
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclo[4]pyrrole with α–β direct linkages†
Yuhua
Sun
a,
Riku
Kitahara
b,
Tomoya
Ichino
 c,
Yuki
Ide
c,
Hisanori
Senboku
a,
Soji
Shimizu
b,
Takayuki
Tanaka
*d and
Yasuhide
Inokuma
*ac
c,
Yuki
Ide
c,
Hisanori
Senboku
a,
Soji
Shimizu
b,
Takayuki
Tanaka
*d and
Yasuhide
Inokuma
*ac
aDivision of Applied Chemistry, Faculty of Engineering, Hokkaido University, Kita 13, Nishi 8, Kita-ku, Sapporo, Hokkaido 060-8628, Japan. E-mail: inokuma@eng.hokudai.ac.jp
bDepartment of Applied Chemistry, Graduate School of Engineering and Centre for Molecular Systems (CMS), Kyushu University, Fukuoka 819-0395, Japan
cInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Kita 21, Nishi 10, Kita-ku, Sapporo, Hokkaido 001-0021, Japan
dDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: tanaka@moleng.kyoto-u.ac.jp
First published on 5th November 2024
Abstract
A cyclo[4]pyrrole bearing pyrrole C(α)–C(β) direct linkages, a contracted porphyrin analogue with no meso-carbon bridge, was synthesized from an oligoketone-related precursor. X-ray crystallography and StrainViz analysis revealed a non-planar structure with a total strain of 20.8 kcal mol−1. The cyclo[4]pyrrole emits fluorescence in the visible region with a quantum yield of 0.026. The NICS calculations indicated a local 6π-aromatic character for each pyrrole unit, and the global π-electronic communication among the four pyrrole units was shown by the frontier orbitals in the neutral form. The cyclo[4]pyrrole underwent reversible stepwise electrochemical two-electron oxidation and the spin density of the radical cation intermediate localized on the 3,2′:5′,3′′-terpyrrole moiety. However, spectroelectrochemical measurements and theoretical calculations indicated the contribution of a triplet diradical dication form with the spin density delocalized across the four pyrrole units and smaller dihedral angles between neighboring aromatic rings. The structural and electrochemical behavior of cyclo[4]pyrrole demonstrated the effects of ring contraction and recombination of the pyrrole–pyrrole linkages on the cyclo[n]pyrrole system.
Introduction
Cyclo[n]pyrroles (1) are porphyrin-related macrocycles composed of n pyrrole units connected by direct C(pyrrole)–C(pyrrole) linkages. Since the synthesis of cyclo[8]pyrrole ([30]octaphyrin(0.0.0.0.0.0.0.0)) by Sessler and co-workers,1 a number of cyclo[n]pyrroles and their furan and thiophene analogues have been reported, with an increasing focus on their anion binding, aromaticity, redox and optical properties.2–6 The typical synthesis of cyclo[n]pyrroles uses fully β-substituted 2,2′-bipyrrole precursors for oxidative C(pyrrole-α)–C(pyrrole-α) coupling reactions, which can facilitate the formation of cyclo[6]– to cyclo[10]pyrroles.1–4 Cyclo[n]pyrroles with n ≥ 5 are classified as expanded porphyrins, and cyclo[4]pyrroles are regarded as contracted porphyrins, because they lack four meso-carbon bridges compared with the formal porphyrin skeleton.7 Recently, there has been growing interest in contracted porphyrins, including corroles,8 norcorroles,9 subporphyrins, and triphyrins(2.1.1),10 because of their distinctive ring-contraction effects on the structural, π-electronic and coordination properties.11 Nevertheless, cyclo[4]pyrroles remain unexplored, with the exception of tetraaza[8]circulene (2),12 in which the β–β-linked cyclo[4]pyrrole structure is embedded as a planar subcomponent of the circulene skeleton (Fig. 1). | ||
| Fig. 1 Cyclo[n]pyrroles and analogous macrocycles. | ||
Cyclo[4]pyrrole has several structural isomers depending on the connectivity of the four pyrrole units. The all α–α-linked isomer, i.e.1 (for n = 4), appears to be synthetically unfeasible because of the large bend angle of the pyrrole-2,5-diyl unit (∼131°).13 Whereas, β–β-linked macrocycles, such as 2, have been investigated owing to the intrinsic interest in their aromaticity derived from the central cyclooctatetraene structure. Among the cyclo[4]pyrrole isomers, the α–β-linked isomer 3 is of interest because of its [2.2](2,5)pyrrolophane structure, which is analogous to 4,14–16 and its expected nonplanar π-conjugated circuit. Pyrrole-containing cyclophane-type macrocycles remain a challenging target in the synthetic chemistry. The development of novel precursors and synthetic methodologies for these target molecules will also contribute to the generation of other novel cyclophane- and porphyrin-related macrocycles. Here, we report the synthesis, structural analysis and optical and electrochemical properties of cyclo[4]pyrrole 3, a ring-contracted porphyrin analogue. X-ray crystallographic analysis of 3 revealed a strained macrocyclic structure with remarkably large deformation angles. Upon electrochemical oxidation, the dihedral angles between neighboring pyrrole units remarkably changed, which interrupted or connected the π-electronic communications among the four pyrrole units, demonstrating the unique ring-contraction effects on the cyclo[n]pyrrole system.
