
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A carbene-stabilized diphosphorus: a triple-bonded diphosphorus (P![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) P) and a bis(phosphinidene) (P–P) transfer agent†
P) and a bis(phosphinidene) (P–P) transfer agent†
Joseph S.
Yoon
,
Mehdi
Abdellaoui
 ,
Milan
Gembicky
and
Guy
Bertrand
*
,
Milan
Gembicky
and
Guy
Bertrand
*
UCSD-CNRS Joint Research Laboratory (IRL 3555), Department of Chemistry and Biochemistry, University of California San Diego, La Jolla, CA 92093-0358, USA. E-mail: gbertrand@ucsd.edu
First published on 2nd September 2024
Abstract
The reaction of the N,N-diisopropyl bromoiminium salt with excess sodium phosphaethynolate (NaPCO) affords a diphospha-urea 2. Under blue light irradiation (450 nm), carbon monoxide is liberated affording the bis(carbene)P2 adduct 3. Photolysis of a benzene solution of 3 at 365 nm gives rise to the carbene dimer, namely the 1,2-bis(diisopropylamino)ethylene as a cis/trans mixture, along with white and red phosphorus. Under the same experimental conditions, but in the presence of excess 2,3-dimethyl-1,3-butadiene, the classical double Diels–Alder adduct of the triple-bonded diphosphorus P![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P was obtained along with the bis(phospholene) formally resulting from a double [4 + 1] reaction of the diene with the bis(phosphinidene) form of P2. A stepwise carbene–carbene exchange reaction also occurs between the monosubstituted aminocarbene of 3 and a cyclic (alkyl)(amino)carbene, possibly involving the transient formation of a diphosphorus analogue of a diazo compound.
P was obtained along with the bis(phospholene) formally resulting from a double [4 + 1] reaction of the diene with the bis(phosphinidene) form of P2. A stepwise carbene–carbene exchange reaction also occurs between the monosubstituted aminocarbene of 3 and a cyclic (alkyl)(amino)carbene, possibly involving the transient formation of a diphosphorus analogue of a diazo compound.
Elemental phosphorus exists in several allotropic forms. Red, violet and black phosphorus are polymeric, whereas white phosphorus is a tetra-atomic molecule. Unlike dinitrogen, discrete diphosphorus (P2) is unstable due to the high energy of the triple bond.1 P2 has been spectroscopically characterized both at high temperatures (>1100 K)2 and in matrices at a few K.3 P2 is also widely seen in the coordination sphere of transition metals.4 However, precursors able to generate P2 in solution and under mild conditions are highly desirable to make this species synthetically useful. This has been achieved by Cummins et al.5 and by Wolf and Goicoechea et al.6 who were able to induce Diels–Alder reactions between transient P2 and dienes, and by Scheer et al.7 who trapped P2 with a molybdenum complex (Fig. 1A). A decade ago, Robinson et al.,8 as well as our group,9 showed that compounds A and B can be viewed as diphosphorus stabilized by N-heterocyclic carbenes (NHCs)10 and cyclic (alkyl)(amino)carbenes (CAACs),11 respectively (Fig. 1B). Herein, we report the synthesis of a compound featuring a P2 fragment capped by two monosubstituted aminocarbenes and show that the diphosphorus unit can be transferred, exhibiting the behaviour of both triple-bonded diphosphorus (P
P) and bis(phosphinidene) (P–P) (Fig. 1C).
 | ||
| Fig. 1 (A) Previously reported examples of generation and trapping of P2. (B) Bis(NHC) and bis(CAAC) adducts of P2. (C) Diphosphorus stabilized by monosubstituted aminocarbenes as triple-bonded diphosphorus and bis(phosphinidene) transfer agent. | ||
Following our discovery that phosphaketenes are the direct precursor of phosphinidenes,12 we envisaged that the reaction of NaPCO13 with a haloiminium salt, followed by deprotonation and loss of CO would lead to an amino phosphaalkyne (R2N–CP), a class of compounds scarcely accessible by known methods.14 Instead, by reacting the N,N-diisopropyl bromoiminium bromide 1 with NaPCO(dioxane)3.5, we observed the surprising formation of 2, the structure of which was ascertained by a single crystal X-ray diffraction study (Fig. 2). We quickly realized that a decarbonylation of 2 would lead to a P2 fragment stabilized by two small monosubstituted aminocarbenes.
