
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrior disulfide bond-mediated Ser/Thr ligation†
Heng
Liu‡
,
Hoi Yee
Chow‡
,
Jiamei
Liu‡
,
Pengfei
Shi‡
and
Xuechen
Li
 *
*
Department of Chemistry, State Key Laboratory of Synthetic Chemistry, The University of Hong Kong, Hong Kong SAR, P. R. China. E-mail: xuechenl@hku.hk
First published on 19th August 2024
Abstract
In this work, we developed a novel strategy, prior disulfide bond-mediated Ser/Thr ligation (PD-STL), for the chemical synthesis of peptides and proteins. This approach combines disulfide bond-forming chemistry with Ser/Thr ligation (STL), converting intermolecular STL into intramolecular STL to effectively proceed regardless of concentrations. We demonstrated the effectiveness of PD-STL under high dilution conditions, even for the relatively inert C-terminal proline at the ligation site. Additionally, we applied this method to synthesize the N-terminal cytoplasmic domain (2-104) of caveolin-1 and its Tyr14 phosphorylated form.
Introduction
Protein chemical synthesis via peptide ligation is a powerful and versatile approach for generating proteins with well-defined structures and properties for various applications in biotechnology, pharmaceutical research, and the study of protein function.1–3 This method involves the chemical synthesis of individual peptide segments through solid-phase peptide synthesis (SPPS), which are then ligated together to form the full-length protein. Chemical protein synthesis offers researchers precise control over the final protein structure, enabling the incorporation of specific modifications, such as post-translational modifications (PTM), unnatural amino acids, or site-specific labels, which can be difficult or impossible to achieve using traditional recombinant protein expression methods.4–9 In this regard, native chemical ligation (NCL),10–12 ketoacid–hydroxylamine (KAHA) ligation,13–15 serine/threonine ligation (STL)16–18 and their variants19,20 have been demonstrated as effective synthetic methods to prepare hundreds of long peptides and proteins over the past few decades. The successful synthesis of functional proteins containing more than 300 amino acid residues21 (e.g., ubiquitin oligomers,22,23 ubiquitinylated α-globin,24 GroEL/ES-dependent protein DapA,25 and mirror image DNA polymerases26–30) showcases the increasing capability of chemical protein synthesis.Despite the ambition and promising achievements to increase the size of synthetic proteins, the synthesis of a protein with more than 200 amino acids can still be very difficult in most cases, due to the following reasons: (1) proteins with large sizes typically contain “difficult peptide” fragments, which have poor solubility and high aggregation propensity in the ligation buffer;31,32 (2) ligation between two peptide fragments usually needs to be conducted at a concentration in the millimolar range, which can be difficult to achieve when peptide fragments are of large size due to solubility issues; (3) the reactivity of the ligating groups at the termini is dramatically affected by the size of the fragment. Facilitating efficient ligation at sub-millimolar concentrations can be a way to avoid solubility and aggregation related adverse issues. As a bimolecular process, the rate of an intermolecular