Results and discussion
Recent research has demonstrated that some ring-contracted porphyrinoids are inaccessible through conventional condensation reactions of pyrrole-based precursors because of unusual ring strain or steric repulsion during macrocyclization.11 To circumvent this problem, alternative methodologies have been developed. For example, calix[3]pyrrole, a highly strained tripyrrolic macrocycle, was synthesized using a cyclic hexaketone precursor via the triple Paal–Knorr pyrrole formation reaction at the 1,4-diketone units.17 The synthesis of a boron-free subporphyrin18 and [2.2](2,5)pyrrolophane 4,14 an o-phenylene linked analogue of 3, has been achieved with Pd-catalyzed cross-coupling reactions. For the synthesis of 3, we combined polyketone-based precursor synthesis and Pd-catalyzed cross-coupling macrocyclization as illustrated in Scheme 1.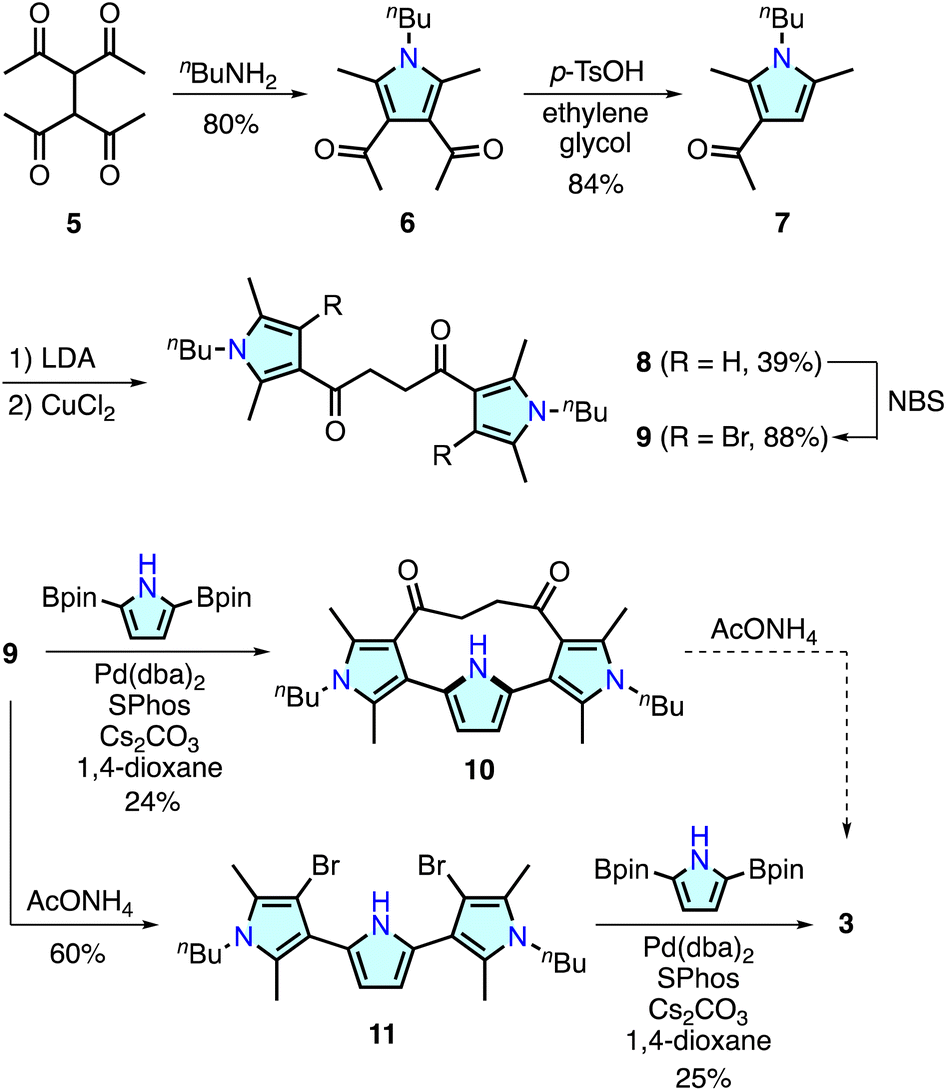 | ||
| Scheme 1 Synthesis of cyclo[4]pyrrole 3. | ||
The important components of 3, namely, 3,2′:5′,3′′-terpyrrole derivatives 10 and 11, were synthesized from 1,1,2,2-tetraacetylethane 5.19 Treatment of 5 with n-butylamine afforded 3,4-diacetylpyrrole analogue 6 in 80% yield. Retro-Friedel–Crafts acylation of 6 with p-toluenesulfonic acid (p-TsOH) and ethylene glycol resulted in the removal of one acyl group to give 7 in 84% yield. A lithium enolate species generated from 7 and lithium diisopropylamide (LDA) was homo-coupled using CuCl2 in dimethylformamide to afford a 3,2′:5′,3′′-terpyrrole synthon 8 in 39% yield. The pyrrole β-CH groups in 8 were further brominated with N-bromosuccinimide (NBS) giving 9 in 88% yield. Suzuki–Miyaura cross-coupling reaction between 9 and 2,5-diborylpyrrole20 at 285 mM concentration afforded macrocycle 10 in 24% yield. In this reaction, a larger [2 + 2] type macrocycle was formed only in <1% yield (see ESI†), whereas a similar macrocyclization reaction for the synthesis of [2.2](2,5)pyrrolophane 4 (ref. 14) required highly diluted conditions (4 mM substrate) for the selective [1 + 1] macrocyclization.
