
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the molecular design principles for efficient diarylethene photoacid and photohydride generators based on the photochemical reaction mechanism†
Yifan
Su
ab,
Dexin
Zheng
ab,
Lingfeng
Ge
ab,
Le
Yu
*c,
David
Lee Phillips
d,
Jiani
Ma
 *ab and
Yu
Fang
ab
*ab and
Yu
Fang
ab
aKey Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710119, China. E-mail: majiani@snnu.edu.cn
bInstitute of New Concept Sensors and Molecular Materials, Shaanxi Normal University, Xi’an 710119, China
cKey Laboratory of Synthetic and Natural Functional Molecule Chemistry of Ministry of Education, College of Chemistry and Materials Science, Northwest University, Xi'an 710127, China. E-mail: yule@nwu.edu.cn
dDepartment of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong SAR, P. R. China
First published on 14th November 2024
Abstract
Photoacid generators (PAGs) and photohydride generators (PHGs) are specific photolabile protecting groups that release acid and hydride, respectively. Over the past decade, great efforts have been devoted to developing novel PAGs and PHGs with advanced efficiency, among which, two of the promising candidates are diarylethene (DAE)-based PAGs and PHGs, which release acids/hydrides during photochromic electrocyclization. The release quantum yield for PAGs is acceptable, while that of PHGs is only 4.2% even after molecular structure modification. In this work, time-resolved transient absorption spectroscopies with femtosecond and nanosecond resolutions along with DFT/TD-DFT calculations were utilized to unravel the detailed photochemical reaction mechanisms of DAE-based PAGs (1o) and PHGs (2o), respectively. The results show that the different photochemical mechanisms are the key that leads to distinctive release quantum yields between 1o and 2o. The factors affecting the release quantum yield are discussed in detail, and several key design principles are proposed to facilitate future rational design of DAE-based PAGs and PHGs.
Introduction
Photolabile protecting groups (PPGs) are currently widely used for photocontrolled chemical and biological processes1–7 for their excellent spatial and kinetic control over the release of caged molecules.8–10 Different leaving groups can be connected to PPGs with specific functions, such as photoCORMs (photoactivatable CO-releasing moieties), photoNORMs (photoactivatable nitric oxide-releasing moieties), and photoactivatable H2S-releasing molecules, as well as photoacid generators (PAGs)11–14 and photohydride generators (PHGs).15PAGs generate acids upon light irradiation and have potential applications in polymer science and biomedicine.16,17 Zhang designed and synthesized BODIPY-based PAGs with visible light activation.18 Fagnoni reported visible-light-driven arylazo sulfone-based PAGs, whose acid release could be tuned by changing the media: a weak acid (sulfinic acid) in nitrogen-purged solutions and a strong acid (sulfonic acid) in oxygen-purged solutions.19 Liao's group investigated merocyanine-based PAGs, which underwent visible-light induced trans–cis isomerization, followed by proton dissociation, and finally underwent a nucleophilic ring closing reaction, which achieves nearly complete proton dissociation and can alter the pH value of the solution.20 Komogortsev et al. reported tosylated derivatives of terarylenes which could serve as UV-induced PAGs with the photoreaction yield exceeding 90%. However, their photogeneration rates were low and a complete transformation required more than 16 hours.21 PHGs refer to molecules that release a hydrogen anion (hydride) upon irradiation,22 and they can be used for photochemical reduction of CO2.23 Glusac's group studied the hydride release mechanism of 10-methyl-9-phenyl-9,10-dihydroacridine and proposed a stepwise electron/hydrogen-atom transfer mechanism.24
DAE derivatives have been used as light-sensitive molecules, and their light-triggered 6π-electrocyclization processes have been studied extensively.25–37 DAE transforms from its initial open-form (o) to a closed-form (c) after photocyclization. Shirinian found that the photochemical transformations of DAEs containing a five-membered heterocyclic ring and phenyl moiety went through tandem photocyclization/[1,9]-sigmatropic rearrangement/heterocyclic ring opening processes.38 They also found that ortho-bromine-substituted DAEs upon UV irradiation lead to bicyclic fused aromatics in tetrahydrofuran (THF), and could release a bromide cation (Lewis acid), which could catalyze electrophilic reactions.39 The Nakashima group and the Kawai group developed a series of PAGs based on DAEs. They first designed self-contained proton releasing PAGs that could quantitatively offer Brønsted acids such as MeOH and MeSO3H40 and later developed one that released CF3SO3H, a super acid.41 They later developed a similar molecule (denoted as 1o, see Scheme 1a) which released a triflate anion with quantum yields up to 50% upon UV irradiation and also generated a carbocation that can act as a Lewis acid.14 When the naphthalene connected to the leaving base group in the DAE-based PAG is replaced by benzothiazole, the molecule may release a hydride ion by forming a new C–N bond. These two research groups designed a benzothiazole-containing terarylene framework as a PHG (denoted as 2o, see Scheme 1b).42 Unfortunately, the release quantum yield is only 4.2%.22 Great efforts have been devoted to improving the efficiency by modifying the three heterocycles, but no significant progress has been made until now.
