
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual active site-mediated Ir single-atom-doped RuO2 catalysts for highly efficient and stable water splitting†
Zhenhua
Tao
a,
Ning
Lv
a,
Hongyu
Zhao
a,
Xu
Luo
a,
Zilan
Li
a,
Jun
Yu
 a,
Lei
Chen
a,
Xupo
Liu
b and
Shichun
Mu
*a
a,
Lei
Chen
a,
Xupo
Liu
b and
Shichun
Mu
*a
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, China. E-mail: msc@whut.edu.cn
bSchool of Materials Science and Engineering, Henan Normal University, Xinxiang, Henan 453007, P. R. China
First published on 13th September 2024
Abstract
The electronic structure modulation through heterogeneous single-atom doping is an effective strategy to improve electrocatalysis performance of catalysts. Here, Ir single-atom doped RuO2 (IrSA/RuO2) is constructed by substituting Ru sites with mono-disperse Ir atoms in RuO2 crystals. The IrSA/RuO2-850 catalyst shows excellent activity for the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) in alkaline media, with overpotentials of only 37 and 234 mV respectively, at a current density of 10 mA cm−2, lower than that of commercial Pt/C (39 mV-HER) and RuO2 (295 mV-OER). Notably, no significant degradation occurs during the 1000 h HER stability test at 500 mA cm−2. Furthermore, IrSA/RuO2-850 also demonstrates superior catalytic activity and stability in acidic media. Theoretical calculations show that the interaction between Ir and RuO2 modulates the electronic structure of both Ru and Ir sites, resulting in the lowest reaction energy barriers of Ru and Ir sites for the HER and OER, respectively, which thermodynamically explains the enhancement of the catalytic activity. Besides, the introduction of Ir atoms also enhances the demetallation energy of Ru atoms and strengthens the structural stability of the crystal, leading to the improved stability of the catalyst. This work provides an effective strategy for construction of high-performing catalysts by precisely controlling the electronic structure and active sites of polymetal atoms.
1. Introduction
Hydrogen energy, as a clean and high-density energy source, occupies an important position in future energy conversion and energy storage.1,2 Electrochemical water splitting is a clean and sustainable way of obtaining hydrogen energy, which avoids environmental pollution and the greenhouse effect caused by fossil energy sources.3,4 However, there are still numerous challenges in the industrialization process of water electrolysis. Currently, platinum (Pt) group noble metal catalysts are widely used in electrolysis, in which Pt and iridium (Ir) based catalysts are often used in the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER), respectively.4–7 However, the limited availability and high cost of Pt and Ir pose a significant constraint on the widespread application of water electrolysis. Additionally, the slow kinetics of oxygen evolution at the anode restricts the overall water splitting (OWS) efficiency.8–10 Meanwhile, the long-term stability of catalysts at high current densities still requires further improvement.11–13 Therefore, the current research objective is to develop low-cost and high-performing catalysts to support industrial applications.Ruthenium (Ru), as an affordable material among the Pt group noble metals, has been widely investigated and shown promising applications due to the moderate reaction energy barrier towards the intermediates in the water electrolysis reaction,14–18 and is expected to replace Pt-based and Ir-based catalysts. Among Ru-based catalysts, ruthenium oxide (RuO2) has been identified as the optimal catalyst for OWS. In comparison to iridium oxide (IrO2), an anodic OER catalyst based on RuO2 is more cost-effective with lower driving voltages.19–21 However, Ru is more prone to undergo oxidation under high voltage conditions, leading to deactivation of catalytically active sites and a decline in catalytic performance.16,22 Moreover, pure RuO2 as a HER catalyst still requires high overpotentials to drive the reaction.19,20,23 Thus, to meet the increasingly stringent requirements under industrial conditions, further in-depth research and optimization of Ru-based catalysts are necessary.
Modulating the electronic structure is an effective modification strategy to optimize the catalytic properties of catalysts,24–28 which includes the construction of heterostructures,19,21,29,30 alloy engineering,31–34 crystal phase engineering,8,35–37 defect engineering,25,28,32,38 and heterogeneous atomic doping strategies.5,15,16,20,27,38,39 Among these, heterogeneous single atom doping can precisely adjust the electronic structure of catalysts,38–41 and synergistically balance the catalytic properties of each component, thereby optimizing the adsorption behavior of catalysts towards reaction intermediates and improving reaction kinetics. It has been reported that Ru atoms as electron acceptors in the RuO2 system can inhibit the transformation of Ru atoms into higher valence states during the reaction process, further enhancing the stability of the material.15,22 Generally speaking, atoms with higher electronegativity in the crystal structure are more prone to gaining electrons. Ir has the same electronegativity as Ru but has more electrons outside the nucleus. In particular, the Ir element possesses special physical properties that make it less susceptible to oxidation even at high potentials.38,42 Furthermore, Ir single-atom doping in RuO2 is of significant importance to construct RuO2-based catalysts, which are expected to exhibit high HER/OER and OWS performance under alkaline conditions.