Although macrocycle 10 was considered one of the most plausible precursors for cyclo[4]pyrrole 3, Paal–Knorr reactions with ammonium acetate under various conditions were unsuccessful. Analysis of the reaction mixture indicated that intramolecular nucleophilic addition of the pyrrolic NH group occurred, as observed during the oxidation reaction of 4 (ESI†).14,21 In contrast, Paal–Knorr reaction of 9 with ammonium acetate gave dibrominated 3,2′:5′,3′′-terpyrrole 11 in 60% yield. Subsequent Pd-catalyzed cross-coupling macrocyclization between 11 and 2,5-diborylpyrrole at 285 mM concentration afforded cyclo[4]pyrrole 3 in 25% yield as a colorless solid. The 1H NMR spectrum of 3 recorded in CDCl3 showed a time-averaged C2h-symmetric signal pattern featuring pyrrolic NH and β–CH protons at 6.34 and 6.21 ppm, respectively (Fig. S45†). High-resolution electrospray ionization time-of-flight mass spectrometry revealed an adduct peak at m/z = 451.2822, which was assigned to a sodium adduct [3 + Na]+ (calculated for C28H36N4Na, m/z 451.2833).
Single crystal X-ray diffraction analysis revealed a non-planar macrocyclic structure of 3 (Fig. 2). The two N-butylated pyrrole moieties were positioned on the same plane, and the other two pyrrole units were arranged almost parallel with a mean interplanar distance of 2.4 Å. The dihedral angle between the two adjacent pyrrole units was 54.78°. The dihedral angle between the pyrrole and benzene planes in the o-phenylene-linked pyrrolophane 4 was slightly larger (57.78°), while both 3 and 4 exhibited C2h-symmetric conformations with two pyrrole-2,5-diyl units slip-stacked in an anti-parallel fashion with a mean distance of 2.4 Å. Diketone-bridged 10 also exhibited a non-planar conformation for the 3,2′:5′,3′′-terpyrrole unit. The dihedral angles between the two adjacent pyrrole units were 53.18° and 68.58°.
