
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A general synthesis of cyclic bottlebrush polymers with enhanced mechanical properties via graft-through ring expansion metathesis polymerization†
Matthew J.
Elardo
 ,
Adelaide M.
Levenson
,
Ana Paula
Kitos Vasconcelos
,
Meredith N.
Pomfret
and
Matthew R.
Golder
*
,
Adelaide M.
Levenson
,
Ana Paula
Kitos Vasconcelos
,
Meredith N.
Pomfret
and
Matthew R.
Golder
*
Department of Chemistry, Molecular Engineering & Science Institute, University of Washington, 36 Bagley Hall, Seattle, WA 98195, USA. E-mail: goldermr@uw.edu
First published on 30th September 2024
Abstract
Bottlebrush polymers represent an important class of macromolecular architectures, with applications ranging from drug delivery to organic electronics. While there is an abundance of literature describing the synthesis, structure, and applications of linear bottlebrush polymers using ring-opening metathesis polymerization (ROMP), there are comparatively less reports on their cyclic counterparts. This lack of research is primarily due to the difficulty in synthesizing cyclic bottlebrush polymers, as extensions of typical routes towards linear bottlebrush polymers (i.e., “grafting-through” polymerizations of macromonomers with ROMP) produce only ultrahigh molar mass cyclic bottlebrush polymers with poor molar mass control. Herein, we report a ring-expansion metathesis polymerization (REMP) approach to cyclic bottlebrush polymers via a “grafting-through” approach utilizing the active pyr-CB6 initiator developed in our lab. The resulting polymers, characterized via GPC-MALS-IV, are shown to have superior molar mass control across a range of target backbone lengths. The cyclic materials are also found to have superior mechanical properties when compared to their linear counterparts, as assessed by ball-mill grinding and compression testing experiments.
Introduction
The manipulation of polymer topology is a major area of interest in synthetic macromolecular chemistry due to the impact of architecture on material proprieties. Recent advances in this area have led to the development of a wide variety of intricate synthetic polymer architectures with defined chain ends, such as star polymers,1 dendrimers,2 and bottlebrush polymers.3–6 Of particular interest, however, are variants without chain ends, namely cyclic polymers, as this specific topology has markedly different properties when compared to linear counterparts, such as higher glass transition temperatures, higher decomposition temperatures, and lower intrinsic viscosities.7–9 Acyclic bottlebrush polymers3,6 are linear polymers with densely grafted macromolecular sidechains; the chemical makeup of the backbone and sidechains intimately control rich solution-state and bulk phenomena including self-assembly10–12 and stimuli-responsiveness in thin films.13 As such, these polymer bottlebrush architectures have found diverse applications from drug delivery14–17 to organic photonics.18–22 There has therefore been intense interest in the development of new linear bottlebrush materials using well-established synthetic paradigms. These strategies can be broken into three general categories: “grafting-to”,23 “grafting-from”,24 and “grafting-through”.25 Of these three distinct synthetic classes, the “grafting-through” method is most desirable because of precise control over features such as grafting density (where high densities up to 100% are easily achievable), brush length, and brush dispersity, but is synthetically challenging due to the steric demands of the polymerizable “macromonomer”.26 For ROMP-derived materials, this problem has been largely solved by the development of advanced olefin metathesis initiators,27,28 which produce bottlebrush polymers with narrow dispersities and good control over molar mass, even when using large and/or branched macromonomers.12,25,29 While examples of linear bottlebrush polymers (BBPs) abound in the literature,6,12–16,18 instances of their cyclic analogues are comparatively sparse. This difference is due to the various challenges associated with cyclic polymer synthesis. One major synthetic approach towards cyclic BBPs, linear ring closure, involves synthesizing a telechelic linear pre-polymer; functional end groups react with each other to form the desired macrocycle. However, this approach inevitably results in linear contaminants from acyclic couplings, requires dilute reaction conditions, and is not suitable for the generation of high molar mass materials.30,31 Nevertheless, this approach allowed access to multiblock cyclic polymers and provided early evidence for unique self-assembly profiles.31 The second major synthetic approach is ring expansion polymerization, of which ring expansion metathesis polymerization (REMP, the cyclic analogue to ROMP) is a common variant.32,33 Early examples of REMP initiators suffer from poor molar mass control and polydisperse products, especially when performing “graft-through” reactions with large macromonomers.34 For instance, Grubbs used this approach to prepare cyclic bottlebrush polymers with macrocyclic alkylidene REMP initiators (e.g., SC-5); however, only ultra-high molar mass materials were reported.34 Due to the inherent difficulty in controlled cyclic bottlebrush polymer growth using “grafting-through”, cyclic BBPs are often prepared via “grafting-from” (Fig. 1A).35–40 | ||
