DOI:
10.1039/D4SC05793G
(Edge Article)
Chem. Sci., 2024,
15, 19991-20001
Dithienonaphthobisthiadiazole synthesized by thienannulation of electron-deficient rings: an acceptor building unit for high-performance π-conjugated polymers†
Received
28th August 2024
, Accepted 22nd October 2024
First published on 18th November 2024
Abstract
The development of building units for π-conjugated polymers is a driving force in advancing the field of organic electronics. In this study, we designed and synthesized dithienonaphthobisthiadiazole (TNT) as a thiophene-fused acceptor (A) building unit and two TNT-based π-conjugated polymers named PTNT2T and PTNT1-F. We found that the microwave-assisted thiophene annulation reaction (thienannulation) of arylethynylated naphthobisthiadiazole (NTz) via C–H functionalization effectively produced TNT moieties. With the π-extended structure of TNT, the polymers had rigid backbones that benefited in-plane and out-of-plane charge carrier transport. Organic field-effect transistors (OFETs) based on PTNT2T exhibited hole mobilities as high as 1.10 cm2 V−1 s−1. Furthermore, organic photovoltaic cells (OPVs) based on PTNT1-F showed high power conversion efficiencies of up to 17.4% when combined with a nonfullerene acceptor. This work provides an efficient method for the thienannulation of electron-deficient rings to access thiophene-fused A building units and shows the great promise of TNT as a building unit for high-performance π-conjugated polymers for organic electronic devices.
Introduction
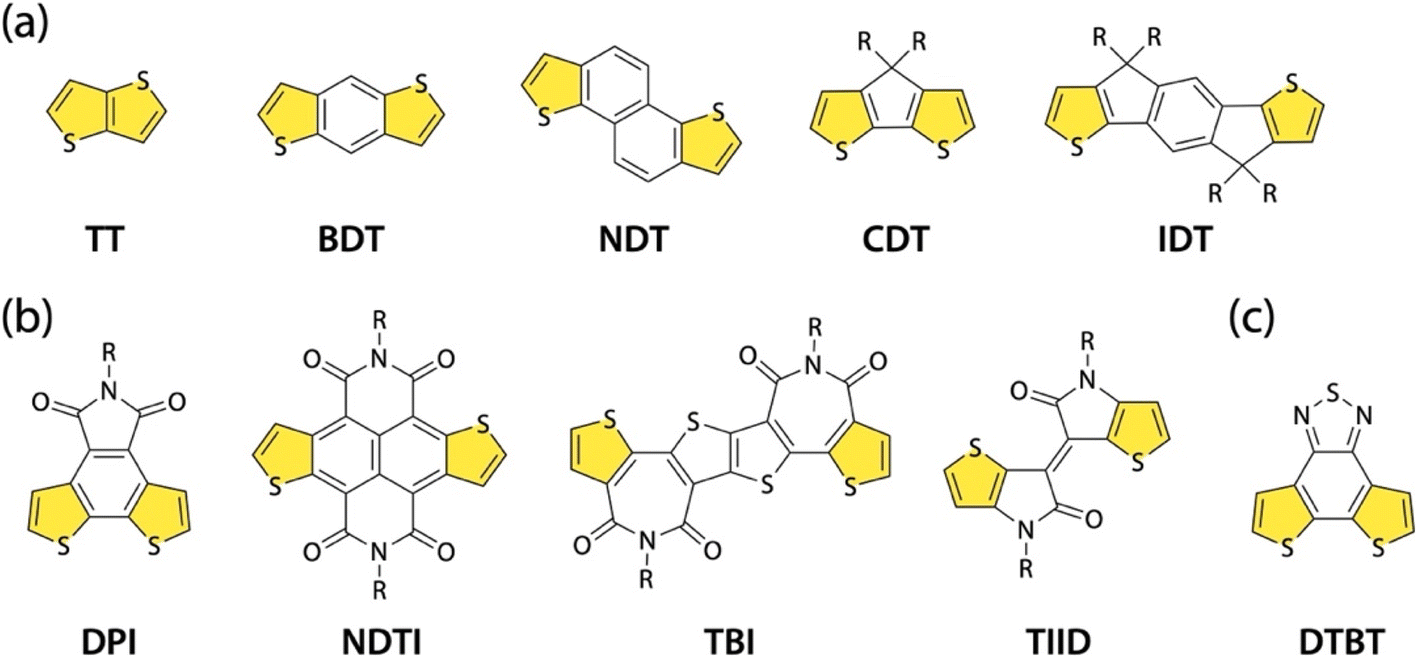
π-Conjugated polymers are an important class of materials in organic electronics.1 This is evidenced by their broad applications in organic field-effect transistors (OFETs),2,3 organic electrochemical transistors (OECTs),4,5 organic photovoltaics (OPVs),6–9 organic photodetectors (OPDs),10 and organic thermoelectrics (OTEs).11 A driving force in advancing this research field is the development of π-conjugated polymers with alternating donor (D)–acceptor (A) building units, that is, D–A polymers.12,13 For instance, they can have small bandgaps owing to the intrachain D–A interaction, which is beneficial for near-IR absorption and thereby OPVs and OPDs.14 In addition, they can form a crystalline structure with close π–π stacking owing to the interchain dipole–dipole interaction, which is beneficial for charge carrier transport.12 Through the judicious choice of the D and A building units, the electronic, optical, and packing structure of the polymers can be precisely controlled to match the needs of the application. Thus, the design of building units is a key issue in the development of D–A polymers. A promising design strategy for the building unit is the fusion of thiophene rings to the end of a heterocyclic structure, which can enhance the backbone rigidity and interchain interaction due to the π-extended structure.15
A number of thiophene-fused rings as the D building unit, such as thienothiophene (TT),16 benzodithiophene (BDT),17 naphthodithiophene (NDT),18,19 cyclopentadithiophene (CDT),20,21 and indacenodithiophene (IDT)22–24 (Fig. 1a), have been investigated for the development of π-conjugated polymers in organic electronics. Key to the synthesis of thiophene-fused rings is the thiophene annulation reaction (thienannulation). TT, BDT, and NDT are typically synthesized via thienannulation.25 On the other hand, thiophene-fused rings as A building units are limited to such imide- and amide-containing compounds as dithienophthalimide (DPI),26,27 naphthodithiophenebisimide (NDTI),28 dithienylthiophenebisimide (TBI),29,30 and thienoisoindigo (TIID)31,32 (Fig. 1b). An example of a nonimide or nonamide thiophene-fused A building unit is dithienobenzothiadiazole (DTBT) (Fig. 1c),33 in which two thiophenes are fused to the end of benzothiadiazole, a well-known A building unit.34 A DTBT-based polymer, D18, exhibits high power conversion efficiencies (PCEs) approaching 20% in nonfullerene (NFA) OPVs.35,36 One possible reason for the limited number of thiophene-fused A building units is the difficulty of applying conventional thienannulation to the A units. Among those thiophene-fused A building units, only NDTI was synthesized using thienannulation. Therefore, in addition to the exploration of new thiophene-fused A building units, the development of a new thienannulation methodology applicable to A units is important for the creation of high-performance D–A polymers.