 | ||
| Fig. 2 X-ray crystal structures of (a) 3 and (b) 10. (Left: top view; right: side view; thermal ellipsoids are set at the 50% probability; N-butyl groups are omitted for clarity in the side views). (c) Deformation angles α and β for the colored pyrrole rings in 3, 4 and 10. α is the averaged deviation angle between the mean planes of N–C(3)–C(4) and N–C(2)–C(3), and β is the averaged deviation angle between the mean plane N–C(2)–C(3) and a C(2)–C(external) bond. | ||
The deformation angles α and β represent the deformation of the pyrrole ring from planarity and the displacement of the carbon atom externally connected to the pyrrole–α position from the bow of the pyrrole ring.22 Cyclo[4]pyrrole 3 showed the largest deformation angle α (5.95°) in the series, while its angle β (17.48°) was comparable to that of 4 (17.55°). Considering deformation angles, cyclo[4]pyrrole 3 is a more strained macrocycle than calix[3]pyrrole (α = 2.52°, β = 9.20°) and related macrocycles.23 In contrast, diketone-linked 10 exhibited notably smaller α and β of 2.09° and 10.01°, respectively.
To further evaluate the macrocyclic ring strain,24 we employed StrainViz analysis25 for compounds 3, 4 and 10 using DFT calculations at the M06-2X26/6-311+G(2d,p) level of theory. This analysis revealed that all the macrocycles exhibited the maximum strain energy around the C–C single bonds between two aromatic rings (Fig. 3). However, the total strain of 3 and 4 was approximately twice than that of 10, and the bond and angle strain energies were similar. The dihedral strain greatly contributed to the total strain of these macrocycles (Fig. S4†). Although cyclo[4]pyrrole 3 exhibited the largest deformation angle α in the crystal structure, the maximum and total strain (1.92 and 20.8 kcal mol−1, respectively) were slightly smaller than that of 4 (2.12 and 22.7 kcal mol−1, respectively). As shown in Fig. S5a,† the steric effect of the methyl group in 3 affected the strain energy and torsional motion, which resulted in less strain. Furthermore, pyrrolophane 4 has a smaller macrocyclic cavity than 3, which resulted in a larger total strain (Fig. S5b†). The large difference in total strain between 3 and 10 is a possible reason for the unsuccessful conversion of 10 to 3 in the Paal–Knorr reaction.27
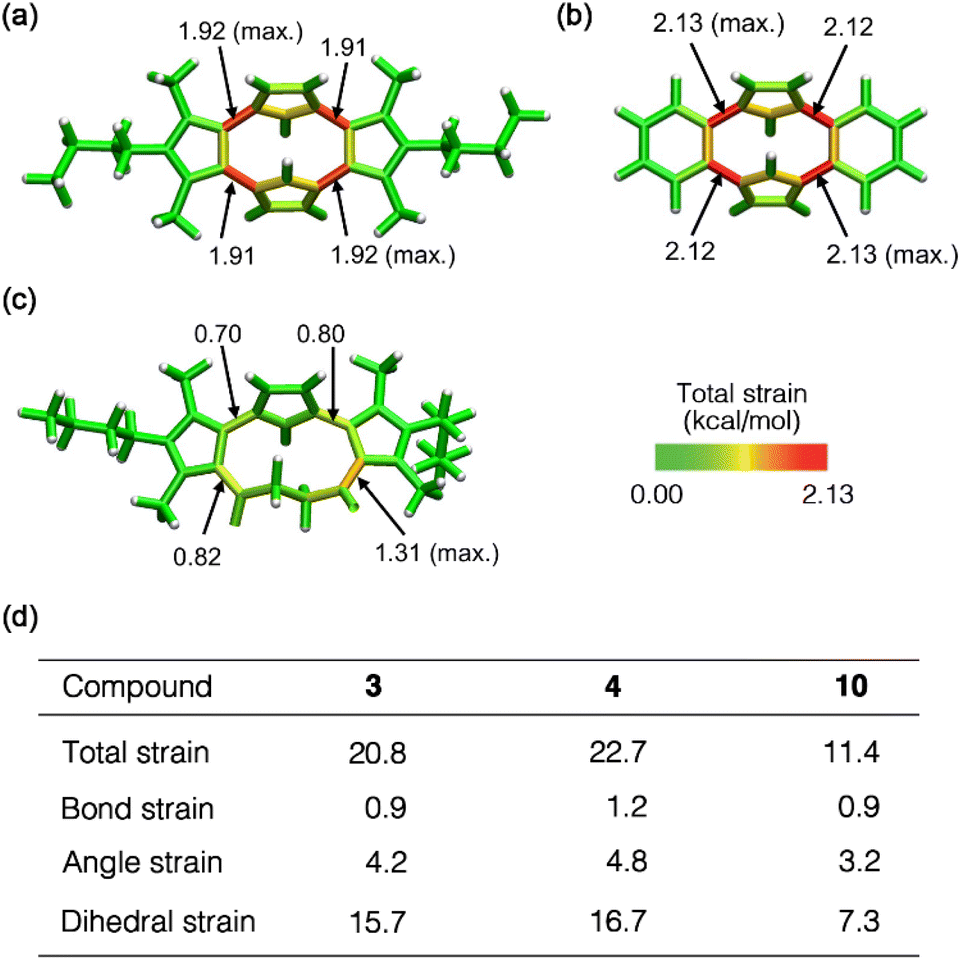 | ||
| Fig. 3 Visualization of the total strain of (a) 3, (b) 4 and (c) 10 using StrainViz analysis at the M06-2X/6-311+G(2d,p) level. (d) Calculated total, bond, angle and dihedral strain energies (in kcal mol−1) of 3, 4 and 10. | ||