| Fig. 1 (A) General paradigm for cyclic bottlebrush polymer synthesis via “grafting-from”; (B) approaches towards cyclic bottlebrush polymers using Ru-mediated REMP. (C) Summary of bulk property enhancements for cyclic PS-BBP and cyclic PDMS-BBP with respect to comparable linear analogues. | ||
Despite their challenging syntheses, cyclic bottlebrush polymers hold promise in enhanced capacity for drug delivery41,42 and self-assembly into a variety of nanostructures, (e.g., supramolecular tubes, rods, plates, spheres, and worm architectures31,43,44). Hence, there is great potential for the development of functional materials using cyclic bottlebrush polymers made with a “grafting-through” approach (Fig. 1B). In this work, we report a controlled synthetic route toward cyclic bottlebrush polymers utilizing the novel REMP initiator pyr-CB6 developed within our research group (Fig. 1B).32,45 We demonstrate excellent control over molar mass with narrow dispersity utilizing macromonomers with disparate thermomechanical properties – “hard” PS-MM (i.e., high Tg, rigid, brittle; PS = polystyrene, Fig. S22–S25†) and “soft” PDMS-MM (i.e., low Tg, flexible, elastomeric; PDMS = polydimethylsiloxane, Fig. S19 and S20†) macromonomers. The resultant polymers are characterized, and their cyclic topologies confirmed, via gel permeation chromatography coupled with multi-angle light scattering, differential viscometry, and refractive index detectors (GPC-MALS-IV-RI; see Fig. S26–S33† for representative DRI traces and Fig. S45–S50† for representative Mark–Houwink–Sakurada intrinsic viscosity plots). The thermal, mechanochemical, and mechanical properties of the bulk materials and crosslinked networks thereof were probed via a combination of differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), ball-mill grinding mechanochemistry (BMG), and compression testing (Fig. 1C). Overall, the efforts described herein encompass a straightforward approach towards cyclic bottlebrush polymers and currently represent the most well-controlled “grafting-through” methodology to access them to date. In other words, this methodology allows for the preparation of cyclic bottlebrush polymers with the same ease as their acyclic counterparts.
Synthesis and solution-state analysis
We began our investigations by probing the activity of less active PCy3-CB6 for PS-MM and PDMS-MM REMP reactions. Interestingly, even when targeting a relatively short backbone degree of polymerization (backbone DP = 25), PDMS-MM afforded material with much higher-than-expected Mn (by nearly two orders of magnitude) and a broad GPC-RI trace with a sizeable low molar mass shoulder; PS-MM afforded material that was too viscous for subsequent analysis (Table 1 and Fig. S40†). Despite PCy3-CB6 providing superior molar mass control relative to those of cyclic alkylidene initiators (e.g., UC-5, UC-6) in our prior REMP studies with norbornene monomers,32 the steric hindrance of macromonomers provide additional kinetic challenges. Fortunately, initial REMP experiments employing the more active pyr-CB6 initiator indicated rapid and complete macromonomer consumption (<1 h) in DCE at 55 °C (Fig. S36 and S37†). Notably, we do not observe any molar mass evolution at extended reaction times following macromonomer conversion (Fig. S36 and S37†), a feature we reported previously which distinguishes pyr-CB6 mechanistically from PCy3-CB6.45 Furthermore, we observe a significant improvement in molar mass control, dispersity, and peak structure when utilizing pyr-CB6 in place of PCy3-CB6 for “grafting-through” REMP towards well-defined cyclic bottlebrush macromolecules. We believe that an increased initiator efficiency combined with relatively slow secondary metathesis (i.e., intermolecular chain transfer) leads to the large disparity in initiator-dependent experimental Mn (Table 1).
We next investigated the ability of our pyr-CB6 initiator to prepare cyclic bottlebrush polymers at a variety of target molar masses (i.e., a range of target backbone DP). A major challenge encountered with cyclic Ru alkylidene initiators utilized by Grubbs is their inability to prepare low DP polymers due to poor initiation efficiency; this shortcoming is especially true when targeting bottlebrush polymers via “grafting-through” of macromonomers.34 We observe good molar mass control over a wide range of target DPs (DP = 10–50) (Table 2) while maintaining low dispersities for both PS-MM (Fig. 2A) and PDMS-MM (Fig. 2B). The result of the improved initiation efficiency is cyclic bottlebrush polymers with Mn < 50 kDa, a significant improvement from previous systems which were limited to ultra-high molar mass polymers in the MDa regime. Hence, our methodology showcases the most powerful examples to date of controlling cyclic bottlebrush backbone DP with “grafting-through” technology.
