
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuadrupolar dinuclear hypervalent tin(IV) compounds with near-infrared emission consisting of Schiff bases based on π-conjugated scaffolds†
Kazuya
Tanimura
 a,
Kento
Tanaka
a,
Masayuki
Gon
ab and
Kazuo
Tanaka
*ab
a,
Kento
Tanaka
a,
Masayuki
Gon
ab and
Kazuo
Tanaka
*ab
aDepartment of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: tanaka@poly.synchem.kyoto-u.ac.jp
bDepartment of Technology and Ecology, Graduate School of Global Environmental Studies, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan
First published on 10th October 2024
Abstract
Since π-conjugated molecules are commonly used as a scaffold for constructing optoelectronic and functional materials, much effort has been devoted to exploring novel molecular scaffolds for obtaining superior properties. This study focuses on dinuclear hypervalent tin(IV) compounds prepared by the ladderization of Schiff bases using hypervalent tin units. The optical measurements found that introducing hypervalent tin atoms can reinforce the D–π–A system. We synthesized two types of dinuclear hypervalent compounds by simple condensation reactions and observed near-infrared (NIR) emission. Also, depending on the direction of the imine bonds, these molecules had different quadrupolar orientations with D–π–A–π–D and A–π–D–π–A systems followed by negative solvatochromism, which is the unique behavior of quadrupolar-derived absorption. Furthermore, the π-conjugated polymers involving dinuclear compounds showed NIR emission in the wavelength range over 800 nm owing to the distinct expansion of π-conjugation. Our findings could be useful not only for constructing electronic structures with narrow energy gaps but also for designing molecules with unique electronic states and environmental responsiveness.
Introduction
Conjugated molecules have attracted significant interest because of their unique optical and electronic properties and wide range of applicability, especially in materials science,1–4 biomedical science5 and environmental sensing.6 Among the various designs of π-conjugated structures, ladder molecules with fused π-conjugated backbones exhibit intriguing optical and physical properties for functional materials because of their rigid structures.7 The ladderization methods have extended beyond basic carbon and have been widely studied using heteroatoms.8 In π-conjugated scaffolds with the ladder-like structures linked by heteroatoms, electronic interactions between the heteroatoms and the π-conjugated system have resulted in unique optical properties and various characteristics derived from the hetero elements that are not present in carbon-based fused rings.9,10 To fulfill the requirements for creating advanced materials with high stability, good processability, and tunable optical properties, it is significant to explore new molecular frameworks for constructing ladder π-conjugated compounds and elucidate their optical and electronic properties.We have been studying the “hypervalent state”11 created by main-group elements for constructing a ladder skeleton based on heteroatoms. The hypervalent states are regarded as multi-coordinate species that formally have electrons exceeding the octet rule, and many researchers have focused on their synthesis, geometry, and reactivity.12–15 Our previous reports have looked at the unique electronic states of the hypervalent state to modify the electronic structure of π-conjugated scaffolds.16 Main group elements like tin,17–21 germanium22,23 and bismuth24 in the hypervalent state are suggested to create coordination position-specific electronic effects on π-conjugated molecules through the intermolecular Lewis D–A interaction at the equatorial position and the polarized bond known as three-center/four-electron (3c–4e) bonds at the apical position. Based on these site-specific electronic properties, we obtained NIR materials without significantly increasing the molecular size and finally obtained polymers that can exhibit NIR absorption and/or emission properties.16,18,22,25 This strategy offers a new way to control the electronic properties of π-conjugated scaffolds by creating unique inductive effects related to the hypervalent state.
In this study, we created a series of hypervalent tin compounds by the ladderization of Schiff bases (also known as azomethines) using hypervalent tin units. Specifically, we observed both negative solvatochromic behaviors and NIR emission with small π-conjugated structures through the process of dinucleation (see Fig. 1). Firstly, as previously reported with mononuclear hypervalent tin derivatives, we also evaluated the electronic effect of the introduction of hypervalent tin into the π-conjugated system by comparing the electronic states of the standard Schiff base Azm and the hypervalent compound TPh. Secondly, as mentioned above, we confirmed that hypervalent tin was able to perturb the electronic state of the π-conjugated scaffolds through different site-specific electronic interactions at the equatorial and axial positions. Furthermore, we also found that the hypervalent tin enhanced the electron-withdrawing ability of the imine moiety, and consequently, charge separation followed by the D–π–A structure was efficiently formed. Next, we synthesized two dinuclear compounds, TPhCN and TPhCN, where the direction of the imine bond is opposite, and evaluated their optical properties. Interestingly, we discovered unique optical properties such as negative solvatochromism originating from “symmetry-breaking”26 in the ground state on the electronic structures of D–π–A–π–D or A–π–D–π–A, depending on the different quadrupolar orientation of the imine bond. We were also able to introduce each dinuclear compound into a π-conjugated polymer and obtain NIR-emitting materials having emission wavelengths over 800 nm by extending the π-conjugation and electronic interactions with the copolymer. Our findings demonstrate that hypervalent tin compounds, π-conjugated molecules bridged by hypervalent atoms, can be a new platform for developing D–A materials.