 | ||
| Fig. 2 Synthesis of diphospha-urea 2 and subsequent photolytic decarbonylation under blue light irradiation to afford bis(carbene)P2 adduct 3. | ||
Photolytic decarbonylation of diphospha-ureas [R2P(CO)PR2] has precedents,15 and gratifyingly, we found that the irradiation of a benzene solution of 2 at 450 nm (blue light) induced the elimination of CO quantitatively, affording the desired compound 3. The X-ray crystal structure of 3 reveals that the two carbene units are coplanar, whereas for the analogous compounds A and B, the bulky carbene units are forced to adopt a twisted conformation to accommodate the steric congestion. The PP bond distance in 3 (2.18 Å) is very similar to those observed for A (2.19 Å) and B (2.18 Å), while the C–P bond lengths in 3 (1.71 Å) are comparable to that of B (1.72 Å) but slightly shorter than in A (1.75 Å). The 31P NMR spectrum of 3 showed a downfield signal at +78.0 ppm akin to that of the bis(CAAC)P2 adduct B (+59.4 ppm), whereas Robinson's bis(NHC)P2 adduct A exhibits a considerably more shielded signal at −73.6 ppm. This is due to the weaker p-accepting ability of NHCs versus CAACs and monosubstituted aminocarbenes.16 These data collectively indicate that the diphosphabutadiene17 resonance structure is significant in 3 as it is in B.
Despite not having a strong bis(carbene)P2 character, we wondered if the carbene units of 3 could be released to generate P2. Upon heating 3 at 80 °C for 6 hours, no reaction was observed. However, upon irradiating a C6D6 solution of 3 (λmax = 349 nm) with 365 nm light, we observed the formation of the carbene dimer, namely the 1,2-bis(diisopropylamino)ethylene 4 as a cis/trans mixture, along with P4 and a red precipitate indicative of red phosphorus (Scheme 1). This was the first evidence for the transient formation of P2, which is known to spontaneously dimerize and polymerize to white and red phosphorus, respectively.18 Based on this encouraging result, we investigated the possibility of intercepting P2 using a 1,3-diene – a reaction well-established in the literature.5,6 Irradiation of a THF solution of 3 at 365 nm in the presence of a large excess of 2,3-dimethyl-1,3-butadiene gave rise to phosphorus containing products 5 and 6 (2/1 ratio), as well as traces of 7, in addition to the carbene dimer 4 as a cis/trans mixture (Scheme 1). The major phosphorus compound 5 is the expected double [4 + 2] Diels–Alder adduct of the triple-bonded diphosphorus, the product observed by Cummins et al.5 and by Wolf and Goicoechea et al.6 More surprisingly, 6 formally results from two [4 + 1] cycloadditions of the diene with P2 acting as a bis(phosphinidene) (P–P), followed by insertion of the carbene dimer 4 into the PP bond. Note that the reaction of phosphinidenes and phosphinidene metal complexes with dienes giving the corresponding phospholenes have precedents,19 as well as the insertion of alkenes into diphosphines.20 We prepared the (bis)phospholene 7 according to a literature procedure,21 added the alkene 4 in THF, and after photolysis at 365 nm, compound 6 was obtained.