 |
| | Fig. 1 Chemical structures of thiophene-fused rings (a) as the D building unit: TT, BDT, NDT, CDT, and IDT; (b) as the A building unit based on imide or amide compounds: DPI, NDTI, TBI, and TIID; and (c) as the A building unit based on the benzothiadiazole compound: DTBT. | |
Naphtho[1,2-c:5,6-c′]bis[1,2,5]thiadiazole (NTz) (Fig. 2a), in which two benzothiadiazoles are fused, is a promising A building unit.37–39 We previously reported that NTz-based polymers such as PNTz4T and PNTz1-F showed good performance in OFETs and/or OPVs (Fig. 2b).40,41 Herein, as a new family of thiophene-fused A building units, we designed and synthesized dithieno[3′,2′:3,4; 3′′,2′′:7,8]naphtho[1,2-c:5,6-c′]bis([1,2,5]thiadiazole) (TNT) (Fig. 2a), in which thiophenes are fused to the ends of NTz. Notably, we found that with the assistance of microwaves in the thienannulation reaction via C–H functionalization reported by Itami and co-workers,42 arylethynylated NTz precursors were converted into TNT derivatives in reasonably high yields. The TNT-based monomers were homopolymerized and copolymerized with a BDT-based monomer, providing π-conjugated polymers PTNT2T and PTNT1-F, which are regarded as the counterparts of PNTz4T and PNTz1-F, respectively (Fig. 2d). To study the impact of thiophene fusion to the ends of NTz, we discuss the properties, thin film structures, charge carrier transport properties, and photovoltaic performances of PTNT2T and PTNT1-F in comparison with those of NTz counterparts PNTz4T and PNTz1-F.
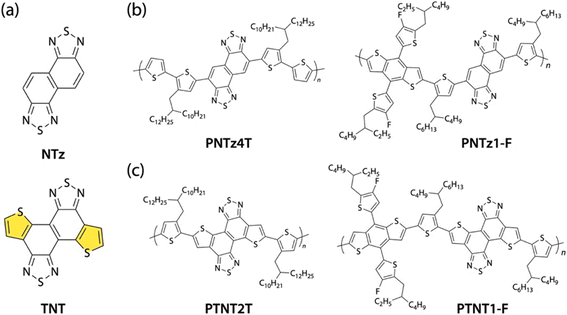 |
| | Fig. 2 Chemical structures of (a) naphtho[1,2-c:5,6-c′]bis[1,2,5]thiadiazole (NTz) and dithieno[3′,2′:3,4; 3′′,2′′:7,8]naphtho[1,2-c:5,6-c′]bis([1,2,5]thiadiazole) (TNT); (b) NTz-based polymers PNTz4T and PNTz1-F; and (c) TNT-based polymers PTNT2T and PTNT1-F. | |
Results and discussion
Synthesis of TNT derivatives via thienannulation
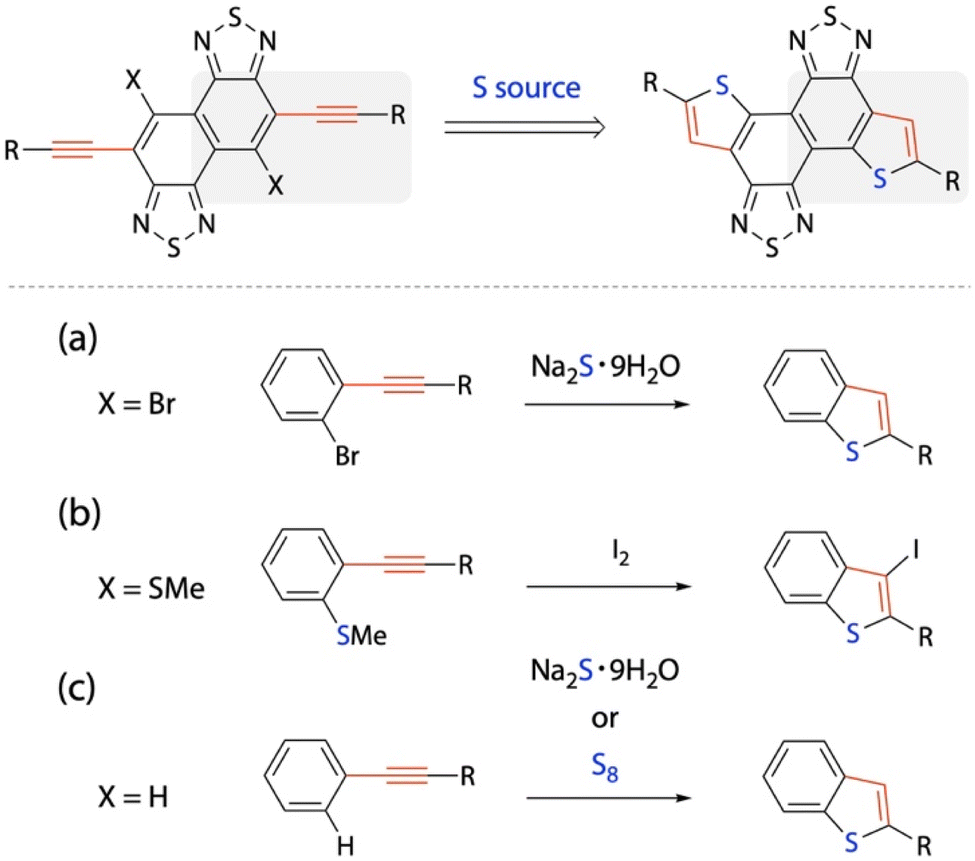
Key to constructing the TNT structure is the thienannulation of NTz via the arylethynylated NTz precursor (Scheme 1). The reaction can be partially regarded as the formation of benzothiophene. Conventional thienannulation reactions use precursors with a halogen or an alkylthio group at the ortho position of the arylethynyl group (Scheme 1a and b).25,43 On the other hand, it has been reported that non-ortho-substituted arylethynylbenzene moieties undergo thienannulation with the use of sodium sulfide hydrate (Na2S·9H2O)28 or elemental sulfur (S8)42 to form benzothiophene moieties (Scheme 1c). As these thienannulation reactions are simpler than the conventional ones for benzothiophene formation using arylethynylbenzene moieties, we exploited these synthetic protocols for the formation of the TNT structure.
