
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDifferentiating carrier protein interactions in biosynthetic pathways using dapoxyl solvatochromism†
Matthew G.
Miyada
 ,
Yuran
Choi
,
Kyle
Rich
,
James J.
La Clair
and
Michael D.
Burkart
*
,
Yuran
Choi
,
Kyle
Rich
,
James J.
La Clair
and
Michael D.
Burkart
*
Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0358, USA. E-mail: mburkart@ucsd.edu
First published on 30th October 2024
Abstract
Carrier protein-dependent synthases are ubiquitous enzymes involved both in primary and secondary metabolism. Biocatalysis within these synthases is governed by key interactions between the carrier protein, substrate, and partner enzymes. The weak and transient nature of these interactions has rendered them difficult to study. Here we develop a useful fluorescent solvatochromic probe, dapoxyl-pantetheinamide, to monitor and quantify carrier protein interactions in vitro. Upon loading with target carrier proteins, we observe dramatic shifts in fluorescence emission wavelength and intensity and further demonstrate that this tool has the potential to be applied across numerous biosynthetic pathways. The environmental sensitivity of this probe allows rapid characterization of carrier protein interactions, with the ability to quantitatively determine inhibition of protein–protein interactions. We anticipate future application of these probes for inhibitor screening and in vivo characterization.
Introduction
Carrier protein (CP)-dependent synthases are enzymatic pathways responsible for the biosynthesis of a wide range of important biomolecules. They can be found in primary metabolic pathways in the case of fatty acid synthases (FAS) and secondary metabolic pathways in the case of polyketide synthases (PKS) and non-ribosomal peptide synthetases (NRPS).1–3 CP-dependent synthases consist of multiple enzymatic domains which catalyze reactions on a growing metabolite as it proceeds through an anabolic pathway. Central to the architecture of these multifunctional synthases is the CP, which carries substrates to and from partner enzymes within the synthase. The shuttling of metabolites to a particular enzyme within a synthase is a process heavily regulated by protein–substrate and protein–protein interactions involving the essential CP.4–6While years of research have been dedicated towards elucidating the exact role of CPs within their respective synthases, progress has been hampered by the fact that CP interactions are inherently transient. As a result, experiments probing CP structure and dynamics often require the placement of CPs in unnatural conditions to capture the protein in a particular conformational state. Moreover, differences in substrate specificity between CP-dependent synthases mean that tools used to study CPs are often only applicable towards a specific synthase or subclass of enzymes. Current techniques which can target a wide range of CP-dependent synthases typically offer little information on the internal organization of the synthases and the types of metabolites they produce. From a proteomic standpoint, it would be extremely useful to develop a tool which can not only examine the activity of multiple CP-dependent synthases of an organism in situ, but also distinguish them based on their biomolecular output. In addition, there would be a benefit for molecular tools that are pathway agnostic, such that interactions could be evaluated on multiple CPs and partner enzymes.
The utility of fluorescence in monitoring enzymatic activity has been well established, owing to facile visualization and minimal perturbance to native biomolecular machinery.7,8 Specifically, solvatochromic dyes exhibit measurable changes in fluorescence emission wavelength and intensity depending on their immediate molecular environment.9,10 The fluorophore 5-(4′′-dimethylaminophenyl)-2-(4′-phenyl)oxazole (dapoxyl) and its solvatochromic properties were first reported in 1997.11 A highly solvent-sensitive dye, the dapoxyl fluorophore featured an emission wavelength difference of over 200 nm and an over 20-fold change in fluorescence intensity between hexanes and aqueous CH3CN. The fluorophore's highly solvatochromic properties, combined with its long emission wavelengths, large Stokes shift, and high quantum yields, have made it a particularly attractive fluorophore in biological applications. Since its development and characterization, dapoxyl derivatives have been applied to quickly visualize pH changes, protein conformations, and macromolecular interactions.12–16
The solvatochromism of dapoxyl fluorophore can be partially attributed to its characteristic single bonds between aromatic rings, which have a profound effect on the energy landscape of electronic states reached by the fluorophore upon excitation. These rotatable bonds allow population of a non-fluorescent twisted intramolecular charge transfer state (TICT) to be populated in polar protic solvents. On the other hand, nonpolar constrained environments favor a locally excited conformation featuring coplanar aromatic rings and high levels of molecular orbital overlap, resulting in increased quantum yields and fluorescence.17
Herein, we present the application of a bifunctional solvatochromic CP-targeting probe, dapoxyl-pantetheinamide (1), as a universal tool to study carrier protein-dependent biosynthesis. Its environmental sensitivity makes it ideal for examining CPs, whose interactions with substrates and partner enzymes are numerous and short-lived. As a substrate mimic, dapoxyl-pantetheinamide was expected to occupy a diverse range of enzymatic active sites and binding pockets when incorporated into CP-dependent biosynthetic pathways. Thus, we anticipated dapoxyl-labeled CPs to be distinguished based on both the types of synthases to which they belong and the proteins with which they interact.
