
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequence-dependent conformational transitions of disordered proteins during condensation†
Jiahui
Wang
a,
Dinesh
Sundaravadivelu Devarajan
 a,
Keerthivasan
Muthukumar
a,
Young C.
Kim
*b,
Arash
Nikoubashman
*cde and
Jeetain
Mittal
*afg
a,
Keerthivasan
Muthukumar
a,
Young C.
Kim
*b,
Arash
Nikoubashman
*cde and
Jeetain
Mittal
*afg
aArtie McFerrin Department of Chemical Engineering, Texas A&M University, College Station, TX 77843, USA. E-mail: jeetain@tamu.edu
bCenter for Materials Physics and Technology, Naval Research Laboratory, Washington, USA. E-mail: youngchan.kim@nrl.navy.mil
cLeibniz-Institut für Polymerforschung Dresden e.V., Hohe Straße 6, 01069 Dresden, Germany. E-mail: anikouba@ipfdd.de
dInstitut für Theoretische Physik, Technische Universität Dresden, 01069 Dresden, Germany
eCluster of Excellence Physics of Life, Technische Universität Dresden, 01062 Dresden, Germany
fDepartment of Chemistry, Texas A&M University, College Station, TX 77843, USA
gInterdisciplinary Graduate Program in Genetics and Genomics, Texas A&M University, College Station, TX 77843, USA
First published on 12th November 2024
Abstract
Intrinsically disordered proteins (IDPs) can form biomolecular condensates through phase separation. It is recognized that the conformation of IDPs in the dense and dilute phases, as well as at the interfaces of condensates, can critically impact their functionality. However, a residue-level understanding of the conformational transitions of IDPs during condensation remains elusive. In this study, we employ a coarse-grained polyampholyte model, comprising an equal number of oppositely charged residues—glutamic acid and lysine—whereby conformations and phase behavior can be tuned by altering the protein sequence. By manipulating sequence patterns from perfectly alternating to block-like, we obtain chains with ideal-like conformations to semi-compact structures in the dilute phase. In the dense phase, however, the chain conformation approaches that of an ideal chain, regardless of the sequence. Simulations across different concentrations reveal that chains transition from small oligomeric clusters in the dilute phase to the dense phase, with a gradual swelling of individual chains. These findings are further validated with naturally occurring protein sequences involved in biological condensate formation. Additionally, we show that chain conformations at the interface display a strong sequence dependence, remaining more collapsed than those in the bulk-like dense phase. This study provides detailed insights into how the conformations of a specific subclass of IDPs (lacking secondary structures) change within condensates and in solution, as governed by their sequences.
Introduction
Liquid–liquid phase separation (LLPS) is crucial for the formation of biomolecular condensates, which in turn play an important role in vital cellular mechanisms such as gene expression, signal transduction, stress response, and the assembly of macromolecular complexes.1–6 Intrinsically disordered proteins (IDPs) and disordered regions within proteins have been identified as a primary driving force for the formation of the condensed phase in many cases. It is recognized that the conformation of IDPs in biomolecular condensates and at their interface can critically impact the resulting functionality. For example, recent studies highlighted that the conformational changes of Dcp2 within P-bodies influence mRNA decapping,7 and conformational expansion of Tau in condensates promotes fibrillization.8 These results emphasize the important role of protein conformations in dictating the function of biomolecular condensates.Extensive research has been conducted to study protein conformations in the dilute phase,9,10 elucidating that the conformation of IDPs can be modulated by factors such as sequence composition,11–16 charge characteristics,17–21 sequence pattern22–26 and solvent environment.27–31 Sequences comprised of charged residues exhibit a globule-to-coil transition with an increase in the net charge per residue.18 Quantitative analyses of charge patterns have demonstrated that enhanced charge segregation in the sequence typically leads to more compact conformations in the dilute phase.22,23 Conformational changes are also modulated by varying electrostatic interactions due to the surrounding solvent environment. Recently, Reddy et al. illustrated that a pH shift from neutral to acidic prompted prothymosin-α to shift from a random coil to a partially collapsed state.32
In the dense phase, it has been observed experimentally that α-synuclein shifts toward more “elongated” conformations during LLPS,33 and Tau K18 exhibits expanded conformations within the droplet phase, in contrast to a compact structural ensemble in the dilute phase.34 A1-LCD protein and its mutated variants have been shown to adopt more extended conformations in the condensates as well through molecular simulations.35 Despite these excellent prior studies on the conformations of IDPs in the dense phase, there is a general lack of understanding of how the protein conformations change when transitioning from the dilute to the condensed phase; are they always more expanded in condensates than in the dilute phase? What are the polymer scaling properties of IDPs in the dense phase and at the interface, and how do these depend on the protein sequence and dilute phase conformations?
