
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGuest modulating the photoactivity of a titanium-oxide cage†
Dexin
Wang‡
a,
Yanshu
Liu‡
a,
Guanyun
Zhang
 *a,
Menghui
Chu
a,
Fangfang
Gao
a,
Guanjie
Chen
a,
Guo
Wang
b,
Chen-Ho
Tung
a and
Yifeng
Wang
*a
*a,
Menghui
Chu
a,
Fangfang
Gao
a,
Guanjie
Chen
a,
Guo
Wang
b,
Chen-Ho
Tung
a and
Yifeng
Wang
*a
aKey Lab for Colloid and Interface Science of Ministry of Education, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, China. E-mail: guanyzhang@sdu.edu.cn; yifeng@sdu.edu.cn
bDepartment of Chemistry, Capital Normal University, Beijing 100048, China
First published on 13th November 2024
Abstract
Two host–guest Ti-oxide clusters, Ti14(NH4)2 and Ti14Cs2, were synthesized and thoroughly characterized. They possess a rarely seen biloculate structure that encapsulates two NH4+ and Cs+ guests, respectively. Interestingly, alkali metal cations can exchange places with NH4+. The ability of the host to capture the guest cations follows the order Cs+ > NH4+ > Rb+ > K+. The guests heavily influence the physiochemical properties and photocatalytic activities of the complexes. Ti14Cs2 exhibits a redshifted visible-light absorption edge, increased charge-separation properties, and enhanced interfacial charge-transfer ability compared to Ti14(NH4)2. It also demonstrates excellent performance in photocatalytic CO2/epoxide cycloaddition reactions regarding the reaction rate, scalability, sunlight usage, catalyst recyclability, and stability. This study presents a novel Ti-oxide-based cage cluster with exchangeable guests and provides insights for enhancing the solar harvesting applications of Ti-oxide cages.
1 Introduction
Since Pedersen, Cram, and Lehn shared the 1987 Nobel Prize in chemistry, the construction of multicomponent supramolecular assemblies has been a topic of extensive investigation. Host–guest compounds represent an important branch of supramolecular assemblies characterized by cage hosts and their enclosed guests.1–3 These complexes play important roles in various applications, including catalysis, separation, drug delivery, sensors, and molecular machines.4–8 Most host–guest complexes are constructed using organic or organometallic hosts such as crown ethers, cyclodextrins, calixarenes, and metallacages.9–13 In contrast, inorganic hosts could have unique characteristics like photoreactivity, electrical conductivity, or magnetic properties.14 Moreover, these properties may be modulated or even controlled by the enclosed guests.15 However, only a few inorganic hosts that can preferentially capture and reversibly exchange their guests have been reported, with {P5W30}15 and {Mo132}3,16 as representative examples. Despite numerous other inorganic cage complexes with encapsulated ions, their guests are structural templates and are not exchangeable.17–20 Exploring new inorganic host–guest compounds and their functionalities remains highly desirable.Titanium-oxide clusters (TOCs) are a class of molecular compounds characterized by their discrete titania cores. They are considered molecular analogues to TiO2 and titanates but have precise molecular structures.21–23 Research on TOCs has been rapidly growing in the past decade due to their photoresponse, photocatalytic properties, and accurate structural information that can be used for elucidating structure–property relationships.14,24–29 Interestingly, a few TOCs were synthesized with encapsulated alkali metal cations like {M@Ti12}28 (M = Cs+, Rb+, K+, H3O+, or NH4+), halide anions like30,31 {I@Ti15Mn2}22 and {I@Ti22},31,32 and organic cations like {amine@Ti8}.33 In these cases, host–guest interactions were determined to be electrostatic following the size complementary rule, just like crown ethers capturing alkali metal cations.28 Moreover, Winpenny et al. and we also reported the use of Ti-oxide rings, e.g., {TixM} (x = 7–9; M = Fe3+, Ga3+, Cr3+, In3+, and Al3+)34,35 and {Ti8},36 to assemble rotaxanes and catenanes. Therefore, TOCs can be a new class of inorganic motifs and can play a significant role in supramolecular chemistry.14 However, it is a pity that there has not been much research done on the photoresponse of TOCs in these complexes, which could be one of the distinguishing characteristics of this class of supramolecular materials.
In this study, two TOCs with the same titanium-oxide host, [Ti14O12Sal10HSal14Ac2]2− (denoted as Ti14), which has a very rarely seen biloculate cage, were synthesized. The guest exchange thermodynamics were studied. We observed that the host–guest interaction not only influenced the dimensions of the Ti14 host but also significantly altered the photo-absorbance, physiochemical properties, and photocatalytic activity of the host. The Cs+-encapsulation endowed the Ti14 host with superior photocatalytic performance compared to that of the NH4+-encapsulation. The former served as a heterogeneous and recyclable photocatalyst to trigger CO2/epoxide cycloaddition reactions. The photocatalytic mechanism and the structure–activity relationship were studied. This study presents a novel metal-oxide host for host–guest chemistry and provides insights for enhancing the solar harvesting applications of such compounds.
2 Results and discussion
2.1 Structural study
The first compound Ti14(NH4)2, with the formula (NH4)2Ti14O12Sal10HSal14AC2 (Sal = salicylate, HSal = monoprotonated salicylate, and AC = acetone), was obtained as orange block crystals through a solvothermal reaction of Ti(OiPr)4, NH4I, and salicylic acid at 80 °C. When caesium acetate (CsAc) was used instead of NH4I under the otherwise same conditions, the red block crystals of Ti14Cs2 were obtained with the formula Cs2Ti14O12Sal10HSal14AC2 (Fig. 1A). Both compounds were prepared in high yields (>30% based on Ti), and gram-scale synthesis is feasible. The structures and compositions were precisely characterized with single-crystal X-ray diffraction (SCXRD) and other complementary techniques, including powder X-ray diffraction (PXRD), FTIR, Raman (Fig. S1†), elemental analysis, and 133Cs NMR analysis. | ||
| Fig. 1 (A) Syntheses and molecular packing of Ti14(NH4)2 and Ti14Cs2. (B) The ball-and-stick views of the Ti14 host. (C) The Ti skeleton of the Ti14 host. Color scheme: green ball, Ti; red ball, O; grey ball, C; pink ball, Cs; orange ball, NH4+; blue bond, Ti–O–C–O–Ti; purple bond, Ti–O–Ti. For clarity, H is omitted from the figures. | ||
According to SCXRD, Ti14(NH4)2 crystallizes in the triclinic crystal system of the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The Ti14(NH4)2 molecule contains a biloculate host with the formula [Ti14O12Sal10HSal14Ac2]2− (denoted as Ti14). Ti14(NH4)2 resembles a centrally symmetric ellipsoidal shape (Fig. 1B), with a long axis of 25 Å and a short axis of 20 Å. Every nine Ti4+ ions are connected by μ2-O and salicylate ions to form a cage with a vast internal cavity. The two cavities of a Ti14 host are separated by a Ti4O2(CO2)2 ring (Fig. 1C). SCXRD analysis reveals that each of the two cavities contains a guest. However, SCXRD cannot distinguish whether the guest is H3O+ or NH4+, and elemental analysis cannot differentiate whether the nitrogen element comes from the guest or the acetonitrile solvent. Therefore, to determine if the guest is H3O+ or NH4+, the colorimetric method was used, and it ascertained that the guests are two NH4+ ions (Fig. S2†).
space group. The Ti14(NH4)2 molecule contains a biloculate host with the formula [Ti14O12Sal10HSal14Ac2]2− (denoted as Ti14). Ti14(NH4)2 resembles a centrally symmetric ellipsoidal shape (Fig. 1B), with a long axis of 25 Å and a short axis of 20 Å. Every nine Ti4+ ions are connected by μ2-O and salicylate ions to form a cage with a vast internal cavity. The two cavities of a Ti14 host are separated by a Ti4O2(CO2)2 ring (Fig. 1C). SCXRD analysis reveals that each of the two cavities contains a guest. However, SCXRD cannot distinguish whether the guest is H3O+ or NH4+, and elemental analysis cannot differentiate whether the nitrogen element comes from the guest or the acetonitrile solvent. Therefore, to determine if the guest is H3O+ or NH4+, the colorimetric method was used, and it ascertained that the guests are two NH4+ ions (Fig. S2†).