Here, the Ir single-atom doped RuO2 (IrSA/RuO2) catalyst is synthesized by solvothermal and subsequent pyrolysis processes. Structure characterization results indicate that Ir atoms replace Ru in the crystal lattice of RuO2 in a single dispersed form. There exists a pronounced interaction between Ir single atoms and RuO2 as a matrix, which alters the electronic structure of both Ru and Ir sites, endowing the electronic structure-modulated IrSA/RuO2 catalysts with enhanced HER/OER and OWS activity and stability, superior to those of commercial Pt/C and RuO2 catalysts. Theoretical calculations further show a significant electron transfer between Ru and Ir atoms, which suggests a strong interaction between Ir and RuO2. The electronic modulation of Ru and Ir sites optimizes the adsorption/desorption behavior of reaction intermediates, providing Ru and Ir sites with the lowest rate-determining step (RDS) reaction energy barriers when they act as active centers for the HER and OER, respectively. Furthermore, the introduction of Ir single-atoms enhances the demetallation energy of Ru atoms, thereby stabilizing the catalyst in electrolytic water.
2. Results and discussion
2.1 Catalyst synthesis
Fig. 1a illustrates the preparation process of catalysts via a two-step method. First, 0.2 g of urea and 0.1 g of ammonium fluoride were added to 40 mL of water and 20 mL of ethanol solution until completely dissolved, followed by the addition of 2 mmol RuCl3 and 0.06 mmol IrCl3 and stirring until completely dissolved; then the above solutions were transferred to a 100 mL Teflon-lined autoclave and maintained at 160 °C for 10 h, and the sample obtained was centrifuged and dried to obtain the precursor. The obtained precursor was then subjected to heat treatment under different conditions (650 °C, 4 h; 750 °C, 4 h; 850 °C, 4 h; and 950 °C, 4 h) to obtain the final catalysts, which were named IrSA/RuO2-650, IrSA/RuO2-750, IrSA/RuO2-850, and IrSA/RuO2-950, respectively. | ||
| Fig. 1 (a) Schematic diagram for a two-step route to prepare IrSA/RuO2; (b) XRD pattern of IrSA/RuO2; (c) TEM image of IrSA/RuO2-850; (d) HAADF-STEM image of IrSA/RuO2-850 and the corresponding elemental mapping images of Ir, O and Ru; (e) AC HAADF-STEM image of IrSA/RuO2-850; (f) corresponding line intensity profile of the area in the yellow box in (e). | ||
2.2 Characterization of the catalyst
First, the phase analysis of the samples was performed using X-ray diffraction (XRD), as depicted in Fig. 1b and S1.† It is evident that all diffraction peaks correspond to the standard card phase of RuO2 (PDF # 40-1290) for pure RuO2 and Ir-containing RuO2, while no discernible peaks related to Ir or its compounds can be observed. The Ir content in the material was determined to be 1.5% by inductively coupled plasma (ICP) analysis, confirming the successful incorporation of Ir. Microscopic morphology and distribution of Ir within the catalyst were characterized using transmission electron microscopy (TEM). Fig. 1c reveals that IrSA/RuO2-850 exhibits a nano-spherical morphology with diameters ranging from 50 to 100 nm. The spacing of the lattice fringe is 0.315 nm, consistent with the (110) crystal face of tetragonal RuO2 (PDF # 40-1290). Furthermore, the morphology of pure RuO2 (Fig. S3†) has no significant alteration compared to Ir-doped RuO2 catalysts. Atomic-level mapping images shown in Fig. 1d indicate a uniform distribution of Ru, Ir and O elements on the catalyst surface, in which the Ir element is present in a mono-dispersed form.For further characterization of the presence and location of Ir, aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) was employed. Fig. 1e reveals a periodic uniformly arranged atomic surface without lattice distortion. Furthermore, numerous isolated bright atoms marked by yellow circles are presented with contrast distinct from the surrounding atoms on the surface, which are attributed to Ir atoms due to higher atomic numbers. Atomic line profiling shown in Fig. 1f demonstrates that the bright spots possess stronger peak intensities and larger atomic diameters, further confirming their attribution to Ir atoms. From this, the element Ir exists in single atomic sites by substitution of Ru in RuO2 crystals.