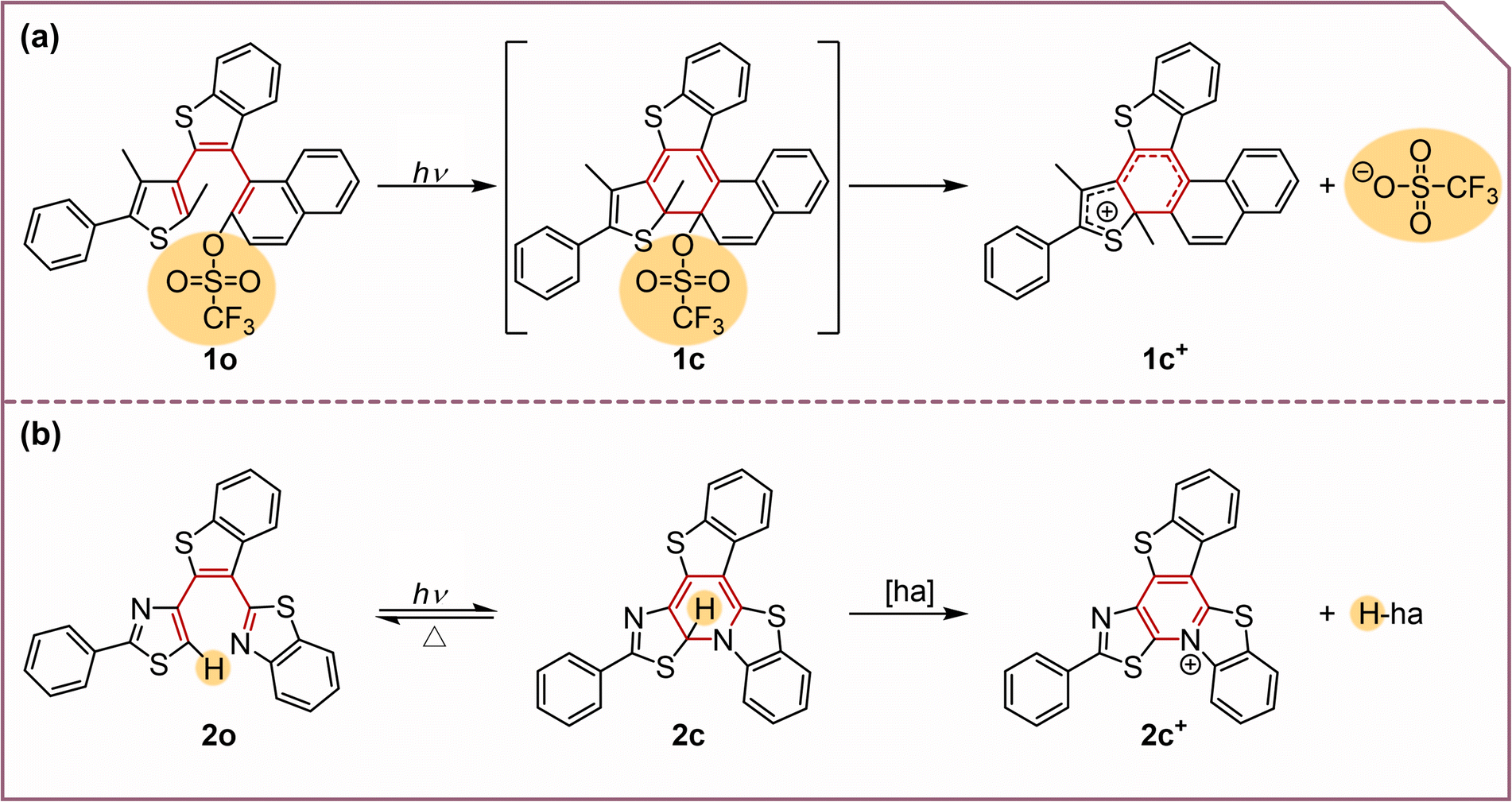 | ||
| Scheme 1 Photochemical reactions of (a) 1o and (b) 2o. “ha” refers to the “hydride acceptor”. | ||
To facilitate the rational design of DAE-based PAGs and PHGs, 1o and 2o were selected as the model compounds of PAGs and PHGs, respectively, for our reaction mechanism study using femtosecond and nanosecond transient absorption (fs-TA and ns-TA) spectroscopy. Excited-state TD-DFT calculations are performed to help analyze the experimental spectra and draw potential energy curves (PECs). With the detailed and explicit reaction mechanisms in hand, key designing principles of efficient PHGs are concluded.
Results and discussion
Both 1o and 2o share the same DAE skeleton as well as a benzothiazole unit, but also have some specific chemical structures that enable them to engage in different photochemical activities, leading to the remarkable different releasing quantum yields.Photochemical reaction mechanism of PAGs (1o)
The steady-state UV-vis spectrum of 1o was recorded (Fig. S5†) after irradiation for a certain time to monitor the overall photochemical reaction. The result is consistent with that reported by Kawai14 where the formed species is assigned to 1c+. The UV-vis spectrum for 1c+ was simulated by employing the M062X functional and 6-311G** basis set, and the good correlation between the experimental spectrum and the calculated one (Fig. S5c†) suggested that the calculation method is appropriate for the molecular system and will be accepted in this work.For the fs-TA data of 1o in acetonitrile (ACN), the singlet excited state of 1o was observed at an early stage (Fig. 1a). The structures of the singlet excited state of both the parallel (P) conformer (denoted as 1o-P(S1)) and the antiparallel (AP) conformer (denoted as 1o-AP(S1)) are shown in Fig. S6.† TD-DFT calculations suggested that both 1o-P(S1) and 1o-AP(S1) exist and contribute to the experimental TA spectrum (Fig. 1e). Later, 1o-AP(S1) decays and 1o-P(S1) blue-shifted from 406 nm to 400 nm, coupled with isosbestic points at 362 and 415 nm (Fig. 1b). Based on the photolysis experiment by observing 1c+, the signals at 340 and 400 nm were assigned to 1c and supported by TD-DFT calculations (Fig. 1f). Subsequently, 1c decreased accompanying with the generation of new signals at 375 and 510 nm and a broad band absorbing above 600 nm (Fig. 1c).