Ti14Cs2 also crystallizes in the triclinic crystal system of the P space group. It possesses the same Ti14 host cage as Ti14(NH4)2. Unlike Ti14(NH4)2, the Ti14 host of Ti14Cs2 encapsulates two Cs+ guest ions inside its two chambers.
The guest cations, NH4+ and Cs+, have electrostatic interactions with the host, analogous to alkali metal cations and crown ethers.37 The distances between the guest and the peripheral O range from 3.05 to 3.32 Å. Since Cs+ has a larger radius of 3.38 Å than NH4+ (2.86 Å), the Ti14 host of Ti14Cs2 is slightly larger than that of Ti14(NH4)2, with the long and short dimensions expanding to 26 and 22 Å, respectively. The Ti–O bond lengths of both compounds are compared in Table S1,† further indicating that Cs+ encapsulation induces an increase in the size of the host. These findings demonstrate that the Ti14 host is flexible, which allows it to change the cavity size according to the guest size, and that the radius of the guest influences the host–guest interactions.
Both compounds can dissolve in DMF solvent. Their structural integrities in DMF solutions were investigated through small-angle X-ray scattering (SAXS) analyses. As illustrated in Fig. S3,† their patterns are comparable and consistent with identical molecular structures. The experimental curves of both compounds closely match the simulated spectra obtained from the crystallographic structure, suggesting that they retain structural integrity in DMF. The pair distance distribution function curve (PDDF; Fig. 2A and discussion in the ESI†) shows peak distributions that agree well with the simulated ones from the crystallographic structure. However, the radii of the second and third peaks in the PDDF curve of Ti14Cs2 are larger than those of Ti14(NH4)2, consistent with the earlier description that Cs+ encapsulation induces an increase in cluster size. The pattern remained unchanged after the solution was stored for three weeks, demonstrating that both compounds are stable in DMF.
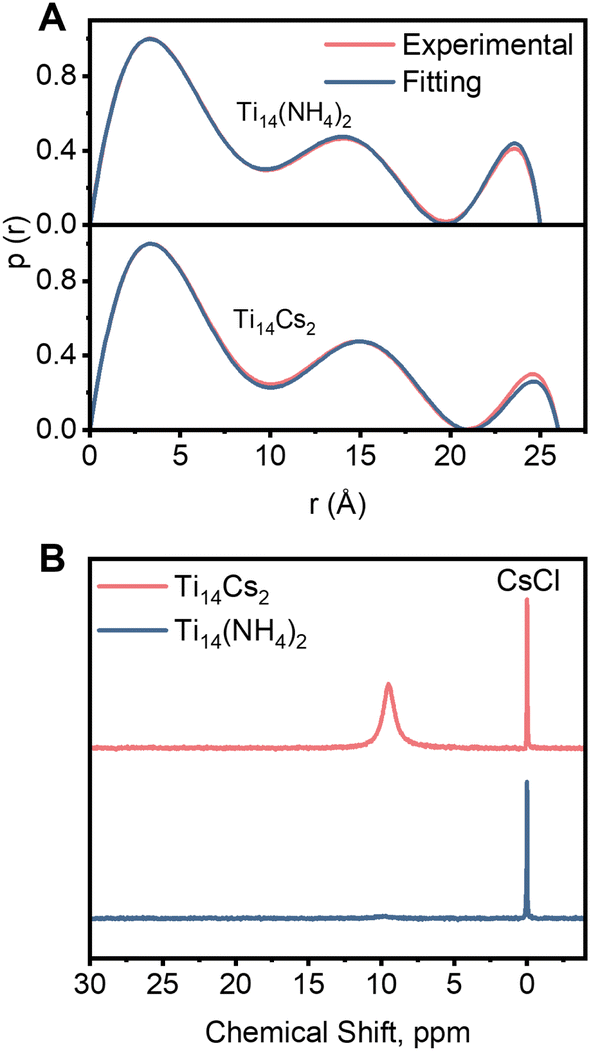 | ||
| Fig. 2 (A) PDDF curves, the probability p(r) vs. radius r of Ti14Cs2 and Ti14(NH4)2. (B) 133Cs NMR of Ti14Cs2 and Ti14(NH4)2 in DMF solvent (the reference is 0.5 M CsCl in D2O). | ||
Next, 133Cs NMR spectra were recorded using 0.5 M aq CsCl in D2O as a standard. Fig. 2B shows no peak in Ti14(NH4)2. In contrast, Ti14Cs2 displays a broad peak at 9.6 ppm, which is assigned to Cs+ located in the cavities of the host. This spectrum and those of previously reported Cs+-enclosing host–guest compounds are compared in Table S2.† The results reveal significant differences in the chemical shifts among the compounds. However, the chemical shift of Ti14Cs2 is comparable to that of a recently reported Ti-salicylate complex, Ti12Cs.38
2.2 Uptake of alkali metal cations
We speculate that the Ti14 host may exhibit different capture abilities for Cs+ and NH4+. To investigate the substitution of Cs+ for NH4+ using 133Cs NMR, solid CsCl was added to 3 mL of DMF solution containing 21.2 μmol of Ti14(NH4)2. As the CsCl solid is entirely insoluble in DMF (solubility below the detection limit of 133Cs NMR), the area of the 9.6 ppm peak could be utilized to determine the quantity of Cs+ captured by the Ti14 host. As shown in Fig. 3A, the peak area in the 133Cs NMR spectrum gradually increased with the added amount of CsCl, indicating the substitution of NH4+ by Cs+ in the solution. Based on the peak area relative to the standard, the amount of captured Cs+ was calculated. From Fig. 3B, the amount of captured Cs+ rose linearly with the added amount of solid CsCl until it reached ca. 42.4 μmol, after which it remained constant. The slope of the linear portion was found to be 0.49 (R2 = 0.999), close to the ideal slope of 0.5, consistent with the assumption that the Ti14 host captured all added CsCl and each host captured two Cs+ ions. | ||
| Fig. 3 (A) 133Cs NMR spectra of replacing NH4+ by Cs+ in the Ti14(NH4)2/DMF solution. (B) Formation of Ti14Cs2 with added Cs+ measured by 133Cs NMR. (C) The variation of the Cs/Ti ratio in the DMF solution with added CsCl measured by ICP-MS. | ||
The above results were further confirmed by ICP-MS analysis. Control experiments demonstrated that after dispersing solid CsCl in DMF and stirring for two days, Cs+ was undetectable in the filtrate by ICP-MS, indicating the insolubility of Cs+ in DMF, consistent with the results of 133Cs NMR. Then, a series of solutions used for 133Cs NMR experiments were analysed using ICP-MS to determine Cs and Ti concentrations and molar ratios. As shown in Fig. 3C, the Cs/Ti molar ratio increased linearly with the amount of CsCl, reaching roughly 42.4 μmol (slope = 0.034 and R2 = 0.998), consistent with the 133Cs NMR results, indicating the capture of Cs+ by the Ti14 host. When the amount of CsCl exceeded 42.4 μmol, the Cs/Ti molar ratio remained constant at 0.142, consistent with the formation of Ti14Cs2 (ideal value of 0.143). The attempt to quantify the released NH4+ was unsuccessful because the TOCs and NH4+ were both in bulk solution and could not be separated. Nonetheless, the results of 133Cs NMR and ICP-MS analyses are consistent, both demonstrating that Cs+ can substitute NH4+ in Ti14(NH4)2. The “titration” curve described above shows that before reaching the endpoint, the yield of Ti14Cs2 and the Cs/Ti molar ratio increased linearly with the addition of CsCl, after which little further change occurred. This suggests that Ti14 should be able to solubilize CsCl in DMF quantitatively, and all dissolved CsCl is located within the Ti14 cavities.