Results and discussion
A dapoxyl labeled pantetheinamide probe
As shown in Fig. 1a, our probe design is a bifunctional probe comprised of a dapoxyl fluorophore attached to the terminus of pantetheine through an amide linkage, referred to as a pantetheinamide. The probe was designed to mimic a natural CP substrate, which is typically comprised of a pantetheine “arm” and thioester-linked acyl substrate. Replacement of the thioester linkage with an amide was intended to prevent hydrolysis of the fluorophore for extended periods in aqueous solution. Critically, CPs accept substrates of highly varied chemical and physical properties, allowing for incorporation of synthetic fluorescent reporters into CP-based metabolomes. Pantetheine-based probes similar to dapoxyl-pantetheinamide (1) have been synthesized to monitor and exploit the promiscuity of CPs, particularly those involved in PK and NRP biosynthesis.18,19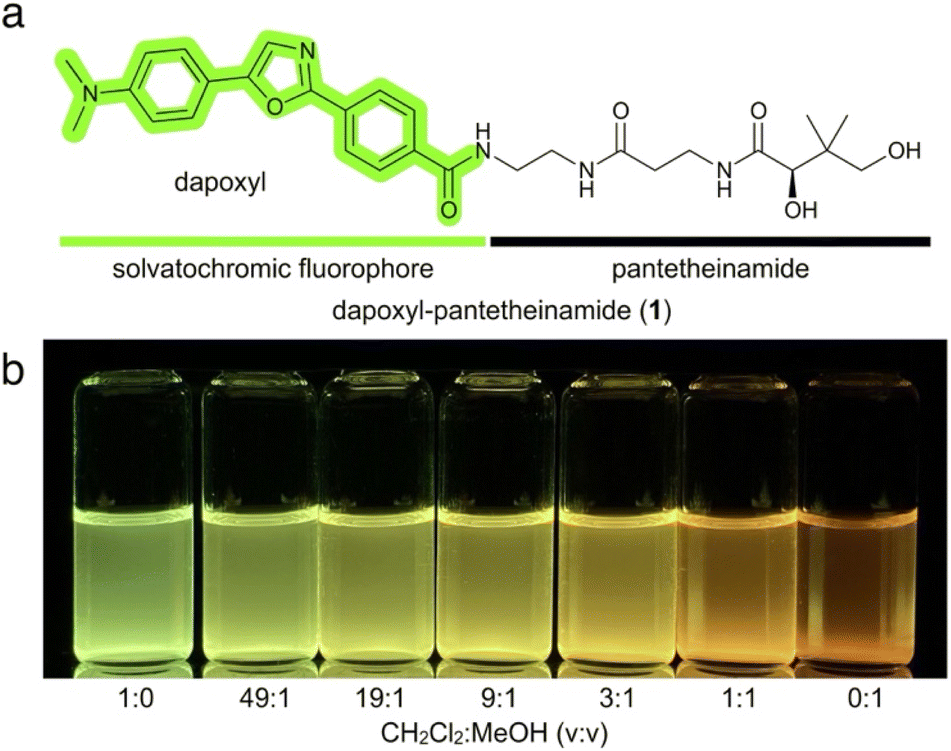 | ||
| Fig. 1 Design and characterization of dapoxyl-pantetheinamide. (a) Dapoxyl-pantetheinamide is a bifunctional probe consisting of a solvatochromic fluorophore and CP-specific substrate analogue. (b) Dapoxyl-pantetheinamide dissolved in solvent mixtures ranging from pure CH2Cl2 to pure MeOH. Visualization under 365 nm UV light. | ||
Dapoxyl-pantetheinamide (1) was synthesized in a nine-step pathway based on the first reported synthesis of dapoxyl (Scheme 1).20 4-Dimethylacetophenone (2) was α-brominated to form 4 using an established two-step procedure.11 The Delépine reaction was employed to convert the α-bromophenone in 4 to the primary α-aminophenone in 6. Coupling with methyl 4-(chloroformyl)benzoate followed by Robinson–Gabriel dehydration of the oxazole ring completed formation of the dapoxyl fluorophore in 8.21 Hydrolysis of the methyl ester, immediately followed by coupling of 9 to p-anisaldehyde acetal protected pantetheinamine 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 and subsequent deprotection of 11 yielded dapoxyl-pantetheinamide (1).