The electronic structure and coordination environment of Ir were further investigated by X-ray absorption spectroscopy (XAS). The X-ray absorption near edge structure (XANES) at the Ir-L3 edge (Fig. 2c) reveals a significant positive shift for the white line of IrSA–RuO2 compared to Ir foil and a weak positive shift compared to the pure IrO2. These results suggest that the valence state of Ir is slightly larger than +4, indicating that Ir exists in the form of single atoms while not in the metal state. In addition, the Ir-L3 edge reveals a positive shift compared to pure RuO2 combined with the negative shift of the Ru 3p XPS spectrum, indicating an electron transfer from Ir to Ru in the crystal structure. This demonstrates that the Ir atom has a pronounced interaction with RuO2 as a matrix. The extended X-ray absorption fine structure (EXAFS) of IrSA/RuO2 (Fig. 2d) presents a prominent peak at 1.2 Å corresponding to the Ir–O first shell coordination, and the peak at 3.2 Å is attributed to the Ir–Ru second shell coordination, and no peaks resemble those of Ir foil. These can prove the absence of Ir–Ir bonds, further confirming the presence of Ir in a single-atom form. Wavelet transformation of IrSA/RuO2-850 (Fig. 2e) reveals two characteristic peaks, in which the peak at R-space 1.5 Å is attributed to the first shell of Ir–O bonds, while the peak at R-space 2.8 Å belongs to the second shell of Ir–Ru bonds, indicating the existence of Ir–O–Ru bonds in the crystal structure. In conclusion, advanced photoelectron spectroscopy techniques confirm the existence of Ir atoms in the form of single atoms within the host crystal structure of RuO2.
 | ||
| Fig. 2 (a) XPS survey spectra, (b) high-resolution Ru 3p XPS spectra of pure RuO2 and IrSA/RuO2-850, (c) XANES spectra, (d) EXAFS fitting curve and (e) EXAFS wavelet transform plots of Ir foil, IrO2 and IrSA/RuO2-850. | ||
2.3 Electrocatalytic performance evaluation
The HER and OER performances of the catalysts were probed by using a three-electrode system under alkaline conditions. Fig. 3a show the linear sweep voltammetry (LSV) curves of the HER for different catalysts in 1 M L−1 KOH. It can be seen that the HER activity of IrSA/RuO2-850 is superior to that of commercial Pt/C and pure RuO2. Moreover, this advantage gradually increases with the increase in current densities. The overpotential of IrSA/RuO2-850 is 37/88 mV at current densities of 10/100 mA cm−2, lower than that of Pt/C (39/105 mV), IrSA/RuO2-650 (64/254 mV), IrSA/RuO2-750 (48/159 mV), IrSA/RuO2-950 (52/205 mV) and pure RuO2 (138/421 mV). The corresponding Tafel slope obtained from the LSV curves (Fig. 3b and S6†) demonstrates that IrSA/RuO2-850 possesses the lowest Tafel value (22.6 mV dec−1), significantly lower than that of Pt/C (27.3 mV dec−1), IrSA/RuO2-650 (66.7 mV dec−1), IrSA/RuO2-750 (48.9 mV dec−1), IrSA/RuO2-950 (51.6 mV dec−1) and RuO2 (218.4 mV dec−1), indicating the fastest reaction kinetics among the catalysts. | ||
| Fig. 3 The performance of catalysts in 1 M L−1 KOH: (a) LSV curves and (b) Tafel slopes of IrSA/RuO2, RuO2 and commercial Pt/C; (c) equivalent circuit and fitted EIS curves of the catalysts; (d) LSV curves, (e) Tafel slopes and (f) equivalent circuit and fitted EIS curves of the catalysts, the black curves is C-RuO2; (g) HER stability test curves of IrSA/RuO2-850 and commercial Pt/C; (h) OER stability test curves of IrSA/RuO2-850, RuO2 and C–RuO2. | ||
The equivalent circuit and fitted electrochemical impedance spectroscopy (EIS) results of catalysts are shown in Fig. 3c, in which IrSA/RuO2-850 exhibits a smaller Rct value (30.3 Ω) than that of IrSA/RuO2-650 (65.3 Ω), IrSA/RuO2-750 (43.8 Ω), IrSA/RuO2-950 (54.7 Ω) and RuO2 (153.5 Ω), which indicates the fastest charge transfer capability, promoting the HER kinetics. Overall, IrSA/RuO2-850 has the fastest reaction kinetics and the smallest transfer resistance of all prepared catalysts, resulting in the optimal HER performance. Moreover, as shown in Fig. 3g, IrSA/RuO2-850 shows no significant degradation (10%) at a larger current density of 500 mA cm−2 even up to 1000 h reactions, indicating better stability than Pt/C whose current density which was initially 100 mA cm−2 decays by 50% after 100 h. This allows IrSA/RuO2 to be a potential HER catalyst for industrial applications.
The LSV curve test results for the catalytic OER performance are depicted in Fig. 3d. It is evident that IrSA/RuO2-850 exhibits an overpotential of 234 mV at 10 mA cm−2, surpassing IrSA/RuO2-650 (271 mV), IrSA/RuO2-750 (256 mV), IrSA/RuO2-950 (264 mV), pure RuO2 (286 mV) and commercial RuO2 (C–RuO2, 295 mV). Calculated Tafel slopes (Fig. 3e) show that IrSA/RuO2-850 exhibits the lowest Tafel value (22.6 mV dec−1), significantly lower than that of IrSA/RuO2-650 (95.3 mV dec−1), IrSA/RuO2-750 (69.4 mV dec−1), IrSA/RuO2-950 (78.5 mV dec−1), RuO2 (99.7 mV dec−1) and commercial-RuO2 (108.2 mV dec−1), which demonstrated that IrSA/RuO2-850 maintained the optimal OER process reaction kinetics. The comparison of overpotentials and Tafel slopes of IrSA/RuO2-850 with those of the reported Ru-based OER catalysts in alkaline media (Table S1†) demonstrates a performance advantage for IrSA/RuO2-850.