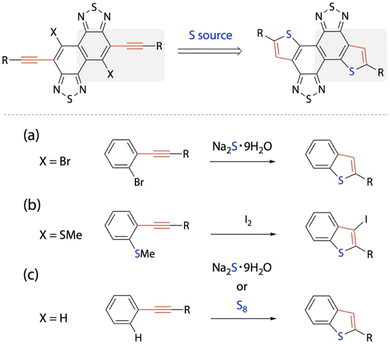 |
| | Scheme 1 Synthetic strategy for the construction of thiophene rings at the ends of NTz. (a and b) Conventional thienannulations of disubstituted benzene and (c) thienannulations of monosubstituted (ethynylated) benzene via C–H functionalization. | |
Scheme 2 shows the synthetic route to TNT derivatives. To examine the thienannulation, we synthesized an ethynylalkylthiophene-flanking NTz derivative (3) using the Sonogashira cross-coupling reaction between dibromo-NTz (1) and 2-ethynyl-3-alkylthiophene (2), where R1 = 2-decyltetradecyl (DT) and R2 = H. It should be noted that the alkyl groups were attached to the 3-position of the thiophene ring to avoid the formation of a double-cyclized byproduct during the thienannulation when S8 was used, such as thienothiophene-flanking NTz derivatives. We first tested the thienannulation of 3 using Na2S·9H2O. It, however, provided a complex mixture without a TNT derivative (4). We then tested the thienannulation of 3 using S8 (2 equivalents) following the reported protocol.42 The use of DMF and DMAc as the solvent at 140 °C afforded 4. In contrast, the use of NMP, DMSO, and DMPU did not afford 4 and likely led to decomposition at the same temperature (Table 1, entries 1–6). However, the yield was limited to 9% (DMAc) and remained so even when the temperature was increased to 180 °C. It has been proposed that this type of thienannulation is initiated by the electrophilic reaction of the substrate with S8. In this regard, it is quite difficult for 3 to undergo an electrophilic reaction owing to the strong electron deficiency of NTz. We decided to use a microwave reactor to accelerate the reaction. As a result, the yield was improved to 16% (entry 7) and 25% when the amount of S8 was increased to 4 equivalents (entry 8) (Table 1). We further hypothesized that the α-position of alkylthiophene could cause some side reactions and decrease the yield. Thus, we synthesized 3 using 2 with the tert-butyldimethylsilyl (TBS) group as R2 (Scheme S1†). Notably, the thienannulation of silyl-protected 2 significantly improved the yield of 4 to 42% (Table 1, entry 9). Moreover, the gram-scale synthesis further improved the yield to 59%. It is worth noting that when the less bulky 2-hexyldecyl (HD), 2-butyloctyl (BO), and dodecyl (C12) groups were attached to 3, the yield was further improved to 46–56% (entries 10–12). When we applied the optimized thienannulation conditions to the BTz derivative, alkylthiophene-flanking DTBT was obtained in a similar yield (50%) to the TNT derivative (Scheme S2†). This methodology was able to more easily access DTBT building units than the conventional methodology.33
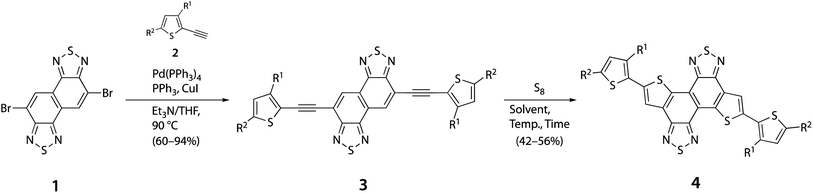 |
| | Scheme 2 Synthesis of TNT derivatives via thienannulation. | |
Table 1 Reaction conditions tested for the thienannulation of 3a
| Entry |
Substituent |
Conditions |
Yield (%) |
| R1 |
R2 |
Solventb |
S8 (equiv.) |
Temp.c (°C) |
Time |
|
DT = 2-decyltetradecyl, HD = 2-hexyldecyl, BO = 2-butyloctyl, C12 = dodecyl, TBS = tert-butyldimethylsilyl.
DMF = N,N-dimethylformamide, NMP = N-methylpyrrolidone, DMSO = dimethyl sulfoxide, DMPU = N,N′-dimethylpropyleneurea, DMAc = N,N-dimethylacetamide.
Following the Arrhenius equation, the total amount of heat for microwave-assisted heating at 200 °C for 45 min is equivalent to that for heating at 140 °C for 48 h.
1.5 g (1.1 mmol) of 3 was used.
|
| 1 |
DT |
H |
DMF |
2 |
140 |
48 h |
2 |
| 2 |
DT |
H |
NMP |
2 |
140 |
48 h |
n.d. |
| 3 |
DT |
H |
DMSO |
2 |
140 |
48 h |
n.d. |
| 4 |
DT |
H |
DMPU |
2 |
140 |
48 h |
n.d. |
| 5 |
DT |
H |
DMAc |
2 |
140 |
48 h |
9 |
| 6 |
DT |
H |
DMAc |
2 |
180 |
48 h |
9 |
| 7 |
DT |
H |
DMAc |
2 |
200 (microwave) |
45 min |
16 |
| 8 |
DT |
H |
DMAc |
4 |
200 (microwave) |
45 min |
25 |
| 9 |
DT |
TBS |
DMAc |
4 |
200 (microwave) |
45 min |
42 (59)d |
| 10 |
HD |
TBS |
DMAc |
4 |
200 (microwave) |
45 min |
46 |
| 11 |
BO |
TBS |
DMAc |
4 |
200 (microwave) |
45 min |
50 |
| 12 |
C12 |
TBS |
DMAc |
4 |
200 (microwave) |
45 min |
56 |
Synthesis of TNT-based polymers
Considering the solubility of the polymer, we selected DT and HD groups as the alkyl groups for the synthesis and evaluation of PTNT2T and PTNT1-F, respectively. The desilylation of 4a (R1 = DT) and 4b (R1 = HD) quantitatively afforded 5a and 5b, and the subsequent dibromination yielded the TNT monomers (6a and 6b) in approximately 90% (Scheme 3). Finally, PTNT2T and PTNT1-F were synthesized by the homopolymerization of 6a (R1 = DT) using hexamethylditin and the copolymerization of 6b (R1 = HD) with a distannylated benzodithiophene derivative (7), respectively, via the Stille cross-coupling reaction. The number-average molecular weight (Mn) and the dispersity (Đ) were 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 2.0 for PTNT2T, and 41000 and 2.1 for PTNT1-F, respectively (Fig. S1 and Table S1†). Interestingly, PTNT2T exhibited similar solubility to PNTz4T even though TNT was a more π-extended building unit than NTz. Both polymer solutions in chlorobenzene (CB) with a concentration of approximately 8 g L−1 remained in solution at room temperature after heating to 100 °C to dissolve the polymers. PTNT1-F showed similar solubility to PNTz1-F and was soluble in chloroform (CF) at around 40–50 °C when its molecular weight was similar to that of PNTz1-F. Thermogravimetric analysis revealed that both polymers were thermally stable up to 400 °C, and differential scanning calorimetry showed no phase transition peak below 350 °C (Fig. S2 and S3†).
000 and 2.0 for PTNT2T, and 41000 and 2.1 for PTNT1-F, respectively (Fig. S1 and Table S1†). Interestingly, PTNT2T exhibited similar solubility to PNTz4T even though TNT was a more π-extended building unit than NTz. Both polymer solutions in chlorobenzene (CB) with a concentration of approximately 8 g L−1 remained in solution at room temperature after heating to 100 °C to dissolve the polymers. PTNT1-F showed similar solubility to PNTz1-F and was soluble in chloroform (CF) at around 40–50 °C when its molecular weight was similar to that of PNTz1-F. Thermogravimetric analysis revealed that both polymers were thermally stable up to 400 °C, and differential scanning calorimetry showed no phase transition peak below 350 °C (Fig. S2 and S3†).