 | ||
| Fig. 1 Chemical structures of the Schiff base Azm, the mononuclear hypervalent tin compound TPh, and the dinuclear hypervalent tin compounds TPhCN and TPhNC. | ||
Results and discussion
Synthesis and characterization
According to the synthetic procedures reported by Peon et al., TPh and TPhBr were prepared, respectively.27 Based on the synthesis of the dinuclear boron azomethine complexes,28 the series of azomethine ligands for dinuclear compounds (NC, NCBr, CN, and CNBr) were prepared through the imine condensation reaction (Scheme 1). The introduction of tin, followed by the formation of the hypervalent state, was achieved by refluxing with the ligands and diphenyltin(IV) oxide in acetone. After reprecipitation, the pure products were obtained in high isolated yields. We confirmed that the expected structures were obtained from characterization data from 1H, 13C{1H}, 119Sn NMR spectroscopy, mass spectrometry (MS), and elemental analysis. From fewer changes in time courses of UV-vis absorption spectra (1.0 × 10−5 M in toluene), it is suggested that TPh, TPhCN, and TPhNC should be stable under ambient conditions (Fig. S1(a)–(c)†). The photostability of each compound under strong light irradiation was also investigated by trans-illuminator (365 nm, 6500 μW cm−2, 60 s) as shown in Fig. S1(d)–(f).† As a result of the absorption measurements, no changes were observed for TPh, whereas photodegradation was partially observed from the dinuclear compounds. | ||
| Scheme 1 Synthesis of mononuclear (TPhBr) and dinuclear (TPhCN, TPhCNBr, TPhNC, and TPhNCBr) hypervalent tin compounds. A chemical structure of the mononuclear compound TPh was also shown. | ||
Next, we synthesized π-conjugated polymers from mononuclear and dinuclear hypervalent tin compounds by the Migita–Kosugi–Stille cross-coupling reaction (Scheme 2). TPhBr, TPhCNBr, and TPhNCBr were polymerized with bithiophene comonomers, and insoluble high molecular weight polymers and low molecular weight oligomers were removed by Soxhlet extraction. Regarding relative molecular weights measured by gel permeation chromatography (GPC) with polystyrene standards, the number of molecular weights (Mn) was estimated to be 4.1 × 103 for P-TPh, 1.8 × 104 for P-TPhCN, and 1.3 × 104 for P-TPhNC. The characterization was carried out by 1H, 13C{1H}, 119Sn NMR spectroscopy, and matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) MS (see the ESI† for more details). The observed changes in absorption spectra over time for each compound did not show any noticeable changes (Fig. S2†). This indicates that each compound is sufficiently stable under the measurement conditions.
 | ||
| Scheme 2 Synthesis of π-conjugated polymers, P-TPh, P-TPhNC, and P-TPhCN. | ||
Structural features of tin compounds
The crystal structures of the mononuclear and dinuclear compounds were determined through single-crystal X-ray structure analysis (Fig. 2, S3 and S4†). Diamantis et al. have already reported the crystal structure of TPh,29 and we referred to their structure to compare the mononuclear and dinuclear compounds. To quantitatively evaluate 5-coordinate geometries in hypervalent states, a geometry index known as the τ5 value is often used (τ5 = (β − α)/60. The coordination center of a molecule can have different shapes depending on its angles. The two largest angles are called β and α. When τ5 is close to 0, the geometry is similar to square pyramidal, while if τ5 is close to 1, the geometry is similar to trigonal bipyramidal).30 The τ5 value of each compound was calculated to be 0.65 for TPh, 0.65 for TPhCN, and 0.40 for TPhNC (TPhNC had asymmetric and disordered structures, and their τ5 values were estimated to be 0.46 and 0.29) (Fig. 2). Thus, TPh and TPhCN formed distorted trigonal bipyramidal geometries, whereas TPhNC displayed a distorted square pyramidal one. Moreover, the torsion angles of C–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C were assessed as 179.7(6)° for TPh, 179(1)° for TPhCN, and 178.3(9)° (178.0(9)°) for TPhNC, indicating that this dinucleation hardly affects the planarity of the molecule. On the contrary, the structural optimization of each compound in quantum chemical calculations with density functional theory (DFT) showed that all the molecules adopted trigonal bipyramidal geometries (τ5 value of TPh: 0.58, TPhCN: 0.57, TPhNC: 0.57) and exhibited highly planar structures (the torsion angles of C–CN–C in TPh: 177.64°, TPhCN: 178.09°, TPhNC: 177.94°) (Fig. 2). From the viewpoint of the packing, the interplane distances of the nearest-neighbor aromatic rings were 4.216 Å for TPhCN and 4.016 Å for TPhNC. These results propose that weak intermolecular interactions could be formed in the crystalline packing (Fig. S3 and S4†). In addition, these data suggest that the structures around the hypervalent tin are flexible and distorted in the crystal due to the dense packing.