The UV-vis absorption spectrum of 3, recorded in acetonitrile, showed an absorption band at 276 nm with an absorption coefficient ε of 4.25 × 104 M−1 cm−1 (Fig. 4). The absorption edge reached to 350 nm, and no absorption band was observed in the visible region. Other 3,2′:5′,3′′-terpyrrole analogues, such as 10 and 11, also exhibited absorption bands in a similar region to 3 (Fig. S9 and S10†). Upon photoexcitation at 270 nm, cyclo[4]pyrrole exhibited fluorescence emission at 434 nm with a fluorescence quantum yield of 2.6%. The fluorescence decay measurement revealed that the emission lifetime of 3 in acetonitrile was 3.05 ns. The cyclic voltammogram of 3 was recorded in dichloromethane containing 0.1 M tetrabutylammonium hexafluorophosphate, and the reversible oxidation waves were observed at −0.20 V and 0.28 V against a ferrocene/ferrocenium (Fc/Fc+) ion couple. Terpyrrole analogues 10 and 11 also showed reversible oxidation waves (Fig. S12 and S13†). While the oxidation potentials of 11 (−0.06 V and 0.43 V) were comparable to those of 3, the oxidation potential of 10 (0.36 V) was rather high because of the electron-withdrawing carbonyl groups.
 | ||
| Fig. 4 (a) UV-vis absorption (black) and fluorescence (red) spectra of 3 in acetonitrile. (b) Cyclic voltammogram of 3 in CH2Cl2. | ||
The molecular orbital (MO) calculation at the B3LYP/6-311G(d,p) level revealed that 3 exhibited relatively high HOMO and HOMO−1 energy levels as compared with 4, consistent with the lower electrochemical oxidation potentials, and the orbital coefficients were well-spread over the four pyrrole units (Fig. 5). Cyclic terpyrrole 10 had a HOMO with large orbital coefficients on the three pyrrole units, but its energy level was lower than that of 3 because of the electron-withdrawing carbonyl groups. Given the reversible two-electron oxidation event of 3 observed by cyclic voltammetry, we further investigated the electrochemical oxidation of 3 using spectroelectrochemical and theoretical analyses.
 | ||
| Fig. 5 Frontier orbitals of 4 (left), 3 (middle) and 10 (right) calculated at the B3LYP/6-311G(d,p) level. | ||
The UV-vis absorption spectra recorded in dichloromethane during electrochemical oxidation at 0.6 V (vs. Ag/Ag+) showed two-step spectral changes (Fig. 6a). At lower oxidation potentials (0.2–0.5 V), new intense bands at 350–500 nm and broad bands covering the 700–1000 nm region appeared, concomitant with a decrease in the absorption at 278 nm, assignable to neutral 3. During the first spectral changes, an isosbestic point was observed at 322 nm. The application of a higher voltage (0.5–0.6 V) led to the appearance of other new absorption bands at 300 nm and 500–600 nm, resulting in a broad absorption spectrum covering from the ultraviolet to the near-infrared region.