The capability of the Ti14 host to capture other alkali metal cations, Rb+ and K+, was further investigated. To this end, two alkali metal chlorides were simultaneously added to a DMF solution of Ti14(NH4)2 and stirred for 24 hours for guest exchange. After removing the insoluble salts, the molar ratio of the captured alkali metal ions was analysed using ICP-MS (the solubility of the salts was also measured). The results can indicate the relative ability of the Ti14 host to capture the two alkali metal ions. As shown in Fig. S4,†Ti14 exhibited different capturing abilities for Cs+, Rb+, and K+, in the order Cs+ > NH4+ > Rb+ > K+. The ionic diameters of Rb+ and K+ are 3.04 and 2.76 Å, respectively, smaller than that of Cs+. Consequently, the capturing ability of Ti14 for K+, Rb+, and Cs+ ions may be associated with the size of the guest, conforming to the size-matching principle.39,40
2.3 Electronic structures and photoresponse
To compare the effects of the guests, Cs+ and NH4+, on the optical absorption properties of the TOCs, UV-vis diffuse reflectance spectroscopy (UV-vis DRS) was used. As depicted in Fig. 4A, the absorption edge of Ti14(NH4)2 is ca. 590 nm, whereas that of Ti14Cs2 is slightly larger at 671 nm. According to the Kubelka–Munk function, the direct bandgap values for Ti14(NH4)2 and Ti14Cs2 are 2.30 and 1.93 eV (Fig. 4B), respectively. Hence, Cs+ enhances the optical absorption performance of the Ti14 host and reduces the bandgap energy.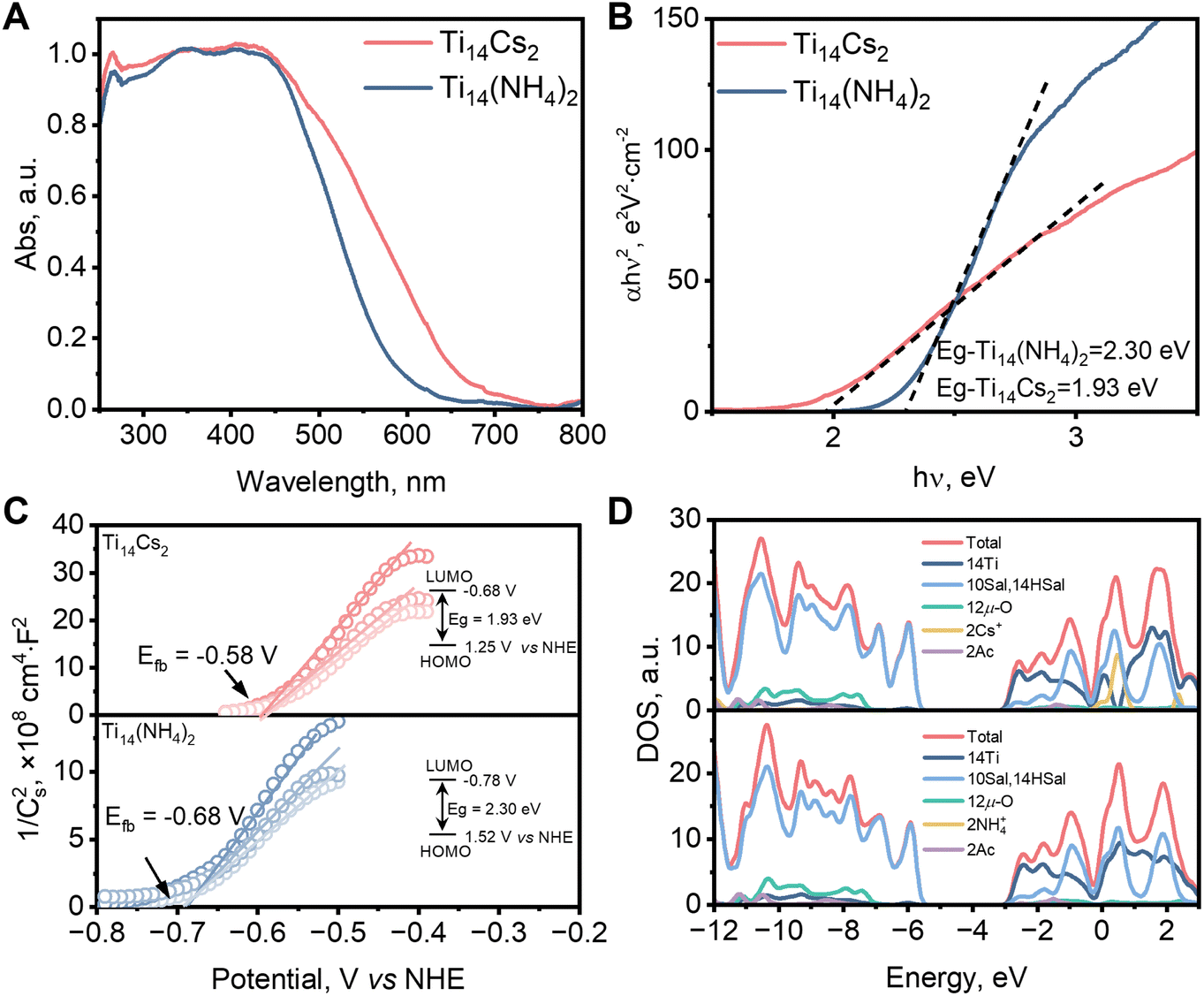 | ||
Fig. 4 (A) UV-vis DRS of Ti14(NH4)2 and Ti14Cs2. (B) Kubelka–Munk function curves. (C) Mott–Schottky plots of Ti14(NH4)2 and Ti14Cs2 at various frequencies, from top to bottom, 100, 200, and 500 Hz. Electrolyte: 0.2 M Na2SO4 solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 v/v ethanol–water). (D) The density of states (DOS) plots of Ti14(NH4)2 and Ti14Cs2. :10 v/v ethanol–water). (D) The density of states (DOS) plots of Ti14(NH4)2 and Ti14Cs2. | ||
Mott–Schottky analyses were used to determine the band potentials. The curves of both compounds exhibit positive slopes, typical of n-type semiconductors (Fig. 4C). According to the curves at different frequencies, the flat-band potentials (Efb) for Ti14(NH4)2 and Ti14Cs2 were determined to be ca. −0.68 and −0.58 V vs. NHE, respectively. Since the conduction band bottom of most n-type semiconductors is ca. 0.10 V lower than Efb,38,41 the lowest unoccupied molecular orbital (LUMO) potentials for Ti14(NH4)2 and Ti14Cs2 are −0.78 and −0.68 V vs. NHE, respectively. Based on their bandgap values, their highest occupied molecular orbital (HOMO) potentials are +1.52 and +1.25 V vs. NHE, respectively. The LUMO and HOMO potentials were also evaluated by electrochemical measurements (Fig. S6†). The values are consistent with those obtained from the combination of Mott–Schottky analyses and UV-vis DRS mentioned above. Notably, the HOMO and LUMO potentials are compatible with the reduction of CO2 and the oxidation of a variety of organic compounds such as epoxides (Fig. S7†), making the TOCs useful in photocatalytic CO2/epoxide cycloaddition and CO2 reduction reactions.