To answer these questions and to decipher the conformational transitions from the dilute to the dense phase during LLPS, we systematically studied a wide range of polyampholyte sequences, which exist in many naturally occurring IDPs.36,37 Further, these polyampholyte sequences are ideal model systems, since their conformations, as quantified by the radius of gyration (Rg), can be readily modulated by altering the charge pattern.22,23,38,39 Herein, we leveraged molecular dynamics simulations applied to the coarse-grained polyampholyte model. We have analyzed the conformational landscape of various sequences both in the dilute phase (single chain) and in the dense phase. By executing simulations across a spectrum of concentrations, we shed light on the transition process from the dilute to the dense phase, insights that are pertinent to the condensation of IDPs. A comparison of the conformations at the interface and within the dense phase revealed the sequence-conformation relationships in different phases of the condensates. Overall, our findings provide a molecular-level understanding of biomolecular conformational shifts during the process of condensation.
Results and discussion
We selected 15 sequences composed of glutamic acid (E) and lysine (K) residues (Fig. S1†). Each E–K variant (EKV) consists of 50 residues with an equal number of E and K residues to maintain a zero net charge. The degree of charge segregation of EKVs was quantified by the sequence charge decoration (SCD) parameter.23,40 To allow for a more straightforward comparison with other IDPs, we normalized the SCD values (nSCD) so that nSCD = 0 for the uniformly charge-patterned sequence and nSCD = 1 for the most charge-segregated diblock sequence (normalization method shown in ESI text†).41 The selected 15 EKVs cover diverse conformations in dilute solutions (single chain) at constant temperature (T = 300 K), ranging from an ideal-like chain (EKV1) to a semi-compact chain (EKV15). This selection allowed us to probe the conformational properties within the dense phase starting with different chain configurations in the dilute phase. First, we analyzed the conformations for EKVs in the dense phase and as a single chain (Fig. 1) by calculating Rg: | (1) |
 | ||
| Fig. 1 Conformational properties of EKVs in the dense phase (red diamonds) and as a single chain (blue circles). (a) Radius of gyration (Rg) as a function of nSCD. (b) Probability distribution of Rg, P(Rg), for EKVs as a single chain. The inset shows P(Rg) for EKVs in the dense phase. Line colors, ranging from purple to red, indicate increasing nSCD. (c) Relative shape anisotropy (κ2) as a function of nSCD. (d) The correlation between end-to-end distance (Re) and Rg for the EKVs compared to an ideal chain (solid line). Snapshots show the conformations in the dense phase (red) and as single chains (blue). The mean values are obtained by dividing the trajectory into 5 independent blocks. Error bars in all panels indicate standard errors about the mean. In some cases, error bars are smaller than the symbol size. | ||
For single chains, Rg decreased with increasing nSCD (Fig. 1a), which is consistent with many previous studies that examined this behavior in detail.22,23,38,42 All error bars in the plots represent the standard error of the mean (detailed calculation provided in the ESI†). To study the conformations in the dense phase, we performed bulk simulations maintaining a constant external pressure (P = 0 atm), allowing the systems to adopt their preferred concentration. For EKV1 (nSCD = 0), no dense phase formed at the investigated temperature (T = 300 K) because the alternating distribution of positively and negatively charged residues resulted in weak interchain attractions.42 For all other EKVs, a dense phase formed, where its concentration increased with increasing nSCD (Fig. S2a†). For these cases, as nSCD increased, we observed a corresponding modest rise in Rg within the dense phase, which amounted to an approximate 12.6% increase compared to the single chain Rg of EKV1. Conversely, for a single chain, there was a marked decrease in Rg, up to 39.3%, from EKV1 to EKV15 (Fig. 1a). Consequently, the disparity in Rg between the dense phase and single chains widened as nSCD increased. Specifically, this difference expanded from 7.4% to 85.6% relative to the Rg of single chains, spanning from EKV2 to EKV15. For single chains, the Rg probability distribution, P(Rg), substantially narrowed with increasing nSCD (Fig. 1b), which reflects the smaller conformational variety of collapsed blocky EKV sequences. In contrast, P(Rg) was much broader in the dense phase and slightly broadened with increasing nSCD. These trends indicate that the dense phase of EKVs exhibits a markedly greater diversity of conformations, which depend only weakly on the specific protein sequence, as compared to their single-chain state. Considering the potential influence of water and counterions on conformational properties, we also performed Martini simulations for the EKVs. Similar to the results from our coarse-grained polyampholyte model, we observed that with increasing nSCD, the polyampholyte concentration in the dense phase increases, though the absolute values are smaller compared to the coarse-grained model due to the presence of water and counterions (Fig. S5a†). The conformational properties also show similar trends as in our coarse-grained model: with increasing nSCD, the single-chain Rg decreases significantly, while the chain conformations in the dense phase remain relatively unchanged. Further, chains in the dense phase have a much larger Rg than in the dilute phase (Fig. S5b†). These results are consistent with prior experiments of Tau proteins, which also found more expanded conformations and enhanced conformational fluctuations for proteins in droplets.34
To characterize the shape of the individual chains, we calculated the average relative shape anisotropy (κ2) using the three eigenvalues (λi) of the gyration tensor:
 | (2) |
 .46 However, Re of EKVs in single-chain state showed notable deviations from this relationship, particularly pronounced in EKVs with high charge segregation.
