed.
- B. Szymański, S. R. Sahoo, R. Valiev, O. Vakuliuk, P. Łaski, K. N. Jarzembska, R. Kamiński, G. Baryshnikov, M. B. Teimouri and D. T. Gryko, New J. Chem., 2024, 48, 2416–2420 RSC.
- H. Görner, M. Megyesi, Z. Miskolczy and L. Biczók, J. Photochem. Photobiol., A, 2010, 214, 188–193 CrossRef.
- K. Rybicka-Jasińska, E. M. Espinoza, J. A. Clark, J. B. Derr, G. Carlos, M. Morales, M. K. Billones, O. O'Mari, H. Ågren, G. V Baryshnikov and V. I. Vullev, J. Phys. Chem. Lett., 2021, 12, 10295–10303 CrossRef PubMed.
- K. Skonieczny, I. Papadopoulos, D. Thiel, K. Gutkowski, P. Haines, P. M. McCosker, A. D. Laurent, P. A. Keller, T. Clark, D. Jacquemin, D. M. Guldi and D. T. Gryko, Angew. Chem., Int. Ed., 2020, 59, 16104–16113 CrossRef CAS PubMed.
- J. S. Siddle, R. M. Ward, J. C. Collings, S. R. Rutter, L. Porrès, L. Applegarth, A. Beeby, A. S. Batsanov, A. L. Thompson, J. A. K. Howard, A. Boucekkine, K. Costuas, J.-F. Halet and T. B. Marder, New J. Chem., 2007, 31, 841–851 RSC.
- M.-C. Chen, Y.-L. Lee, Z.-X. Huang, D.-G. Chen and P.-T. Chou, Chem.–Eur. J., 2020, 26, 7124–7130 CrossRef CAS PubMed.
- W. Rodríguez-Córdoba, L. Gutiérrez-Arzaluz, F. Cortés-Guzmán and J. Peon, Chem. Commun., 2021, 57, 12218–12235 RSC.
- B. Sadowski, M. Kaliszewska, Y. M. Poronik, M. Czichy, P. Janasik, M. Banasiewicz, D. Mierzwa, W. Gadomski, T. D. Lohrey, J. A. Clark, M. Łapkowski, B. Kozankiewicz, V. I. Vullev, A. L. Sobolewski, P. Piatkowski and D. T. Gryko, Chem. Sci., 2021, 12, 14039–14049 RSC.
- Y. M. Poronik, B. Sadowski, K. Szychta, F. H. Quina, V. I. Vullev and D. T. Gryko, J. Mater. Chem. C, 2022, 10, 2870–2904 RSC.
- S. Saha and A. Samanta, J. Phys. Chem. A, 1998, 102, 7903–7912 CrossRef CAS.
- M.-C. Chen, D.-G. Chen and P.-T. Chou, Chempluschem, 2021, 86, 11–27 CrossRef CAS PubMed.
- G. Micheletti, L. Iannuzzo, M. Calvaresi, S. Bordoni, D. Telese, E. Chugunova and C. Boga, RSC Adv., 2020, 10, 34670–34680 RSC.
- G. Rauhut, A. A. Jarzecki and P. Pulay, J. Comput. Chem., 1997, 18, 489–500 CrossRef CAS.
- F. Eckert, G. Rauhut, A. R. Katritzky and P. J. Steel, J. Am. Chem. Soc., 1999, 121, 6700–6711 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the synthesis, spectroscopic characterisation, crystallographic and computational data. CCDC 2288872 and 2359982–2359987. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07275h |
| This journal is © The Royal Society of Chemistry 2025 |