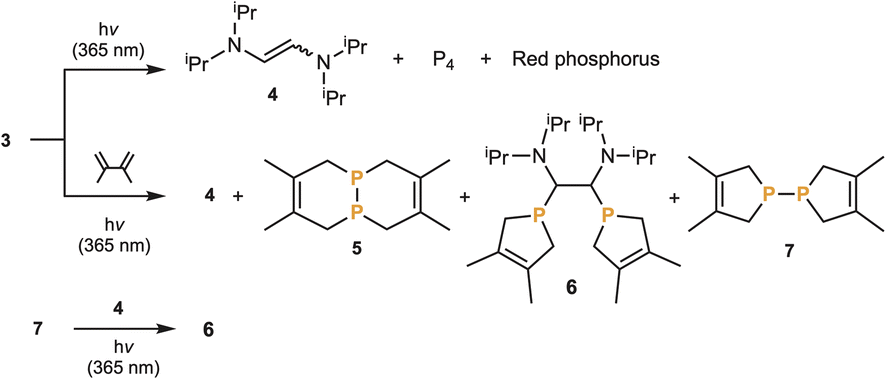 | ||
| Scheme 1 Irradiative cleavage of carbene fragments from 3 and trapping of diphosphorus with 2,3-dimethylbutadiene to give double Diels–Alder products 5 and bis(phospholene)-alkene adduct 6, with traces of 7. Independent synthesis of 6 through photolysis of a mixture of 7 and 4. | ||
The formation of 6 and 7 is intriguing and suggests that the elimination of the two aminocarbenes 8 occurs stepwise. The first carbene elimination leads to the hitherto unknown diphosphorus analogue 9 of a diazo compound, which is trapped via a [4 + 1] cycloaddition with dimethylbutadiene giving 10. Then a second carbene elimination gives a phosphinidene, which also reacts with the diene affording 7 (Scheme 2).
 | ||
| Scheme 2 Proposed intermediates in the formation of 7, via stepwise elimination of carbene fragments from 3. | ||
In 2016, we reported that a stable singlet phosphinidene12 quickly reacted with carbenes to give the corresponding adduct.22 Therefore, in an attempt to trap P2 as a (bis)phosphinidene, we added five equivalents of cyclic (alkyl)(amino)carbene 11,11a to a THF solution of 3. To our surprise, without any irradiation, a clean reaction occurred giving rise to the bis(CAAC)P2 adduct 12,9 along with the mixed carbene dimer 13. Repeating the experiment, with only two equivalents of CAAC 11, we observed the formation of 14, along with 13, clearly demonstrating the stepwise nature of the reaction (Scheme 3). These carbene–carbene exchange reactions are similar to those observed with the stable phosphinidene, for which DFT calculations predicted an associative mechanism.23 The formation of the mixed carbene dimer 13 confirms the prediction. Note that no exchange reaction occurred with imidazole-2-ylidenes.
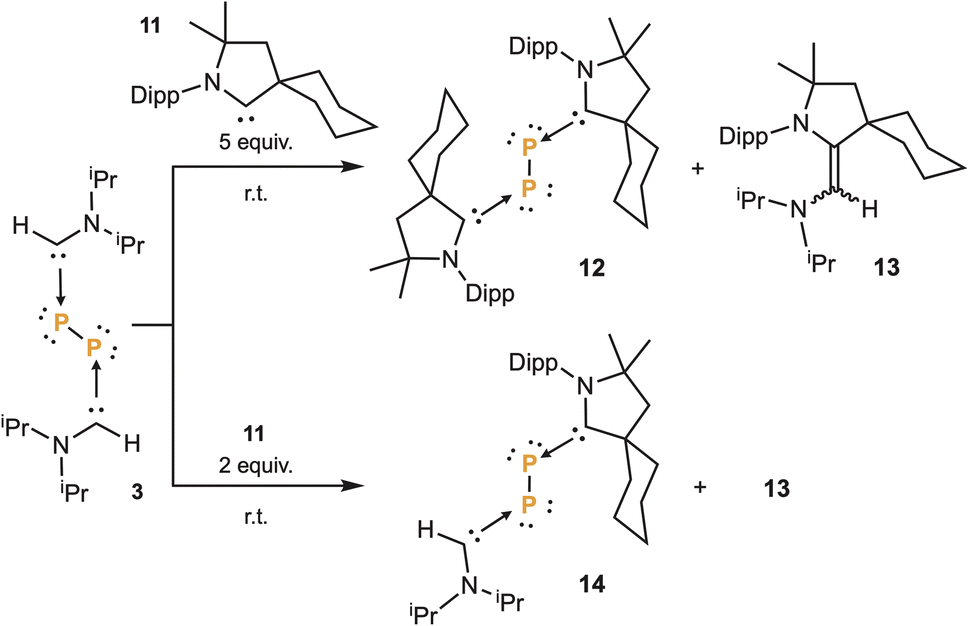 | ||
| Scheme 3 Double and mono substitution of aminocarbenes at diphosphorus with CAAC 11, affording 12 and 14, with concomitant formation of mixed carbene dimer 13. | ||
Conclusions
In summary, we disclose a new synthetic route to bis(carbene)P2 adducts. Under irradiation at 365 nm, the bis(monosubstituted aminocarbene)P2 adduct 3 is a generator of P2, under the classical triple-bonded form (PP), but 3 also acts as a bis(phosphinidene) (P–P) synthetic equivalent. These findings provide new insights into the stabilization and reactivity of diphosphorus species, offering promising avenues for future synthetic applications.