.46 However, Re of EKVs in single-chain state showed notable deviations from this relationship, particularly pronounced in EKVs with high charge segregation.
These results demonstrate that the conformations observed in the dense phase, as well as those of the sequences with low to moderate degree of charge segregation in the dilute phase, are closely akin to the conformation of an ideal chain. These observed conformational properties can be understood by considering the attractive interactions between monomers. In the dense phase, chains are surrounded by other chains, allowing monomers from a chain to interact with neighboring chains, leading to the observed chain expansion. In contrast, in the dilute phase, monomers can only form intramolecular contacts, thereby leading to a collapsed conformation to achieve a state of minimum free energy. Within the dense phase, the chain conformations nearly mirror the random-walk characteristics of an ideal chain, which maximizes the conformational entropy (and thus minimizes the free energy).46 As a result, in the dense phase, conformations of EKVs exhibited a minor sequence-dependent variation. Conversely, the conformation of an isolated chain in a poor solvent is determined by the intrachain interactions that involve an equilibrium between long-range electrostatic repulsion and attraction, which is substantially modulated by the sequence.22
Having established the conformations within the dense and dilute phases, we proceeded to analyze the conformational transitions between these two phases by simulating the EKVs across a series of fixed concentrations, from 0.2 mg ml−1 to 100 mg ml−1. This was achieved by maintaining a constant number of chains (N = 500) and modifying the volume of the simulation box (Fig. 2a). When the concentration exceeds the saturation concentration csat, some chains spontaneously assemble into a droplet so that the system phase separates into a dilute and a dense phase. To provide a more detailed account of this transition, we have selected the sequence EKV5 as a representative case (Fig. 2a and b). This sequence has a csat of 0.68 mg ml−1, which lies within the concentration range we explored. At concentrations c ≤ 0.4 mg ml−1 < csat, the number of clusters in the entire system is roughly equal to the number of chains, i.e., 500, indicating that the system is indeed below its saturation concentrations (refer to the ESI† for the technical specifics of the cluster analysis). In alignment with this behavior, the distributions of the Rg were consistent with those observed in our single-chain simulations (Fig. S4†).
 | ||
| Fig. 2 Concentration scan for the EKVs. (a) Tie line for EKV5 at T = 300 K indicating coexisting dense (cdense) and dilute (csat) phase concentrations along with snapshots at different concentrations. (b) Rg (black, left y-axis) and number of clusters (green, right y-axis) of EKV5 as functions of concentration. The red and blue dashed horizontal lines represent the Rg in the bulk dense phase and of a single chain, respectively, with shaded horizontal areas indicating the corresponding error bars. Note that the error bars are smaller than the line width and may not be visible. The scatter points represent the average Rg for each chain across the analyzed frames, colored according to the probability from high (red) to low (purple). (c) Normalized Rg as a function of normalized concentration for the EKVs. The black solid line is the fitted curve for all simulation data (symbols). The mean values are obtained by dividing the trajectory into 5 independent blocks. Error bars in panels (b and c) indicate standard errors about the mean. In some cases, error bars are smaller than the symbol size. | ||
Intriguingly, in the concentration range csat < c < 4 mg ml−1, we observe a clear decrease in the number of clusters, marking the gradual aggregation of the chains; yet, the number of clusters is still much larger than unity, demonstrating that a lot of chains are still dispersed, accompanied by small clusters comprising 2 to 10 chains (Fig. S6a†). The local concentration within these clusters is comparable to that of the dense phase concentrations found in our bulk simulations (Fig. S6b†). These findings imply that phase separation is taking place above csat, despite the absence of macroscopic phase separation, which may be due to the limited size of the simulated systems. At a concentration around c = 4.0 mg ml−1, the manifestation of a dense phase is more pronounced, marked by the emergence of a droplet (Fig. 2a). At this concentration, the chain Rg, averaged over the whole system, has a rather wide distribution, and lies between the Rg in the dense phase and that of a single chain (Fig. 2b). As the concentration was increased further, the number of clusters diminishes to one, signifying that all chains formed a single condensate. Correspondingly, Rg continued to increase, approaching the value of the dense bulk phase, Rdenseg. Note that the small discrepancy between the Rg measured in our condensate simulations and Rdenseg likely originates from slightly collapsed chains located at the condensate interface.47 These findings reveal a gradual increase in average chain size as the system transitions from the dilute phase to the dense phase. To validate the observed increasing trend, we compared the distribution of individual chain sizes with that of the entire system, demonstrating that the average chain size serves as a reliable indicator of the overall system properties (Fig. S7†). Additionally, the analysis of unique interaction partners confirms the quality of the sampling and reinforces the robustness of the observed trend (Fig. S8†). To further validate the observed trends, we tested different initial configurations for EKV5 by starting simulations from an equilibrated dense-phase droplet containing all chains (Fig. S9†). We then adjusted the overall concentration by resizing the simulation box, finding that the average Rg gradually decreased from the dense phase to the dilute phase. The results differed only at 2 mg ml−1, which can be attributed to kinetic barriers that prevent achieving the same equilibrium state. Nevertheless, these results indicate that our conclusions are not significantly influenced by the initial configuration.