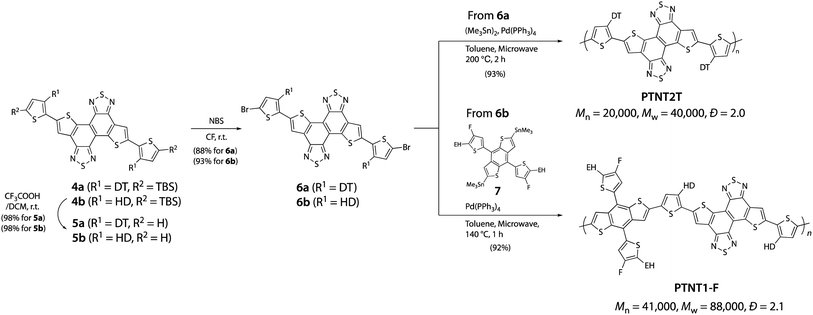 |
| | Scheme 3 Synthetic route to TNT-based polymers. | |
Comparison of electronic properties between NTz and TNT
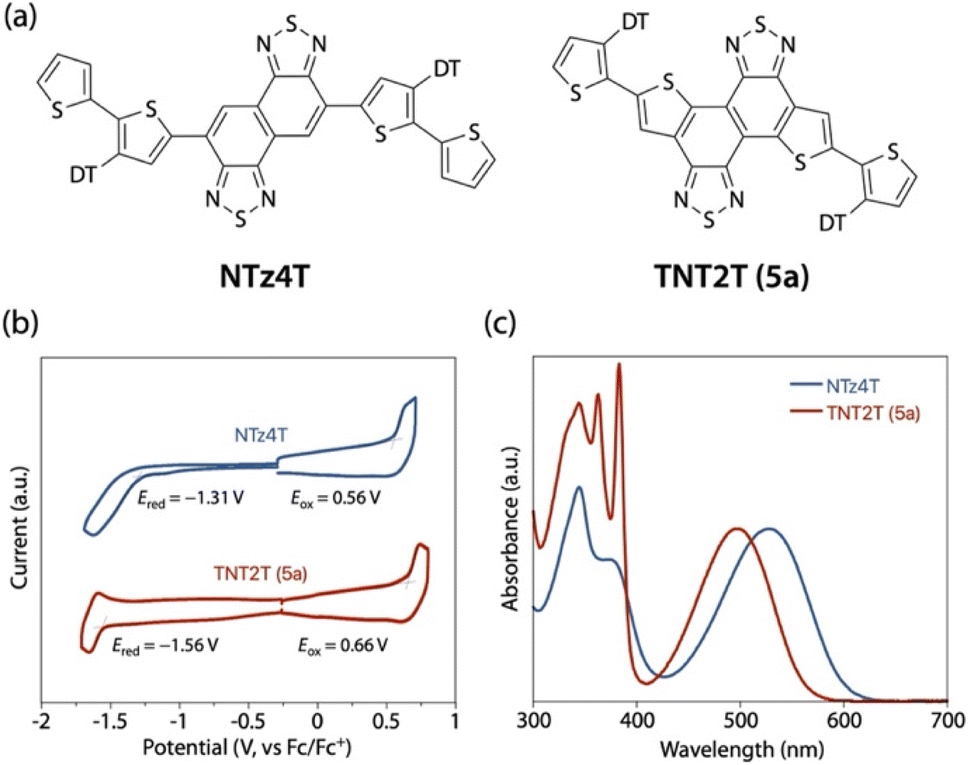
To examine the effect of thiophene-fusion on the electronic properties, we carried out the electrochemical and optical measurements for 5a (TNT2T) in comparison with an NTz counterpart, NTz4T (Fig. 3a). Using cyclic voltammetry (CV), we determined the HOMO and LUMO energy levels (EHOMO and ELUMO, respectively), or ionization potential (IP) and electron affinity (EA), respectively, of the compounds (Fig. 3b). Based on the onset oxidation and reduction potentials (Eox and Ered, respectively), the EHOMO and ELUMO of NTz4T were estimated to be −5.36 eV and −3.49 eV and those of TNT2T were −5.46 and −3.24 eV, respectively. Thus, the EHOMO and ELUMO of TNT2T were deeper and shallower by 0.10 and 0.25 eV than those of NTz4T, respectively. The lower EHOMO of TNT2T than that of NTz4T is most likely due to the weaker electron-rich nature of the alkylthiophene moieties for TNT2T than the alkylbithiophene moieties for NTz4T. The shallower ELUMO of TNT2T than that of NTz4T can be explained by the weaker electron-poor nature of the TNT moiety than the NTz moiety, because, in TNT, two electron-rich thiophenes are fused to NTz.
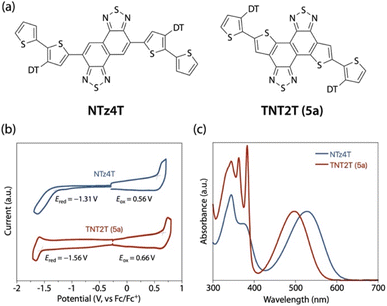 |
| | Fig. 3 (a) Chemical structures of NTz4T and TNT2T (5a) that correspond to the repeat unit for PNTz4T and PTNT2T. (b) Cyclic voltammograms and (c) UV-vis absorption spectra of NTz4T and TNT2T in solution. | |
Fig. 3c depicts the absorption spectra of the compounds in CB solution. The absorption band for TNT2T was blue-shifted by 30–40 nm compared to that for NTz4T; the absorption maxima (λmax) and onset (λonset) for NTz4T were 528 nm and 600 nm and those for TNT2T were 497 nm and 561 nm, respectively. The blue-shift in TNT2T relative to that in NTz4T can be explained by the reduced intramolecular D–A interaction (push–pull effect); again, the alkylthiophene moieties as the D unit for TNT2T are less electron-rich than the alkylbithiophene moieties for NTz4T, and the TNT moiety as the A unit for TNT2T is less electron-poor than the NTz moiety for NTz4T. These electronic properties are consistent with those predicted by the DFT calculation (Fig. S4†).
Polymer properties
The EHOMO and ELUMO of polymers were also evaluated by CV using the thin films (Fig. 4a). Based on the onset redox potentials, the EHOMO and ELUMO of PTNT2T were estimated to be −5.42 and −3.18 eV, respectively, which were deeper by 0.17 eV and shallower by 0.15 eV than those of PNTz4T (EHOMO = −5.25, ELUMO = −3.33 eV), respectively (Table 2). The EHOMO and ELUMO of PTNT1-F were estimated to be −5.53 and −3.17 eV, which were deeper by 0.15 eV and shallower by 0.14 eV than those of PNTz1-F (EHOMO = −5.38 eV, ELUMO = −3.31 eV), respectively.
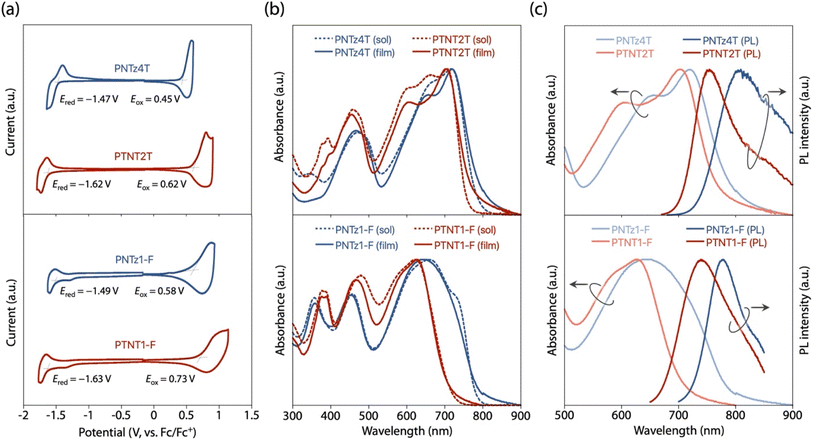 |
| | Fig. 4 (a) Cyclic voltammograms, (b) UV-vis absorption spectra in both solution and film, and (c) PL spectra along with UV-vis absorption spectra in film for PNTz4T and PTNT2T (upper panel), and PNTz1-F and PTNT1-F (lower panel). | |
Table 2 Electrochemical and optical properties of the polymers
| Compounds |
E
HOMO/IPa (eV) |
E
LUMO/EAa (eV) |
λ
max
(nm) |
λ
onset
(nm) |
E
optgd (eV) |
λ
emmax
(nm) |
ΔESSf (eV) |
| Sol |
Film |
Sol |
Film |
|
Estimated using redox potentials determined from cyclic voltammograms. Solution samples were used for NTz2T and TNT2T and the thin film samples were used for the polymers. All the potentials were calibrated with the half-wave potential of the ferrocene/ferrocenium redox couple measured under identical conditions. HOMO and LUMO energy levels were estimated using the following equations, HOMO (eV): −4.80 − Eonsetox, LUMO (eV): −4.80 − Eonsetred.