The electronic structures were then determined by density functional theory (DFT) calculations. For each cluster, the HOMO is primarily composed of salicylate groups, while the LUMO is predominantly composed of Ti 3d orbitals, with some contribution from salicylate (Fig. 4D). This indicates that photoexcitation causes ligand-to-metal charge transfer (LMCT) and ligand-to-ligand charge transfer (LLCT).42–47 The calculated bandgap energy of Ti14Cs2 is slightly smaller than that of Ti14(NH4)2, corresponding to a redshift of ca. 7 nm in the absorption band edge. While this is consistent with the trend of the experimental results, the calculated bandgap energy difference is very small, presumably owing to the errors of the DFT calculations.48
Steady-state and transient photoluminescence (PL) spectra were recorded to compare the two TOCs' charge-separation efficiency. Ti14Cs2 and Ti14(NH4)2 exhibit prominent PL peaks at 340–420 nm and smaller peaks ranging from 450 to 500 nm (Fig. 5A and S5†), with the peak intensity of Ti14Cs2 being smaller than that of Ti14(NH4)2. Moreover, the PL lifetime of Ti14Cs2, i.e., 21.8 ns, is slightly longer than that of Ti14(NH4)2, i.e., 18.1 ns (Fig. 5B). Steady-state and transient PL spectra suggest that Ti14Cs2 has a higher charge-separation efficiency than Ti14(NH4)2. Ti14Cs2 has a smaller arc radius than Ti14(NH4)2, which suggests a reduced charge transfer resistance, according to the electrochemical impedance spectra (EIS; Fig. 5C). Fig. 5D shows that when exposed to visible light (λ ≥ 400 nm), Ti14Cs2 generated a transient photoanodic current that is roughly 1.5 times greater than that of Ti14(NH4)2. During this process, the sacrificial reagent in the electrolyte, which is isopropanol in this study, was oxidized by the photogenerated holes while the photogenerated electrons were eliminated by the bias when a TOC was photoexcited. Ti14Cs2 exhibited a higher photoanodic current, indicating a higher photo-electric conversion capacity, which is associated with its lower charge-transfer resistance, higher charge-separation efficiency, and stronger absorption of visible light. Overall, Ti14Cs2 has better photoinduced electron–hole separation and interfacial charge-transfer capability compared to Ti14(NH4)2, which are beneficial for sunlight harvesting and particularly photocatalysis.
 | ||
| Fig. 5 (A) steady-state PL spectra under 300 nm excitation. (B) PL decay spectra (λ = 395 nm) under 280 nm excitation. (C) Electrochemical impedance spectra (inset shows the equivalent circuit). (D) Transient photoanodic current at a bias of +1.0 V vs. NHE under visible light irradiation. | ||
2.4 Comparison of the photocatalytic activity
The addition of CO2 to epoxides, with 100% atom efficiency, is extremely promising for CO2 capture and storage and the production of value-added fine chemicals, e.g., cyclic carbonates and carbonate polymers.49,50 Currently, the catalysts used for cycloaddition reactions mainly consist of solid materials containing Lewis acidic or basic sites.51–54 The reaction requires high temperature and pressure. Moreover, the complexity of the catalyst's surface structures makes it difficult to study the structure–activity relationship. Due to the photocatalytic properties and well-defined structures, TOCs have recently been utilized in CO2/epoxide cycloaddition reactions. This has enabled the synergistic effects of Lewis acid and solar energy, providing exceptional catalytic performance under mild conditions.24,38,41,43–45,55A typical aliphatic epoxide, 1,2-hexene oxide (1a), was used as the model substrate in an investigation into the CO2/epoxide cycloaddition reaction to compare the catalytic activity of Ti14Cs2 and Ti14(NH4)2. The catalytic reaction was conducted under neat conditions of 1 bar CO2 and room temperature, with 1a serving as both the solvent and the reactant (Scheme 1). Interestingly, Ti14(NH4)2 dissolves in both 1a and the product 1b, while Ti14Cs2 is entirely insoluble in both liquids. Therefore, Ti14Cs2 represents a recyclable photocatalyst, offering advantages over Ti14(NH4)2 in this reaction.
 | ||
| Scheme 1 The model CO2 cycloaddition reaction. | ||
Complete conversion of 1a into 1b took 54 and 90 hours, respectively, at room temperature and in the dark, using Ti14(NH4)2 and Ti14Cs2 as catalysts (Fig. 6A). Fig. S6 and S7† present FTIR, PXRD, and small-angle X-ray diffraction (SAXS) data that demonstrate that Ti14(NH4)2 and Ti14Cs2 were the true catalysts since they both remained stable throughout the reactions. Note that the crystalline Ti14Cs2 sample has a low surface area, <3 m2 g−1, implying a low utilization efficiency of the catalytic sites. For comparison, the estimated theoretical surface area of individual Ti14(NH4)2 molecules is 1868 m2 g−1, far larger than the crystalline Ti14Cs2 sample (see the ESI† for the calculations). Consequently, the experimental catalytic activity of Ti14Cs2 was underestimated, which may be the reason for the slower catalytic reaction. Fig. 6A also shows that simulated sunlight and visible light significantly accelerated the reactions. To quantify the effects of light on the catalysis, the initial rates were measured (Fig. S10†). For Ti14Cs2, the initial rate of the dark reaction was 1.2 × 10−4 mol h−1, while under visible light and simulated sunlight irradiation, the rates were 3.4 and 6.6 × 10−4 mol h−1, respectively. The enhancement factors are 2.8 and 5.5, respectively (Fig. 6B). For Ti14(NH4)2, the initial rate under dark conditions is 2.5 × 10−4 mol h−1, and the enhancement factors are 1.5 and 2.4 for visible light and simulated sunlight, respectively. It can be seen from the enhancement factors that light was more important in the Ti14Cs2 system than in the Ti14(NH4)2 system. Under simulated sunlight irradiation, Ti14Cs2 reached a slightly larger rate than Ti14(NH4)2, despite achieving a lower rate in the dark. Furthermore, given the poor catalytic site utilization efficiency of Ti14Cs2, the real activity of Ti14Cs2 under simulated sunlight irradiation ought to be significantly higher than that of Ti14(NH4)2 under otherwise identical conditions.