N–C were assessed as 179.7(6)° for TPh, 179(1)° for TPhCN, and 178.3(9)° (178.0(9)°) for TPhNC, indicating that this dinucleation hardly affects the planarity of the molecule. On the contrary, the structural optimization of each compound in quantum chemical calculations with density functional theory (DFT) showed that all the molecules adopted trigonal bipyramidal geometries (τ5 value of TPh: 0.58, TPhCN: 0.57, TPhNC: 0.57) and exhibited highly planar structures (the torsion angles of C–CN–C in TPh: 177.64°, TPhCN: 178.09°, TPhNC: 177.94°) (Fig. 2). From the viewpoint of the packing, the interplane distances of the nearest-neighbor aromatic rings were 4.216 Å for TPhCN and 4.016 Å for TPhNC. These results propose that weak intermolecular interactions could be formed in the crystalline packing (Fig. S3 and S4†). In addition, these data suggest that the structures around the hypervalent tin are flexible and distorted in the crystal due to the dense packing.
 | ||
| Fig. 2 (a) ORTEP drawings of the hypervalent tin compounds, TPhCN and TPhNC (50% probability for thermal ellipsoids). Hydrogens and disordered atoms were omitted to clarify. All crystallographic data are shown in ESI.† (b) Geometrical optimized structures of TPhCN and TPhNC (gray carbon atoms; red oxygen atoms; blue atom nitrogen; pale green tin atoms; hydrogen atoms are omitted for clarity) (B3LYP/6-31G(d) for C, H, N, O and LanL2DZ for Sn). | ||
Formation of hypervalent states
The formation of hypervalent states can be confirmed by the distribution of molecular orbitals (MOs) obtained from DFT calculations. In general, it is known that hypervalent states consist of three types of orbitals in apical positions: bonding, non-bonding, and antibonding orbitals, originating from a 3c–4e bond. Correspondingly, according to the distribution of the MOs for TPh, it was found that bonding, non-bonding, and anti-bonding orbitals should be assigned to HOMO−10, HOMO−7, and LUMO+11 and +13, respectively (Fig. S5†). Furthermore, natural bonding orbital (NBO) calculations also supported the presence of the significant bond between tin and nitrogen atoms and the corresponding orbitals constituting the 3c–4e bond on each atom (Fig. S6†). From the above data, it is expected that the tin in TPh forms the hypervalent state, which should have a coordination position-specific electronic effect on the π-conjugated scaffolds.To assess the electronic effects of the formation of the hypervalent state, UV-vis absorption spectra of TPh and the model compound Azm were measured in toluene (1.0 × 10−5 M), as shown in Fig. 3a. The absorption band of TPh was bathochromically shifted compared to that of Azm. The DFT calculations were performed to gain deep insight into the origin of the electronic states in the hypervalent tin derivatives (Fig. 3b and Tables S3–S6†). We evaluated the electronic effects of the hypervalent state on the π-conjugated scaffolds by comparing model compounds Azm and AzmOMe. According to the MOs of Azm, AzmOMe, and TPh, the LUMO energy levels were significantly stabilized by the tin coordination compared with the HOMO energy levels (Azm: HOMO = −5.90 eV, LUMO = −1.55 eV, AzmOMe: HOMO = −5.22 eV, LUMO = −1.17 eV, TPh: HOMO = −5.40 eV, LUMO = −2.21 eV). This result suggests that the MO is stabilized by electron-accepting nitrogen–tin (N–Sn) coordination at the equatorial position. Moreover, the stabilization of the HOMO energy level is relatively suppressed by the electron-donating 3c–4e bond, as described in our previous report (Fig. 3c).17 The hypervalent states can exhibit electronic effects specific to their coordination positions, irrespective of ligand structures.