|
|
|||||
|---|---|---|---|---|---|
| Monomera | Theoretical DPb | Measured DPc | Theoretical Mnb (kDa) | Measured Mnc (kDa) | Đ |
| a PS-MM M n = 4.6 kDa; PDMS-MMMn = 6.3 kDa. b Calculated by [MM]0/[I]0, where [MM] is the molar amount of macromonomer and [I] is the molar amount of CB6 (plus 32 eq. pyridine). c Determined by GPC-MALS. | |||||
| PS-MM | 10 | 18 | 46.0 | 83.0 | 1.1 |
| PS-MM | 20 | 28 | 90.0 | 130 | 1.1 |
| PS-MM | 30 | 37 | 140 | 170 | 1.2 |
| PS-MM | 40 | 43 | 180 | 200 | 1.2 |
| PS-MM | 50 | 52 | 230 | 240 | 1.3 |
| PDMS-MM | 10 | 41 | 63.0 | 260 | 1.1 |
| PDMS-MM | 20 | 81 | 130 | 510 | 1.1 |
| PDMS-MM | 30 | 114 | 190 | 720 | 1.1 |
| PDMS-MM | 40 | 151 | 250 | 950 | 1.2 |
| PDMS-MM | 50 | 175 | 320 | 1100 | 1.2 |

 | ||
| Fig. 2 Solution-state characterization of (A) PS-MM and (B) PDMS-MM REMP by GPC-MALS-RI for target DP = 10–50. | ||
Another challenge with preparing cyclic bottlebrush polymers via “grafting-through” REMP is that the polymerizations do not work well even with modest sized macromonomers. For instance, in the seminal report of this methodology from Grubbs using SC-5 and SC-6, they observe only 65% conversion of PS-MM (Mn = 6.6 kDa).34 Here, we demonstrate that analogous REMP utilizing pyr-CB6 works well for even larger macromonomers; increasing the brush length (PS-MM, Mn = 8.3 kDa) does not have a detrimental impact on molar mass control and only modestly increases the dispersity of the resulting cyclic bottlebrush polymer (Fig. S38 and Table S3†). Although more dilute conditions are required to maintain molar mass control (ca. 40 mg mL−1, versus ca. 90 mg mL−1 for the lower molar mass PS-MM), the reaction is nearly quantitative with >90% monomer conversion after 3.5 hours as assessed by GPC-RI (Fig. S39†). We next probed the topology of the putative cyclic bottlebrush polymers in dilute solution via GPC with in-line multi-angle light scattering, differential viscometry, and refractive index detectors (GPC-MALS-IV-RI). To determine absolute molar masses, we directly measured the specific refractive index increment (i.e., dn/dc) for representative ROMP and REMP bottlebrush polymers (Fig. S51–S58 and Table S4†). Interestingly, cyclic bottlebrush polymers derived from both PS-MM and PDMS-MM had lower magnitude dn/dc values relative to linear analogs in CHCl3; topology dependent refractive indices have been observed in other organic polymer scaffolds as well.46–48 In general, we observe longer retention times in the GPC analysis of REMP bottlebrush polymers when compared to ROMP bottlebrush polymers of similar molar masses prepared using Grubbs 3 initiator (see ESI† for Experimental details) for both PS-MM (Fig. 3A) and PDMS-MM (Fig. 3B) REMP reactions. Likewise, plots of molar mass vs. GPC retention time indicate higher molar masses for the REMP polymers across all elution volumes (Fig. S43 and S44†). These results collectively indicate that the REMP polymers are more compact in solution than their ROMP counterparts of similar molar mass, a feature that is characteristic of cyclic polymers due to their lack of chain ends.7–9
 | ||
| Fig. 3 Solution-state characterization of cyclic and linear (A) PS-BBP and (B) PDMS-BBP by GPC-MALS-RI. | ||
Similarly, Mark–Houwink–Sakurada (MHS) analyses of the REMP BBPs reveal significantly lower intrinsic viscosities than for ROMP BBPs of similar molar mass ([η]cyclic/[η]linear = ca. 0.57–0.63) for both PS (Fig. 4A) and PDMS (Fig. 4B).7–9 As the bottlebrush polymers presented in this study have comparatively short backbone DPs relative to prior “grafting-through” REMP studies,34 dilute solution-state behavior as assessed by intrinsic viscosity measurements suggests deviation from Flory–Fox behavior (i.e., [η] ∼ M0.7).49 Specifically, only a small increase in intrinsic viscosity with increasing molar mass is observed with a Mark–Houwink parameter, α, between ca. 0.3–0.4 (Fig. S45–S50†). These data suggest that the bottlebrush polymers in this study, all with relatively short backbone DPs, behave more like star-polymers in solution.49 Nonetheless, importantly both ROMP and REMP polymers have similar Mark–Houwink parameters and therefore differences in the observed intrinsic viscosities are due to molecular architecture (i.e., linear versus cyclic) rather than differences in backbone structure and/or conformation.33
 | ||
| Fig. 4 Mark–Houwink–Sakurada plots of cyclic and linear (A) PS-BPP and (B) PDMS-BBP as measured by GPC-MALS-IV-RI. | ||
We also used the collective analytical approaches described above to qualitatively probe the efficiency of polymer backbiting by comparing the GPC-RI traces and MHS plots of quenched (i.e., ethyl vinyl ether, EVE, was added at the end of the polymerization reaction) and unquenched aliquots from the same samples (Fig. S34, S35, S47, S50 and Tables S1, S2†). If backbiting is incomplete and Ru remains in the cyclic polymer backbone, quenching with EVE produces linear polymers following macrocycle opening via cross-metathesis. This process would result in a shift to shorter retention times and higher intrinsic viscosities in the GPC-RI traces and MHS plots, respectively. We see no significant difference in either of these parameters between quenched and unquenched aliquots from our reaction mixtures, indicating that backbiting is operative insofar as we can measure.