 | ||
| Fig. 1 (a–d) fs-TA spectra of 1o in ACN (λex = 266 nm). (e) The comparison of the computed electronic absorption spectrum of (magenta line) 1o-AP(S1) and (black line) 1o-P(S1) with (green line) the fs-TA spectrum recorded at 222 fs. (f) The comparison of (magenta line) the computed electronic absorption spectrum of 1c with (brown line) the fs-TA spectrum recorded at 3.5 ps. [TD-M062X/6-311G**/SMD (ACN) with a scale factor of 1.02 and a half-width of 1500 cm−1]. | ||
ns-TA spectra were collected for 1o under analogous experimental conditions, and the same signals were obtained as seen in the late fs-TA stage (Fig. 2a). These signals were quenched under oxygen quenching conditions, where the decay time constant at 510 nm was 631 ns under open air and 290 ns under Ar-saturated conditions, respectively (Fig. 2b). Simulated UV-vis spectra of the triplet states of 1o-P and 1o-AP (denoted as 1o-P(T1) and 1o-AP(T1), respectively) exhibited reasonable similarity with the ns-TA spectrum (Fig. 2c). Thereafter, the species probed from hundreds of ps to 1.1 μs can be attributed to 1o-P(T1) and 1o-AP(T1).
 | ||
| Fig. 2 (a) ns-TA spectra of 1o in ACN (λex = 266 nm). (b) The kinetics and the fitting plots are shown at 510 nm under open air and Ar-saturated conditions in ACN. (c) Comparison of the computed electronic absorption spectrum of (magenta line) 1o-AP(T1) and (black line) 1o-P(T1) in ACN with (red line) the ns-TA spectrum recorded at 100 ns. [TD-M062X/6-311G**/SMD (ACN) with a scale factor of 1.02 and a half-width of 1500 cm−1]. | ||
Combining the experimental and DFT/TD-DFT results, it is concluded that 1o-P and 1o-AP undergo different pathways. The structures of 1o-P(S0) and 1o-AP(S0) are shown in the ESI as Fig. S6.† As shown in the schematic energy diagram (Fig. 3a), after 1o-AP(S0) is excited to the Franck–Condon (FC) region of the S1 state (denoted as 1o-AP-FC(S1)), it converts to the semi-ring-closed S1 intermediate, denoted as 1o-AP-CI(S1), following an antisymmetrical twisting pathway. At this point, the energies of S0 (62.0 kcal mol−1) and S1 (63.3 kcal mol−1) are quasi-degenerated, indicating the presence of a nearby conical intersection (CI). According to the barrierless PEC connecting 1o-AP-FC(S1) and 1o-AP-CI(S1) obtained by a relaxed scan towards the C12–C31 bond distance within 1o, the relaxation process on the S1 state could be accomplished at an ultrafast timescale (Fig. S7†). After internal conversion (IC) to the S0 state, 1o-AP-CI(S0) undergoes a synergistic ring-closing and triflate anion departure process, generating 1c+ and releasing 58.8 kcal mol−1 energy. At 1o-AP-FC(S1), the energy difference between S1 and T4 states is only 3.5 kcal mol−1 with spin orbit coupling matrix elements (SOCME) of 4.5 cm−1, indicating the presence of the feasible intersystem crossing (ISC) pathway. However, the ISC process is a minor pathway as it cannot compete with the IC pathway that follows a steep descending PEC. On the other hand, 1o-P(S0) is excited to 1o-P-FC(S1) upon irradiation and quickly evolves to the nearby S1 state minimum (denoted as 1o-P(S1)), which subsequently switches to the triplet state via ISC. Among the possible ISC pathways, S1 → T3 might be the most efficient one for the small energy gap (3.2 kcal mol−1) and large SOCME (4.0 cm−1). Additionally, due to the repulsion between the thiophene and naphthalene moieties, 1o-P could not convert to the ring-closed form and is therefore a non-reactive conformer. Additionally, the Gibbs free energy of 1o-AP(S0) is 2.3 kcal mol−1 lower than that of 1o-P(S0) (Table S1†), and thus the 1o-AP(S0) would have 95% population according to Boltzmann distribution at room temperature. The conversion barrier of 1o-AP(S0) to 1o-P(S0) is 3.0 kcal mol−1 (Fig. S8†). The stable presence of 1o-AP is one of the reasons for the high triflate anion release yield.