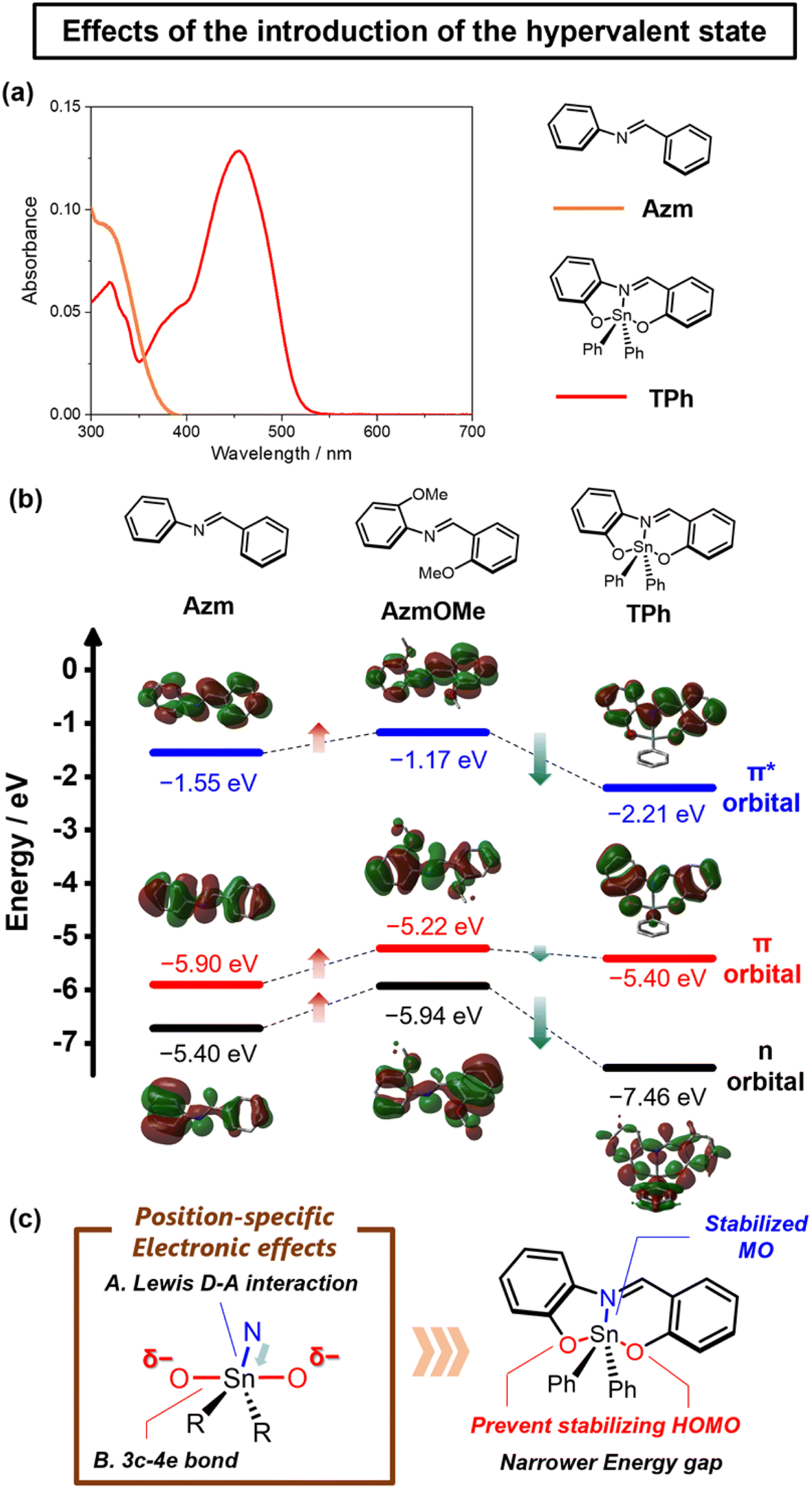 | ||
| Fig. 3 (a) UV-vis absorption spectra of Azm and TPh in toluene (1.0 × 10−5 M). (b) Energy diagrams selected MOs of Azm, AzmOMe, and TPh obtained with TD-DFT calculations at TD-B3LYP/6-31G(d) level for C, H, N, O, and LanL2DZ for Sn (isovalue = 0.02). (c) Schematic illustration of the electronic effects of hypervalent tin. | ||
The distributions of electric charges and dipole moments before and after introducing the hypervalent tin were also examined using DFT calculations. From Fig. S7,† it was found that the unsymmetrical distribution of electric charges was observed from the surface of molecular electrostatic potential (MEP) of TPh compared with Azm and AzmOMe, and a larger degree of negative charge was observed from the benzene ring of the N-side than that of the C-side, like D–π–A molecules. These results represent that the introduction of the hypervalent tin atom should induce charge separation and reinforce the inductive effect of the imine bond. This is because of the enhanced electron-withdrawing ability by the coordination of the tin atom, which works as the Lewis acid. In addition, the dipole moment of Azm was 1.431 debye along the direction of the imine bond, while that of TPh was 2.778 debye, indicating that the asymmetric electronic state should be realized (Fig. S7†). These data suggest that the different electronic states should be realized in both dinuclear compounds where only the nitrogen position is different.