The equivalent circuit and fitted EIS results of the catalysts are shown in Fig. 3f. IrSA/RuO2-850 possesses a smaller Rct value of 43.7 Ω than that of IrSA/RuO2-650 (74.2 Ω), IrSA/RuO2-750 (54.3 Ω), IrSA/RuO2-950 (66.5 Ω), RuO2 (128.7 Ω) and C–RuO2 (153.3 Ω), suggesting that the incorporation of Ir into RuO2 does accelerate the charge transfer. Double layer capacitance (Cdl) of IrSA/RuO2-850 and C–RuO2 was tested by using CV curves in the non-faradaic region (Fig. S7a and b†). As shown in Fig. S7c,† IrSA/RuO2-850 possesses a larger Cdl value of 4.15 mF cm−2 than C–RuO2 (1.96 mF cm−2). The electrochemical active surface area (ECSA) was deduced from the Cdl, and the results are displayed in Fig. S7d,† in which the ECSA of IrSA/RuO2-850 and C–RuO2 is 69.2 and 32.6, respectively, indicating that IrSA/RuO2-850 has a higher density of active sites, favorable for the adsorption/desorption of more reactive intermediates, therefore enhancing the catalytic activity. As shown in Fig. S8,† the mass activity of IrSA/RuO2-850 (0.223 mA mg Ru−1) increases by 2.75 times compared with that of C–RuO2 (0.081 mA mg Ru−1), indicating that the introduction of Ir improves the intrinsic activity of Ru sites.
The stability test results (Fig. 3h) obtained by a chronoamperometry method show that IrSA/RuO2-850 has a significant improvement in the tolerance of the OER, and the current density decreases by only 15% at 100 h constant voltage, surpassing both C–RuO2 and pure RuO2 catalysts, with the current density decreasing by 70% and 20%, respectively, after a 50 h chronoamperometry test. To further explore the stability, the dissolved amount of Ru in the electrolyte for IrSA/RuO2-850 was obtained by ICP testing at different time intervals. As shown in Fig. S9,† the dissolution rate of Ru is small for the initial 60 h, and elevated for the next 60–100 h; the trend is similar to the attenuation of the stability test curve (Fig. 3h). After 100 h of stability testing, the amount of Ru dissolved is about 8.8%, indicating no occurrence of significant dissolution of Ru in the catalyst.
After the OER stability test, the performance degradation of IrSA/RuO2-850 was assessed by using LSV curves. It can be found from Fig. S10† that, after the stability test, the overpotential of IrSA/RuO2-850 is 248 and 327 mV at the current densities of 10 and 50 mA cm−2, respectively, which attenuates only 19 and 28 mV from the initial state, demonstrating that IrSA/RuO2-850 can still maintain good catalytic activity after the stability test.
The OER performance test of all prepared samples and C–RuO2 was further carried out in acidic media (0.5 M H2SO4) to better evaluate the OER catalytic activity under broad pH conditions. As shown in Fig. S11a and b,† when the current density reaches 10/50 mA cm−2, IrSA/RuO2-850 reveals a lower overpotential (195/249 mV) than C–RuO2 (246/335 mV), IrSA/RuO2-650 (228/303 mV), IrSA/RuO2-750 (211/272 mV), IrSA/RuO2-950 (216/291 mV) and RuO2 (237/312 mV). This also indicates the high OER activity for IrSA/RuO2-850 in acidic media. Besides, it exhibits the lowest Tafel slope and the smallest Rct as shown in Fig. S11c and d,† indicating superior reaction kinetics. These properties result in its excellent OER performance in acidic media. Note that, compared with previously reported RuO2 catalysts tested in acidic media (Table S2†), the as-fabricated IrSA/RuO2-850 catalyst exhibits outstanding performance in terms of overpotential and Tafel slope values toward the OER. The chronoamperometry test results (Fig. S11e†) demonstrate that IrSA/RuO2-850 can operate stably in acidic media for 100 h (only a decrease of 13%), exhibiting superior stability compared to C–RuO2 (decrease of 70% for 60 h).