 | ||
| Fig. 3 The calculated singlet and triplet state energy level diagram for (a) 1o-AP(S0) and (b) 1o-P(S1). Inset: PEC of 1o-AP: blue line for the S0 state, red line for the S1 state, and the green dotted line represents the path of the photochemical reaction (energy in kcal mol−1) (the ground state energy of 1o-AP(S0) is used as zero for obtaining relative energies). [(TD)M062X/6-311G**/SMD (ACN)]. | ||
Photochemical reaction mechanism of PHGs (2o)
The fs-TA and ns-TA results of 2o in THF are displayed in Fig. S9a–d.† The signal of 550 nm was seen upon irradiation and gradually decreased, and then two bands emerged at 385 and 412 nm, with an isosbestic point at 490 nm. Therefore, the attenuation signal at 550 nm is attributed to the FC state of the AP configuration (the structure is shown in Fig. 5a). The electronic absorption spectra of the singlet excited states of 2o-P and 2o-AP (denoted as 2o-P(S1) and 2o-AP(S1), respectively) are simulated (Fig. S9e†) and help to make the assignment of 385 nm to 2o-AP(S1) and 412 nm to 2o-P(S1). The two intermediates then decayed, and the absorbances at 420 and 500 nm appeared. The simulated UV-vis spectra of the triplet 2o-AP (denoted as 2o-AP(T1)) and 2c are in good agreement with the experimental results (Fig. S9f†). The signal at 380 nm can be recognized more obviously in the late ns-TA stage, which is attributed to the triplet 2o-P (denoted as 2o-P(T1)) (Fig. S10†).The fs-TA and ns-TA spectra were collected for 2o with the addition of methylene blue as a hydride acceptor (Fig. 4a–e). Compared to the situation without methylene blue, the remarkable difference was that a band at 393 nm was produced after 10 μs in ns-TA spectra (Fig. 4e). Methylene blue stabilizes the hydride ion and therefore facilitates the generation of the oxidation product 2c+. The comparison between the experimental spectrum at 15 μs and the calculated electronic absorption spectrum confirms the signal at 393 nm to be 2c+ (Fig. 4f). The TA data of 2o in THF with addition of NH4PF6 (Fig. S11†) unravelled that 2c+ decreased in the late stage of ns-TA. This is explained by the fact that the added NH4PF6 can react with the oxidation products to form ion pairs (Fig. S12†).
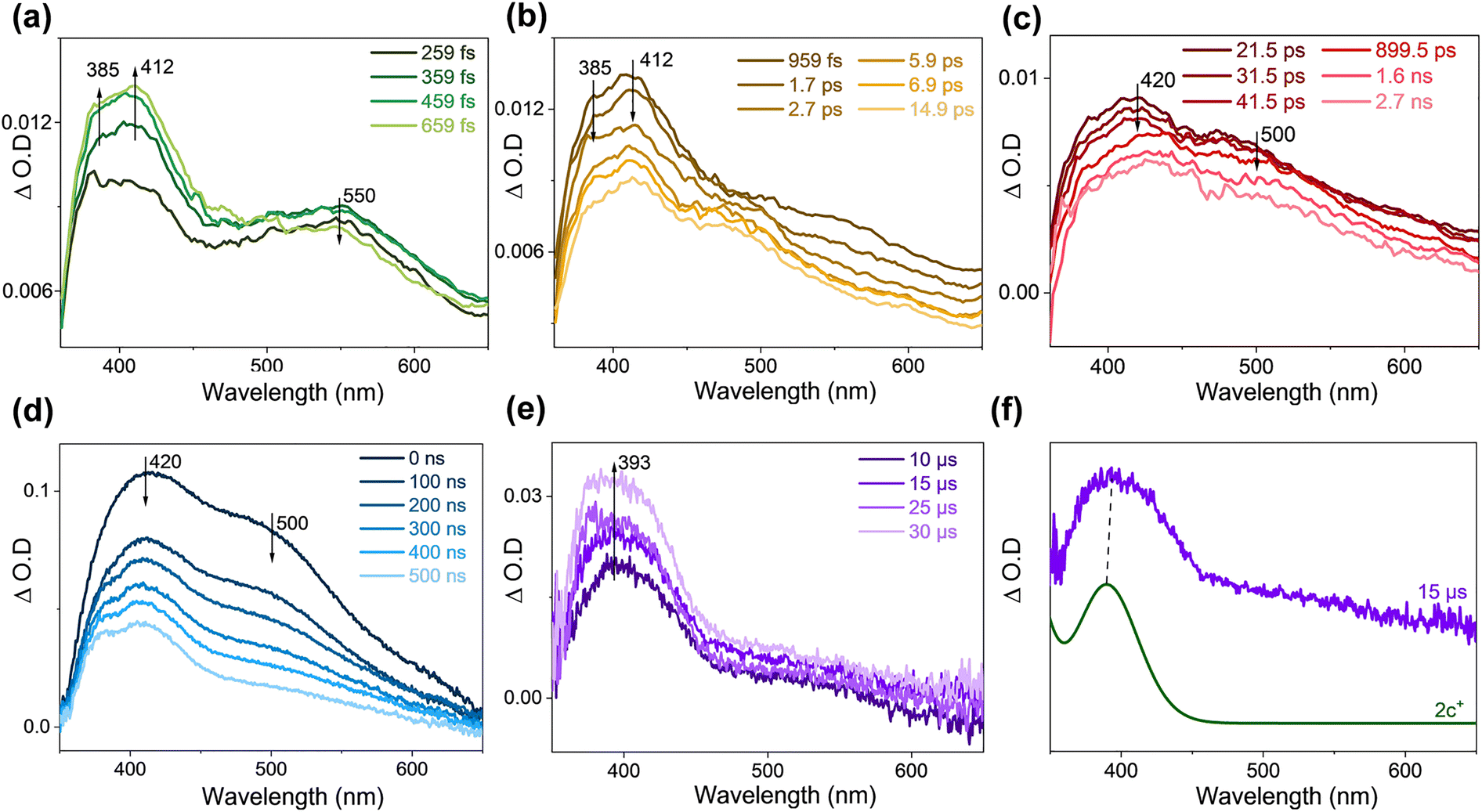 | ||
| Fig. 4 (a–c) fs-TA spectra and (d and e) ns-TA spectra of 2o in THF with addition of methylene blue (λex = 355 nm). (f) Comparison of (green line) the computed electronic absorption spectrum of 2c+ with (purple line) the ns-TA spectrum recorded at 15 μs. [TD-M062X/6-311G**/SMD (THF) with a scale factor of 1.02 and a half-width of 1500 cm−1]. | ||