To substantiate the generality of these conclusions, we analyzed all EKVs (Fig. S10 and S11†) and normalized the Rg and concentrations (Fig. 2c). We normalized the Rg to span between the value for a single chain, Rsingleg, and the value of a chain in the dense bulk phase, Rdenseg. We normalized the concentrations by the csat values. We note that the csat values for EKV10 to EKV15 were extrapolated based on the data from sequences with lower nSCD, owing to the scarcity of chains in the dilute phase during slab co-existence simulations (Fig. S2b†). These extrapolated csat values did not affect the overall trend, as all the corresponding Rg values were similar to Rdenseg. For EKVs with a low nSCD value (i.e., EKV2 to EKV7), Rg closely approximated Rsingleg at concentrations below or slightly above the saturation concentration. At higher concentrations, where a distinct condensate formed, Rg increased until it almost reached Rdenseg. For the EKVs with higher nSCD values, the smallest concentration that we explored in our simulations is much larger than their saturation concentration, hence we did not observe a Rg value close to that of a single chain. However, with an increase in concentration, we still noted a rise in Rg until it plateaued at a value marginally smaller than Rdenseg. The normalized Rg values for all EKVs collapse onto a single sigmoidal curve (R2 = 0.95), exhibiting a uniform trend of increase from the dilute to the dense phase. This convergence demonstrates that the gradual increase in Rg from the dilute phase to the dense phase is a universal attribute across all EKVs.
Having established that for EKVs bearing zero net charge with diverse charge patterns, Rg progressively increases from the dilute phase to the dense phase, we next aimed to test whether the observations made for the EKVs translate to natural IDPs. We selected four proteins, previously demonstrated to undergo LLPS in vitro, namely the low-complexity (LC) domain of FUS,48 the disordered C-terminal domain of TDP-43,49 the LC domain of hnRNPA2 (ref. 50) and the N-terminal disordered RGG domain of LAF-1 (LAF-1 RGG).51 Following previous research on the phase behavior of these IDPs,52 we conducted simulations at T = 300 K for the first three proteins, and at T = 260 K for LAF-1 RGG. We maintained a constant total monomer count of approximately 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 while varying the box volume to explore a range of concentrations from 0.4 mg ml−1 to 400 mg ml−1. For all four sequences, the average Rg of the entire system initially remained nearly constant when the proteins remained dispersed in solution, and the number of clusters was close to the number of chains. As the dense phase formed (indicated by the decreasing number of clusters shown in Fig. 3), the average Rg gradually increased, approaching the Rg that is characteristic of the bulk dense phase (Fig. 3 and S12†). The progression of Rg with increasing concentration is similar to those observed for the EKVs, with Rg gradually increasing as the system evolves from the dilute phase into the dense phase. To evaluate the impact of the initial configuration, we performed additional simulations starting from a condensed droplet (as with the EKVs) and found no significant differences in the results (Fig. S13 and S14†). These findings suggest that during the phase separation process, IDPs undergo a transition from a dilute state to an oligomeric state, and ultimately to a dense state, accompanied by a gradual chain expansion.
000 while varying the box volume to explore a range of concentrations from 0.4 mg ml−1 to 400 mg ml−1. For all four sequences, the average Rg of the entire system initially remained nearly constant when the proteins remained dispersed in solution, and the number of clusters was close to the number of chains. As the dense phase formed (indicated by the decreasing number of clusters shown in Fig. 3), the average Rg gradually increased, approaching the Rg that is characteristic of the bulk dense phase (Fig. 3 and S12†). The progression of Rg with increasing concentration is similar to those observed for the EKVs, with Rg gradually increasing as the system evolves from the dilute phase into the dense phase. To evaluate the impact of the initial configuration, we performed additional simulations starting from a condensed droplet (as with the EKVs) and found no significant differences in the results (Fig. S13 and S14†). These findings suggest that during the phase separation process, IDPs undergo a transition from a dilute state to an oligomeric state, and ultimately to a dense state, accompanied by a gradual chain expansion.