Inspired by the excellent HER and OER performance of IrSA/RuO2-850, it was assembled into a two-electrode cell as both the anode and cathode for OWS. The LSV results depicted in Fig. 4a reveal the outstanding alkaline OWS performance of the IrSA/RuO2-850 ‖ IrSA/RuO2-850 couple, with a potential of 1.504 V at a current density of 10 mA cm−2, lower than that of a commercial Pt/C ‖ RuO2 couple (1.568 V). Stability evaluations as shown in Fig. 4c demonstrate the 100 h stability at 50 mA cm−2 for the IrSA/RuO2-850 ‖ IrSA/RuO2-850 couple, with only 10% degradation, outperforming the commercial Pt/C ‖ RuO2 couple. Additionally, hydrogen and oxygen gases (H2/O2) were collected in an H-type electrolytic cell equipped with a proton exchange membrane, and the volumes were measured via the gas drainage method (Fig. 4b). The ratio of H2 to O2 emitted over a range of time intervals was approximately 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating an OWS faradaic efficiency close to 100% for the catalyst. The above results demonstrate that the IrSA/RuO2-850 catalyst possesses a notable reduction in the overall potential for water electrolysis and an enhancement in the stability, which holds significant implications for industrial catalysis towards electrolytic water splitting.
:1, indicating an OWS faradaic efficiency close to 100% for the catalyst. The above results demonstrate that the IrSA/RuO2-850 catalyst possesses a notable reduction in the overall potential for water electrolysis and an enhancement in the stability, which holds significant implications for industrial catalysis towards electrolytic water splitting.
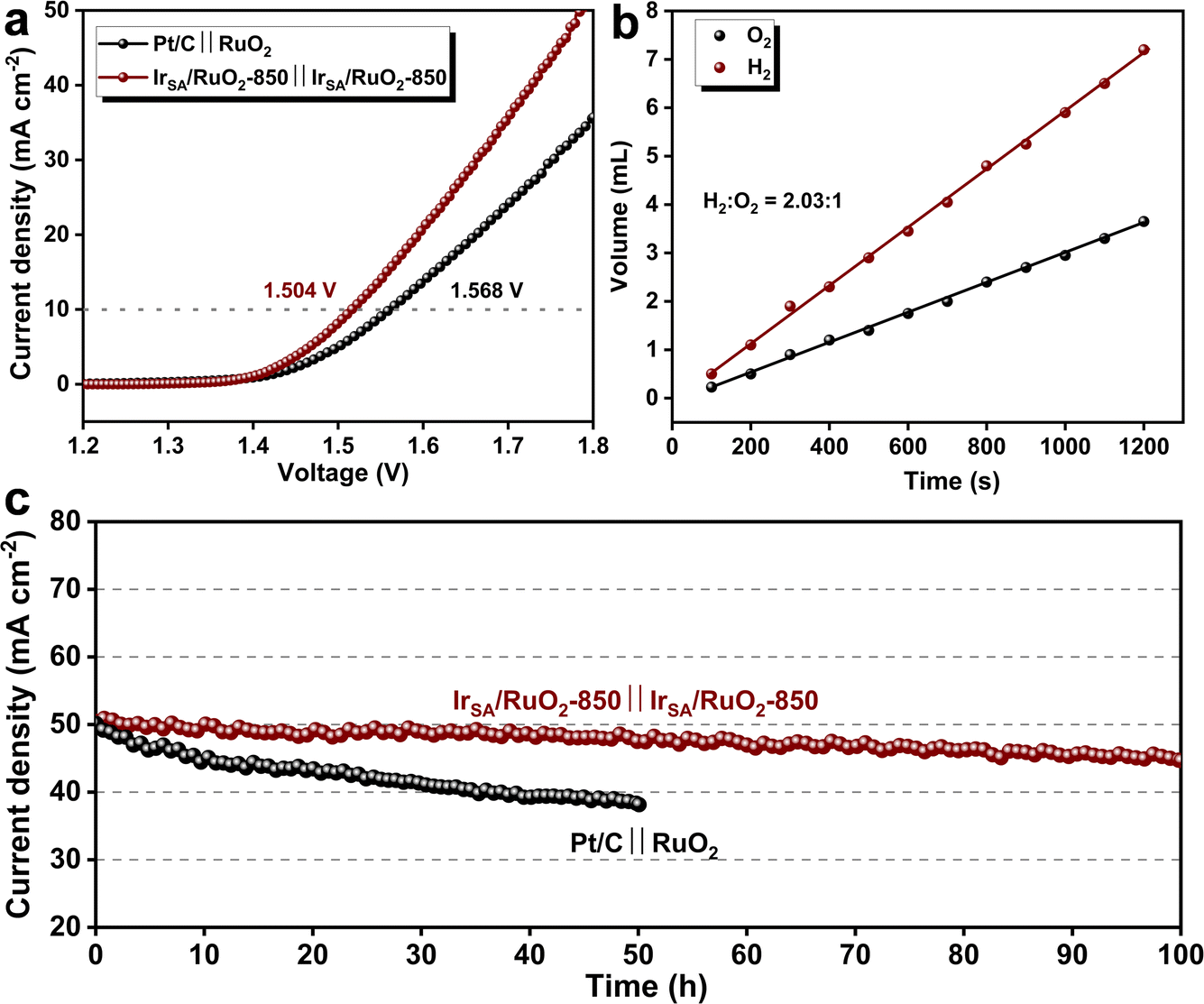 | ||
| Fig. 4 (a) LSV curves and (c) IT curves of two electrode OWS devices with IrSA/RuO2-850 ‖ IrSA/RuO2-850 and commercial Pt/C ‖ RuO2 catalysts; (b) volume of collected H2 and O2versus time. | ||
2.4 Density functional theory calculation
The optimization of catalyst performance can be elucidated by density functional theory (DFT) calculations. Fig. S13a and b† show the optimized geometric models for RuO2 and Ir single atom doped RuO2. The charge density difference and two-dimensional slice (Fig. S13c†) prove that the introduction of Ir atoms results in a rearrangement of the electronic structure of IrSA/RuO2, indicating a strong interaction between Ir and RuO2. Further Bader charge analysis (Fig. 5c) shows that the valence state of Ru+1.27 in IrSA/RuO2 is lower than that of Ru+1.51 in pure RuO2 as well as the Ir+1.32 in the same structure, suggesting that the Ir acts as an electron donor to transfer electrons to the Ru. This is consistent with the XANES analysis result that the spectral line of IrSA/RuO2 slightly shifts towards higher energy compared to those of IrO2, with a negative shift in Ru 3p spectra of IrSA/RuO2-850 compared to pure RuO2. In addition, the decrease in the average valence state of Ru is favorable for the enhancement of the OER stability.12,17 This electronic structure-modulated IrSA/RuO2 catalyst can severely affect the adsorption/desorption behavior of reaction intermediates. | ||
| Fig. 5 The Gibbs free energy diagram of (a) the HER; (b) the OER at the Ir or Ru site of IrSA/RuO2, RuO2 and the IrO2 system; (c) Bader charge of IrSA/RuO2 and RuO2. (d) The calculated demetallation energy of IrSA/RuO2 and RuO2; (e) schematic of the IrSA/RuO2 catalyst's unique two-site catalysis. | ||
To investigate the effect of catalysts with a modulated electronic structure on catalytic performance, the Gibbs free energy of different reaction sites during the HER process to reflect the adsorption intermediates was first calculated. By comparing the RDS barrier of different reaction sites in the HER process (Fig. 5a), it can be seen that the RDS of the Ru site on IrSA/RuO2 is OH* + H* to H* with an energy barrier of 0.36 eV, lower than that of the hydrolysis of Ru sites on RuO2 (0.51 eV) and the barrier of the reaction intermediates OH* + H* to H* of Ir sites on IrSA/RuO2 (0.56 eV), suggesting that the introduction of Ir enables the Ru site on IrSA/RuO2 to drive the HER with a lower overpotential.