22 and subsequent deprotection of 11 yielded dapoxyl-pantetheinamide (1).
 | ||
| Scheme 1 Synthesis of dapoxyl-pantetheinamide (1) from 4-dimethylaminoacetophenone (2) and p-anisaldehyde acetal protected pantetheinamine 10. | ||
The solvatochromic properties of 1 were immediately evident, as dissolution of the probe in a range of solvent mixtures produced a range of fluorescent colors and intensities (Fig. 1b). As expected, levels of fluorescence were minimal in polar protic solvents such as MeOH, while nonpolar aprotic solvents including CH2Cl2 induced a pronounced fluorescence yield increase and blueshift.
Characterization of fluorescently labeled carrier proteins
We immediately studied the ability of 1 to report on CPs in vitro by using a previously described one-pot reaction.23 As shown in Scheme 2, this is a two-step process that begins with conversion to 1a by the action of three coenzyme A biosynthetic enzymes (CoaA, CoaD, CoaE). Coenzyme A analogue 1a is then loaded on to a CP by the action of Sfp, a 4′-phosphopantetheinyl transferase (PPTase), forming a fluorescently labeled crypto-CP as depicted by 1b in Scheme 2. | ||
| Scheme 2 One-pot chemoenzymatic probe-loading reaction. This process involves two discrete steps run in tandem. The first serves to convert the dapoxyl-pantetheinamide 1 into the corresponding coenzyme A analogue 1a, which is then loaded on to an apo-carrier protein (CP) to form a fluorescent crypto-CP 1b. The most fluorescent state occurs upon protein loading in 1b. | ||
Our studies began with AcpP, a component of Escherichia coli type II fatty acid synthase. This CP was chosen as the initial candidate because of its reported ability to sequester a tethered substrate within the hydrophobic interior of its core four-helical bundle.6 Several hypotheses have been proposed to explain this phenomenon, including protection of the nascent metabolite thioester linkage from hydrolysis and the induction of allosteric conformational changes to control partner enzyme recognition and catalysis.22,23
We therefore expected that, once tethered onto AcpP, the synthetic dapoxyl substrate would induce an observable fluorescence shift compared to its fluorescence alone in solution. Indeed, the bright green dapoxyl-AcpP exhibited a 150 nm blueshift and 15-fold increase in intensity as compared to the weakly red fluorescent dapoxyl-CoA (Fig. 2a and c). Complete conversion of apo- (unmodified) AcpP to dapoxyl-AcpP was verified using conformationally-sensitive urea-PAGE (Fig. 2b).
 | ||
| Fig. 2 Application to E. coli AcpP. (a) Reaction mixtures of 1, 1a and loading onto AcpP as crypto-1b-AcpP (Scheme 2). (b) 15% urea-PAGE analysis of reaction mixtures visualized by Coomassie Blue stain (left) and 365 nm UV light (right). (c) Fluorescence emission spectra of reaction mixtures. Each reaction is denoted by colored squares which match the color of the plots in (c). | ||
Interestingly, we observed higher fluorescence levels for dapoxyl-pantetheinamide (1) versus the CoA analogue (1a) as well as a maximum emission wavelength comparable to that of dapoxyl-AcpP. This is attributed to the low solubility of dapoxyl-pantetheinamide in aqueous solution and its formation of highly fluorescent precipitates. We expect that if the same amount of dapoxyl-pantetheinamide were fully water soluble, it would display similar fluorescence properties to its CoA counterpart.