Optical and electronic properties of dinuclear compounds
UV-vis absorption and photoluminescence spectra were measured in toluene (1.0 × 10−5 M) to compare the electronic structures of dinuclear hypervalent tin compounds TPhNC and TPhCN. Fig. 4a, b and Table 1 illustrate that TPh exhibited the absorption band around 400–500 nm, whereas TPhNC and TPhCN displayed the absorption bands around 550–650 nm. In addition, the emission bands of dinuclear hypervalent tin compounds were bathochromically shifted compared to that of TPh. This implies that the dinucleation contributes to extending the π-conjugation through the imine moieties. Compared TPhNC with TPhCN, the maximum absorption wavelength of TPhCN (λmaxabs = 604 nm) was longer than that of TPhNC (λmaxabs = 632 nm), and the molar extinction coefficient of TPhCN (εabs = 2.68 × 104 cm−1 M−1) was larger than that of TPhNC (εabs = 1.61 × 104 cm−1 M−1). Moreover, both maxima in the fluorescence wavelengths were reached in the NIR region, and TPhCN had a longer wavelength of photoluminescence and lower quantum yield (QY) than TPhNC (TPhCN: λFLmax = 777 nm, ΦFL = 1.0%, TPhNC: λFLmax = 768 nm, ΦFL = 3.1%). The bathochromic wavelength shifts in absorption and PL spectra originating from the extension of the π-conjugated system were achieved by the π-extension of TPh with a narrow energy gap owing to the electronic effects of hypervalent tin. This suggests that hypervalent tin has the same electronic effects on the π-conjugated system in the dinuclear compounds for narrowing the energy gap as observed when the mononuclear compound is laddered. In the previous research by Peon et al.,27TPh derivatives are known to form bending structures by photoexcitation. In contrast, our dinuclear hypervalent compounds hardly showed such bending behaviors compared to the optimized structures in the ground and excited states (Fig. S8†). Therefore, the symmetric structures of the dinuclear compounds should contribute to the suppression of the structural relaxation by photoexcitation. In addition, the structural differences were hardly observed between the optimized structures of TPhCN and TPhNC in the ground and excited states (Fig. S8†). It is, therefore, implied that the difference in the Stokes shift could be attributed to the difference in the electronic state of each compound. These findings indicate that the transitional characteristics in MO exhibit different responses depending on the positions of dinucleation in the ladder structure. | ||
| Fig. 4 (a) UV-vis absorption and (b) fluorescence spectra of TPh, TPhCN, and TPhNC in toluene (1.0 × 10−5 M). (c) Energy diagrams selected MOs and oscillator strengths (f) of selected transition bands of TPh, TPhCN, and TPhNC obtained with DFT and TD-DFT calculations at TD-B3LYP/6-31G(d)//B3LYP/6-31G(d) level for C, H, N, O, and LanL2DZ for Sn (isovalue = 0.02). | ||
The electronic states of π-conjugated molecules can be influenced by the direction of imine bonds and the positions of nitrogen atoms in forming the hypervalent state. We explored the electronic states and the impact of dinucleation by comparing the mononuclear and dinuclear hypervalent tin compounds (Fig. 4c, S9 and Tables S7, S8†). As depicted in Fig. 4c, TPhCN and TPhNC exhibited narrower energy gaps than TPh because of the extension of their π-conjugated scaffolds. Focusing on the range of changes in the MO levels compared to TPh, the HOMO of TPhCN is destabilized more significantly than that of TPhNC (TPhCN: −4.87 eV, TPhNC: −5.25 eV), and the LUMO of TPhNC is stabilized more dramatically than that of TPhCN (TPhCN: −2.54 eV, TPhNC: −2.97 eV). We considered that these are derived from the direction of the imine bonds. In TPhCN, the electron-donating effect of the two imine bonds contributed to the central benzene ring, which is expected to destabilize the HOMO by increasing the electron density. In TPhNC, on the other hand, the electron-withdrawing effect of the two imine bonds should stabilize the LUMO by promoting electron deficiency of the central benzene ring. Moreover, we assessed the MO energy levels using cyclic voltammetry (CV), as illustrated in Fig. S10.† The HOMO and LUMO energy levels of TPh, TPhCN, and TPhNC determined from the cyclic voltammograms closely mirrored the trends observed in the results from the DFT calculations. Notably, it was concluded that the positions of dinucleation in the ladder structure strongly affected the optical and electronic properties.