Absorption maximum.
Absorption onset.
Optical band gap calculated by using the following equation: Eoptg (eV) = 1240/λedge (nm).
Maximum emission excited at λmax.
Stokes shift for polymer thin films in energy.
|
|
NTz4T
|
−5.36 |
−3.49 |
528 |
— |
600 |
— |
— |
— |
— |
|
TNT2T (5a) |
−5.46 |
−3.24 |
497 |
— |
561 |
— |
— |
— |
— |
|
PNTz4T
|
−5.25 |
−3.33 |
717 |
720 |
784 |
800 |
1.55 |
809 |
0.19 |
|
PTNT2T
|
−5.42 |
−3.18 |
703 |
705 |
758 |
777 |
1.60 |
754 |
0.11 |
|
PNTz1-F
|
−5.38 |
−3.31 |
658 |
636 |
780 |
793 |
1.56 |
778 |
0.36 |
|
PTNT1-F
|
−5.53 |
−3.17 |
620 |
629 |
710 |
711 |
1.74 |
739 |
0.29 |
The UV-vis absorption spectra of the polymers in the CB solution and the thin films are shown in Fig. 4b, and the corresponding parameters are summarized in Table 2. In solution, the λmax and λonset for PTNT2T were 703 and 758 nm, respectively, which were blue-shifted by ca. 20 nm compared to those for PNTz4T (λmax = 717 nm and λonset = 784 nm). PTNT1-F exhibited λmax and λonset of 620 and 710 nm, which were significantly blue-shifted compared to those for PNTz1-F (λmax = 658 nm and λonset = 780 nm). It should be noted that the main absorption bands in the thin films were similar to those in solution for each of the polymers, suggesting partial aggregation derived from the coplanar backbone. The λmax and λonset in the thin film were similar to those in solution for all the polymers: PTNT2T (λmax = 705 nm and λonset = 777 nm), PNTz4T (λmax = 720 nm and λonset = 800 nm), PNTz1-F (λmax = 636 nm and λonset = 793 nm), and PTNT1-F (λmax = 629 nm and λonset = 711 nm). Accordingly, the Eoptg calculated from the λonset for PTNT2T was Eoptg = 1.60 eV, wider than that for PNTz4T (Eoptg = 1.55 eV). Similarly, PTNT1-F had a wider Eoptg (1.74 eV) than PNTz1-F (Eoptg = 1.56 eV). This agrees with the experimental result that the TNT-based polymers had deeper EHOMO and shallower ELUMO than the NTz-based polymers, as described above.
The variations in the energy levels and the absorption spectra between the NTz- and TNT-based polymers were similar to those observed between NTz4T and TNT2T. As discussed above, TNT is expected to have a weaker electron-poor nature than NTz, and hence the TNT-based polymers have reduced intrachain D–A interaction compared to the NTz-based polymers. This would result in larger Eoptgs (blue-shifted absorption) and thus the shallower ELUMOs and deeper EHOMOs for the TNT-based polymers than for the NTz-based polymers. These polymer properties were consistent with the computation using the DFT method at the B3LYP/6-31g(d) level (Fig. S5†).
We also studied the PL spectra of the polymers in the thin film (Fig. 4c and Table 2). The emission maxima (λemmax) of PTNT2T and PNTz4T were λemmax = 754 and 809 nm, respectively. Consequently, the Stokes shift value (ΔESS) for PTNT2T (ΔESS = 0.11 eV) was smaller than that for PNTz4T (ΔESS = 0.19 eV). Similarly, the λemmax of PTNT1-F and PNTz1-F were λemmax = 739 and 778 nm, respectively, and therefore, ΔESS for PTNT1-F (ΔESS = 0.29 eV) was smaller than that for PNTz1-F (ΔESS = 0.36 eV). These results suggest that the TNT-based polymers had higher backbone rigidity than their NTz-based counterparts, most likely due to the π-extended structure of TNT along the polymer backbone.
We further investigated the temperature dependence (20–100 °C) of the absorption spectra in the CB solution. In PNTz4T (Fig. 5a), the absorption spectrum largely blue-shifted and the absorption shape became featureless upon heating to 100 °C. By contrast, in PTNT2T (Fig. 5b), the absorption spectrum did not show such a large blue-shift and it still had vibronic structures even when heated to 100 °C. In PNTz1-F (Fig. 5c), as similar to the case in PNTz4T, the absorption spectrum was significantly blue-shifted and became structureless upon heating. In PTNT1-F (Fig. 5d), the spectrum changed only slightly with increasing temperature; the change was even less than that in PTNT2T. These results also validate that TNT-based polymers have more rigid backbones than NTz-based polymers.
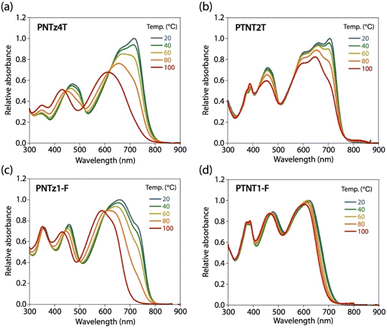 |
| | Fig. 5 Temperature-dependence of the absorption spectra in the CB solution for (a) PNTz4T, (b) PTNT2T, (c) PNTz1-F, and (d) PTNT1-F. | |
OFET properties
We tested OFET devices with a top-gate/bottom-contact architecture, in which a patterned Au electrode was pretreated with octanethiol, and CYTOP was employed as the dielectric layer.44 The polymer layer was deposited by spin-coating from a CB solution and then annealed at 200 °C for 30 min. Fig. 6a and c depict transfer curves and Fig. 6b and d depict output curves of the OFET devices based on these polymers. All polymers exhibited unipolar p-type characteristics with relatively low threshold voltages of around −10 V and high current on and off ratios of ∼105. PTNT2T showed a field-effect hole mobility (μFET) of 1.10 cm2 V−1 s−1, which was higher than that of PNTz4T (μFET = 0.81 cm2 V−1 s−1) (Table 3). PTNT1-F showed a μFET of 0.18 cm2 V−1 s−1, which was also higher than its NTz-based counterpart PNTz1-F (μFET = 0.004 cm2 V−1 s−1) (Table 3). Thus, thiophene fusion improved the in-plane charge carrier transport.