ligation is highly dependent on the substrate concentration. Thus, transforming intermolecular ligation into an intramolecular process by linking the N-terminal and C-terminal fragments together, both covalently and non-covalently, is a promising way to overcome the obstacle. In Kemp's pioneering work reported in 1981, a dibenzofuran-based template was developed to covalently link N-terminal and C-terminal fragments via phenolic ester and disulfide, respectively.33,34 In 1999, Dawson reported the conformationally assisted NCL.35 The N-terminal and C-terminal ligation partners of the chymotrypsin inhibitor 2 protein folded into a 3D structure before ligation via a non-covalent interaction, where the ligating Cys40 and Asp39 thioester were located in proximity. Since then, several templated ligation strategies have been developed.36,37 Unfortunately, the peptide assembly assisted ligation38 and RNA catalyzed ligation39 are less general due to the critical sequence dependency. Meanwhile, difficult removal of peptide or PNA tags in NCL templated by coiled coil peptides,40,41 proteins42 or DNA,43–45 renders these methods more efficient for protein labelling rather than synthesis. As recent examples, Kay46 and Liu47 reported the templated NCL which was facilitated by introducing cycloalkyne/azide and split intein CfaN/CfaC pairs to the ligation partners through removable Lys modification and backbone Hmb installation, respectively (Fig. 1a). The click reaction and intein excision led to covalent prior capture formation, which allowed efficient NCL at low concentrations (2–50 μM). Both methods have been successfully applied for the synthesis of the E. coli 50S ribosome subunit L32,46 D-form extracellular domain of TIGIT,47 and D-form Ig-like C2-type domain of tropomyosin kinase C.47
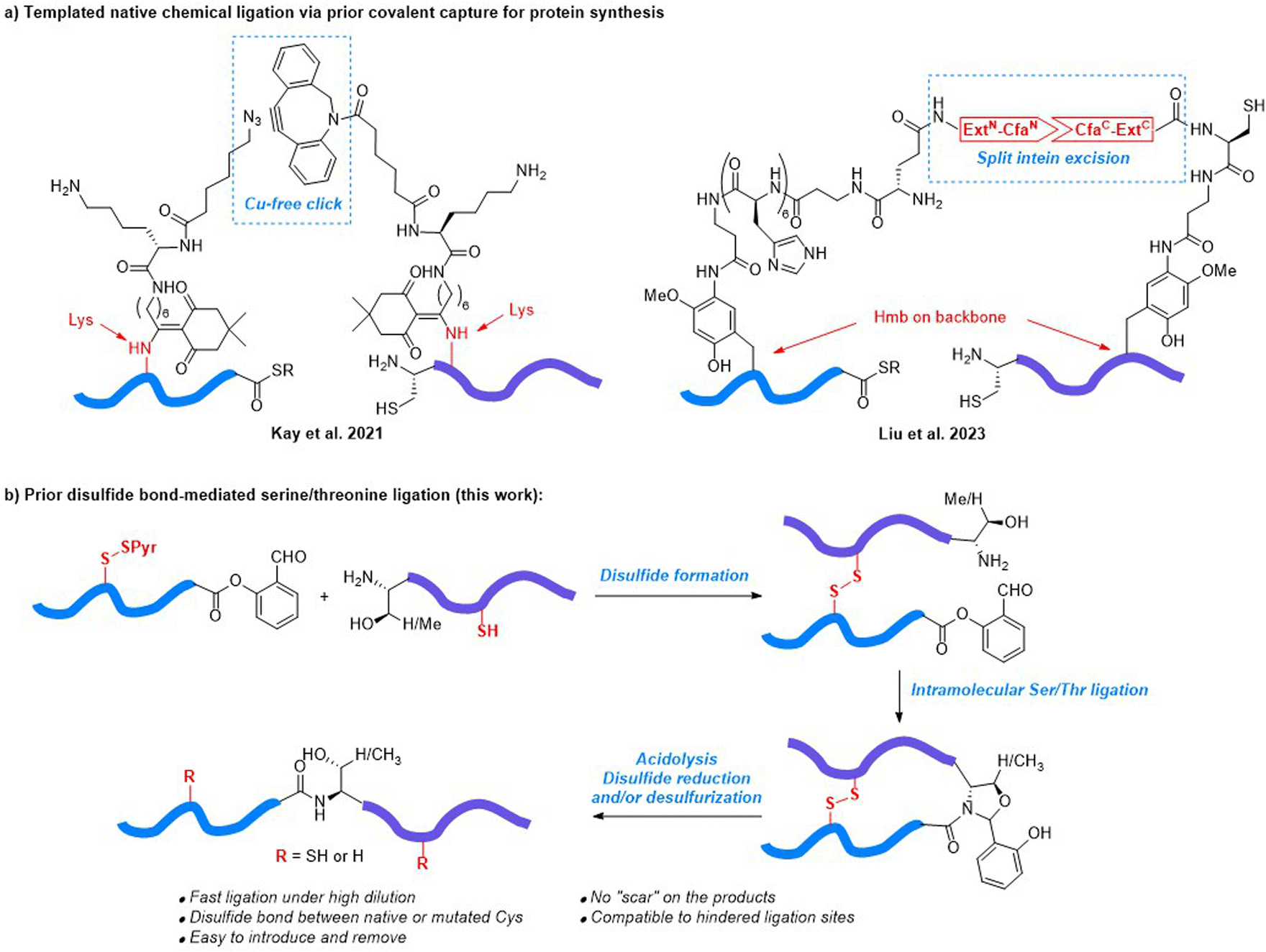 | ||
| Fig. 1 Reported templated native chemical ligation strategies and the design of prior disulfide bond-mediated serine/threonine ligation. (a) Templated native chemical ligation via prior covalent linkage in protein synthesis. (b) Prior disulfide bond-mediated serine/theronine ligation (this work). | ||
Disulfide is the native conformation restricting element in proteins and peptide hormones. The intrachain and interchain disulfide networks play key roles in the stabilization of secondary and tertiary structures. We hypothesized that the disulfide linkage between native or mutated Cys residues could be leveraged to facilitate prior capture-mediated STL at low concentrations, without the need for incorporating complex sidechain/backbone modifications. In this prior disulfide bond-mediated Ser/Thr ligation (PD-STL), a peptide C-terminal salicylaldehyde (SAL) ester and a peptide with an N-terminal serine or threonine residue, were first joined together by a disulfide linkage, followed by an intramolecular STL to form the ligated product (Fig. 1b). The non-reducing conditions of STL perfectly fit the demand for disulfide linkage, and the available disulfide formation48 and desulfurization methods49–51 allow the disulfide template installation with site flexibility. Herein, we report the development of PD-STL and its application in the synthesis of the N-terminal cytoplasmic domain of caveolin-1 (Cav-1) and the Tyr14 phosphorylated form.