Thermal and mechanical properties
With two classes of cyclic bottlebrush polymers in hand, we next turned our attention to evaluating the impact of topology on bulk properties of our bottlebrush polymers. We began by probing the thermal properties of linear and cyclic PS bottlebrush polymers (Mn = 129 kDa), by thermal gravimetric analysis (TGA) and differential scanning calorimetry. Interestingly, we found no significant differences in the thermal stability via TGA (Td = 374 °C and 368 °C for linear and cyclic PS-BBPs, respectively) or thermal transition temperature via DSC (Tg = 100 °C and 95 °C for linear and cyclic PS-BBPs, respectively) between the linear and cyclic samples (Fig. S59–S62†). While cyclic polymers are known to exhibit higher decomposition temperatures (Td) and glass transition (Tg) temperatures, we surmise that the high mass percentage of polymeric brush in each sample obfuscates subtle topology-dependent thermal differences. Because of the similar thermal properties of our PS bottlebrush polymers, we next decided to probe the mechanical stability of the bulk PS brushes by subjecting them to ball-mill grinding (BMG) conditions. While separate studies have been conducted by Peterson, Kim, Hwang, and Choi probing the relative independent stability of cyclic50 and bottlebrush51 polymers to linear analogues under ball-milling conditions, no work has been done exploring the relative stability of cyclic bottlebrush polymers under these conditions. Since it has been previously demonstrated that the degradation rates of linear polymers under BMG conditions scale linearly with increasing Tg,52 a feature not observed in either cyclic50 or bottlebrush systems,51 and that cyclic polymers tend to degrade slower than linear polymers with of comparable Mn under these conditions,50 we hypothesized that the cyclic brush polymers may have unique stability to mechanochemical degradation in the solid state. Indeed, we observed that a linear PS-BBP (Mn = 200 kDa) degraded approximately 30% faster than a cyclic PS-BBP (Mn = 250 kDa) (Fig. 5, S41 and S42; see ESI Section 8† for Experimental details and Tables S5 and S6† for raw data). Thus, we have demonstrated the ability to create materials with enhanced mechanical stability without perturbing thermal properties. This finding, in conjunction with previous findings demonstrating the improved wear resistance and shear stability of cyclic polymer brushes,53 may be desirable for applications in polymer coatings such as antifouling materials, where abrasion and impact damage shorten the material's lifespan.54 | ||
| Fig. 5 Degradation kinetics for cyclic (purple) and linear (gold) PS-BBPs under ball-mill grinding conditions. The inverse of the number-average molar mass was determined for each polymer at each timepoint, and linear regressions were performed to determine the line of best fit (dashed lines). Rate constants were determined by multiplying the slope of each trendline by the number average molar mass of PS-MM. See ESI Section 8† for details. | ||
We next turned our attention to the PDMS-BBP samples for bulk characterization. These studies were inspired by the universal bottlebrush polymer crosslinking methodology developed by Bates55 previously used to prepare supersoft elastomers from PDMS bottlebrush polymers.56 In this work, bifunctional benzophenone BisBP-PDMS initiator facilitated indiscriminate brush C–H abstraction and subsequent curing under photochemical irradiation (Fig. 6A). We reasoned that these mild crosslinking conditions would allow us to explore the impact of bottlebrush polymer topology on the resulting materials' mechanical properties. While we are the first to explore the mechanical properties of crosslinked cyclic bottlebrush networks, previous independent work from Tew, Sun, and Veige and Sumerlin have found that cyclic polymer networks have greater compressibility57 and stretchability,58 greater swelling ratios in organic solvent,57,59 and increased toughness58 when compared to networks prepared from linear analogues. Therefore, we reasoned that such phenomena would extend to our cyclic PDMS-BBP networks and hypothesized that they should have superior mechanical properties compared to networks made from linear PDMS-BBP at the same crosslinking density. We accordingly synthesized crosslinked networks from high molar mass linear (ROMP, Mn = 1.73 MDa) and cyclic (REMP, Mn = 3.49 MDa) PDMS-BBP samples with a crosslinking density (χ) of 1 mol BisBP-PDMS per individual brush in the bottlebrush polymer (i.e., for χ = 1.00, ncrosslinker = nbottlebrush × DPbottlebrush). It should be noted that while there is a slight mismatch between bottlebrush polymer absolute molar masses, because cyclic polymers are more compact than their linear counterparts, the PDMS-BBP precursors to these network samples are actually quite comparable due to similar radii of gyration (Rg = ca. 20–25 nm). Furthermore, in their work with cyclic polymer gels, Tew found no change in mechanical properties upon doubling the molar mass of the linear polymer precursor,57 and Sumerlin and Veige reported mechanical data for cyclic polymers ca. 1.5 times larger than their linear analogues.59 We thus reasoned that the slightly higher molar mass of our cyclic BBPs was unlikely to obfuscate any difference in mechanical properties between the samples. Consistent with reports on the aforementioned cyclic polymer networks (vide supra), our cyclic PDMS-BBP demonstrated significantly (p < 0.001) higher swelling ratios (191%) in ethyl acetate than their linear analogues (148%) (Table S7†). Upon compression testing of freshly cured cylindrical specimens (Fig. 6B), indeed while both networks had similar Young's moduli (762 and 721 kPa, respectively), networks prepared from cyclic PDMS-BBP demonstrated statistically significant increases in compressive strength, strain at break, and toughness (p < 0.01 for all parameters, n = 3) compared to those prepared from linear PDMS-BBP at χ = 1.00 (Fig. 6C; see Fig. S63–S67† for statistical analysis results and Table S7† for numerical values).