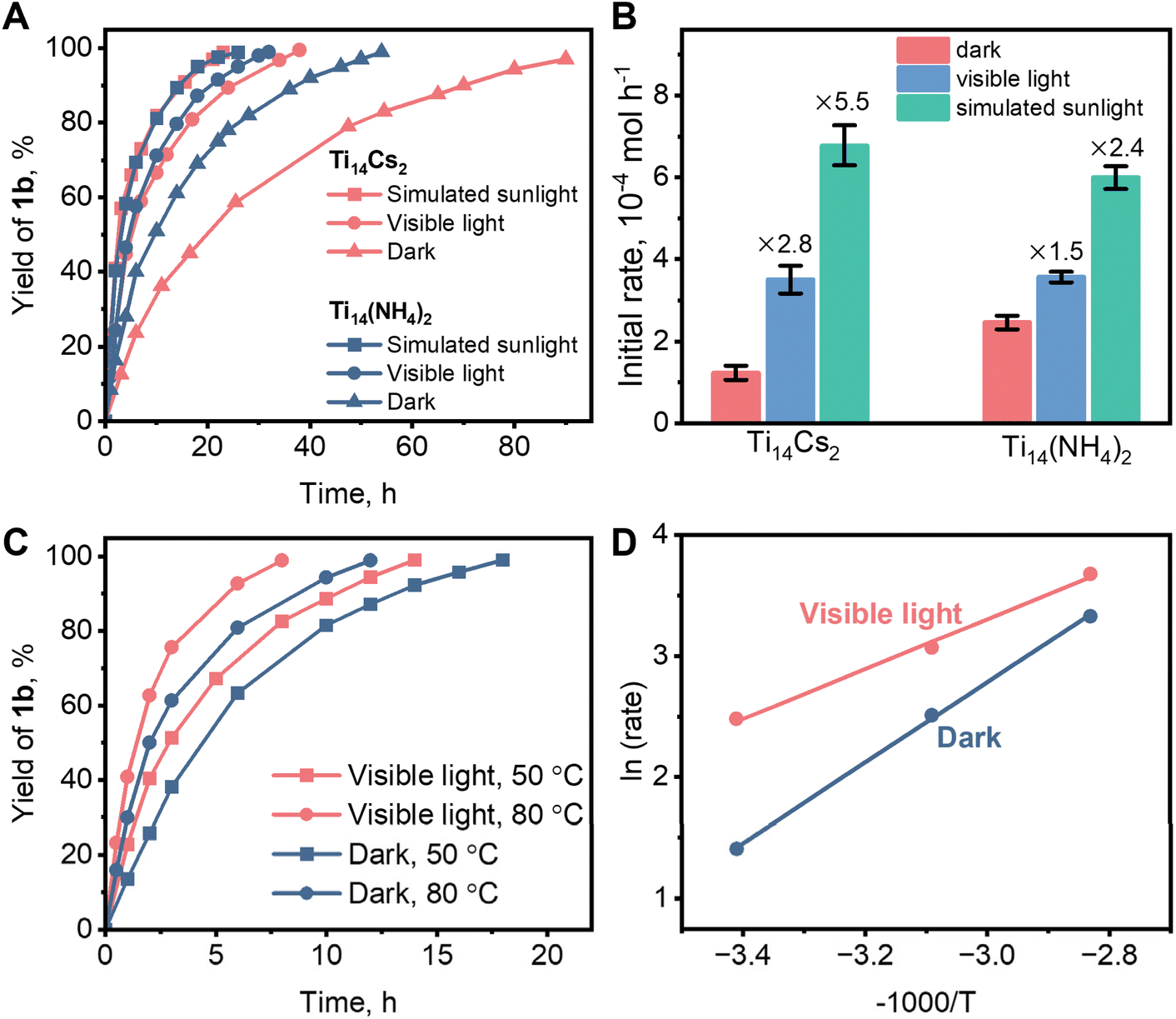 | ||
| Fig. 6 (A) The kinetics of Ti14(NH4)2 and Ti14Cs2 catalyzed or photocatalyzed model reactions (Scheme 1) at room temperature. (B) The formation rate of 1b in the early stage of the cycloaddition reactions (see Fig. S10† for the calculations). (C) The reaction kinetics at 50 and 80 °C photocatalyzed by Ti14Cs2 under visible-light irradiation. (D) Arrhenius plots of the dark and the light reactions. The unspecified conditions are shown in Scheme 1. | ||
According to their UV-vis DRS, Ti14Cs2 exhibits much better absorption properties than Ti14(NH4)2 in the 600–700 nm range (recall Fig. 4A). Consequently, it was expected that the effects of the light wavelength on the reaction kinetics of the two systems would also be distinct. We then used a 605 nm monochromatic LED to trigger the reaction. The data in Fig. S11† show that 605 nm light had a greater effect on the Ti14Cs2 system than on the Ti14(NH4)2 system. Therefore, light is indeed an important driving force in the CO2/epoxide cycloaddition reaction in the current systems.
Because of its heterogeneous nature and better photoresponse, Ti14Cs2 was chosen to study the photocatalytic cycloaddition mechanism. The effect of temperature on the catalytic activity was first investigated. As shown in Fig. 6C, raising the temperature to 50 and 80 °C reduced the time needed for complete conversion under dark conditions from 90 to 18 and 14 h, respectively. Visible light further accelerated the reaction rates at any temperature. For instance, the time for complete conversion at 50 and 80 °C was reduced to 12 and 8 h, respectively. This suggests the presence of a photo-thermal synergistic effect in the catalytic process. Namely, temperature can greatly affect the reaction rate, and light can multiply the effects of temperature. The apparent activation energies were calculated based on the initial rates at various temperatures and the Arrhenius equation (Fig. 6D). The values are 27.6 and 17.1 kJ mol−1 under dark and visible-light conditions, respectively. It is evident that light significantly reduced the apparent activation energy, thereby enhancing the reaction rate.
The outcomes of multiple control trials carried out for ten hours at 20 °C under visible light are displayed in Fig. S12.† The poor yield of 1b in the absence of Ti14Cs2 suggests that Ti14Cs2 was the photocatalyst. Conversely, if the cocatalyst TBAB was absent, 1a converted slowly without 1b formation. Using TBAC or TBAI to replace TBAB caused a decrease in yield. Hence, both the photocatalyst and the cocatalyst were essential for the success of the photocatalytic cycloaddition reaction. Furthermore, with the addition of hole scavengers like isopropanol and methanol and electron scavengers like 1,4-benzoquinone and AgNO3, the yields of 1b decreased significantly (see Fig. S13† and the discussion). Given that the catalytic activity of Ti14Cs2 in the cycloaddition reaction under dark conditions was attributed to its Lewis acidic Ti4+ sites, the enhancement of the reaction rate by light must indicate that both photo-generated electrons and holes served as additional active species, consistent with our previously proposed mechanism.24,38,55
2.5 The potential of Ti14Cs2 in CO2/epoxide cycloaddition
The potential of Ti14Cs2 in the CO2/epoxide cycloaddition reaction was explored. Ti14Cs2 remained insoluble during the photocatalytic processes, maintaining its solid state. It could be recycled by centrifugation. In the cycling experiments, the catalytic activity of Ti14Cs2 remained nearly constant (Fig. S8A†). The slight decrease in the product yield may be attributed to the loss of the catalyst during recycling. Moreover, the FTIR (Fig. S8B†) and PXRD (Fig. S8C†) spectra and scanning electron microscopy micrographs (Fig. S8D†) of the recycled Ti14Cs2 are nearly identical to those of the freshly synthesized sample, indicating that the molecular structure and crystal structure remained stable. Therefore, Ti14Cs2 is a recyclable photocatalyst with high stability in CO2/epoxide cycloaddition.To estimate the catalytic potential of Ti14Cs2, a scaleup experiment was performed, increasing the amount of 1a to 30 mmol while maintaining the other parameters constant. Under visible-light irradiation at 50 °C, it took 74 h for complete conversion of 1a, and the 1b yield was close to 100%, with a TON value of 1422 and a TOF value of 19.2 h−1 (Fig. S14A†). Given that Ti14Cs2 has excellent sunlight absorption properties, a 3 mmol scaleup experiment was also performed under natural sunlight irradiation at ambient temperature, and a 100% yield of 1b was obtained after a 40 h reaction (Fig. S14B†).
The scope of epoxides in the cycloaddition reaction was investigated. As indicated in Table 1, Ti14Cs2 had a high conversion and yield for all the epoxides. Epoxides with common alkyl groups (entries 1–3) and halogen groups (entries 4–5) reacted effectively, demonstrating that the catalytic system can handle both groups. Surprisingly, the high steric groups of phenyl epoxides (entry 6) could also be tolerated, with high conversion and almost 100% yield, suggesting that this approach is suitable for substituted aromatic epoxides. An internal epoxide with a large steric hindrance could also be wholly converted (entry 7) but at a slower rate.