 | ||
| Fig. 3 Concentration scans for the disordered domains of natural proteins: (a) FUS LC, (b) TDP-43, (c) hnRNPA2, and (d) LAF-1 RGG. Rg (black lines, left y-axis) and number of clusters (green lines, right y-axis) as functions of concentration. The red and blue dashed horizontal lines represent the Rg in the bulk dense phase and of a single chain, respectively, with shaded horizontal areas indicating the corresponding error bars. Note that some error bars are smaller than the line width and may not be visible. The scatter points represent the average Rg for each chain across the analyzed frames, colored according to the probability from high (red) to low (purple). The mean values are obtained by dividing the trajectory into 5 independent blocks. Error bars in all panels indicate standard errors about the mean. In some cases, error bars are smaller than the symbol size. | ||
Upon condensation, an interfacial region emerges between the dilute and dense phases, which likely plays an important role in the functionality and stability of MLOs.53–55 Previously, our group elucidated the conformation of homopolymer chains and select IDPs at the interface of condensate droplets, and here we conducted a similar analysis for EKVs.47 To eliminate the effects of (local) curvature of the condensate interface in a droplet geometry, we performed slab simulations to study conformations at interfaces. Taking EKV5 as an illustrative example (data for the other sequences are presented in the ESI†), a dense phase was observed, with occasional appearances of several chains in the dilute phase (Fig. 4a). Interface boundaries were determined by fitting the concentration profile relative to the distance from the condensate center-of-mass to monomer in the z direction (dzCOM) using a hyperbolic tangent function:56
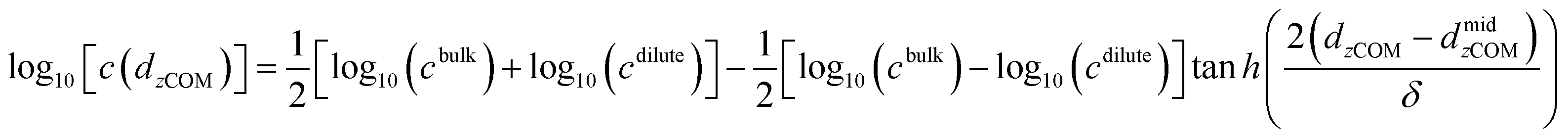 | (3) |
 | ||
| Fig. 4 Analysis of EKV5 conformations at the interface via slab simulation. (a) Snapshot depicting one side of the interface of a condensate from the slab simulation. (b) Concentration profiles with respect to the distance from the condensate center-of-mass in the z-direction, dzCOM. The solid line is the fitted curve for the simulation data (symbols). (c) Average Rg with respect to the distance from the condensate center-of-mass to the chain's center-of-mass in the z direction, dzCOM–COM. The red and blue dashed horizontal lines represent the Rg in the bulk dense phase and of a single chain, respectively, with shaded horizontal areas indicating the corresponding error bars. Note that the error bars are smaller than the line width and may not be visible. The mean values are obtained by dividing the trajectory into 5 independent blocks. Error bars in panel (b) indicate standard errors about the mean. Error bars in panel (c) are calculated based on the frames where chains are present at specific positions. In some cases, error bars are smaller than the symbol size. | ||
Upon identifying the interfacial region (Fig. 4b), we analyzed the local Rg with respect to the distance along the z-direction between a chain's center-of-mass and the center-of-mass of the condensate, dzCOM–COM. In the dense region of the slab, Rg overlapped with the value obtained in the bulk dense phase, Rdenseg. Within the interface region, Rg decreased and remained smaller than those observed for the chains within the bulk phase. Within the dilute phase, Rg exhibited pronounced fluctuations around the size of a single chain, Rsingleg. These fluctuations can be attributed to the limited statistical data available due to the low concentration of chains in the coexisting dilute phase. Importantly, all EKVs exhibit similar qualitative behavior, with conformations in the interfacial region being more compact than those in the dense interior and less compact than the dilute phase (Fig. S15 and S16†). To validate our findings and ensure consistency across different simulation methodologies, we compared simulation results from droplets in cubic simulation boxes (4 mg ml−1, droplet formed) and from slab geometries for EKV5 (Fig. S17†). Although the (local) curvature of the droplet interface may lead to some deviation, both approaches consistently demonstrated that the chains gradually collapse as they transition from the dense phase into the dilute phase.
Conclusions
In this simulation study, we investigated the conformational properties of proteins during phase separation by simulating model sequences composed of an equal number of oppositely charged monomers arranged in different patterns (referred to as EKVs) and naturally occurring disordered proteins. Despite extensive previous research on phase separation,1,2 the description of conformational transitions underlying the condensation process remains largely qualitative. Due to the heterogeneous and dynamic nature of protein ensembles, it is difficult to tackle this question experimentally, which makes the simulation approaches used here an attractive avenue for gaining a comprehensive and quantitative understanding of sequence-dependent changes in conformational properties.We decipher the self-assembly process itself by conducting simulations with increasing protein concentration. As expected, proteins initially stay as individual molecules in the dilute phase at low concentrations but then start to assemble into larger clusters at concentrations only above their saturation concentration. The cluster size grows with increasing protein concentration and eventually a single protein droplet forms with all proteins incorporated in it at very high concentrations. Importantly, ensemble conformations progressively shift from single-chain to bulk dense-phase and follow a sequence-independent universal behavior as a function of protein concentration normalized by the saturation concentration.