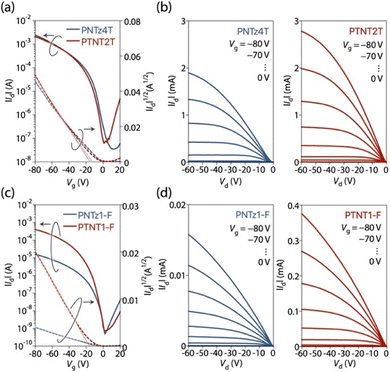 |
| | Fig. 6 (a and c) Transfer and (b and d) output curves of OFET devices: (a and b) PNTz4T and PTNT2T and (c and d) PNTz1-F and PTNT1-F. | |
Table 3 OFET and OPV properties
| Polymer |
OFET |
OPVb |
|
μ
FET [μFETave]a (cm2 V−1 s−1) |
J
SC [JEQESC]c (mA cm−2) |
V
OC (V) |
FF |
PCE [PCEave]d (%) |
|
μ
FET: maximum field-effect hole mobility. μFETave : average field-effect hole mobility of more than 5 different devices.
Y6 was used as the acceptor.
J
EQESC: JSC calculated from the EQE spectrum.
PCE: maximum power conversion efficiency. PCEave: average power conversion efficiency of more than 10 different cells.
|
|
PNTz4T
|
0.81 [0.65] |
22.8 [22.9] |
0.70 |
0.69 |
11.1 [10.8] |
|
PTNT2T
|
1.10 [0.77] |
22.5 [22.8] |
0.80 |
0.74 |
13.4 [13.1] |
|
PNTz1-F
|
0.004 [0.003] |
24.5 [24.4] |
0.79 |
0.69 |
13.3 [12.6] |
|
PTNT1-F
|
0.18 [0.13] |
27.1 [27.0] |
0.86 |
0.74 |
17.4 [17.0] |
OPV properties
OPV cells with a conventional structure (ITO/PEDOT:PSS/polymer:Y6/PNDI-F3N-Br/Ag) were fabricated. Fig. 7a and b show the current density–voltage (J–V) curves and the external quantum efficiency (EQE) spectra of the PTNT2T and PNTz4T cells and Table 3 summarizes the OPV properties. The PTNT2T cell exhibited a short-circuit current density (JSC) of 22.5 mA cm−2, which was similar to that of the PNTz4T cell (JSC = 22.8 mA cm−2). Both cells exhibited EQEs of approximately 70–80% in the polymer and Y6 absorption regions. The JSC values calculated from the EQE spectra (JEQESC) were consistent with the JSC values obtained in the J–V measurement (Table 3). Meanwhile, the PTNT2T cell exhibited a higher open-circuit voltage (VOC) (VOC = 0.80 V) than the PNTz4T cell (VOC = 0.70 V), which can be explained by the lower EHOMO of PTNT2T than that of PNTz4T. Furthermore, the PTNT2T cell provided a higher fill factor (FF) of 0.74 than the PNTz4T cell (FF = 0.69). As a result, the PTNT2T cell exhibited a PCE of 13.4%, which was higher than that of the PNTz4T cell (PCE = 11.1%). Fig. 7c and d show the J–V curves and the EQE spectra of the PTNT1-F and PNTz1-F cells. The PTNT1-F cell exhibited a PCE of 17.4%, which is reasonably high for binary blend OPVs, with a JSC of 27.1 mA cm−2, a VOC of 0.86 V, and an FF of 0.74 (Table 3). The performance of the PTNT1-F cell was markedly higher than those for the PNTz1-F cell (PCE = 13.3%, JSC = 24.5 mA cm−2, VOC = 0.79 V, FF = 0.69) (Table 3). The higher JSC was consistent with the higher EQE values in the PTNT1-F cell than in the PNTz1-F cell, particularly in the 450–650 nm polymer absorption region. The higher VOC in the PTNT1-F cell than in the PNTz1-F cell is also ascribed to the deeper EHOMO of PTNT1-F than that of PNTz1-F.
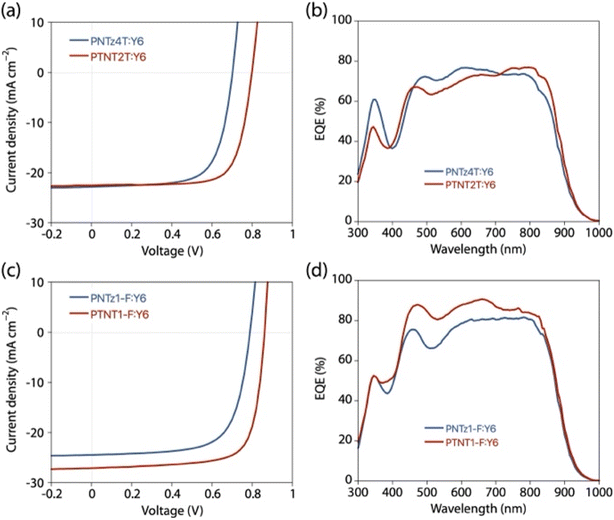 |
| | Fig. 7 (a and c) J–V curves and (b and d) EQE spectra of polymer:Y6 cells. (a and b) PTNT2T and PNTz4T cells and (c and d) PTNT1-F and PNTz1-F cells. | |
We evaluated out-of-plane hole mobility (μSCLC) based on the space-charge-limited current (SCLC) model (Fig. S6†). Notably, the μSCLC of 1.7 × 10−3 cm2 V−1 s−1 for PTNT2T:Y6 blend film was markedly higher than that for PNTz4T:Y6 (μSCLC = 2.8 × 10−4 cm2 V−1 s−1). Furthermore, the PTNT1-F:Y6 blend film exhibited a μSCLC of 8.0 × 10−4 cm2 V−1 s−1, which was also higher than that for PNTz1-F:Y6 (μSCLC = 1.5 × 10−4 cm2 V−1 s−1). The trend in μSCLC correlated well with the photovoltaic performance, e.g., JSC and FF.
Polymer order in thin films
We conducted two-dimensional grazing incidence wide-angle X-ray diffraction (2D GIXD) measurements for the polymer neat and blend films on a glass substrate (Fig. 8a, b, e, f, i and j). PNTz4T and PTNT2T neat films showed diffraction corresponding to the (0 1 0) π–π stacking structures and the (h 0 0) lamellar structures along the qxy and ∼qz axes, respectively, indicating that the backbone orientation was predominantly edge-on for both polymers (Fig. 8a and e). The d-spacing of 3.67 Å (qxy = 1.71 Å−1) for the (0 1 0) π–π stacking structure (dπ) of PTNT2T was wider than that of PNTz4T (dπ = 3.56 Å, qxy = 1.76 Å−1) (Fig. 8i). Moreover, the coherence length (Lπ) calculated using the Scherrer equation for the π–π stacking structure in PTNT2T (Lπ = 17 Å) was significantly shorter than that in PNTz4T (49 Å). Thus, although somewhat surprising, the crystallinity of PTNT2T diminished compared to that of PNTz4T even though the former incorporated a more π-extended core structure than the latter. This was because the alkyl groups were substituted on the thiophene rings at the near side of the TNT moiety in PTNT2T. In contrast, the alkyl groups were substituted on the thiophene rings at the far side of the NTz moiety in PNTz4T (Fig. 9). As indicated by the computational finding that the TNT–alkylthiophene linkage had a larger dihedral angle than the NTz–alkylthiophene linkage (Fig. S5†), the polymer backbone of PTNT2T can be more twisted than that of PNTz4T. Meanwhile, PNTz1-F and PTNT1-F neat films exhibited diffraction corresponding to the (0 1 0) π–π stacking structures and the (1 0 0) lamellar structures along the ∼qz and qxy axes, respectively, indicating a predominantly face-on orientation (Fig. 8b and f). PTNT1-F and PNTz1-F showed similar dπ values of around 3.7 Å and similar Lπ values of 13–14 Å (Fig. 8j), which sharply contrast with the trend observed in PTNT2T and PNTz4T. This is probably because the relatively wide dπ and relatively short Lπ values of PTNT1-F and PNTz1-F are limited by the intermolecular steric hindrance of the alkylthienyl substituents on the BDT unit. Based on the fact that the TNT-based polymers had lower and/or similar crystallinity compared to the NTz-based polymers, the higher μFET observed in the TNT-based polymers than those observed in the NTz-based polymers possibly originated from the rigid polymer backbone that could facilitate intramolecular charge carrier transport.