 | ||
| Fig. 6 (a) UV-vis absorption spectra during the electrochemical oxidation of 3 in CH2Cl2. (b) Spin density distribution plots for 3˙+ (left) and the triplet state of 32+ (right) with an isovalue of 0.001. | ||
To analyze the observed stepwise oxidation events of 3, we expanded the scope of calculations for oxidized species. The optimized structure of 3˙+ showed increased dihedral angles around one pyrrole-2,5-diyl unit (>65°), while the other three pyrrole units underwent coplanarization with dihedral angles of ca. 38°. These structural features suggest that the π-electronic conjugation in 3˙+ is roughly divided across the 3,2′:5′,3′′-terpyrrole and single pyrrole units. Indeed, the calculated spin density is mainly spread over the terpyrrole segment (Fig. 6b). For the dication 32+, when the structural optimization was conducted at the RB3LYP/6-311G(d,p) level, the structure changed to the terpyrrole-type structure similar to 3˙+ with the dihedral angles between the two adjacent pyrrole units of 32° and 67°. The bond length alternation indicates quinoid-type π-electronic conjugation (Fig. S18†). However, optimization at the UB3LYP/6-311G(d,p) level revealed that the singlet or triplet diradical dication states have essentially similar structures to neutral 3.
Unexpectedly, the total energy comparison indicates that the triplet diradical dication state is 0.50 kcal mol−1 more stable than the closed-shell dication state. The open-shell singlet diradical dication state is much higher in energy (+1.64 kcal mol−1 relative to the triplet state). The order of the energy levels is independent of the calculation levels (Table S1†) and a similar tendency was observed when the calculations were performed using solvation (CH2Cl2) models (Table S2†). The time-dependent density functional theory (TDDFT) calculation well-reproduced the absorption profiles of 3 and 3˙+, while those of 32+ could not be assigned to the experimental spectra (Fig. S19–22†). It is conceivable that the conformational changes between the quinoidal form in the closed-shell state and the cyclophane form in the open-shell triplet state are fast under ambient conditions, meaning the obtained absorption spectra derive from both states. The balance between the two states is susceptible to the spin delocalization in the triplet state (Fig. 6b) as well as the local aromaticity in the pyrrole segments. The aromatic nature of the pyrrole segments is compared using the ACID28,29 and NICS(0)30,31 calculations (Fig. S23 and S24†). Accordingly, the terpyrrole-like segment in the dication state is only weakly aromatic and the pyrrol-2,5-diyl segment remains aromatic. In the triplet state, the aromatic character of the pyrrole segments is slightly weakened. Although NMR and ESR detection of chemically generated dication species 32+ using oxidants such as magic blue was unsuccessful (Fig. S25–S27†), the electrochemical absorption spectra in Fig. 6a are best interpreted as a contribution from both the triplet diradical and closed-shell forms of 32+.
Conclusions
In summary, we synthesized α–β directly linked cyclo[4]pyrrole 3, a ring-contracted congener of the cyclo[n]pyrrole family, using polyketone-based precursors and a Pd-catalyzed cross-coupling reaction. As a result of the ring-contraction effect, 3 exhibited a non-planar and highly strained macrocyclic structure, unlike larger cyclo[n]pyrroles 1. The conformational restriction in 3 led to unique optical spectral changes during electrochemical oxidation. In the radical cation form 3˙+, the dihedral angles around the pyrrol-2,5-diyl segment increased, which interrupted the global π-conjugation, resulting in the formation of a 3,2′:5′,3′′-terpyrrole-localized radical cation. In contrast, dication 32+ largely consisted of the triplet diradical dication form with the spin density delocalized onto the four pyrrole units bearing lower dihedral angles. The current results demonstrate the impact of both the ring contraction and introduction of the α–β linkage on the structural and π-electronic properties of cyclo[n]pyrroles. Further investigation into novel isomers of cyclo[n]pyrroles (n ≥ 4) would greatly contribute to the porphyrin-related chemistry.Data availability
All the data related to this manuscript are available in the ESI.†Author contributions
Y. Inokuma designed the study and supervised the project together with T. T. Synthesis and characterization of new compounds were mainly performed by Y. S. and Y. Ide. Electrochemical measurements and discussions were performed by Y. S., R. K., H. S. and S. S. Strain calculations were performed by T. I. and other computational studies were performed by T. T.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the JSPS Grant-in-Aid for Scientific Research (B) (No. 22H02058 and 23K17942), the Asahi Glass Foundation and the JST FOREST Program (No. JPMJFR211H), of which Y. Inokuma is the principal investigator. The Institute for Chemical Reaction Design and Discovery (ICReDD) was established by World Premier International Research Initiative (WPI), MEXT, Japan.Notes and references
- D. Seidel, V. Lynch and J. L. Sessler, Cyclo[8]pyrrole: A Simple-to-Make Expanded Porphyrin with No Meso Bridges, Angew. Chem., Int. Ed., 2002, 41, 1422–1425 CrossRef PubMed.