Even though the EKVs and natural proteins exhibit pronounced sequence-dependent conformational characteristics in the dilute phase (Fig. S18a†),38 the conformation within the dense phase demonstrates only a very weak sequence dependence (Fig. 5). In fact, the relationship between the protein's Rg and Re within the condensates is consistent with theoretical predictions for an ideal chain 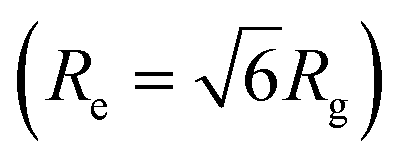 and their intramolecular distance (Rij) closely mirrors the scaling expected for an ideal chain (Fig. S18b†). These analyses strongly suggest that the protein conformations within the dense phase are akin to that of an ideal chain, which is characterized by being entropy-driven and not strongly influenced by the protein sequence or dilute-phase conformational properties.46 We note that our results are consistent with the limited experimental data available in the literature.8,33,35,57
and their intramolecular distance (Rij) closely mirrors the scaling expected for an ideal chain (Fig. S18b†). These analyses strongly suggest that the protein conformations within the dense phase are akin to that of an ideal chain, which is characterized by being entropy-driven and not strongly influenced by the protein sequence or dilute-phase conformational properties.46 We note that our results are consistent with the limited experimental data available in the literature.8,33,35,57
 | ||
| Fig. 5 Correlation between Rgsingle and Rginterface (green squares, left y-axis) or Rgdense (red diamonds, right y-axis) for all investigated EKVs. The mean values are obtained by dividing the trajectory into 5 independent blocks. Error bars indicate standard errors about the mean. In some cases, error bars are smaller than the symbol size. | ||
For conformational characteristics of proteins at the droplet interface, we do observe a significant dependence on the protein sequence, and hence their dilute-phase properties (Fig. 5). Importantly, the protein chains at the interface are always more compact than the chains in the dense phase but remain more extended as compared to those in the dilute phase, independent of their sequence.
The results presented here provide insight into the conformational properties, quantified by Rg, of a specific subclass of IDPs (without secondary structures) during phase separation. Our findings highlight the conformational changes these IDPs undergo as they transition from the dilute phase to the dense phase, including their behavior at the interface. These results improve our understanding of how IDP conformational transitions contribute to the formation of biomolecular condensates and may influence their functional and pathological roles. Future research is needed to assess whether these findings extend to other disordered proteins, particularly those with secondary structures, which may exhibit distinct behaviors and preferences during phase separation.
Data availability
Details on the MD simulations and cluster analysis are provided in the ESI.†Author contributions
Jiahui Wang: data curation, formal analysis, investigation, methodology, visualization, writing – original draft; Dinesh Sundaravadivelu Devarajan: data curation, formal analysis, investigation, writing – original draft; Keerthivasan Muthukumar: data curation, formal analysis, investigation; Young C. Kim: project administration, supervision, writing – review & editing; Arash Nikoubashman: project administration, supervision, writing – review & editing; Jeetain Mittal: conceptualization, funding acquisition, project administration, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based on the research supported by the National Institute of General Medical Science (NIGMS) of the National Institutes of Health under the grant R01GM136917, R35GM153388 and the Welch Foundation under the grant A-2113-20220331. The data on TDP-43 disordered domain were generated as part of a project supported by NINDS and NIA grant R01NS116176. A. N. acknowledges funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through Project 470113688. Y. C. K. is supported by the Office of Naval Research via the U.S. Naval Research base program. We gratefully acknowledge the computational resources provided by the Texas A&M High Performance Research Computing (HPRC) to complete this work.References
- A. A. Hyman, C. A. Weber and F. Jülicher, Annu. Rev. Cell Dev. Biol., 2014, 30, 39–58 CrossRef CAS PubMed.
- S. F. Banani, H. O. Lee, A. A. Hyman and M. K. Rosen, Nat. Rev. Mol. Cell Biol., 2017, 18, 285–298 CrossRef CAS PubMed.
- H. Falahati and A. Haji-Akbari, Soft Matter, 2019, 15, 1135–1154 Search PubMed.
- S. Alberti, A. Gladfelter and T. Mittag, Cell, 2019, 176, 419–434 CrossRef CAS PubMed.
- G. L. Dignon, R. B. Best and J. Mittal, Annu. Rev. Phys. Chem., 2020, 71, 53–75 CrossRef CAS PubMed.
- A. S. Lyon, W. B. Peeples and M. K. Rosen, Nat. Rev. Mol. Cell Biol., 2021, 22, 215–235 CrossRef CAS PubMed.