The photoluminescence spectra of TPh derivatives in the solid state were observed and compared with those in the solution state (Fig. S11†). Focusing on the emission wavelengths, the change was hardly observed in TPh (λmaxFL,solid = 576 nm, ΦFL,solid = 18.4%). This slight wavelength change should be due to the weak intermolecular interactions. On the other hand, the emission bands of both dinuclear compounds TPhCN and TPhNC were shifted to the longer wavelength regions, leading to their solid-state NIR luminescence (TPhCN: λmaxFL,solid = 836 nm, ΦFL = 0.5%, TPhNC: λmaxFL,solid = 796 nm, ΦFL,solid = 2.4%). Considering these results and the optimized structures in the ground and excited states, their symmetric structures should restrict structural relaxation. Consequently, we speculate that the observed emission in the longer wavelength region can be attributed to intermolecular interactions. The dinuclear compounds also exhibited aggregation-caused quenching properties that reduced the quantum yield.31 These results suggest that the construction of symmetric structures affects not only the electronic state of the molecules but also their mobility.
We draw the MEP surface to examine the charge distribution of these hypervalent compounds (Fig. 5). TPh showed the unsymmetrical charge distribution, and the benzene ring of the N-side had a larger negative charge than that of the C-side, as described previously (Fig. 5a). In addition, reflecting the charge distribution of TPh, TPhCN had a large negative charge in the center benzene ring (Fig. 5b), while TPhNC had that in the side benzene rings (Fig. 5c). Therefore, this molecular design of dinucleation provided charge separation and quadrupolar molecules in the ground state, and TPhCN and TPhNC constituted the ring-fused A–π–D–π–A and the D–π–A–π–D systems, respectively.
 | ||
| Fig. 5 Solvent-dependent UV-vis absorption and fluorescence spectra of (a) TPh, (b) TPhCN, and (c) TPhNC in each solvent (1.0 × 10−5 M, toluene/each solvent = 1/99 v/v). Inset: Molecular electrostatic potential (MEP) surface of each compound with DFT calculations at TD-B3LYP/6-31G(d)/level for C, H, N, O, and LanL2DZ for Sn (isovalue = 0.02). | ||
The absorption spectra were measured in dichloromethane (CH2Cl2), chloroform (CHCl3), toluene, and cyclohexane (c-hex) to gain insights into the solvent effects on these compounds. Surprisingly, both TPhCN and TPhNC exhibited negative solvatochromism in the only absorption property regardless of their neutral and symmetric structures. Meanwhile, the emission wavelengths hardly changed depending on those solvents (Fig. 5). In general, negative solvatochromism is caused by photoexcitation changes in the dipole moment.32 However, the DFT calculations suggested that the dipole moments in TPhCN and TPhNC are almost identical (Fig. S12†). On the other hand, there are few examples of negative solvatochromism in the only absorption property because of “Symmetry breaking” in the ground state. Terenziani et al. predicted the existence of molecules with negative solvatochromism only in the absorption spectrum (referred to as “Class III” in the literature).26,33,34 They proved that polymethine derivatives fall into this category.33 These reports showed that large quadrupolar dyes caused negative solvatochromism in the only absorption property because the ground state was bistable (D–π–A−–π–D+ or D+–π–A−–π–D) and the excited state was stable as nonpolar. Therefore, we estimated the dinuclear compounds TPhCN and TPhNC have a large quadrupolar in the only ground state owing to the electronic effects unique to the hypervalent state and their imine moiety. We further observed the solvent effects in the DFT calculations. We reproduced the results, that is, the same as the tendency of optical measurements (Fig. S13,† the calculation was performed against CH2Cl2, CHCl3, toluene, and c-hex). These findings propose that the electronic effects of hypervalent states can be exploited to create a novel D–A system in small molecules beyond the conventional D–A linking method.