Additionally, the Gibbs free energy of IrSA/RuO2, RuO2 and IrO2 catalysts at different reaction sites reflecting the OER intermediates was also calculated. Fig. 5b demonstrates that the RDS of these four reaction sites is all the formation of *OOH from *O, in which the Ir site on IrSA/RuO2 acting as the active center has the lowest thermodynamic energy barrier (1.8 eV), lower than that of Ru sites on IrSA/RuO2 (2.12 eV) and RuO2 (2.1 eV), and Ir sites on IrO2 (1.94 eV). This reduction in the reaction energy barrier thermodynamically corroborates the enhancement of OER performance.
To explore the improvement in catalyst stability, the demetallation energy of Ru atoms on IrSA/RuO2 and pure RuO2 was calculated. demonstrates that the demetallation energy barrier of IrSA/RuO2 (8.33 eV) is higher than that of RuO2 (6.53 eV), suggesting that the introduction of Ir enhances the crystal stability of RuO2, which in turn boosts the stability in catalytic reactions. Fig. 5e shows the schematic diagram of the unique two-site catalysts in the HER and OER, in which the Ru and Ir sites on the catalyst surface act as the active center for the HER and OER, respectively.
3. Conclusions
Here, Ir single atom doped RuO2 (IrSA/RuO2) catalysts were successfully constructed for water electrolysis under alkaline conditions. A series of characterization results show that the interaction between Ir and the RuO2 matrix simultaneously changes the electronic structures of Ru and Ir atoms as dual active sites for the hydrogen/oxygen evolution reactions (HER/OER), respectively, resulting in excellent catalytic activity and stability. The overpotential of the IrSA/RuO2-850 catalyst at 10 mA cm−2 is 37 mV for the HER and 234 mV for the OER, superior to that of commercial Pt/C (39 mV-HER) and RuO2 (295 mV-OER), with steady operation for 1000 h at higher current density (500 mA cm−2) during the HER. A two-electrode cell with IrSA/RuO2-850 demonstrates superior overall water splitting performance compared to those assembled with commercial Pt/C and RuO2. Also, IrSA/RuO2-850 demonstrates excellent OER activity (195 mV at 10 mA cm−2) and stability in acidic media. Theoretical calculations unveil that the strong interaction between Ir–RuO2 alters the electronic structure and coordination environment of both Ir and Ru sites, optimizing the adsorption/desorption behavior of the reaction intermediates, with the lowest rate-determining step reaction energy barriers when Ru and Ir sites act as the catalytically active centers of the HER and OER, respectively. Additionally, the enhanced demetallation energy of the Ru atom due to single atom Ir insertion is believed to be a crucial factor contributing to the improved stability of the catalyst. This work provides a strategy for designing highly active and stable catalysts and offers insights for the research and development of catalysts under industrial conditions.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Zhenhua Tao: conceptualization, methodology, writing – original draft, data curation, visualization. Ning LV: methodology, formal analysis. Hongyu Zhao: software. Xu Luo: investigation. Jun Yu: supervision. Lei Chen: project administration. Xupo Liu: validation. Shichun Mu: writing – review & editing, funding acquisition, resources.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially sponsored by the National Natural Science Foundation of China (Grant No. 22379117 and 22179104) and the State Key Laboratory of Advanced Technology for Materials Synthesis and Processing (Wuhan University of Technology) (2023-ZT-1).Notes and references
- T. Da Silva Veras, T. S. Mozer, D. Da Costa Rubim Messeder Dos Santos and A. Da Silva César, Int. J. Hydrogen Energy, 2017, 42, 2018–2033 CrossRef.