| Entry | Substrate | Product | Time (h) | Yield (%) |
|---|---|---|---|---|
| a Reaction conditions: epoxide (3 mmol), Ti14Cs2 (100 mg), TBAB (0.5 mmol), CO2 (1 bar), biphenyl (50 mg), visible light, and 50 °C. | ||||
| 1 |
|
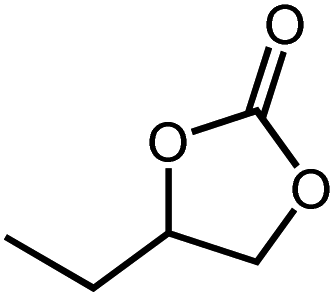
|
20 h | >99 |
| 2 |

|
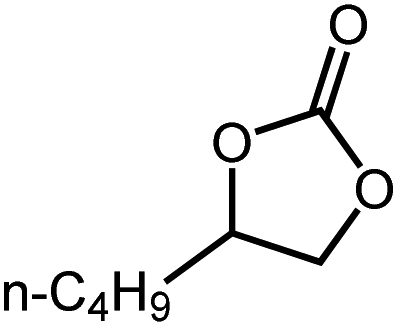
|
14 h | >99 |
| 3 |

|

|
24 h | >99 |
| 4 |
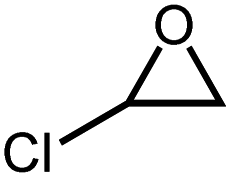
|

|
30 h | >99 |
| 5 |

|
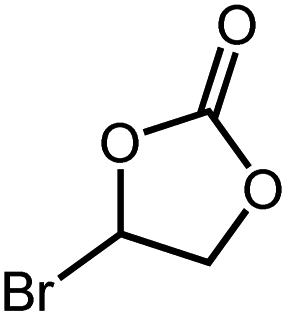
|
26 h | >99 |
| 6 |

|
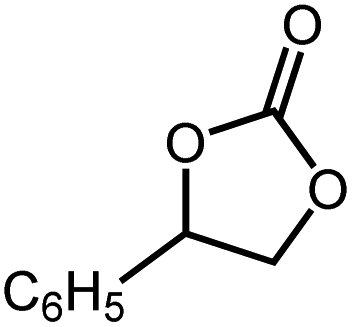
|
30 h | >99 |
| 7 |

|

|
40 h | 95 |
The performance of Ti14Cs2 was compared to that of state-of-the-art photocatalysts, particularly TOCs. As shown in Table S3,† the activity of Ti14Cs2 is comparable to their activity. The performance of Ti14Cs2 is also much better than that of TiO2 P25 (with a yield of 31% after 10 h, Fig. S12†), a benchmark commercial photocatalyst. Given the high activity, heterogeneous nature, high stability, scalable synthesis, and broad substrate scope, Ti14Cs2 has considerable potential for CO2 storage and synthesis of cyclocarbonates.
2.6 A brief discussion on guest controlled photocatalysis of Ti14
Both NH4+ and Cs+ do not undergo charge transfer with the Ti14 host because they are non-reducible cations. As a result, their impact on the properties of Ti14 primarily manifests in altering its geometric structure brought about by host–guest interactions, which affect Ti–O bond lengths, cavity sizes, and overall dimensions.Compared to NH4+, Cs+ reduces the solubility of Ti14 in epoxides and cyclic carbonates, making Ti14Cs2 a recyclable heterogeneous photocatalyst. Indeed, we also noticed that the solubility of Ti14Cs2 in DMF was much lower than that of Ti14(NH4)2, with approximately 35 and 152 g of solubility in 1 L of DMF at room temperature (26 °C), respectively. The following could be used to explain why Ti14Cs2 and Ti14(NH4)2 solubilize differently in organic solvents. Since ammonium halides are generally soluble in organic solvents, NH4+ can reach the bulk solution. The weak host–guest interaction of Ti14(NH4)2 relative to that of Ti14Cs2 might also facilitate the access of NH4+ for the bulk solution. Ti14(NH4)2 is therefore more soluble in 1a and DMF. In comparison, the host–guest interaction of Ti14Cs2 is stronger and cesium halide is insoluble in these solvents. Both can inhibit the solvation and lead to the low solubility of Ti14Cs2 in organic solvents.
According to various characterization techniques, the guest ion also influences the photophysical and photocatalytic properties of the host–guest TOCs. First, Ti14Cs2 exhibits better light absorption, charge separation, and interfacial charge transfer properties compared to Ti14(NH4)2. These are generally beneficial for solar light absorption and photocatalytic quantum efficiency. On the other hand, CO2 is reductively activated by photogenerated electrons, and epoxide is oxidatively activated by photogenerated holes during CO2/epoxide cycloaddition reactions.24,43,56 This mechanism was proposed previously and is also supported by the control experimental results herein (recall Fig. S11†). In this regard, a more negative CB potential and a more positive VB potential are both favorable for photocatalytic CO2/epoxide cycloaddition reactions. This means the band potentials of Ti14(NH4)2 should be better than those of Ti14Cs2 in photocatalytic CO2/epoxide cycloaddition reactions. The above paradox and the catalysis results suggest that the former must dominate the current catalytic reactions under light irradiation. Namely, the enhancement of light absorption, charge transfer, and interfacial charge transfer, due to the incorporation of Cs+ within the cavity of Ti14, effectively overcomes the drawbacks of the decrease in reducing and oxidizing power.
3 Conclusion
Two host–guest TOCs, namely Ti14(NH4)2 and Ti14Cs2, which possess a novel biloculate Ti14 host cage that contains two adjacent chambers, are reported. The ability to take up monovalent cations by the Ti14 host follows the order Cs+ > NH4+ > Rb+ > K+. The host–guest interaction not only influences the dimensions of the host but also alters the photophysical properties and photocatalytic activity of the host. Cs+ endows the host with better visible-light absorption, response, and charge-separation properties than Ti14(NH4)2. Cs+ also renders the compound insoluble in epoxides and cyclocarbonates, and thus Ti14Cs2 is a recyclable heterogeneous photocatalyst for CO2/epoxide cycloaddition reactions. Ti14Cs2 showed superior photocatalytic activity in these valuable conversions regarding TOF, stability, recyclability, sunlight utilization, and substrate scope. This study presents novel biloculate, Ti-oxide-based clusters with exchangeable guests and provides insights for enhancing the solar harvesting applications of such compounds.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for Ti14Cs2 and Ti14(NH4)2 have been deposited at the CCDC under 2324222 and 2324223 and can be obtained from https://www.ccdc.cam.ac.uk/.Author contributions
G. Z. and Y. W. designed the study and interpreted the data. D. W., Y. L., M. C., F. G., G. C., and C. T. carried out data processing. G. W. and Y. W. carried out the DFT simulation. D. W., Y. L., G. Z., and Y. W. wrote the manuscript and created all of the figures, with edits and assistance from all other contributing authors.Conflicts of interest
There is no conflict to declare.Acknowledgements
The authors gratefully acknowledge the National Natural Science Foundation of China (Grant No. 22276112 and 22306110), the Natural Science Foundation of Shandong Province (Grant No. ZR2023QB100), the Shandong Excellent Young Scientists Fund (Grant No. 2023HWYQ-053), and the Taishan Scholar Program of Shandong Province (Grant No. tsqn20230655) for their financial support.Notes and references
- P. B. Crowley, Origins of the Host–Guest Terminology, Cryst. Growth Des., 2023, 23, 8469–8473 Search PubMed.
- C. Schäffer, H. Bögge, A. Merca, I. A. Weinstock, D. Rehder, E. T. K. Haupt and A. Müller, A Spherical 24 Butyrate Aggregate with a Hydrophobic Cavity in a Capsule with Flexible Pores: Confinement Effects and Uptake–Release Equilibria at Elevated Temperatures, Angew. Chem., Int. Ed., 2009, 48, 8051–8056 CrossRef PubMed.
- A. Ziv, A. Grego, S. Kopilevich, L. Zeiri, P. Miro, C. Bo, A. Müller and I. A. Weinstock, Flexible Pores of a Metal Oxide-Based Capsule Permit Entry of Comparatively Larger Organic Guests, J. Am. Chem. Soc., 2009, 131, 6380–6382 CrossRef CAS PubMed.
- D. Xia, P. Wang, X. Ji, N. M. Khashab, J. L. Sessler and F. Huang, Functional Supramolecular Polymeric Networks: The Marriage of Covalent Polymers and Macrocycle-Based Host–Guest Interactions, Chem. Rev., 2020, 120, 6070–6123 CrossRef CAS PubMed.
- E. Haviv, B. Chen, R. Carmieli, L. Houben, H. Cohen, G. Leitus, L. Avram and R. Neumann, Guest Transition Metals in Host Inorganic Nanocapsules: Single Sites, Discrete Electron Transfer, and Atomic Scale Structure, J. Am. Chem. Soc., 2020, 142, 14504–14512 Search PubMed.
- Q. Shi, X. Wang, B. Liu, P. Qiao, J. Li and L. Wang, Macrocyclic Host Molecules with Aromatic Building Blocks: the State of the Art and Progress, Chem. Commun., 2021, 57, 12379–12405 RSC.