Electronic properties of conjugated polymers using TPh, TPhCN and TPhNC
Optical measurements with polymers showed that P-TPhCN and P-TPhNC exhibited absorption and emission bands in longer wavelength regions than P-TPh (Fig. 6a and Table 2). It should be noted that the emission maximum wavelengths in P-TPhCN and P-TPhNC exceeded 800 nm (P-TPh: λmaxabs = 518 nm, λmaxFL = 621 nm, ΦFL = 35.8%, P-TPhCN: λmaxabs = 658 nm, λmaxFL = 828 nm, ΦFL = 0.3%, P-TPhNC: λmaxabs = 683 nm, λmaxFL = 801 nm, ΦFL = 1.2%, 1.0 × 10−5 M in CHCl3). Since the conjugated system of the dinuclear compound can also be extended by polymerization, followed by the bathochromic shifts of optical bands, it is shown that the dinuclear compounds are a promising building block for constructing NIR materials. The solvent effects on the optical properties were also investigated with the polymers (Fig. S14 and Table S10†). Shoulders, possibly due to aggregation, were observed from all polymers in cyclohexane. The change in the PL spectrum was observed only from P-TPh, and similar spectra were obtained from P-TPhCN and P-TPhNC. Moreover, similar shifts in the absorption wavelength were observed in P-TPhCN and P-TPhNC. These results imply that electronic structures of the monomers could be maintained in the polymer main-chains. In the film state, bathochromic wavelength shifts in the absorption and PL spectra, and the aggregation-caused quenching were observed from all polymers comparing to those of the solution state, possibly due to interactions between polymer chains (Fig. S15 and Table S11†). | ||
| Fig. 6 (a) UV-vis absorption and fluorescence spectra of P-TPh, P-TPhCN, and P-TPhNC in CHCl3 (1.0 × 10−5 M per repeating unit). (b) Molecular orbitals of P-TPhCN, and P-TPhNC (tri-units) at TD-B3LYP/6-31G(d)/level for C, H, N, O, S and LanL2DZ for Sn (isovalue = 0.02). | ||
The DFT calculations were performed to assess the extension of conjugation in mononuclear and dinuclear compounds with two and three units (Fig. 6b and S16–S21†). In the mononuclear compound, the HOMO and LUMO expanded as the number of units increased, whereas expansion of π-conjugation in the entire molecule was hardly observed, which might be attributed to the twisted bithiophene unit. In the A–π–D–π–A dinuclear compound TPhCN, as the number of units increased, the LUMO was expanded because of its high planarity, while the HOMO was localized in the dinuclear compound owing to the electron-rich moiety of the center benzene ring. On the other hand, in the D–π–A–π–D dinuclear compound TPhNC, the HOMO was extended throughout the main chain, and the LUMO was localized on the dinuclear compound unit because of the centered electron-deficient benzene ring. These results suggest that the charge distribution induced by each direction of dinucleation (TPhCN or TPhNC) leads to different orbitals in the extended MOs, altering the energy level of each MO. As described above, polymerization of polarized D–π–A–π–D or A–π–D–π–A type molecules is suggested to induce specific orbital extensions, and the polarized state in π-conjugated scaffolds can be featured by constructing conjugated polymers.
Conclusion
By employing the electronic effects of hypervalent tin, we realized unique electronic structures and functions based on the ladderization of the π-conjugated Schiff base scaffold. In the mononuclear compound, the tin coordination enhanced the electron-withdrawing character in the inductive effect from the imine bond. As a result, the D–π–A structures were built without incorporating electron-donating units. This design strategy is useful in that it is simple and different from the usual D–A linkage and will be able to create additional molecular diversity by ligands and main group elements. Moreover, in the dinuclear compounds, we succeeded in synthesizing two kinds of compounds with NIR luminescence properties in the facile two-step procedure. These molecules showed the two types of quadrupolar, A–π–D–π–A and D–π–A–π–D, derived from two imine bonds and hypervalent tin in the molecule. Negative solvatochromism was observed in the absorption property originating from strong quadrupolar structures. Finally, conjugated polymerization of each molecule not only achieved luminescence in the NIR wavelength region over 800 nm because of the extended conjugation but also observed different orbital distributions on the polymer main chain originating from the electric charge distribution of the monomer. Our findings propose that the hypervalent atoms should be a promising tool for modulating the electronic structures of π-conjugated scaffolds followed by optical properties. This approach can be further developed by designing new ligands and diversifying the main group elements, which might be a platform for novel functional materials.Data availability
ESI† available. Crystallographic data for TPhCN and TPhNC has been deposited at the CCDC under 2373875 and 2373876.Author contributions
K. T. (Kazuya Tanimura) wrote the original draft, conducted the data analysis, and performed additional experiments. K. T. (Kento Tanaka) synthesized the compounds, performed the experiments, conducted the data analysis, and performed additional experiments. M. G. and K. T. (Kazuo Tanaka) supervised the project. All the authors discussed the results and contributed to editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Hirose Foundation (for M. G.), a Grant-in-Aid for Scientific Research (B) (for M. G.) (JP22H02130 and JP23K23398) and (for K. T.) (JP24K01570).References
- A. Haque, K. M. Alenezi, M. S. Khan, W.-Y. Wong and P. R. Raithby, Chem. Soc. Rev., 2023, 52, 454–472 RSC.
- Z. Zhang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 4422–4433 CrossRef CAS PubMed.
- D. Luo, W. Jang, D. D. Babu, M. S. Kim, D. H. Wang and A. K. K. Kyaw, J. Mater. Chem. A, 2022, 10, 3255–3295 RSC.
- D. Meng, R. Zheng, Y. Zhao, E. Zhang, L. Dou and Y. Yang, Adv. Mater., 2022, 34, 2107330 CrossRef CAS PubMed.
- G. Feng, G.-Q. Zhang and D. Ding, Chem. Soc. Rev., 2020, 49, 8179–8234 RSC.