The molecule 2o is composed of three groups, the phenylthiazole (PT) and benzothiazole (BTZ) groups are linked to the central benzothiophene (BTP) via a single bond, and the freestanding or synergistic twisting of PT and BTZ gives rise to various 2o-AP and 2o-P conformers. Only one enantiomer within each chiral pair is discussed (the relative energy, Gibbs free energy and thermal weight of all enantiomers are listed in Table S2†), including two AP conformers (2o-APa(S0) and 2o-APb(S0)) and three P conformers (2o-Pa(S0), 2o-Pb(S0) and 2o-Pc(S0)). Different from 1o, the AP conformers of 2o are a minority (25% population in total), while, among the majority non-reactive 2o-P conformers, 2o-Pa(S0) is the most stable one. The energy (free energy) difference between 2o-APa(S0) and 2o-APb(S0) is only 0.0(4) (0.6) kcal mol−1 (Table S2†), and they could interconvert via a barrier of 4.7 kcal mol−1 (Fig. 5). According to the ground state relaxed scan PEC along the C22–N11 distance (rC–N) in Fig. S13,†2o-APb(S0) could accomplish the cyclization process by overcoming a 30.7 kcal mol−1 barrier (2o-TS(S0)) and yield the hydrogenated intermediate 2c(S0) (Fig. 5a). The ring-closing reaction of 2o could also take place upon photoexcitation (PEC given in Fig. S14†): 2o-FCa(S1) and 2o-FCb(S1) quickly evolve into the stable S1 state intermediate 2o(S1) (rC–N = 1.8 Å) via synergistic twisting of PT and BTZ. Along the S1 ring-closing pathway, 2o(S1) could sequentially evolve as 2o-CI(S1) (rC–N = 1.7 Å) and 2c(S1) (rC–N = 1.4 Å) by absorbing energies of 1.7 and 2.4 kcal mol−1. If IC takes place at the CI region, 2o-CI(S1) would convert to 2c(S0) immediately. The entire reaction process of 2o follows a ring-closing pathway on both S1 and S0 states. Since 2c is a stable intermediate, the dehydrogenation process can only occur with assistance of a hydride acceptor to generate 2c+, implying a stepwise route for the photoinduced dehydrogenation of 2o. The triplet ring-closing pathway of 2o is negligible. At 2o(S1), the only possible ISC channel is S1 → T1 with an energy gap of 4.8 kcal mol−1, where the ISC rate would be inefficient due to rather small SOCME (0.5 cm−1). Even the ISC takes place, the intermediate could transform into 2o(T1) (36.7 kcal mol−1) or 2c(T1) (25.9 kcal mol−1), which was located at each end of the triplet ring-closing pathway of 2o. Alternatively, when the three nonreactive 2o-P(S0) conformers are excited to the S1 state, the dominant relaxation pathway for all of them is S1 → T2 ISC as shown in Fig. 5b. The involvement of triplet states in photo-relaxation of 2o is in agreement with TA experimental studies by detecting 2o-P(T1) (Fig. S9d and S10†).