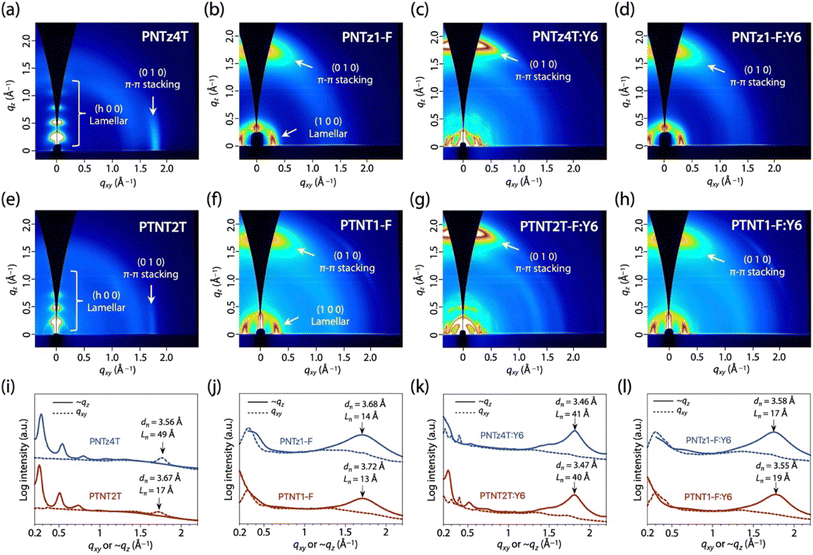 |
| | Fig. 8 (a–h) 2D GIXD patterns of polymer neat and blend films. (i–l) Cross-sectional diffraction profiles cut from the 2D GIXD patterns along the quasi-qz (solid line) and qxy (dotted line) axes. (a, e and i) PNTz4T and PTNT2T, (b, f and j) PNTz1-F and PTNT1-F, (c, g and k) PNTz4T:Y6 and PTNT2T:Y6, and (d, h and l) PNTz1-F:Y6 and PTNT1-F:Y6. | |
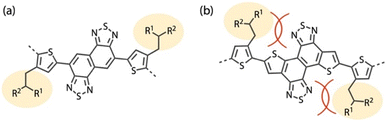 |
| | Fig. 9 Schematic illustration of plausible steric hindrance for (a) NTz-based polymers and (b) TNT-based polymers. | |
We also conducted 2D GIXD measurements for polymer:Y6 blend films on ITO/PEDOT:PSS substrates to clarify the packing order in the OPV cells (Fig. 8c, d, g, h, k and l). All the blend films showed diffractions corresponding to the (0 1 0) π–π stacking structures in the wide-angle region along the qz axis, indicating a predominantly face-on orientation that is favorable for charge carrier transport in OPVs. The PNTz4T:Y6 and PTNT2T:Y6 blend films showed similar dπ values shorter than 3.5 Å with the similar Lπ values of around 40 Å (Fig. 8c, g and k). In addition, the PTNT1-F:Y6 and PNTz1-F:Y6 blend films also showed similar π–π stacking diffractions and dπ and Lπ values of around 3.6 Å and 17–19 Å, respectively (Fig. 8d, h and l). These diffractions resulted from the superposition of the π–π stacking diffractions of the polymer and Y6, and the diffraction of Y6 was likely dominant considering the qz values (Fig. S7 and S8†). Thus, we did not observe significant differences in the packing order between the PNTz4T:Y6 and PTNT2T:Y6 blend films and that between the PTNT1-F:Y6 and PNTz1-F:Y6 blend films. In addition, atomic force microscopy indicated no apparent difference in the film morphology of the cells (Fig. S9†). Thus, the high μSCLC in the TNT-based polymers resulted from the rigid polymer backbone that originated from the π-extended structure of TNT, which plausibly improved the intrachain charge carrier transport.
Conclusion
TNT, a thiophene-fused A building unit for π-conjugated polymers, was designed and synthesized for the first time by a microwave-assisted thienannulation reaction of arylethynylated NTz precursors via C–H functionalization. This synthetic methodology can be applied to various electron-deficient rings to afford thiophene-fused A building units. Two polymer systems were synthesized using the TNT building units: PTNT2T and PTNT1-F. These TNT-based polymers had a shallower ELUMO and a deeper EHOMO than their NTz counterparts (PNTz4T and PNTz1-F), most likely because of the weaker electron deficiency of TNT than that of NTz. An important structural feature of TNT-based polymers is their rigid backbones due to the π-extended TNT building unit, which are beneficial for charge carrier transport. Indeed, the TNT-based polymers showed higher hole mobilities in both in-plane and out-of-plane directions than the NTz-based polymers. Furthermore, the TNT-based polymers also showed higher photovoltaic performance than the NTz-based polymers. We believe that TNT has great potential as a building unit for high-performance π-conjugated polymers.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
To. M. and S. S. synthesized the monomers and polymers and carried out the UV-vis, CV, DFT calculations, and 2D GIXD experiments. M. H. supported the synthesis of the monomers. Y. S. and H. O. carried out PL measurements. Ts. M. conducted fabrication and measurements of OFET devices. Ts. M., To. M., and S. S. conducted fabrication and measurements of OPV cells. Ts. M. and I. O. prepared the manuscript, and Ts. M., To. M., S. S., and M. H. prepared the ESI.† All authors discussed and commented on the manuscript. Ts. M. and I. O. directed the project.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the MIRAI-Program (Grant No. JPMJMI20E2) from the Japan Science and Technology Agency, the project JPNP20015 subsidized by the New Energy and Industrial Technology Development Organization (NEDO), and KAKENHI from the Japan Society for the Promotion of Science (Grant No. 22K14745). 2D GIXD experiments were performed at BL46XU of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2024A1666). The authors thank Dr T. Koganezawa (JASRI) for support in 2D GIXD measurements.
References
-
J. R. Reynolds, B. C. Thompson and T. A. Skotheim, Handbook of Conducting Polymers, CRC Press, Boca Raton, 4th edn, 2019 Search PubMed.
- A. F. Paterson, S. Singh, K. J. Fallon, T. Hodsden, Y. Han, B. C. Schroeder, H. Bronstein, M. Heeney, I. McCulloch and T. D. Anthopoulos, Adv. Mater., 2018, 30, 1801079 CrossRef.
- J. Chen, J. Yang, Y. Guo and Y. Liu, Adv. Mater., 2022, 34, e2104325 CrossRef.
- M. Berggren, X. Crispin, S. Fabiano, M. P. Jonsson, D. T. Simon, E. Stavrinidou, K. Tybrandt and I. Zozoulenko, Adv. Mater., 2019, 31, e1805813 CrossRef.