- R. W. Tibble, A. Depaix, J. Kowalska, J. Jemielity and J. D. Gross, Nat. Chem. Biol., 2021, 17, 615–623 Search PubMed.
- J. Wen, L. Hong, G. Krainer, Q.-Q. Yao, T. P. J. Knowles, S. Wu and S. Perrett, J. Am. Chem. Soc., 2021, 143, 13056–13064 CrossRef CAS PubMed.
- A. S. Holehouse and B. B. Kragelund, Nat. Rev. Mol. Cell Biol., 2023, 25, 187–211 Search PubMed.
- S. Rekhi, D. Sundaravadivelu Devarajan, M. P. Howard, Y. C. Kim, A. Nikoubashman and J. Mittal, J. Phys. Chem. B, 2023, 127, 3829–3838 Search PubMed.
- H. S. Ashbaugh and H. W. Hatch, J. Am. Chem. Soc., 2008, 130, 9536–9542 CrossRef CAS PubMed.
- A. Vitalis and R. V. Pappu, J. Comput. Chem., 2009, 30, 673–699 Search PubMed.
- H. Hofmann, A. Soranno, A. Borgia, K. Gast, D. Nettels and B. Schuler, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16155–16160 CrossRef CAS PubMed.
- C. M. Miller, Y. C. Kim and J. Mittal, Biophys. J., 2016, 111, 28–37 CrossRef CAS PubMed.
- A. S. Holehouse and R. V. Pappu, Annu. Rev. Biophys., 2018, 47, 19–39 Search PubMed.
- U. Baul, D. Chakraborty, M. L. Mugnai, J. E. Straub and D. Thirumalai, J. Phys. Chem. B, 2019, 123, 3462–3474 CrossRef CAS PubMed.
- S. Müller-Späth, A. Soranno, V. Hirschfeld, H. Hofmann, S. Rüegger, L. Reymond, D. Nettels and B. Schuler, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14609–14614 CrossRef PubMed.
- A. H. Mao, S. L. Crick, A. Vitalis, C. L. Chicoine and R. V. Pappu, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8183–8188 Search PubMed.
- H. S. Samanta, D. Chakraborty and D. Thirumalai, J. Chem. Phys., 2018, 149, 163323 Search PubMed.
- J. A. Marsh and J. D. Forman-Kay, Biophys. J., 2010, 98, 2383–2390 Search PubMed.
- Z. M. Jedlinska and R. A. Riggleman, Soft Matter, 2023, 19, 7000–7010 RSC.
- R. K. Das and R. V. Pappu, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13392–13397 CrossRef CAS PubMed.
- L. Sawle and K. Ghosh, J. Chem. Phys., 2015, 143, 085101 CrossRef PubMed.
- W. Zheng, G. Dignon, M. Brown, Y. C. Kim and J. Mittal, J. Phys. Chem. Lett., 2020, 11, 3408–3415 CrossRef CAS PubMed.
- A. M. Rumyantsev, A. Johner and J. J. de Pablo, ACS Macro Lett., 2021, 10, 1048–1054 CrossRef CAS PubMed.
- M. Kumar Hazra and Y. Levy, Phys. Chem. Chem. Phys., 2020, 22, 19368–19375 RSC.
- H. T. Tran, A. Mao and R. V. Pappu, J. Am. Chem. Soc., 2008, 130, 7380–7392 CrossRef CAS PubMed.
- H. Kang, P. A. Pincus, C. Hyeon and D. Thirumalai, Phys. Rev. Lett., 2015, 114, 068303 CrossRef PubMed.
- J. Huihui, T. Firman and K. Ghosh, J. Chem. Phys., 2018, 149, 085101 CrossRef PubMed.
- S. Wohl, M. Jakubowski and W. Zheng, J. Phys. Chem. Lett., 2021, 12, 6684–6691 CrossRef CAS PubMed.
- X. Yuan, H. W. Hatch, J. C. Conrad, A. B. Marciel and J. C. Palmer, Soft Matter, 2023, 19, 4333–4344 RSC.
- L. Baidya and G. Reddy, J. Phys. Chem. Lett., 2022, 13, 9589–9598 CrossRef CAS PubMed.
- D. Ubbiali, M. Fratini, L. Piersimoni, C. H. Ihling, M. Kipping, I. Heilmann, C. Iacobucci and A. Sinz, Angew. Chem., 2022, 134, e202205726 CrossRef.
- A. Majumdar, P. Dogra, S. Maity and S. Mukhopadhyay, J. Phys. Chem. Lett., 2019, 10, 3926–3936 CrossRef PubMed.
- D. J. Bauer, L. S. Stelzl and A. Nikoubashman, J. Chem. Phys., 2022, 157, 154903 CrossRef CAS PubMed.
- J. J. Madinya, L.-W. Chang, S. L. Perry and C. E. Sing, Mol. Syst. Des. Eng., 2020, 5, 632–644 RSC.