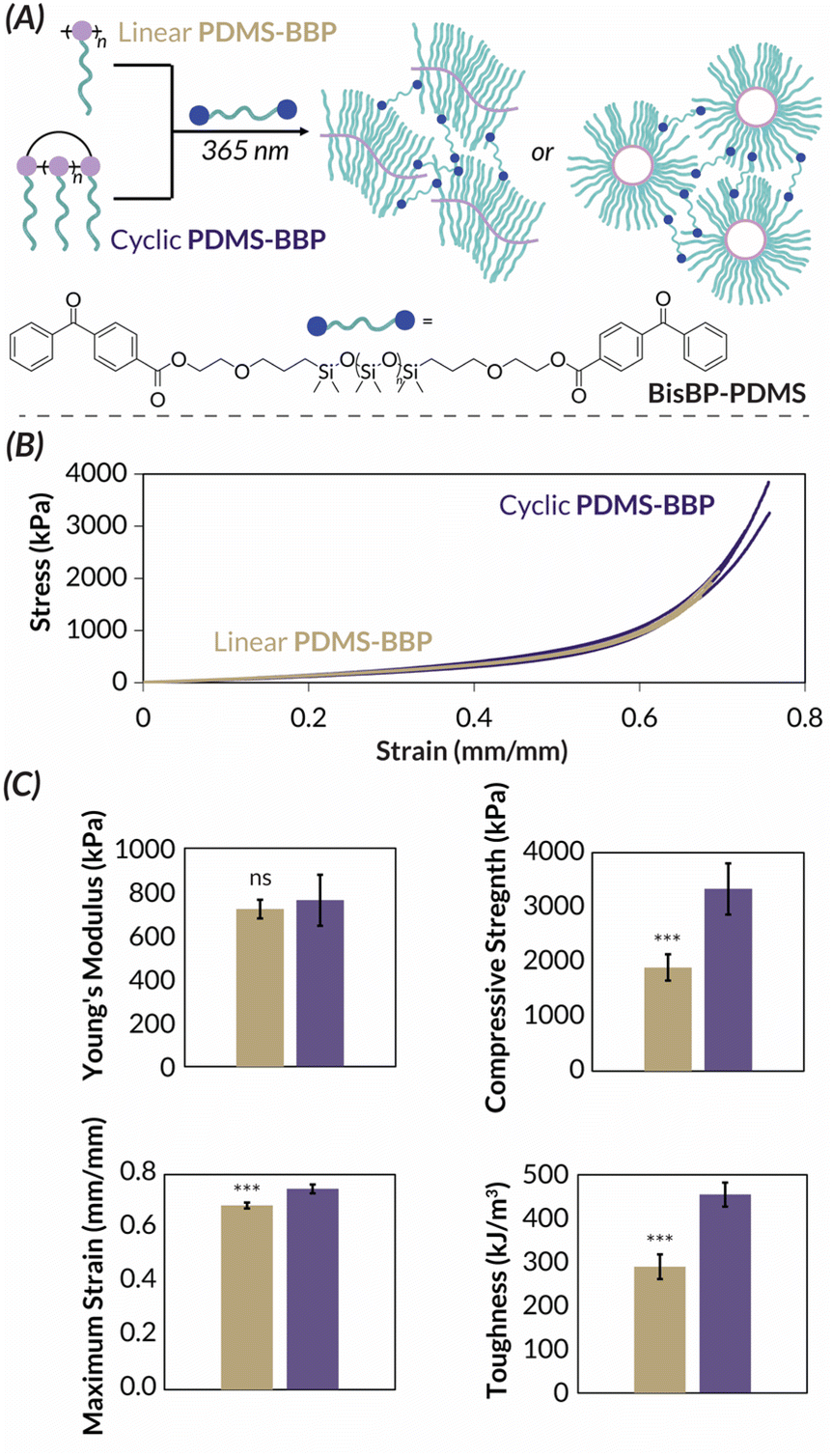 | ||
| Fig. 6 (A) Cartoon demonstrating cross-linking chemistry. (B) Stress–strain curve for linear (gold) and cyclic (purple) PDSM-BBP networks in compression tests. (C) Selected mechanical properties of linear (gold) and cyclic (purple) PDMS-BBP networks. *** p < 0.01 for n = 3. ns: no significance (p > 0.05) for n = 3. | ||
With such broad utility for PDMS-based bottlebrush networks across applications in soft robotics,60 sensors,56,61 and electronics,62 it is significant that polymer topology alone can enhance mechanical properties. More generally, we anticipate these collective findings to advance the molecular engineering of mechanically robust bottlebrush polymers networks63 where backbone topology, rather than chemical composition, controls bulk behavior.