- T. Köhler, D. Seidel, V. Lynch, F. O. Arp, Z. Ou, K. M. Kadish and J. L. Sessler, Formation and Properties of Cyclo[6]pyrrole and Cyclo[7]pyrrole, J. Am. Chem. Soc., 2003, 125, 6872–6873 CrossRef PubMed.
- T. Okujima, C. Ando, S. Agrawal, H. Matsumoto, S. Mori, K. Ohara, I. Hisaki, T. Nakae, M. Takase, H. Uno and N. Kobayashi, Template Synthesis of Decaphyrin without Meso-Bridges: Cyclo[10]pyrrole, J. Am. Chem. Soc., 2016, 138, 7540–7543 CrossRef PubMed.
- H. Matsumoto, T. Okujima, S. Mori, A. C. C. Bacilla, M. Takase, H. Uno and N. Kobayashi, Cyclo[9]pyrrole: Selective Synthesis of [34]Nonaphyrin(0.0.0.0.0.0.0.0.0), Org. Lett., 2021, 23, 3442–3446 CrossRef PubMed.
- S. V. Mulay, O. Dishi, Y. Fang, M. R. Niazi, L. J. W. Shimon, D. F. Perepichka and O. Gidron, A macrocyclic oligofuran: synthesis, solid state structure and electronic properties, Chem. Sci., 2019, 10, 8527–8532 RSC.
- J. Krömer, I. Rios-Carreras, G. Fuhrmann, C. Musch, M. Wunderlin, T. Debaerdemaeker, E. Mena-Osteritz and P. Bäuerle, Synthesis of the First Fully α-Conjugated Macrocyclic Oligothiophenes: Cyclo[n]thiophenes with Tunable Cavities in the Nanometer Regime, Angew. Chem., Int. Ed., 2000, 39, 3481–3486 CrossRef.
- J. L. Sessler and S. J. Weghorn, Expanded, Contracted, and Isomeric Porphyrins, Pergamon Press, New York, 1997 Search PubMed.
- R. Orłowski, D. Gryko and D. T. Gryko, Synthesis of Corroles and Their Heteroanalogs, Chem. Rev., 2017, 117, 3102–3137 CrossRef PubMed.
- T. Ito, Y. Hayashi, S. Shimizu, J.-Y. Shin, N. Kobayashi and H. Shinokubo, Gram-Scale Synthesis of Nickel(II) Norcorrole: The Smallest Antiaromatic Porphyrinoid, Angew. Chem., Int. Ed., 2012, 51, 8542–8545 CrossRef CAS.
- S. Shimizu, Recent Advances in Subporphyrins and Triphyrin Analogues: Contracted Porphyrins Comprising Three Pyrrole Rings, Chem. Rev., 2017, 117, 2730–2784 CrossRef CAS.
- K. Watanabe, N. N. Pati and Y. Inokuma, Contracted porphyrins and calixpyrroles: synthetic challenges and ring-contraction effects, Chem. Sci., 2024, 15, 6994–7009 RSC.
- F. Chen, Y. S. Hong, S. Shimizu, D. Kim, T. Tanaka and A. Osuka, Synthesis of a Tetrabenzotetraaza[8]circulene by a “Fold-in” Oxidative Fusion Reaction, Angew. Chem., Int. Ed., 2015, 54, 10639–10642 CrossRef CAS.
- F. Wang, Y. Zhang, W. Zhou, Z. Long, S. Xiong, Q. Zhang, J. L. Sessler and Q. He, “One-pot” synthesis of oligopyrrolic dialdehydes, New J. Chem., 2024, 48, 8153–8157 RSC.