- J. Andrews and B. Shabani, Int. J. Hydrogen Energy, 2012, 37, 1184–1203 CrossRef CAS.
- Z. Yu, Y. Duan, X. Feng, X. Yu, M. Gao and S. Yu, Adv. Mater., 2021, 33, 2007100 CrossRef CAS.
- H. Sun, X. Xu, H. Kim, W. Jung, W. Zhou and Z. Shao, Energy Environ. Mater., 2023, 6, e12441 CrossRef CAS.
- J. Wang, H. Yang, F. Li, L. Li, J. Wu, S. Liu, T. Cheng, Y. Xu, Q. Shao and X. Huang, Sci. Adv., 2022, 8, eabl9271 CrossRef CAS PubMed.
- S. Zhang, L. Yin, Q. Li, S. Wang, W. Wang and Y. Du, Chem. Sci., 2023, 14, 5887–5893 RSC.
- Y. Song, Y. Zhang, W. Gao, C. Yu, J. Xing, K. Liu and D. Ma, Chem. Sci., 2024, 15, 9851–9857 RSC.
- H. Zhao, J. Yin and P. Xi, Trans. Tianjin Univ., 2023, 29, 395–405 CrossRef.
- X. Liu, Z. Xing, M. Ajmal, C. Shi, R. Gao, L. Pan, Z. Huang, X. Zhang and J. Zou, Trans. Tianjin Univ., 2023, 29, 247–253 CrossRef.
- J.-W. Zhao, Z.-X. Shi, C.-F. Li, L.-F. Gu and G.-R. Li, Chem. Sci., 2021, 12, 650–659 RSC.
- N. Wang, S. Song, W. Wu, Z. Deng and C. Tang, Adv. Energy Mater., 2024, 14, 2303451 CrossRef.
- M. Chen, N. Kitiphatpiboon, C. Feng, A. Abudula, Y. Ma and G. Guan, eScience, 2024, 3, 100111 CrossRef.
- G. Chen, J. Zhang, W. Chen, R. Lu, C. Ma, Z. Wang and Y. Han, Chem. Sci., 2024, 10, 1039 Search PubMed.
- Z. Tao, H. Zhao, N. Lv, X. Luo, J. Yu, X. Tan and S. Mu, Adv. Funct. Mater., 2024, 34, 2312987 CrossRef.
- D. Zhang, M. Li, X. Yong, H. Song, G. I. N. Waterhouse, Y. Yi, B. Xue, D. Zhang, B. Liu and S. Lu, Nat. Commun., 2023, 14, 2517 CrossRef PubMed.
- Y. Wang, R. Yang, Y. Ding, B. Zhang, H. Li, B. Bai, M. Li, Y. Cui, J. Xiao and Z.-S. Wu, Nat. Commun., 2023, 14, 1412 CrossRef.
- J. Ying, J.-B. Chen, Y.-X. Xiao, S. I. Cordoba De Torresi, K. I. Ozoemena and X.-Y. Yang, J. Mater. Chem. A, 2023, 11, 1634–1650 RSC.
- X. Zhou, M. Mukoyoshi, K. Kusada, T. Yamamoto, T. Toriyama, Y. Murakami, S. Kawaguchi, Y. Kubota, O. Seo, O. Sakata, T. Ina and H. Kitagawa, Chem. Sci., 2024, 15, 7560–7567 RSC.
- F. Ren, J. Xu and L. Feng, Nano Res., 2024, 17, 3785–3793 CrossRef.
- W. Li, R. Liu, G. Yu, X. Chen, S. Yan, S. Ren, J. Chen, W. Chen, C. Wang and X. Lu, Small, 2024, 20, 2307164 CrossRef.
- W. Jia, X. Cao, X. Chen, H. Qin, L. Miao, Q. Wang and L. Jiao, Small, 2024, 20, 2310464 CrossRef.