- N. A. Kukhta, A. Marks and C. K. Luscombe, Chem. Rev., 2022, 122, 4325–4355 CrossRef CAS.
- M. Riede, D. Spoltore and K. Leo, Adv. Energy Mater., 2021, 11, 2002653 CrossRef CAS.
- G. Zhang, F. R. Lin, F. Qi, T. Heumüller, A. Distler, H.-J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K.-Y. Jen and H.-L. Yip, Chem. Rev., 2022, 122, 14180–14274 CrossRef CAS PubMed.
- S. Guan, Y. Li, C. Xu, N. Yin, C. Xu, C. Wang, M. Wang, Y. Xu, Q. Chen, D. Wang, L. Zuo and H. Chen, Adv. Mater., 2024, 36, e2400342 CrossRef PubMed.
- Y. Jiang, S. Sun, R. Xu, F. Liu, X. Miao, G. Ran, K. Liu, Y. Yi, W. Zhang and X. Zhu, Nat. Energy, 2024, 9, 975–986 CrossRef CAS.
- F. P. G. de Arquer, A. Armin, P. Meredith and E. H. Sargent, Nat. Rev. Mater., 2017, 2, 16100 CrossRef.
- B. Russ, A. Glaudell, J. J. Urban, M. L. Chabinyc and R. A. Segalman, Nat. Rev. Mater., 2016, 1, 16050 CrossRef CAS.
- K. Müllen and W. Pisula, J. Am. Chem. Soc., 2015, 137, 9503–9505 CrossRef.
- M. Kim, S. U. Ryu, S. A. Park, K. Choi, T. Kim, D. Chung and T. Park, Adv. Funct. Mater., 2019, 10, 1904545 Search PubMed.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
- M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036–3140 CrossRef CAS PubMed.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328–333 CrossRef CAS PubMed.
- H. Pan, Y. Li, Y. Wu, P. Liu, B. S. Ong, S. Zhu and G. Xu, J. Am. Chem. Soc., 2007, 129, 4112–4113 CrossRef CAS.
- I. Osaka, T. Abe, S. Shinamura and K. Takimiya, J. Am. Chem. Soc., 2011, 133, 6852–6860 CrossRef CAS PubMed.
- I. Osaka, T. Kakara, N. Takemura, T. Koganezawa and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 8834–8837 CrossRef CAS.
- M. Zhang, H. N. Tsao, W. Pisula, C. Yang, A. K. Mishra and K. Müllen, J. Am. Chem. Soc., 2007, 129, 3472–3473 CrossRef CAS PubMed.
- H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, D. Y. Yoon, R. Graf, W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133, 2605–2612 CrossRef CAS.
- W. Zhang, J. Smith, S. E. Watkins, R. Gysel, M. McGehee, A. Salleo, J. Kirkpatrick, S. Ashraf, T. Anthopoulos, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2010, 132, 11437–11439 CrossRef CAS.
- X. Zhang, H. Bronstein, A. J. Kronemeijer, J. Smith, Y. Kim, R. J. Kline, L. J. Richter, T. D. Anthopoulos, H. Sirringhaus, K. Song, M. Heeney, W. Zhang, I. McCulloch and D. M. DeLongchamp, Nat. Commun., 2013, 4, 2238 CrossRef PubMed.
- D. Venkateshvaran, M. Nikolka, A. Sadhanala, V. Lemaur, M. Zelazny, M. Kepa, M. Hurhangee, A. J. Kronemeijer, V. Pecunia, I. Nasrallah, I. Romanov, K. Broch, I. McCulloch, D. Emin, Y. Olivier, J. Cornil, D. Beljonne and H. Sirringhaus, Nature, 2014, 515, 384–388 CrossRef CAS PubMed.
- K. Takimiya, M. Nakano, M. J. Kang, E. Miyazaki and I. Osaka, Eur. J. Org Chem., 2012, 2013, 217–227 CrossRef.
- H. Wang, Q. Shi, Y. Lin, H. Fan, P. Cheng, X. Zhan, Y. Li and D. Zhu, Macromolecules, 2011, 44, 4213–4221 CrossRef CAS.
- L. Li, F. Meng, M. Zhang, Z. Zhang and D. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202206311 CrossRef CAS PubMed.
- Y. Fukutomi, M. Nakano, J.-Y. Hu, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 11445–11448 CrossRef CAS.
- M. Saito, I. Osaka, Y. Suda, H. Yoshida and K. Takimiya, Adv. Mater., 2016, 28, 6921–6925 CrossRef CAS PubMed.
- Y. Wang, Z. Yan, H. Guo, M. A. Uddin, S. Ling, X. Zhou, H. Su, J. Dai, H. Y. Woo and X. Guo, Angew. Chem., Int. Ed., 2017, 56, 15304–15308 CrossRef CAS PubMed.
- G. W. P. van Pruissen, F. Gholamrezaie, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem., 2012, 22, 20387–20393 RSC.
- G. Kim, S.-J. Kang, G. K. Dutta, Y.-K. Han, T. J. Shin, Y. Y. Noh and C. Yang, J. Am. Chem. Soc., 2014, 136, 9477–9483 CrossRef CAS.
- F. A. Arroyave, C. A. Richard and J. R. Reynolds, Org. Lett., 2012, 14, 6138–6141 CrossRef CAS PubMed.
- P. Cong, Z. Wang, Y. Geng, Y. Meng, C. Meng, L. Chen, A. Tang and E. Zhou, Nano Energy, 2023, 105, 108017 CrossRef CAS.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS.
- H. Lu, W. Liu, G. Ran, Z. Liang, H. Li, N. Wei, H. Wu, Z. Ma, Y. Liu, W. Zhang, X. Xu and Z. Bo, Angew. Chem., Int. Ed., 2023, 62, e202314420 CrossRef CAS PubMed.
- M. Wang, X. Hu, P. Liu, W. Li, X. Gong, F. Huang and Y. Cao, J. Am. Chem. Soc., 2011, 133, 9638–9641 CrossRef CAS PubMed.
- I. Osaka, M. Shimawaki, H. Mori, I. Doi, E. Miyazaki, T. Koganezawa and K. Takimiya, J. Am. Chem. Soc., 2012, 134, 3498–3507 CrossRef CAS.
- I. Osaka and K. Takimiya, Adv. Mater., 2017, 29, 1605218 CrossRef.
- V. Vohra, K. Kawashima, T. Kakara, T. Koganezawa, I. Osaka, K. Takimiya and H. Murata, Nat. Photonics, 2015, 9, 403–408 CrossRef CAS.
- L.-H. Chou, T. Mikie, M. Saito, C.-L. Liu and I. Osaka, ACS Appl. Mater. Interfaces, 2022, 14, 14400–14409 CrossRef CAS PubMed.
- L. Meng, T. Fujikawa, M. Kuwayama, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2016, 138, 10351–10355 CrossRef CAS PubMed.
- D. Yue and R. C. Larock, J. Org. Chem., 2002, 67, 1905–1909 CrossRef CAS.
- A. J. Kronemeijer, V. Pecunia, D. Venkateshvaran, M. Nikolka, A. Sadhanala, J. Moriarty, M. Szumilo and H. Sirringhaus, Adv. Mater., 2014, 26, 728–733 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Tomokazu
Morioku
a,
Shota
Suruga
a,
Momoka
Hada
*ab,
Tomokazu
Morioku
a,
Shota
Suruga
a,
Momoka
Hada