Conclusions
In conclusion, we demonstrate in this report an efficient and general methodology for the synthesis of macrocyclic bottlebrush polymers via “grafting-through”. Our approach, which takes advantage of the superior initiation efficiency of our cyclic benzylidene REMP initiator pyr-CB6, produces densely grafted macrocyclic bottlebrush polymers with good control over molar mass and dispersity. We report a significant improvement over previous cyclic Ru alkylidene initiators to this end, especially with respect to preparing lower molar mass (<100 kDa) backbones and polymers with long brush lengths (>5 kDa). We demonstrate that the methodology is general, with the ability to tune the parent macromonomer composition and resultant backbone length easily and independently. The cyclic topology of these bottlebrush polymers was interrogated using solution-state analyses (GPC-MALS-IV-RI). Moreover, the bulk thermal and mechanical properties of the BBPs were probed. It was found that despite similar thermal properties, the cyclic PS-BBP materials are more stable to mechanochemical degradation than their linear counterparts under ball-mill grinding conditions. Furthermore, we found significant enhancements to the mechanical properties of crosslinked PDMS-BBP elastomers with cyclic topologies. Compared to networks prepared from linear PDMS-BBP, our elastomers prepared from cyclic PDMS-BBP had statistically significant higher compressive strength, strain at break, toughness, and swelling ratio. We envision this methodology finding broad appeal in the development of well-defined macrocyclic bottlebrush polymers for applications spanning porous materials, mechanically resilient coatings/lubricants, tougher soft robotics, and self-assembled nanoarchitectures.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
M. J. E., A. M. L., and M. R. G. conceived of the idea. M. J. E., A. M. L., and M. N. P. conducted synthetic experiments and analyzed physical properties. M. J. E. conducted ball-milling experiments. A. P. K. V. performed mechanical testing experiments. M. J. E. and M. R. G. wrote the manuscript; all authors discussed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation under CHE-2142922 and generous start-up funds from the University of Washington. M. J. E. acknowledges the National Science Foundation Graduate Research Fellowship Program (DGE-2140004) for a graduate research fellowship. A. M. L. and M. N. P. each acknowledge the University of Washington Clean Energy Institute for a graduate research fellowship. This material is based in part upon work supported by the state of Washington through the University of Washington Clean Energy Institute. NMR spectroscopy resources are supported under NIH S10 OD030224-01A1. We would also like to thank Morgan Skala for her assistance with setting up ball-mill grinding experiments.Notes and references
- A. Mandal and A. F. M. Kilbinger, ACS Macro Lett., 2023, 12, 1372–1378 Search PubMed.
- H. Liu, Y. Wang, W. Mo, H. Tang, Z. Cheng, Y. Chen, S. Zhang, H. Ma, B. Li and X. Li, Adv. Funct. Mater., 2020, 30, 1910275 Search PubMed.
- S. S. Sheiko, B. S. Sumerlin and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 759–785 Search PubMed.
- S. C. Radzinski, J. C. Foster, R. C. Chapleski Jr, D. Troya and J. B. Matson, J. Am. Chem. Soc., 2016, 138, 6998–7004 Search PubMed.
- S. J. Scannelli, M. Alaboalirat, D. Troya and J. B. Matson, Macromolecules, 2023, 56, 3838–3847 Search PubMed.
- R. Verduzco, X. Li, S. L. Pesek and G. E. Stein, Chem. Soc. Rev., 2015, 44, 2405–2420 Search PubMed.
- F. M. Haque and S. M. Grayson, Nat. Chem., 2020, 12, 433–444 Search PubMed.
- H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 251–284 Search PubMed.
- T.-W. Wang and M. R. Golder, Polym. Chem., 2021, 12, 958–969 Search PubMed.
- T.-P. Lin, A. B. Chang, S.-X. L. Luo, H.-Y. Chen, B. Lee and R. H. Grubbs, ACS Nano, 2017, 11, 11632–11641 Search PubMed.
- R. Liang, Q. Song, R. Li, A. N. Le, X. Fu, Y. Xue, X. Ji, W. Li and M. Zhong, Angew. Chem., Int. Ed., 2022, 61, e202210067 Search PubMed.