Synthase-based CP fluorescence profiling
We continued to profile fluorescence changes induced by CP loading by applying identical visualization techniques to CPs from an array of synthases. While CP-dependent synthases are categorized based on the types of metabolites they produce (FAS, PKS or NRPS), they may also be classified as long polypeptide chains consisting of multiple catalytic domains (type I) or discrete enzymes which coordinate with each other during biosynthesis (type II).1 While type II FAS and PKS CPs typically sequester their cargo, their counterparts in type I synthases and type II NRPSs do not display the same behavior.24We therefore predicted CPs of the latter classifications (type I FAS, type I PKS, and type II NRPS) to display decreased fluorescence due to the comparative increase in environment polarity and looser constraints on probe movement. As depicted in Fig. 3a–c, dapoxyl-loaded actACP (type II PKS) and AcpP (type II FAS) displayed the highest intensities in the green wavelength range (500–550 nm), while hACP (type I FAS), DEBS Acp4 (type I PKS), EntB (type II NRPS), and PltL (type II NRPS) produced a more subdued orange (550–600 nm) fluorescence response (purity of proteins provided in ESI Fig. S1†). These results align exactly with our hypothesis that the observed fluorescence changes are correlated with the individual abilities of each CP to sequester their cargo.
 | ||
| Fig. 3 Dapoxyl fluorescence profiling of various CPs. (a) Reaction mixtures of 1, 1a and loading onto CPs as their crypto-1b-CP state (Scheme 2). AcpP, E. coli fatty acid synthase acyl carrier protein; hACP, human fatty acid synthase acyl carrier protein; DEBS Acp4, 6-deoxyerythronolide B synthase module 4 acyl carrier protein; actACP, actinorhodin polyketide synthase acyl carrier protein; EntB, enterobactin nonribosomal peptide synthase peptidyl carrier protein; PltL, pyoluteorin nonribosomal peptide synthase peptidyl carrier protein. (b) 15% urea-PAGE analysis of reaction mixtures visualized by Coomassie Blue stain (left) and 365 nm UV light (right). (c) Fluorescence emission spectra of reaction mixtures. Each reaction is denoted by colored squares which match the color of the plots in (c). | ||
Interaction-based CP fluorescence profiling
In addition to cargo sequestration, type II FAS CPs are expected to participate in a mechanism termed “chain-flipping”, in which the cargo is transferred from the hydrophobic interior of the CP to the active site of its partner enzyme.25 We expected dapoxyl-pantetheinamide, a fluorescent environment-sensitive substrate analogue, to indirectly capture these protein–protein interactions. Again, we turned to E. coli FAS and its CP, AcpP, as a model system (Fig. 4a), monitoring fluorescence upon addition of one equivalent of various partner enzymes expected to participate in chain-flipping. For most partner enzymes, a 1.5 to 5-fold fluorescence increase was observed, excepting NAD(P)H-dependent reductases FabG and FabI, which resulted in 3-fold and 1.5-fold decreases in fluorescence respectively. Most notably, addition of FabF resulted in a 50 nm blueshift in fluorescence wavelength, changing the color of the green reaction mixture to a distinct blue hue (Fig. 4b and c). | ||
| Fig. 4 (a) Type II fatty acid biosynthesis in E. coli. The fatty acyl substrate is tethered to carrier protein AcpP via a thioester linkage on the terminal thiol of Ser36-4′-phosphopantetheine. AcpP shuttles the growing fatty acyl substrate to various partner enzymes to catalyze biosynthesis. Partner enzymes include malonyl-CoA-AcpP acyltransferase FabD; ketosynthases FabH, FabF, and FabB; ketoreductase FabG; dehydratases FabZ and FabA; and enoyl reductase FabI. (b) Reaction mixtures of crypto-1b-AcpP (50 μM) loading reactions with an added 1:1 equivalent of fatty acid synthase partner protein visualized with 365 nm UV. (c) Fluorescence emission spectra of crypto-1b-AcpP interacting with partner proteins. Each mixture in (b) is denoted by colored squares which match the color of the plots in (c). | ||
We expect each of these fluorescence responses to reflect the varied nature of sequestration, constraint, and intermolecular interactions experienced by the dapoxyl fluorophore within the active sites of each partner enzyme. In particular, the intensity decreases observed upon addition of FabG and FabI are hypothesized to result from the increased solvent accessibility and overall space in the active sites of these enzymes. On the other hand, the large fluorescence increase and blueshift exhibited by ketosynthase FabF in Fig. 4c may indicate an increased hydrophobicity and rigidity experienced by the fluorescent substrate analogue. This was further supported by the fact that a mutation (FabF R206A) shown to cause a significant effect on FabF activity26 underwent 50% loss in fluorescence upon addition of AcpP loaded with 1 (ESI Fig. S2†).