- G. Yu, K. Jie and F. Huang, Supramolecular Amphiphiles Based on Host–Guest Molecular Recognition Motifs, Chem. Rev., 2015, 115, 7240–7303 Search PubMed.
- D.-H. Qu, Q.-C. Wang, Q.-W. Zhang, X. Ma and H. Tian, Photoresponsive Host–Guest Functional Systems, Chem. Rev., 2015, 115, 7543–7588 Search PubMed.
- X. Jiang, H. Yu, J. Shi, Q. Bai, Y. Xu, Z. Zhang, X.-Q. Hao, B. Li, P. Wang, L. Wu and M. Wang, From Mechanically Interlocked Structures to Host–Guest Chemistry Based on Twisted Dimeric Architectures by Adjusting Space Constraints, CCS Chem., 2022, 4, 2127–2139 Search PubMed.
- M. Mohamadhoseini and Z. Mohamadnia, Supramolecular Self-Healing Materials via Host-Guest Strategy between Cyclodextrin and Specific Types of Guest Molecules, Coord. Chem. Rev., 2021, 432, 213711 Search PubMed.
- X.-Z. Li, Y.-L. Liang, L.-P. Zhou, L.-X. Cai, Q.-Y. Zhu, Z. Wang, X.-Q. Guo, D.-N. Yan, S.-J. Hu, S.-C. Li, S.-Y. Wu, S.-L. Han, R. Chen, P.-M. Cheng, K. Cheng, X.-S. Feng, T.-P. Sheng, C. He, F.-R. Dai and Q.-F. Sun, Angew. Chem., Int. Ed., 2021, 61, e202202450 Search PubMed.
- J. Wankar, N. G. Kotla, S. Gera, S. Rasala, A. Pandit and Y. A. Rochev, Recent Advances in Host–Guest Self-Assembled Cyclodextrin Carriers: Implications for Responsive Drug Delivery and Biomedical Engineering, Adv. Funct. Mater., 2020, 30, 1909049 Search PubMed.
- A. Müller, H. Reuter and S. Dillinger, Supramolecular Inorganic Chemistry: Small Guests in Small and Large Hosts, Angew. Chem., Int. Ed., 1995, 34, 2328–2361 Search PubMed.
- C. Liu and Y. Wang, Supramolecular Chemistry of Titanium Oxide Clusters, Chem.–Eur. J., 2021, 27, 4270–4282 CrossRef CAS PubMed.
- J. A. Fernández, X. López, C. Bo, C. d. Graaf, E. J. Baerends and J. M. Poblet, Polyoxometalates with Internal Cavities: Redox Activity, Basicity, and Cation Encapsulation in [Xn+P5W30O110](15-n)- Preyssler Complexes, with X = Na+, Ca2+, Y3+, La3+, Ce3+, and Th4+, J. Am. Chem. Soc., 2007, 129, 12244–12253 Search PubMed.
- O. Petina, D. Rehder, E. T. K. Haupt, A. Grego, I. A. Weinstock, A. Merca, H. Bögge, J. Szakács and A. Müller, Guests on Different Internal Capsule Sites Exchange with Each Other and with the Outside, Angew. Chem., Int. Ed., 2011, 50, 410–414 Search PubMed.
- S. Yang, T. Wei and F. Jin, When Metal Clusters Meet Carbon Cages: Endohedral Cluster Fullerenes, Chem. Soc. Rev., 2017, 46, 5005–5058 Search PubMed.
- Q.-M. Wang, Y.-M. Lin and K.-G. Liu, Role of Anions Associated with the Formation and Properties of Silver Clusters, Acc. Chem. Res., 2015, 48, 1570–1579 Search PubMed.
- J. Zhang, S. Stevenson and H. C. Dorn, Trimetallic Nitride Template Endohedral Metallofullerenes: Discovery, Structural Characterization, Reactivity, and Applications, Acc. Chem. Res., 2013, 46, 1548–1557 CrossRef CAS PubMed.
- P. Gouzerh and A. Proust, Main-Group Element, Organic, and Organometallic Derivatives of Polyoxometalates, Chem. Rev., 1998, 98, 77–111 Search PubMed.
- Y. Yan, C. Li, Y. Wu, J. Gao and Q. Zhang, From Isolated Ti-Oxo Clusters to Infinite Ti-oxo Chains and Sheets: Recent Advances in Photoactive Ti-based MOFs, J. Mater. Chem. A, 2020, 8, 15245–15270 Search PubMed.
- P. Coppens, Y. Chen and E. Trzop, Crystallography and Properties of Polyoxotitanate Nanoclusters, Chem. Rev., 2014, 114, 9645–9661 Search PubMed.
- L. Rozes and C. Sanchez, Titanium Oxo-Clusters: Precursors for a Lego-like Construction of Nanostructured Hybrid Materials, Chem. Soc. Rev., 2011, 40, 1006–1030 Search PubMed.
- C. Y. Liu, H. H. Niu, D. X. Wang, C. Gao, A. Said, Y. S. Liu, G. Wang, C. H. Tung and Y. F. Wang, S-Scheme Bi-oxide/Ti-oxide Molecular Hybrid for Photocatalytic Cycloaddition of Carbon Dioxide to Epoxides, ACS Catal., 2022, 12, 8202–8213 CrossRef CAS.
- M.-Y. Fu, H.-Y. Wang, H.-L. Zhai, Q.-Y. Zhu and J. Dai, Assembly of a Titanium-Oxo Cluster and a Bismuth Iodide Cluster, a Single-Source Precursor of a p–n-Type Photocatalyst, Inorg. Chem., 2021, 60, 9589–9597 Search PubMed.
- C. Liu, J. Hu, W. Liu, F. Zhu, G. Wang, C.-H. Tung and Y. Wang, Binding Modes of Salicylic Acids to Titanium Oxide Molecular Surfaces, Chem.–Eur. J., 2020, 26, 2666–2674 Search PubMed.
- Y.-J. Liu, W.-H. Fang, L. Zhang and J. Zhang, Recent Advances in Heterometallic Polyoxotitanium Clusters, Coord. Chem. Rev., 2020, 404, 213099 Search PubMed.
- G. Zhang, W. Li, C. Liu, J. Jia, C.-H. Tung and Y. Wang, Titanium-Oxide Host Clusters with Exchangeable Guests, J. Am. Chem. Soc., 2018, 140, 66–69 CrossRef CAS PubMed.
- P. D. Matthews, T. C. King and D. S. Wright, Structure, Photochemistry and Applications of Metal-doped Polyoxotitanium Alkoxide Cages, Chem. Commun., 2014, 50, 12815–12823 RSC.
- S. Ilic, A. M. May, P. M. Usov, H. D. Cornell, B. Gibbons, P. Celis-Salazar, D. R. Cairnie, J. Alatis, C. Slebodnick and A. J. Morris, An Aluminum-Based Metal–Organic Cage for Cesium Capture, Inorg. Chem., 2022, 61, 6604–6611 Search PubMed.
- Y. Lv, J. Willkomm, A. Steiner, L. Gan, E. Reisner and D. S. Wright, Encapsulation of a ‘Naked’ Br− Anion in a Polyoxotitanate Host, Chem. Sci., 2012, 3, 2470–2473 RSC.
- J. Hou, J. Hu, Q. Sun, G. Zhang, C.-H. Tung and Y. Wang, A Post-Functionalizable Iso-Polyoxotitanate Cage Cluster, Inorg. Chem., 2016, 55, 7075–7078 CrossRef CAS PubMed.
- X. Cui, H. Li, X. Li, Y.-X. Wang, T. Li, J. Dong, Z.-N. Chen and X.-M. Zhang, A Systematic Crystallographic Study of the Host–Guest Interactions between Amines and Ti8 Polyoxotitanium Clusters, Cryst. Growth Des., 2024, 24, 851–858 CrossRef CAS.