- V. K. Praveen, B. Vedhanarayanan, A. Mal, R. K. Mishra and A. Ajayaghosh, Acc. Chem. Res., 2020, 53, 496–507 CrossRef CAS PubMed.
- J. Lee, A. J. Kalin, T. Yuan, M. Al-Hashimi and L. Fang, Chem. Sci., 2017, 8, 2503–2521 RSC.
- A. Fukazawa and S. Yamaguchi, Chem.–Asian J., 2009, 4, 1386–1400 CrossRef CAS PubMed.
- M. Stolar and T. Baumgartner, Chem. Commun., 2018, 54, 3311–3322 RSC.
- T. Delouche, M. Hissler and P.-A. Bouit, Coord. Chem. Rev., 2022, 464, 214553 CrossRef CAS.
- J. I. Musher, Angew. Chem., Int. Ed., 1969, 8, 54–68 CrossRef CAS.
- R. Kumar, T. Dohi and V. V Zhdankin, Chem. Soc. Rev., 2024, 53, 4786–4827 RSC.
- K. Tamao, J. Synth. Org. Chem., 1990, 48, 457–462 CrossRef CAS.
- K. Akiba, Heteroat. Chem., 2009, 20, 232–234 CrossRef.
- J. Hyvl, Dalton Trans., 2023, 52, 12597–12603 RSC.
- M. Gon, S. Ito, K. Tanaka and Y. Chujo, Bull. Chem. Soc. Jpn., 2021, 94, 2290–2301 CrossRef CAS.
- M. Gon, K. Tanaka and Y. Chujo, Chem.–Eur. J., 2021, 27, 7561–7571 CrossRef CAS PubMed.
- M. Gon, K. Tanimura, M. Yaegashi, K. Tanaka and Y. Chujo, Polym. J., 2021, 53, 1241–1249 CrossRef CAS.
- M. Gon, Y. Morisaki, K. Tanimura, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2023, 7, 1345–1353 RSC.
- M. Gon, T. Kato, K. Tanimura, C. Hotta and K. Tanaka, RSC Mechanochemistry, 2024, 1, 322–327 RSC.
- M. Gon, Y. Morisaki, K. Tanimura and K. Tanaka, Dalton Trans., 2024, 53, 11858–11866 RSC.
- M. Gon, M. Yaegashi, K. Tanaka and Y. Chujo, Chem.–Eur. J., 2023, 29, e202203423 CrossRef CAS PubMed.
- M. Gon, M. Yaegashi and K. Tanaka, Bull. Chem. Soc. Jpn., 2023, 96, 778–784 CrossRef CAS.
- K. Tanimura, M. Gon and K. Tanaka, Inorg. Chem., 2023, 62, 4590–4597 CrossRef CAS PubMed.
- M. Gon, K. Tanaka and Y. Chujo, Polym. J., 2023, 55, 723–734 CrossRef CAS.
- F. Terenziani, A. Painelli, C. Katan, M. Charlot and M. Blanchard-Desce, J. Am. Chem. Soc., 2006, 128, 15742–15755 CrossRef CAS PubMed.
- J. S. Zugazagoitia, M. Maya, C. Damián-Zea, P. Navarro, H. I. Beltran and J. Peon, J. Phys. Chem. A, 2010, 114, 704–714 CrossRef CAS PubMed.
- S. Ohtani, M. Nakamura, M. Gon, K. Tanaka and Y. Chujo, Chem. Commun., 2020, 56, 6575–6578 RSC.
- A. A. Diamantis, J. M. Gulbis, M. Manikas and E. R. T. Tiekink, Phosphorus, Sulfur Silicon Relat. Elem., 1999, 150, 251–259 Search PubMed.
- A. G. Blackman, E. B. Schenk, R. E. Jelley, E. H. Krenske and L. R. Gahan, Dalton Trans., 2020, 49, 14798–14806 RSC.
- S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765–768 CrossRef CAS PubMed.
- S. Nigam and S. Rutan, Appl. Spectrosc., 2001, 55, 362A–370A CrossRef CAS.
- F. Terenziani, O. V. Przhonska, S. Webster, L. A. Padilha, Y. L. Slominsky, I. G. Davydenko, A. O. Gerasov, Y. P. Kovtun, M. P. Shandura, A. D. Kachkovski, D. J. Hagan, E. W. Van Stryland and A. Painelli, J. Phys. Chem. Lett., 2010, 1, 1800–1804 CrossRef CAS.
- C. Sissa, F. Delchiaro, F. Di Maiolo, F. Terenziani and A. Painelli, J. Chem. Phys., 2014, 141, 164306 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details “bisTPh_SI.pdf”. CCDC 2373875 and 2373876. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05006a |
| This journal is © The Royal Society of Chemistry 2024 |