Data availability
All experimental procedures and characterization data can be found in the ESI.†Author contributions
J. Y. performed synthetic experiments. J. Y. and M. A. carried out the UV-vis studies. M. G. performed X-ray crystallographic analyses. G. B. supervised and designed the project. The manuscript was written by J. Y., M. A., and G. B.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF (CHE-2246948). We would like to thank Dr Ying Kai Loh and Dr Rodolphe Jazzar for fruitful discussions. We also wish to thank the UCSD Molecular Mass Spectrometry and NMR facilities.Notes and references
- (a) M. Regitz and O. J. Scherer, Multiple Bonds and Low Coordination in Phosphorus Chemistry, Thieme, Stuttgart, Germany, 1990 Search PubMed; (b) K. B. Dillon, F. Mathey and J. F. Nixon, Phosphorus: The Carbon Copy, Wiley, Chichester, UK, 1998 Search PubMed.
- O. J. Scherer, Angew. Chem., Int. Ed., 2000, 39, 1029–1030 CrossRef CAS.
- A. Kornath, A. Kaufmann and M. Torheyden, J. Chem. Phys., 2002, 116, 3323–3326 CrossRef CAS.
- (a) O. J. Scherer, H. Sitzmann and G. Wolmershäuser, Angew. Chem., Int. Ed., 1984, 23, 968–969 CrossRef; (b) L. N. Grant, B. Pinter, B. C. Manor, R. Suter, H. Grützmacher and D. J. A. Mindiola, Chem.–Eur. J., 2017, 23, 6272–6276 CrossRef CAS PubMed; (c) S. Wang, J. D. Sear, C. E. Moore, A. L. Rheingold, M. L. Neidig and J. S. Figueroa, Science, 2022, 375, 1393–1397 CrossRef CAS; (d) E. Peresypkina, A. Virovets and M. Scheer, Coord. Chem. Rev., 2021, 446, 213995 CrossRef CAS; (e) B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164–4177 CrossRef CAS PubMed; (f) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed.
- (a) D. Tofan and C. C. Cummins, Angew. Chem., Int. Ed., 2010, 49, 7516–7518 CrossRef CAS PubMed; (b) N. A. Piro, J. S. Figueroa, J. T. McKellar and C. C. Cummins, Science, 2006, 313, 1276–1279 CrossRef CAS PubMed; (c) A. Velian, M. Nava, M. Temprado, Y. Zhou, R. W. Field and C. C. Cummins, J. Am. Chem. Soc., 2014, 136, 13586–13589 CrossRef CAS PubMed.
- (a) G. Hierlmeier, A. Hinz, R. Wolf and J. M. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 431–436 CrossRef CAS PubMed; (b) G. Hierlmeier and R. Wolf, Eur. J. Inorg. Chem., 2022, 10, e202101057 CrossRef PubMed.
- C. Resinger, F. Dielmann, R. szlosek, A. V. Virovets and M. Scheer, Angew. Chem., Int. Ed., 2023, 62, e202218828 CrossRef.
- Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970–14971 CrossRef CAS.
- O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 5530–5533 CrossRef CAS.
- (a) A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS; (b) P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem., 2021, 5, 711–725 CrossRef CAS.
- (a) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS; (b) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed; (c) S. K. Kushvaha, A. Mishra, H. W. Roesky and K. C. Mondal, Chem. Asian J., 2022, 17, e202101301 CrossRef.
- L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, Chem, 2016, 1, 147–153 CAS.
- (a) J. M. Goicoechea and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 16968–16994 CrossRef CAS; (b) F. F. Puschmann, D. Stein, D. Heift, C. Hendriksen, Z. A. Gal, H. F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2011, 50, 8420–8423 CrossRef CAS PubMed.
- (a) R. Appel and M. Poppe, Angew. Chem., Int. Ed., 1989, 28, 53–54 CrossRef; (b) J. Grobe, D. Le Van, B. Lüth and M. Hegemann, Chem. Ber., 1990, 123, 2317–2320 CrossRef CAS; (c) J. Grobe, D. Le Van, M. Hegemann, B. Krebs and M. Läge, Chem. Ber., 1992, 125, 411–414 CrossRef CAS; (d) F. E. Hahn, D. Le Van, M. C. Moyes, T. von Fehren, R. Fröhlich and E. U. Würthwein, Angew. Chem., Int. Ed., 2001, 40, 3144–3148 CrossRef CAS; (e) M. Regitz, Chem. Rev., 1990, 90, 191–213 CrossRef CAS.
- (a) K. M. Szkop, A. R. Jupp, H. Razumkov, M. Xu and D. W. Stephan, Chem.–Eur. J., 2019, 25, 10084–10087 CrossRef CAS; (b) R. Appel and W. Paulen, Chem. Ber., 1983, 116, 109–113 CrossRef CAS; (c) V. Plack, J. R. Goerlich and R. Schmutzler, Z. Anorg. Allg. Chem., 1999, 625, 919–922 CrossRef CAS.
- R. Nakano, R. Jazzar and G. Bertrand, Nat. Chem., 2018, 10, 1196–1200 CrossRef CAS.
- (a) E. S. Yang, D. W. N. Wilson and J. M. Goicoechea, Angew. Chem., Int. Ed., 2023, 62, e2022180 Search PubMed; (b) V. D. Romanenko, L. S. Kachkovskaya and L. N. Markovskii, Zh. Obshch. Khim., 1985, 55, 2140–2141 CAS.
- H. W. Melville and S. C. Gray, Trans. Faraday Soc., 1936, 32, 271–285 RSC.
- (a) A. Velian and C. C. Cummins, J. Am. Chem. Soc., 2012, 134, 13978–13981 CrossRef CAS PubMed; (b) A. Marinetti, F. Mathey, J. Fischer and A. Mitschler, J. Am. Chem. Soc., 1982, 104, 4484–4485 CrossRef CAS.
- (a) K. Hirano and M. Miura, Tetrahedron Lett., 2017, 58, 4317–4322 CrossRef CAS; (b) A. B. Burg, J. Am. Chem. Soc., 1961, 83, 2226–2231 CrossRef CAS; (c) Y. Sato, S. I. Kawaguchi, A. Nomoto and A. Ogawa, Chem.–Eur. J., 2019, 25, 2295–2302 CrossRef CAS; (d) K. W. Morse and J. G. Morse, J. Am. Chem. Soc., 1973, 95, 8469–8470 CrossRef CAS; (e) I. Hajdók, F. Lissner, M. Nieger, S. Strobel and D. Gudat, Organometallics, 2009, 28, 1644–1651 CrossRef.
- L. D. Quin and J. Szewczyk, Phosphorus, Sulfur, 1984, 21, 161–170 CrossRef CAS.
- M. M. Hansmann, R. Jazzar and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 8356–8359 CrossRef CAS.
- M. M. Hansmann and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 15885–15888 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2367862–2367865. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05091f |
| This journal is © The Royal Society of Chemistry 2024 |