- K. Kawamoto, M. Zhong, K. R. Gadelrab, L.-C. Cheng, C. A. Ross, A. Alexander-Katz and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 11501–11504 Search PubMed.
- X. Li, S. L. Prukop, S. L. Biswal and R. Verduzco, Macromolecules, 2012, 45, 7118–7127 Search PubMed.
- J. A. Johnson, Y. Y. Lu, A. O. Burts, Y. Xia, A. C. Durrell, D. A. Tirrell and R. H. Grubbs, Macromolecules, 2010, 43, 10326–10335 Search PubMed.
- M. L. Ohnsorg, P. C. Prendergast, L. L. Robinson, M. R. Bockman, F. S. Bates and T. M. Reineke, ACS Macro Lett., 2021, 10, 375–381 Search PubMed.
- F. Vohidov, L. E. Milling, Q. Chen, W. Zhang, S. Bhagchandani, H. V.-T. Nguyen, D. J. Irvine and J. A. Johnson, Chem. Sci., 2020, 11, 5974–5986 Search PubMed.
- J. Liu, A. O. Burts, Y. Li, A. V. Zhukhovitskiy, M. F. Ottaviani, N. J. Turro and J. A. Johnson, J. Am. Chem. Soc., 2012, 134, 16337–16344 Search PubMed.
- A. L. Liberman-Martin, C. K. Chu and R. H. Grubbs, Macromol. Rapid Commun., 2017, 38, 1700058 Search PubMed.
- A. L. Liberman-Martin, A. B. Chang, C. K. Chu, R. H. Siddique, B. Lee and R. H. Grubbs, ACS Macro Lett., 2021, 10, 1480–1486 Search PubMed.
- G. M. Miyake, V. A. Piunova, R. A. Weitekamp and R. H. Grubbs, Angew. Chem., Int. Ed., 2012, 51, 11246–11248 Search PubMed.
- G. M. Miyake, R. A. Weitekamp, V. A. Piunova and R. H. Grubbs, J. Am. Chem. Soc., 2012, 134, 14249–14254 Search PubMed.
- B. R. Sveinbjörnsson, R. A. Weitekamp, G. M. Miyake, Y. Xia, H. A. Atwater and R. H. Grubbs, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14332–14336 Search PubMed.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 6633–6639 Search PubMed.
- M. Xie, J. Dang, H. Han, W. Wang, J. Liu, X. He and Y. Zhang, Macromolecules, 2008, 41, 9004–9010 Search PubMed.
- Y. Xia, B. D. Olsen, J. A. Kornfield and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 18525–18532 Search PubMed.
- N. Hadjichristidis, M. Pitsikalis, H. Iatrou and S. Pispas, Macromol. Rapid Commun., 2003, 24, 979–1013 Search PubMed.
- O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 Search PubMed.
- T.-L. Choi and R. H. Grubbs, Angew. Chem., Int. Ed., 2003, 42, 1743–1746 Search PubMed.
- Z.-H. Guo, A. N. Le, X. Feng, Y. Choo, B. Liu, D. Wang, Z. Wan, Y. Gu, J. Zhao, V. Li, C. O. Osuji, J. A. Johnson and M. Zhong, Angew. Chem., Int. Ed., 2018, 57, 8493–8497 Search PubMed.
- R. Liénard, J. De Winter and O. Coulembier, J. Polym. Sci., 2020, 58, 1481–1502 Search PubMed.
- M. Schappacher and A. Deffieux, Science, 2008, 319, 1512–1515 Search PubMed.
- T.-W. Wang, P.-R. Huang, J. L. Chow, W. Kaminsky and M. R. Golder, J. Am. Chem. Soc., 2021, 143, 7314–7319 Search PubMed.
- C. W. Bielawski, D. Benitez and R. H. Grubbs, Science, 2002, 297, 2041–2044 Search PubMed.
- Y. Xia, A. J. Boydston and R. H. Grubbs, Angew. Chem., Int. Ed., 2011, 50, 5882–5885 Search PubMed.
- K. Zhang and G. N. Tew, ACS Macro Lett., 2012, 1, 574–579 Search PubMed.
- D. Pal, Z. Miao, J. B. Garrison, A. S. Veige and B. S. Sumerlin, Macromolecules, 2020, 53, 9717–9724 Search PubMed.
- Z. Miao, S. A. Gonsales, C. Ehm, F. Mentink-Vigier, C. R. Bowers, B. S. Sumerlin and A. S. Veige, Nat. Chem., 2021, 13, 792–799 Search PubMed.
- A. Narumi, M. Yamada, Y. Unno, J. Kumaki, W. H. Binder, K. Enomoto, M. Kikuchi and S. Kawaguchi, ACS Macro Lett., 2019, 8, 634–638 Search PubMed.
- D. Pal, J. B. Garrison, Z. Miao, L. E. Diodati, A. S. Veige and B. S. Sumerlin, Macromolecules, 2022, 55, 7446–7453 Search PubMed.