- H. Lyons, R. T. Veettil, P. Pradhan, C. Fornero, N. De La Cruz, K. Ito, M. Eppert, R. G. Roeder and B. R. Sabari, Cell, 2023, 186, 327–345 CrossRef CAS PubMed.
- D. S. Devarajan, S. Rekhi, A. Nikoubashman, Y. C. Kim, M. P. Howard and J. Mittal, Macromolecules, 2022, 55, 8987–8997 CrossRef CAS PubMed.
- D. S. Devarajan, J. Wang, A. Nikoubashman, Y. C. Kim and J. Mittal, Nat. Commun., 2024, 15, 1912 CrossRef PubMed.
- T. Firman and K. Ghosh, J. Chem. Phys., 2017, 148, 123305 CrossRef PubMed.
- U. Rana, C. P. Brangwynne and A. Z. Panagiotopoulos, J. Chem. Phys., 2021, 155, 125101 CrossRef CAS PubMed.
- Y.-H. Lin and H. S. Chan, Biophys. J., 2017, 112, 2043–2046 CrossRef CAS PubMed.
- J. Rudnick and G. Gaspari, Science, 1987, 237, 384–389 CrossRef CAS PubMed.
- O. Jagodzinski, E. Eisenriegler and K. Kremer, J. Phys., 1992, 2, 2243–2279 CAS.
- R. I. Dima and D. Thirumalai, J. Phys. Chem. B, 2004, 108, 6564–6570 CrossRef CAS.
- M. Rubinstein and R. H. Colby, Polymer physics, Oxford University Press, New York, 2003 Search PubMed.
- J. Wang, D. S. Devarajan, A. Nikoubashman and J. Mittal, ACS Macro Lett., 2023, 12, 1472–1478 CrossRef CAS PubMed.
- A. Patel, H. O. Lee, L. Jawerth, S. Maharana, M. Jahnel, M. Y. Hein, S. Stoynov, J. Mahamid, S. Saha, T. M. Franzmann, A. Pozniakovski, I. Poser, N. Maghelli, L. A. Royer, M. Weigert, E. W. Myers, S. Grill, D. Drechsel, A. A. Hyman and S. Alberti, Cell, 2015, 162, 1066–1077 CrossRef CAS PubMed.
- A. E. Conicella, G. H. Zerze, J. Mittal and N. L. Fawzi, Structure, 2016, 24, 1537–1549 CrossRef CAS PubMed.
- V. H. Ryan, G. L. Dignon, G. H. Zerze, C. V. Chabata, R. Silva, A. E. Conicella, J. Amaya, K. A. Burke, J. Mittal and N. L. Fawzi, Mol. Cell, 2018, 69, 465–479 CrossRef CAS PubMed.
- S. Elbaum-Garfinkle, Y. Kim, K. Szczepaniak, C. C.-H. Chen, C. R. Eckmann, S. Myong and C. P. Brangwynne, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7189–7194 CrossRef CAS PubMed.
- G. L. Dignon, W. Zheng, R. B. Best, Y. C. Kim and J. Mittal, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9929–9934 CrossRef CAS PubMed.
- B. Gouveia, Y. Kim, J. W. Shaevitz, S. Petry, H. A. Stone and C. P. Brangwynne, Nature, 2022, 609, 255–264 CrossRef CAS PubMed.
- T. J. Böddeker, K. A. Rosowski, D. Berchtold, L. Emmanouilidis, Y. Han, F. H. T. Allain, R. W. Style, L. Pelkmans and E. R. Dufresne, Nat. Phys., 2022, 18, 571–578 Search PubMed.
- W. P. Lipiński, B. S. Visser, I. Robu, M. A. A. Fakhree, S. Lindhoud, M. M. A. E. Claessens and E. Spruijt, Sci. Adv., 2022, 8, eabq6495 Search PubMed.
- I.-F. W. Kuo, C. J. Mundy, B. L. Eggimann, M. J. McGrath, J. I. Siepmann, B. Chen, J. Vieceli and D. J. Tobias, J. Phys. Chem. B, 2006, 110, 3738–3746 CrossRef CAS PubMed.
- A. Joshi, A. Walimbe, A. Avni, S. K. Rai, L. Arora, S. Sarkar and S. Mukhopadhyay, Nat. Commun., 2023, 14, 7331 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The details of the model and simulations, method of nSCD calculation, method for cluster analysis, amino acid code for the model and natural proteins, dense phase and saturation concentrations, probability distribution of κ2, cluster size distributions, effect of concentration scan on probability distribution of Rg, number of clusters, and average Rg, concentration profile and Rg with respect to the distance from the condensate's center of mass for the EKVs, interresidue distance analysis for the EKVs. See DOI: https://doi.org/10.1039/d4sc05004e |
| This journal is © The Royal Society of Chemistry 2024 |