A solvatochromic protein–protein interaction inhibitor assay
Lastly, we assessed whether the solvatochromism of the dapoxyl fluorophore would be able to indirectly detect inhibition of protein–protein interactions. Cerulenin is a potent active site inhibitor of ketosynthases FabB and FabF, forming a covalent linkage with a catalytic cysteine residue. We expected addition of cerulenin to a 1:1 dapoxyl-AcpP/FabB or FabF mixture to disrupt chain-flipping of the solvatochromic substrate, returning its fluorescence profile to levels observed prior to the addition of FabB and FabF. Indeed, upon addition of cerulenin, the emission maximum decreased in wavelength and decreased in intensity, resembling the spectra of dapoxyl-AcpP alone (Fig. 5a, b, d and e).
 | ||
| Fig. 5 (a) Loading reactions of crypto-1b-AcpP (50 μM) with added FabB (50 μM) and cerulenin (various concentrations) visualized with 365 nm UV. (b) Fluorescence emission spectra of crypto-1b-AcpP (50 μM) loading reactions with added FabB (50 μM) and cerulenin (various concentrations). (c) Dose response curve of maximum AcpP–FabB fluorescence emission in response to increasing concentrations of cerulenin. (d) Loading reactions of crypto-1b-AcpP (50 μM) loading reactions with added FabF (50 μM) and cerulenin (various concentrations) visualized with 365 nm UV. (e) Fluorescence emission spectra of crypto-1b-AcpP (50 μM) loading reactions with added FabF (50 μM) and cerulenin (various concentrations). (f) Dose response curve of maximum AcpP–FabF fluorescence emission in response to increasing concentrations of cerulenin. | ||
Furthermore, we were able to extrapolate approximate IC50 values of 22.5 μM and 21.1 μM for the inhibition of AcpP–FabB and AcpP–FabF interactions, respectively, by cerulenin (Fig. 5c and f). These values are higher than 3 μM and 20 μM IC50 values reported for cerulenin against FabB and FabF, respectively.27 These differences may be attributed to significant differences in assay conditions: previous studies measured the effects of cerulenin on FabB/F activity in isolation, while the assay described here measures its effect on interactions with a carrier protein. Moreover, the lack of a stark difference between FabB and FabF IC50 values may be rationalized by the former study's use of myristoyl-AcpP as a substrate, which is known to have reduced activity for FabB compared to FabF.28
Conclusions
In summary, we have developed a bifunctional solvatochromic probe which selectively labels CPs across a range of biosynthetic contexts. Dapoxyl-pantetheinamide exhibits dramatic changes in fluorescence while proceeding through the chemoenzymatic CoA-based pathway which loads it onto its target protein. Even post-loading, the fluorescent properties of the probe can vary to reflect CP-involved protein–substrate and protein–protein interactions. The emissive properties of the sensitive dapoxyl fluorophore span an order-of-magnitude range of intensities and a 100 nm range of wavelengths, which surpasses other fluorophores we have evaluated to date in the context of CPs.We anticipate that the observed fluorescence changes are based upon the conformations adopted by the dapoxyl fluorophore in these varied contexts. The rotatable sigma bonds between the dimethylamino-donor and carbonyl-acceptor supply the molecule with several conformers, each with a unique fluorescence profile. Deducing the structural conformation and local environment of a molecule based on its fluorescent properties poses a difficult challenge. For example, the Lippert–Mataga equation29 tells us that the Stokes shift of a solvatochromic fluorophore is correlated with the overall polarizability of the surrounding solvent. In this context, we assume that shorter emission wavelengths, such as the blueshift observed upon addition of FabF to dapoxyl-AcpP (Fig. 4b), correspond to occupation of nonpolar local environments (i.e. nonpolar neighboring residues). While models such as Lippert–Mataga theory can be used to deduce isolated parameters of the space occupied by the fluorophore, the experimentally collected spectra are a result of multiple competing factors including polarizability, hydrogen bonding, and viscosity, which cannot be readily deconvoluted. Future photophysical and structure-based studies may shed more light on the reality of these assumptions.