 | ||
| Fig. 5 The calculated singlet and triplet state energy level diagram for (a) 2o-AP and (b) 2o-P. Inset: PEC of 2o-AP: blue line for the S0 state, red line for the S1 state, and the green dotted line represents the path of the photochemical reaction (energy in kcal mol−1) (the ground state energy of 2o-APa(S0) is used as zero for obtaining relative energies). [(TD)M062X/6-311G**/SMD (THF)]. | ||
Inspired by the distinguished photoreleasing efficiency of 1o, we propose the following ideas to improve the quantum yield of 2o based on the above mentioned experimental and theoretical results. First, compared with thermo-dynamically favorable 1o-AP(S0), 2o-AP(S0) is minority among all 2o conformers at room temperature. To stabilize 2o-AP(S0) and increase its thermal population, enhancing the rigidity of the skeleton is a possible strategy, e.g. inhibiting the rotation of PT and BTZ groups via a single bond by introducing steric hindrance or an intramolecular hydrogen bond. Second, as screened by steady-state UV-vis experiments, 1c releases a triflate anion and then spontaneously generates 1c+, while 2c+ can only be observed in the presence of a hydride acceptor. As well, the energy barrier for the reverse ring-opening process of 2c (15.5 kcal mol−1) is much lower than that of 1c+ (58.8 kcal mol−1). Thus, the 2c intermediate would revert to 2o-AP(S0) thermally rather than yielding 2c+. It is expected that increasing the dehydrogenate capability of 2c by incorporating electron-donating groups would promote the 2c → 2c+ efficiency. Third, according to the potential energy profiles, the relaxation from 1o-AP-FC(S1) to 1o-AP-CI is barrierless, thus inhibiting ISC to triplet states. However, on the S1 surface of 2o-AP, the minimum energy intermediate 2o(S1) has a slightly lower energy than 2o-CI(S1), so the process of approaching the CI region or the ring-closed conformation along the reaction pathway is endothermic. This leads to a decrease in the IC quantum yield because it cannot compete with the energetically favored ISC pathway. Fourth, the N and S-containing heterocycles usually give rise to enhanced SOC strength and are beneficial for promoted ISC efficiency.43–46 As a fact, the SOCME for the ISC channel of PHGs is larger than that of PAGs. Additionally, the extended conjugating system lowers the energy of triplet states and offers more possible ISC channels for low-lying singlet states. Since the triplet pathway is a competing side-reaction with respect to the cyclization pathway in the singlet states, reducing the ISC efficiency is a feasible way to improve the release quantum yield of PHGs.
Experimental
Synthesis of compounds
1o and 2o were prepared following reported methods.14,42The 1H NMR results are shown in Fig. S1 and S2,† respectively.
Photolysis experiments
The photolysis experiment was conducted in a Rayonet RPR 100 containing 16 lamps (266 nm). Cooling was achieved with an internal cold finger. The UV-vis spectra were used to monitor the photolysis process of 1o.fs-TA experiment
A femtosecond regenerative amplified Ti:sapphire laser system was applied to carry out the fs-TA experiments. A white continuum light (330–800 nm) was selected as the probe pulse, which was generated in a CaF2 crystal by about 5% of the amplified 800 nm output obtained from the laser system. The probe pulse was divided into two beams; one beam would pass through the sample solution sealed in a 2 mm path-length cuvette, and the other was used as a reference to monitor the stability of the probe pulse. A 266 nm laser beam was employed to excite 1o, and a 355 nm laser beam was employed to excite 2o.ns-TA experiment
ns-TA experiments were performed on a laser flash spectrometer (LP-980, Edinburgh Instruments). A 266 nm pulse from the fourth harmonic output and a 355 nm pulse from the third harmonic output of the Nd:YAG laser were used as the pump beam, and a 150-W xenon lamp was used as the probe light source. Sample solutions of 1o and 2o with absorbance of 0.6 at 266 nm and 355 nm respectively were prepared and placed in a 1 cm quartz cuvette.DFT calculations
The optimized geometries for the ground state intermediates associated with the reaction pathways of 1o and 2o were determined by DFT calculations using the M062X functional with the 6-311G** basis set. The excited state intermediates' geometries and UV-vis spectra were computed by TD-DFT/M062X/6-311G** calculations. For all optimized geometries, vibrational frequency calculations were carried out to verify whether is a minimum (zero imaginary frequency) or transition state (TS) (only one imaginary frequency). Additionally, the transition states (TS) were also confirmed through intrinsic reaction coordinate (IRC) calculations. The PECs along the corresponding reaction coordinate on the S1 or S0 state were recorded by a relaxed scan approach, and geometry optimization was performed at each step with the scanned variable (internal coordinate) maintained constant. All the theoretical calculations mentioned above were performed using the Gaussian 16 program.47 The spin–orbit coupling matrix elements (SOCME) were calculated at the TD-DFT/M062X/6-311G** level of theory with spin–orbit mean-field (SOMF) methods employing the ORCA 5.0.3 program,48 and the solvent effects of ACN (ε = 37.5) and THF (ε = 7.52) were accounted for by the SMD model.49 The Cartesian coordinates, energies and free energies for all intermediates in the S0, S1, and T1 states involved in the reaction paths are listed in the ESI†. Multiwfn software was applied to analyze the excitation characteristics and atomic charge distributions.50Conclusions
In order to help the rational design of PHGs with improved quantum yield, the photochemical reaction mechanisms of 1o and 2o as PAGs and PHGs models are extensively investigated by utilizing time-resolved spectroscopy and DFT/TD-DFT calculations. It is unravelled that 1o and 2o follow different reaction pathways upon photoexcitation. The spontaneous cyclization process of 1o takes place immediately after pumped to the singlet excited state, generating the semi-closing intermediate (1c) which lies in the vicinity of the CI region. After decay to the S0 state, the release of the triflate anion from 1o follows a synergistic pattern, that is, the ring-closing and de-acidification occur simultaneously. The entire photoinduced reaction process of 1o completes within a few ps. On the other hand, the ring closure and hydride release of 2o occur stepwise in the presence of a hydride receptor. Being excited to the FC region of the singlet excited state, 2o first relaxes to the S1 minimum, which decays to S0 by climbing up the barrier approaching the CI region and forming the closed-form intermediate 2c. Generation of 2c+ can only take place with the aid of a hydride acceptor. Based on the detailed reaction mechanisms, several design principles for efficient PHGs are proposed. The current work unveils the structure–reactivity relationship for rational design of novel versatile photoacid and photohydride generators, as well as presenting general suggestions for developing advanced PHG based optical materials.Data availability
The data that support the findings of this study are available in the ESI.†Author contributions
Yifan Su: investigation, methodology, data curation, visualization, software and writing – original draft. Dexin Zheng and Lingfeng Ge: investigation, methodology and writing – review & editing. Le Yu: theoretical calculations, supervision and writing – review & editing. Jiani Ma: resources, validation, investigation, data curation, supervision and writing – review & editing. David Lee Phillips and Yu Fang: investigation, supervision and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by grants from the National Science Fund of China for Excellent Young Scholars (22322301), the Fundamental Research Funds for the Central Universities (1301032383, GK202207001) to Jiani Ma. Le Yu would like to thank the support from the Fund of Education Department of Shaanxi Provincial Government (23JP172). The authors acknowledge the technical support of HongZhi-Wei Technology (Shanghai) Co. LTD.References
- M. J. Hansen, W. A. Velema, M. M. Lerch, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2015, 44, 3358–3377 RSC.