- Y. Morimoto, Y. H. Koo, K. Otsubo, H. Kitakado, S. Seki, A. Osuka and T. Tanaka, Dibenzodiazapyracylenes: Doubly N-Doped Cyclopenta-Fused Polycyclic Molecules That Exhibit High Carrier Mobility, Angew. Chem., Int. Ed., 2022, 61, e202200341 CrossRef CAS.
- J. F. Haley Jr and P. M. Keehn, Cyclophanes IV: The synthesis of [2.2] (2,5)pyrrolophane, Tetrahedron Lett., 1975, 16, 1675–1678 CrossRef.
- J. F. Haley Jr, S. M. Rosenfeld and P. M. Keehn, Cyclophanes. 10. Synthesis and conformational behavior of [2.2](2,5)pyrrolophanes, J. Org. Chem., 1977, 42, 1379–1386 CrossRef.
- Y. Inaba, Y. Nomata, Y. Ide, J. Pirillo, Y. Hijikata, T. Yoneda, A. Osuka, J. L. Sessler and Y. Inokuma, Calix[3]pyrrole: A Missing Link in Porphyrin-Related Chemistry, J. Am. Chem. Soc., 2021, 143, 12355–12360 CrossRef CAS.
- L. Liu, J. Kim, L. Xu, Y. Rao, M. Zhou, B. Yin, J. Oh, D. Kim, A. Osuka and J. Song, Synthesis of Subporphyrin Free Bases, Angew. Chem., Int. Ed., 2022, 61, e202214342 CrossRef CAS PubMed.
- R. G. Charles, Tetraacetylethane, Org. Synth., 1959, 39, 61 CrossRef CAS.
- M. E. Hoque, R. Bisht, C. Haldar and B. Chattopadhyay, Noncovalent Interactions in Ir-Catalyzed C–H Activation: L-Shaped Ligand for Para-Selective Borylation of Aromatic Esters, J. Am. Chem. Soc., 2017, 139, 7745–7748 CrossRef CAS PubMed.
- Y. Morimoto, A. Osuka and T. Tanaka, Synthesis, properties and reactivity of an ortho-phenylene-cyclopentene-bridged tetrapyrrole, J. Porphyrins Phthalocyanines, 2021, 25, 937–943 CrossRef CAS.
- Y. Tobe, K. Ueda, K. Kakiuchi, Y. Odaira, Y. Kai and N. Kasai, Synthesis, structure and reactivities of [6]paracyclophanes, Tetrahedron, 1986, 42, 1851–1858 CrossRef CAS.
- Y. Inaba, Y. Kakibayashi, Y. Ide, J. Pirillo, Y. Hijikata, T. Yoneda and Y. Inokuma, Strain-Induced Ring Expansion Reactions of Calix[3]pyrrole-Related Macrocycles, Chem. Eur. J., 2022, 28, e202200056 CrossRef CAS PubMed.
- K. Watanabe, K. Shibata, T. Ichino, Y. Ide, T. Yoneda, S. Maeda and Y. Inokuma, Strain-based design, direct macrocyclization, and metal complexation of thiazole-containing calix[3]pyrrole Analogues, Inorg. Chem. Front., 2024, 11, 3548–3554 RSC.
- C. E. Colwell, T. W. Price, T. Stauch and R. Jasti, Strain visualization for strained macrocycles, Chem. Sci., 2020, 11, 3923–3930 RSC.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- T. Sano, Y. Sun, T. Mukai, Y. Inaba, T. Yoneda, Y. Ide, J. Pirillo, Y. Hijikata and Y. Inokuma, Toward calix[2]-type macrocycles: Synthesis and structural analysis of cyclic tetraketone and highly strained furanophane, J. Porphyrins Phthalocyanines, 2023, 27, 1067–1073 CrossRef CAS.
- R. Herges and D. Geuenich, Delocalization of Electrons in Molecules, J. Phys. Chem. A, 2001, 105, 3214–3220 CrossRef CAS.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Anisotropy of the Induced Current Density (ACID), a General Method To Quantify and Visualize Electronic Delocalization, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion, Chem. Rev., 2005, 105, 3842–3888 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, theoretical calculations and copies of NMR spectra. CCDC 2382889 and 2382890. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06670g |
| This journal is © The Royal Society of Chemistry 2024 |