- G. A. Timco, A. Fernandez, A. K. Kostopoulos, C. A. Muryn, R. G. Pritchard, I. Strashnov, I. J. Vitorica-Yrezebal, G. F. S. Whitehead and R. E. P. Winpenny, An Extensive Family of Heterometallic Titanium(IV)–Metal(III) Rings with Structure Control through Templates, Angew. Chem., Int. Ed., 2017, 56, 13629–13632 Search PubMed.
- G. A. Timco, A. Fernandez, A. K. Kostopoulos, J. F. Charlton, S. J. Lockyer, T. R. Hailes, R. W. Adams, E. J. L. McInnes, F. Tuna, I. J. Vitorica-Yrezabal, G. F. S. Whitehead and R. E. P. Winpenny, Hybrid Organic-Inorganic Rotaxanes, Including a Hetero-Hybrid [3]Rotaxane Featuring Two Distinct Heterometallic Rings and a Molecular Shuttle, Angew. Chem., Int. Ed., 2018, 57, 10919–10922 CrossRef CAS PubMed.
- C. Liu, C. Gao, A. Said, H. Niu, D. Wang, C. H. Tung and Y. Wang, Assembly of Interlocked Superstructures with a Titanium Oxide Molecular Ring in Water, Inorg. Chem., 2021, 60, 14520–14524 CrossRef CAS PubMed.
- J. W. Steed, First- and Second-Sphere Coordination Chemistry of Alkali Metal Crown Ether Complexes, Coord. Chem. Rev., 2001, 215, 171–221 Search PubMed.
- Y. Liu, G. Zhang, D. Wang, G. Chen, F. Gao, C.-H. Tung and Y. Wang, A Cryptand-like Ti-Coordination Compound with Visible-Light Photocatalytic Activity in CO2 Storage, Dalton Trans., 2024, 53, 1989–1998 Search PubMed.
- I.-H. Chu, H. Zhang and D. V. Dearden, Macrocyclic Chemistry in the Gas Phase: Intrinsic Cation Affinities and Complexation Rates for Alkali Metal Cation Complexes of Crown Ethers and Glymes, J. Am. Chem. Soc., 1993, 115, 5736–5744 Search PubMed.
- Z. Jing, Y. Zhou, T. Yamaguchi, K. Ohara, J. Pan, G. Wang, F. Zhu and H. Liu, Alkali Metal Ion Recognition by 18-Crown-6 in Aqueous Solutions: Evidence from Local Structures, J. Phys. Chem. B, 2023, 127, 4858–4869 CrossRef CAS PubMed.
- D. Wang, A. Said, Y. Liu, H. Niu, C. Liu, G. Wang, Z. Li, C.-H. Tung and Y. Wang, Cr–Ti Mixed Oxide Molecular Cages: Synthesis, Structure, Photoresponse, and Photocatalytic Properties, Inorg. Chem., 2022, 61, 14887–14898 Search PubMed.
- C.-Y. Luo, L.-J. Ma, W. Liu, Y.-C. Tan, R.-N. Wang, J.-L. Hou and Q.-Y. Zhu, Topotactic Conversion of Titanium-Oxo Clusters to a Stable TOC-Based Metal–Organic Framework with the Selective Adsorption of Cationic Dyes, Inorg. Chem., 2024, 63, 5961–5971 CrossRef CAS PubMed.
- A. Said, C. Liu, C. Gao, D. Wang, H. Niu, Y. Liu, G. Wang, C. H. Tung and Y. Wang, Lead-Decorated Titanium Oxide Compound with a High Performance in Catalytic CO2 Insertion to Epoxides, Inorg. Chem., 2023, 62, 1901–1910 Search PubMed.
- A. Said, G. Y. Zhang, D. X. Wang, G. J. Chen, Y. S. Liu, F. F. Gao, C. H. Tung and Y. F. Wang, Divalent Heterometal Doped Titanium-Oxide Cluster Polymers: Structures, Photoresponse, and Photocatalysis, Inorg. Chem., 2023, 62, 13476–13484 Search PubMed.
- D. Wang, Y. Liu, G. Chen, F. Gao, G. Zhang, G. Wang, C.-H. Tung and Y. Wang, Ligation of Titanium-oxide and {Mo2} Units for Solar CO2 Storage, Inorg. Chem., 2023, 62, 21074–21082 Search PubMed.
- H.-C. Su, Y.-Y. Wu, J.-L. Hou, G.-L. Zhang, Q.-Y. Zhu and J. Dai, Dye Molecule Bonded Titanium Alkoxide: A Possible New Type of Dye for Sensitized Solar Cells, Chem. Commun., 2016, 52, 4072–4075 RSC.
- H.-L. Zhai, J.-L. Hou, C.-Y. Luo, L.-J. Ma, Q.-Y. Zhu and J. Dai, Photocurrent and Gelation Properties of Polyphenol-Modified Titanium-Oxo Compounds, Inorg. Chem., 2022, 61, 13191–13198 CrossRef CAS PubMed.
- Y. Lv, J. Cheng, A. Steiner, L. Gan and D. S. Wright, Dipole-induced Band-gap Reduction in an Inorganic Cage, Angew. Chem., Int. Ed., 2014, 53, 1934–1938 Search PubMed.
- X. Wu, C. Chen, Z. Guo, M. North and A. C. Whitwood, Metal-and Halide-Free Catalyst for the Synthesis of Cyclic Carbonates from Epoxides and Carbon Dioxide, ACS Catal., 2019, 9, 1895–1906 Search PubMed.
- Z. Huang, F. Li, B. Chen and G. Yuan, Cycloaddition of CO2 and Epoxide Catalyzed by Amino-and Hydroxyl-Rich Graphitic Carbon Nitride, Catal. Sci. Technol., 2016, 6, 2942–2948 Search PubMed.
- G. Zhai, Y. Liu, L. Lei, J. Wang, Z. Wang, Z. Zheng, P. Wang, H. Cheng, Y. Dai and B. Huang, Light-Promoted CO2 Conversion from Epoxides to Cyclic Carbonates at Ambient Conditions over a Bi-Based Metal–Organic Framework, ACS Catal., 2021, 11, 1988–1994 CrossRef CAS.
- T. Zhang, H. Chen, S. Liu, H. Lv, X. Zhang and Q. Li, Highly Robust {Ln4}-Organic Frameworks (Ln= Ho, Yb) for Excellent Catalytic Performance on Cycloaddition Reaction of Epoxides with CO2 and Knoevenagel Condensation, ACS Catal., 2021, 11, 14916–14925 CrossRef CAS.
- Y. Rachuri, J. F. Kurisingal, R. K. Chitumalla, S. Vuppala, Y. Gu, J. Jang, Y. Choe, E. Suresh and D.-W. Park, Adenine-based Zn(II)/Cd(II) Metal–organic Frameworks as Efficient Heterogeneous Catalysts for Facile CO2 Fixation into Cyclic Carbonates: a DFT-supported Study of the Reaction Mechanism, Inorg. Chem., 2019, 58, 11389–11403 CrossRef CAS PubMed.
- K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, Mg−Al mixed Oxides as Highly Active Acid−Base Catalysts for Cycloaddition of Carbon Dioxide to Epoxides, J. Am. Chem. Soc., 1999, 121, 4526–4527 Search PubMed.
- A. Said, G. Chen, G. Zhang, D. Wang, Y. Liu, F. Gao, G. Wang, C.-H. Tung and Y. Wang, Enhancing the Photocatalytic Performance of a Rutile Unit Featuring a Titanium-Oxide Cluster by Pb2+ Doping, Dalton Trans., 2024, 53, 3666–3674 Search PubMed.
- G. Zhai, Y. Liu, Y. Mao, H. Zhang, L. Lin, Y. Li, Z. Wang, H. Cheng, P. Wang, Z. Zheng, Y. Dai and B. Huang, Improved Photocatalytic CO2 and Epoxides Cycloaddition via the Synergistic Effect of Lewis Acidity and Charge Separation over Zn Modified UiO-bpydc, Appl. Catal., B, 2022, 301, 120793 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2324222 and 2324223. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04983g |
| ‡ D. Wang and Y. Liu have equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2024 |