- M. Klausen and M. Blanchard-Desce, J. Photochem. Photobiol., C, 2021, 48, 100423 CrossRef CAS.
- G. C. R. Ellis-Davies, Angew. Chem., Int. Ed., 2023, 62, e202206083 CrossRef CAS PubMed.
- C. C. Romão, W. A. Blättler, J. D. Seixas and G. J. L. Bernardes, Chem. Soc. Rev., 2012, 41, 3571–3583 RSC.
- M. Klimezak, J. Chaud, A. Brion, F. Bolze, B. Frisch, B. Heurtault, A. Kichler and A. Specht, Adv. Healthcare Mater., 2024, 13, 2400354 CrossRef CAS PubMed.
- C. A. Hammer, K. Falahati, A. Jakob, R. Klimek, I. Burghardt, A. Heckel and J. Wachtveitl, J. Phys. Chem. Lett., 2018, 9, 1448–1453 CrossRef CAS PubMed.
- Y. A. Jézéquel, F. Svěrák, A. Ramundo, V. Orel, M. Martínek and P. Klán, J. Org. Chem., 2024, 89, 4888–4903 CrossRef PubMed.
- A. Sikder, M. Banerjee, T. Singha, S. Mondal, P. K. Datta, A. Anoop and N. D. P. Singh, Org. Lett., 2020, 22, 6998–7002 CrossRef CAS PubMed.
- S. Banala, M. C. Arvin, N. M. Bannon, X.-T. Jin, J. J. Macklin, Y. Wang, C. Peng, G. Zhao, J. J. Marshall, K. R. Gee, D. L. Wokosin, V. J. Kim, J. M. McIntosh, A. Contractor, H. A. Lester, Y. Kozorovitskiy, R. M. Drenan and L. D. Lavis, Nat. Methods, 2018, 15, 347–350 CrossRef CAS PubMed.
- C. C. Warford, C.-J. Carling and N. R. Branda, Chem. Commun., 2015, 51, 7039–7042 RSC.
- C. Berton, D. M. Busiello, S. Zamuner, E. Solari, R. Scopelliti, F. Fadaei-Tirani, K. Severin and C. Pezzato, Chem. Sci., 2020, 11, 8457–8468 RSC.
- Y. Liao, Acc. Chem. Res., 2017, 50, 1956–1964 CrossRef CAS PubMed.
- Y. Cheng, X. Ma, J. Zhai and X. Xie, Chem. Commun., 2023, 59, 1805–1808 RSC.
- R. Mizutsu, R. Asato, C. J. Martin, M. Yamada, Y. Nishikawa, S. Katao, M. Yamada, T. Nakashima and T. Kawai, J. Am. Chem. Soc., 2019, 141, 20043–20047 CrossRef CAS PubMed.
- R. Weinstain, T. Slanina, D. Kand and P. Klán, Chem. Rev., 2020, 120, 13135–13272 CrossRef CAS PubMed.
- T. Sun, L. Kang, H. Zhao, Y. Zhao and Y. Gu, Adv. Sci., 2024, 11, 2302875 CrossRef CAS PubMed.
- N. Zivic, P. K. Kuroishi, F. Dumur, D. Gigmes, A. P. Dove and H. Sardon, Angew. Chem., Int. Ed., 2019, 58, 10410–10422 CrossRef CAS PubMed.
- K. Sambath, Z. Wan, Q. Wang, H. Chen and Y. Zhang, Org. Lett., 2020, 22, 1208–1212 CrossRef CAS PubMed.
- L. D. Terlizzi, A. Martinelli, D. Merli, S. Protti and M. Fagnoni, J. Org. Chem., 2023, 88, 6313–6321 CrossRef PubMed.
- Z. Shi, P. Peng, D. Strohecker and Y. Liao, J. Am. Chem. Soc., 2011, 133, 14699–14703 CrossRef CAS PubMed.
- A. N. Komogortsev, C. V. Milyutin, B. V. Lichitsky and V. G. Melekhina, Tetrahedron, 2022, 114, 132780 CrossRef CAS.