- L. Xiao, L. Qu, W. Zhu, Y. Wu, Z. Liu and K. Zhang, Macromolecules, 2017, 50, 6762–6770 Search PubMed.
- C. E. Wang, H. Wei, N. Tan, A. J. Boydston and S. H. Pun, Biomacromolecules, 2016, 17, 69–75 Search PubMed.
- X.-Y. Tu, C. Meng, Y.-F. Wang, L.-W. Ma, B.-Y. Wang, J.-L. He, P.-H. Ni, X.-L. Ji, M.-Z. Liu and H. Wei, Macromol. Rapid Commun., 2018, 39, 1700744 Search PubMed.
- J. Yang, R. Wang and D. Xie, Macromolecules, 2019, 52, 7042–7051 Search PubMed.
- C. Chen, M. K. Singh, K. Wunderlich, S. Harvey, C. J. Whitfield, Z. Zhou, M. Wagner, K. Landfester, I. Lieberwirth, G. Fytas, K. Kremer, D. Mukherji, D. Y. W. Ng and T. Weil, Nat. Commun., 2021, 12, 3959 Search PubMed.
- A. Levenson, C. Morrison, P.-R. Huang, T.-W. Wang, Z. Carter-Schwendler and M. Golder, ACS Macro Lett., 2023, 12, 1286–1292 Search PubMed.
- R. W. Clarke, M. R. Caputo, L. Polo Fonseca, M. L. McGraw, L. T. Reilly, K. A. Franklin, A. J. Müller and E. Y.-X. Chen, J. Am. Chem. Soc., 2024, 146, 4930–4941 Search PubMed.
- M. L. McGraw, R. W. Clarke and E. Y.-X. Chen, J. Am. Chem. Soc., 2021, 143, 3318–3322 Search PubMed.
- C. Hu, E. Louisy, G. Fontaine and F. Bonnet, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3175–3179 Search PubMed.
- S. Dutta, M. A. Wade, D. J. Walsh, D. Guironnet, S. A. Rogers and C. E. Sing, Soft Matter, 2019, 15, 2928–2941 Search PubMed.
- J. Noh, M. B. Koo, J. Jung, G. I. Peterson, K. T. Kim and T.-L. Choi, J. Am. Chem. Soc., 2023, 145, 18432–18438 Search PubMed.
- J. Noh, G. I. Peterson and T.-L. Choi, Angew. Chem., Int. Ed., 2021, 60, 18651–18659 Search PubMed.
- G. I. Peterson, W. Ko, Y.-J. Hwang and T.-L. Choi, Macromolecules, 2020, 53, 7795–7802 Search PubMed.
- Y. Liu, Y. Wu and F. Zhou, Langmuir, 2023, 39, 37–44 Search PubMed.
- Z. Wang, L. Scheres, H. Xia and H. Zuilhof, Adv. Funct. Mater., 2020, 30, 1908098 Search PubMed.
- S. Mukherjee, R. Xie, V. G. Reynolds, T. Uchiyama, A. E. Levi, E. Valois, H. Wang, M. L. Chabinyc and C. M. Bates, Macromolecules, 2020, 53, 1090–1097 Search PubMed.
- V. G. Reynolds, S. Mukherjee, R. Xie, A. E. Levi, A. Atassi, T. Uchiyama, H. Wang, M. L. Chabinyc and C. M. Bates, Mater. Horiz., 2020, 7, 181–187 Search PubMed.
- K. Zhang, M. A. Lackey, J. Cui and G. N. Tew, J. Am. Chem. Soc., 2011, 133, 4140–4148 Search PubMed.
- Y. Chen, Z. Fan, Y. Liu, Y. Wang, Z. Wang, R. Zhang, B. Ma, Y. Liu, W. Zhang, L. Tang and T. Sun, Giant, 2023, 15, 100177 Search PubMed.
- D. Konar, V. K. Jakhar, K. A. Stewart, D. W. Lester, A. S. Veige and B. S. Sumerlin, Macromolecules, 2024, 57, 1779–1787 Search PubMed.
- Y. Adeli, F. Owusu, F. A. Nüesch and D. M. Opris, ACS Appl. Mater. Interfaces, 2023, 15, 20410–20420 Search PubMed.
- X. Yu, Y. Wang, M. Li, Y. Zhang, Y. Huang, Q. Qian, Y. Zheng, Q. Hou and X. Fan, ACS Appl. Polym. Mater., 2023, 5, 2750–2759 Search PubMed.
- P. Xu, S. Wang, A. Lin, H.-K. Min, Z. Zhou, W. Dou, Y. Sun, X. Huang, H. Tran and X. Liu, Nat. Commun., 2023, 14, 623 Search PubMed.
- B. R. Clarke, C. L. Witt, M. Ilton, A. J. Crosby, J. J. Watkins and G. N. Tew, Angew. Chem., Int. Ed., 2024, 63, e202318220 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06050d |
| This journal is © The Royal Society of Chemistry 2024 |