- C. Roy, R. R. Rao, K. A. Stoerzinger, J. Hwang, J. Rossmeisl, I. Chorkendorff, Y. Shao-Horn and I. E. L. Stephens, ACS Energy Lett., 2018, 3, 2045–2051 CrossRef.
- Y. Gao, D. Zheng, Q. Li, W. Xiao, T. Ma, Y. Fu, Z. Wu and L. Wang, Adv. Funct. Mater., 2022, 32, 2203206 CrossRef.
- T. Tang, X. Bai, Z. Wang and J. Guan, Chem. Sci., 2024, 15, 5082–5112 RSC.
- Y. Zhang, H. Liu, S. Zhao, C. Xie, Z. Huang and S. Wang, Adv. Mater., 2023, 35, 2209680 CrossRef.
- C. Chen, H. Jin, P. Wang, X. Sun, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Chem. Soc. Rev., 2024, 53, 2022–2055 RSC.
- R. Li, S. Guo, X. Wang, X. Wan, S. Xie, Y. Liu, C. Wang, G. Zhang, J. Cao, J. Dai, M. Ge and W. Zhang, Chem. Sci., 2024, 15, 10084–10091 RSC.
- Y. Zhu, X. Liu, S. Jin, H. Chen, W. Lee, M. Liu and Y. Chen, J. Mater. Chem. A, 2019, 7, 5875–5897 RSC.
- Q. Xu, J. Zhang, H. Zhang, L. Zhang, L. Chen, Y. Hu, H. Jiang and C. Li, Energy Environ. Sci., 2021, 14, 5228–5259 RSC.
- X. Wang, X. Wan, X. Qin, C. Chen, X. Qian, Y. Guo, Q. Xu, W.-B. Cai, H. Yang and K. Jiang, ACS Catal., 2022, 12, 9437–9445 CrossRef CAS.
- H. Zhu, S. Sun, J. Hao, Z. Zhuang, S. Zhang, T. Wang, Q. Kang, S. Lu, X. Wang, F. Lai, T. Liu, G. Gao, M. Du and D. Wang, Energy Environ. Sci., 2023, 16, 619–628 RSC.
- Y. Yang, Z. Jia, Q. Wang, Y. Liu, L. Sun, B. Sun, J. Kuang, S. Dai, J. He, S. Liu, L. Duan, H. Tang, L.-C. Zhang, J. J. Kruzic, J. Lu and B. Shen, Energy Environ. Sci., 2024, 17, 5854–5865 RSC.
- Y. Lu, L. Tang, P. Wang, M. He, C. Yang and Z. Li, ACS Catal., 2023, 13, 13804–13815 CrossRef.
- Q. Liu, Z. Yan, J. Gao and E. Wang, ACS Sustainable Chem. Eng., 2022, 10, 14926–14934 CrossRef.
- H. Chen, M. Zhang, Y. Wang, K. Sun, L. Wang, Z. Xie, Y. Shen, X. Han, L. Yang and X. Zou, Nano Res., 2022, 15, 10194–10217 CrossRef.
- S. Zhou, H. Jang, Q. Qin, L. Hou, M. G. Kim, S. Liu, X. Liu and J. Cho, Angew. Chem., Int. Ed., 2022, 61, e202212196 CrossRef CAS PubMed.
- K. Chen, Z. Liu, S. Zhu, Y. Liu, Y. Liu, L. Wang, T. Xia, Z. Zhao, H. Gao, S. Cheng and H. Guo, Adv. Funct. Mater., 2024, 2406259 CrossRef.
- F.-F. Zhang, C.-Q. Cheng, J.-Q. Wang, L. Shang, Y. Feng, Y. Zhang, J. Mao, Q.-J. Guo, Y.-M. Xie, C.-K. Dong, Y.-H. Cheng, H. Liu and X.-W. Du, ACS Energy Lett., 2021, 1588–1595 CrossRef CAS.
- G. F. S. R. Rocha, M. A. R. Da Silva, A. Rogolino, G. A. A. Diab, L. F. G. Noleto, M. Antonietti and I. F. Teixeira, Chem. Soc. Rev., 2023, 52, 4878–4932 RSC.
- Y. Shi, Z.-R. Ma, Y.-Y. Xiao, Y.-C. Yin, W.-M. Huang, Z.-C. Huang, Y.-Z. Zheng, F.-Y. Mu, R. Huang, G.-Y. Shi, Y.-Y. Sun, X.-H. Xia and W. Chen, Nat. Commun., 2021, 12, 3021 CrossRef PubMed.
- A. Shankar, S. Marimuthu and G. Maduraiveeran, J. Mater. Chem. A, 2024, 12, 121–127 RSC.
- J. Guan, D. Li, R. Si, S. Miao, F. Zhang and C. Li, ACS Catal., 2017, 7, 5983–5986 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04909h |
| This journal is © The Royal Society of Chemistry 2024 |