As demonstrated by our final experiments, we expect dapoxyl-pantetheinamide to be applicable in the context of drug discovery, namely ACP-protein active site inhibitors. Protein–protein interactions within bacterial CP-dependent pathways provide a significantly underexplored target for therapeutic design.30 AcpP, the fatty acid synthase CP from E. coli, is known to interact with upwards of 25 partner enzymes, all involved in essential metabolic pathways.31 The labeling of AcpP with dapoxyl-pantetheinamide could allow exploration of several of these interactions with a single fluorescent tool. Moreover, the sensitivity of the dapoxyl fluorophore allows for quantitative analysis to be performed on these interactions and their inhibitors. These experiments lay the groundwork for development of fluorescence-based high-throughput screening methods for drug discovery.
Studies from our group have also demonstrated the uptake of fluorescent pantetheinamides by bacteria, paving the way for microscopy and cytometry-based studies.32–34 We therefore anticipate future applications of dapoxyl-pantetheinamide in vivo to allow similar experiments and further visualize CP-dependent synthases in their native contexts. Overall, our novel bifunctional solvatochromic probe provides several opportunities to probe CPs, their substrates, and the enzymes with which they interact.
Data availability
Copies of NMR spectra of the synthetic intermediates and probes have been provided in the ESI.† Copies of uncropped gels are available in ESI Fig. S3.† Raw NMR and XLS data files as well all plots are openly free upon request. ESI Fig. S4–S7† provides an alternate-colored version of Fig. 2–5 to enhance accessibility.Author contributions
M. G. M., J. J. L. and M. D. B. conceived the project. M. G. M. and K. R. conducted chemical synthesis. M. G. M. and Y. C. conducted fluorometric studies and all other experimental data. M. G. M. and J. J. L. analysed the data. The manuscript was written by M. G. M., J. J. L. and M. D. B. with contributions and proofreading from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NIH R01 AI168993. M. G. M. was supported by the Chemistry-Biology Interfaces Training Grant, NIH Grant T32GM146648. We thank X. Huang, A. Mrse, and Y. Su for assistance with acquisition of NMR (X. H. and A. M.) and mass spectral (Y. S.) data.Notes and references
- J. R. Lai, A. Koglin and C. T. Walsh, Biochemistry, 2006, 45, 14869–14879 CrossRef PubMed.
- D. M. Byers and H. Gong, Biochem. Cell Biol., 2007, 85, 649–662 CrossRef PubMed.
- A. C. Mercer and M. D. Burkart, Nat. Prod. Rep., 2007, 24, 750–773 RSC.
- C. Nguyen, R. W. Haushalter, D. J. Lee, P. R. L. Markwick, J. Bruegger, G. Caldara-Festin, K. Finzel, D. R. Jackson, F. Ishikawa, B. O'Dowd, J. A. McCammon, S. J. Opella, S. C. Tsai and M. D. Burkart, Nature, 2013, 505, 427–431 CrossRef.
- J. T. Mindrebo, A. Patel, W. E. Kim, T. D. Davis, A. Chen, T. G. Bartholow, J. J. La Clair, J. A. McCammon, J. P. Noel and M. D. Burkart, Nat. Commun., 2020, 11, 1–15 CrossRef.
- A. Roujeinikova, W. J. Simon, J. Gilroy, D. W. Rice, J. B. Rafferty and A. R. Slabas, J. Mol. Biol., 2007, 365, 135–145 CrossRef PubMed.
- E. A. Specht, E. Braselmann and A. E. Palmer, Annu. Rev. Physiol., 2017, 79, 93–117 CrossRef PubMed.
- S. B. Wagh, V. A. Maslivetc, J. J. La Clair and A. Kornienko, ChemBioChem, 2021, 22, 3109–3139 CrossRef.
- A. S. Klymchenko, Acc. Chem. Res., 2017, 50, 366–375 CrossRef.
- G. S. Loving, M. Sainlos and B. Imperiali, Trends Biotechnol., 2010, 28, 73–83 CrossRef PubMed.