- C. J. Martin, J. P. Calupitan, M. Minamide, R. Asato, Y. Goto, G. Rapenne, T. Nakashima and T. Kawai, J. Photochem. Photobiol., A, 2020, 397, 112594 CrossRef CAS.
- A. McSkimming and S. B. Colbran, Chem. Soc. Rev., 2013, 42, 5439–5488 RSC.
- X. Yang, J. Walpita, D. Zhou, H. L. Luk, S. Vyas, R. S. Khnayzer, S. C. Tiwari, K. Diri, C. M. Hadad, F. N. Castellano, A. I. Krylov and K. D. Glusac, J. Phys. Chem. B, 2013, 117, 15290–15296 CrossRef CAS PubMed.
- A. G. Lvov, E. K. Kouame and M. M. Khusniyarov, Chem.–Eur. J., 2023, 29, e202301480 CrossRef CAS PubMed.
- J. Volarić, W. Szymanski, N. A. Simeth and B. L. Feringa, Chem. Soc. Rev., 2021, 50, 12377–12449 RSC.
- O. Galangau, S. Delbaere, N. Ratel-Ramond, G. Rapenne, R. Li, J. P. D. C. Calupitan, T. Nakashima and T. Kawai, J. Org. Chem., 2016, 81, 11282–11290 CrossRef CAS PubMed.
- A. G. Lvov, V. Z. Shirinian, V. V. Kachala, A. M. Kavun, I. V. Zavarzin and M. M. Krayushkin, Org. Lett., 2014, 16, 4532–4535 CrossRef CAS PubMed.
- A. V. Zakharov, E. B. Gaeva, A. G. Lvov, A. V. Metelitsa and V. Z. Shirinian, J. Org. Chem., 2017, 82, 8651–8661 CrossRef CAS PubMed.
- R. Murata, T. Yago and M. Wakasa, J. Phys. Chem. A, 2015, 119, 11138–11145 CrossRef CAS PubMed.
- K. Tani, Y. Ishibashi, H. Miyasaka, S. Kobatake and M. Irie, J. Phys. Chem. C, 2008, 112, 11150–11157 CrossRef CAS.
- M. T. Indelli, S. Carli, M. Ghirotti, C. Chiorboli, M. Ravaglia, M. Garavelli and F. Scandola, J. Am. Chem. Soc., 2008, 130, 7286–7299 CrossRef CAS PubMed.
- S. Fredrich, T. Morack, M. Sliwa and S. Hecht, Chem.–Eur. J., 2020, 26, 7672–7677 CrossRef CAS PubMed.
- A. V. Yadykov, A. G. Lvov, M. M. Krayushkin, A. V. Zakharov and V. Z. Shirinian, J. Org. Chem., 2021, 86, 10023–10031 CrossRef CAS PubMed.
- P. Ravat, T. Šolomek, D. Häussinger, O. Blacque and M. Juríček, J. Am. Chem. Soc., 2018, 140, 10839–10847 CrossRef CAS PubMed.
- Y. Nakakuki, T. Hirose, H. Sotome, M. Gao, D. Shimizu, R. Li, J. Hasegawa, H. Miyasaka and K. Matsuda, Nat. Commun., 2022, 13, 1475 CrossRef CAS PubMed.
- H. Hamamoto, D. Shimizu and K. Matsuda, Chem.–Eur. J., 2024, 30, e202401353 CrossRef CAS PubMed.
- A. G. Lvov, V. Z. Shirinian, A. V. Zakharov, M. M. Krayushkin, V. V. Kachala and I. V. Zavarzin, J. Org. Chem., 2015, 80, 11491–11500 CrossRef CAS PubMed.
- A. V. Zakharov, A. V. Yadykov, E. B. Gaeva, A. V. Metelitsa and V. Z. Shirinian, J. Org. Chem., 2021, 86, 16806–16814 CrossRef CAS PubMed.
- T. Nakashima, K. Tsuchie, R. Kanazawa, R. Li, S. Iijima, O. Galangau, H. Nakagawa, K. Mutoh, Y. Kobayashi, J. Abe and T. Kawai, J. Am. Chem. Soc., 2015, 137, 7023–7026 CrossRef CAS PubMed.
- R. Li, T. Nakashima and T. Kawai, Chem. Commun., 2017, 53, 4339–4341 RSC.
- C. J. Martin, M. Minamide, J. P. D. C. Calupitan, R. Asato, J. Kuno, T. Nakashima, G. Rapenne and T. Kawai, J. Org. Chem., 2018, 83, 13700–13706 CrossRef CAS PubMed.
- T. Yang, Y. Li, Z. Zhao and W. Z. Yuan, Sci. China Chem., 2023, 66, 367–387 CrossRef CAS.
- N. Aizawa, A. Matsumoto and T. Yasuda, Sci. Adv., 2021, 7, eabe5769 CrossRef CAS PubMed.
- L. Ma, Y. Liu, H. Tian and X. Ma, JACS Au, 2023, 3, 1835–1842 CrossRef CAS PubMed.
- Y. Tu, J. Liu, H. Zhang, Q. Peng, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2019, 58, 14911–14914 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, rev. A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- F. Neese, Software update: The ORCA program system-Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06202g |
| This journal is © The Royal Society of Chemistry 2024 |