- Z. Diwu, Y. Lu, C. Zhang, D. H. Klaubert and R. P. Haugland, Photochem. Photobiol., 1997, 66, 424–431 CrossRef.
- Z. Diwu, C.-S. Chen, C. Zhang, D. H. Klaubert and R. P. Haugland, Chem. Biol., 1999, 6, 411–418 CrossRef PubMed.
- Q. Wang and D. S. Lawrence, J. Am. Chem. Soc., 2005, 127, 7684–7685 CrossRef.
- J. S. Salafsky, J. Chem. Phys., 2006, 125, 74701 CrossRef.
- J. Min, J. W. Lee, Y. H. Ahn and Y. T. Chang, J. Comb. Chem., 2007, 9, 1079–1083 CrossRef.
- N. Kikuchi, A. Reed, Y. V. Gerasimova and D. M. Kolpashchikov, Anal. Chem., 2019, 91, 2667–2671 CrossRef PubMed.
- S. Sasaki, G. P. C. Drummen and G. Konishi, J. Mater. Chem. C, 2016, 4, 2743 Search PubMed.
- J. L. Meier and M. D. Burkart, Methods Enzymol., 2009, 458, 219–254 Search PubMed.
- J. L. Meier, A. D. Patel, S. Niessen, M. Meehan, R. Kersten, J. Y. Yang, M. Rothmann, B. F. Cravatt, P. C. Dorrestein, M. D. Burkart and V. Bafna, J. Proteome Res., 2011, 10, 320–329 CrossRef.
- Z. Diwu, C. Beachdel and D. H. Klaubert, Tetrahedron Lett., 1998, 39, 4987–4990 CrossRef.
- B. M. Krasovitskii, V. M. Shershukov and V. L. Volkov, Chem. Heterocycl. Compd., 1986, 22, 1022–1025 CrossRef.
- J. Beld, H. Cang and M. D. Burkart, Angew. Chem., Int. Ed., 2014, 53, 14456–14461 CrossRef PubMed.
- A. S. Worthington and M. D. Burkart, Org. Biomol. Chem., 2006, 4, 44–46 RSC.
- E. Płoskoń, C. J. Arthur, S. E. Evans, C. Williams, J. Crosby, T. J. Simpson and M. P. Crump, J. Biol. Chem., 2008, 283, 518–528 CrossRef.
- J. E. Cronan, Biochem. J., 2014, 460, 157–163 CrossRef PubMed.
- J. T. Mindrebo, L. E. Misson, C. Johnson, J. P. Noel and M. D. Burkart, Biochemistry, 2020, 59, 3626–3638 CrossRef PubMed.
- A. C. Price, K. Choi, R. J. Heath, Z. Li, S. W. White and C. O. Rock, J. Biol. Chem., 2000, 276, 6551–6559 CrossRef PubMed.
- P. Edwards, J. Sabo Nelsen, J. G. Metz and K. Dehesh, FEBS Lett., 1997, 402, 62–66 CrossRef CAS PubMed.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1955, 28, 690–691 CrossRef CAS.
- A. L. Osterman and T. P. Begley, in Progress in Drug Research, Birkhäuser, Basel, 2007, vol. 64, pp. 131–170 Search PubMed.
- K. Charov and M. D. Burkart, ACS Infect. Dis., 2019, 5, 1518–1523 CrossRef CAS PubMed.
- K. M. Clarke, A. C. Mercer, J. J. La Clair and M. D. Burkart, J. Am. Chem. Soc., 2005, 127, 11234–11235 CrossRef CAS PubMed.
- C. P. Reyes, J. J. La Clair and M. D. Burkart, Chem. Commun., 2009, 46, 8151–8153 RSC.
- M. Džunková, J. J. La Clair, T. Tyml, D. Doud, F. Schulz, S. Piquer-Esteban, D. Porcel Sanchis, A. Osborn, D. Robinson, K. B. Louie, B. P. Bowen, R. M. Bowers, J. Lee, V. Arnau, W. Díaz-Villanueva, R. Stepanauskas, T. Gosliner, S. V. Date, T. R. Northen, J. F. Cheng, M. D. Burkart and T. Woyke, Microbiome, 2023, 11, 1–24 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05499g |
| This journal is © The Royal Society of Chemistry 2024 |