Results and discussion
As a simple but effective model to test the effect of PD-STL, we chose a peptide with a C-terminal proline for ligation. Peptides with C-terminal proline thioesters are known to be difficult to ligate to N-terminal cysteine peptides by native chemical ligation.52 Moreover, Ser/Thr ligation barely proceeds with peptides carrying C-terminal proline SAL esters at low concentrations.53 Thus, we started our model study using C-terminal proline SAL esters to demonstrate the rate-increasing effect of disulfide linkage. As a control experiment, peptides 1a and 2a were dissolved in pyridine/AcOH buffer to trigger the ligation process with a final concentration of 0.25 mM. As expected, it took a long time (3 days) to fully convert to the ligation intermediate, based on liquid chromatography-mass spectrometry (LC-MS) analysis (Table 1, entry 1). Increasing the ligation concentration to 10 mM led to faster conversion, but 6 hours were still needed for the reaction to be completed (Table 1, entry 3). Similar results were observed using peptide 2b as the N-terminal Thr peptide, where the location of Cys was moved away from the ligation site (Table 1, entries 2 and 4).| Entry | SAL esters | Thr peptides | Ligation conc. | Time to finish |
|---|---|---|---|---|
| a For prior disulfide bond-mediated STL, peptides 1b (1.0 equiv.) and 2a/2b (1.5 equiv.) were dissolved in MeOH containing 0.1% TFA with a concentration of 10 mM (peptide SAL esters) to form disulfide. After removing 2-thiopyridone, the crude product together with excess peptides 2a/2b was dissolved in pyridine/HOAc 1/1 v/v for STL. b For STL without disulfide bond capture, peptides 1a (1.0 equiv.) and 2a/2b were also dissolved in MeOH containing 0.1% TFA for 4 hours to mimic the prior capture cases, followed by STL in pyridine/HOAc 1/1 v/v buffer. | ||||
| 1 | Fmoc-GPARGP-OSAL ester 1a | TCLVGSQKAPSEVPTAGF 2a | 0.25 mM | >3 days |
| 2 | Fmoc-GPARGP-OSAL ester 1a | TALVGSQKCPSEVPTAGF 2b | 0.25 mM | >3 days |
| 3 | Fmoc-GPARGP-OSAL ester 1a | TCLVGSQKAPSEVPTAGF 2a | 10 mM | 6 h |
| 4 | Fmoc-GPARGP-OSAL ester 1a | TALVGSQKCPSEVPTAGF 2b | 10 mM | 3 h |
| 5 | Fmoc-GPC(SPyr)RGP-OSAL ester 1b | TCLVGSQKAPSEVPTAGF 2a | 0.25 mM | <12 min |
| 6 | Fmoc-GPC(SPyr)RGP-OSAL ester 1b | TALVGSQKCPSEVPTAGF 2b | 0.25 mM | <78 min |
Alternatively, peptide 1b with a side chain of cysteine temporarily protected by S-pyridine (SPyr) was synthesized, where Cys(SPyr) functions as an effective precursor for hetero-disulfide bond formation.54 As a result, peptides 1b (1.0 equiv.) and 2a (1.5 equiv.) efficiently formed a heterodimer via a disulfide bond when dissolved in MeOH containing 0.1% TFA with a concentration of 10 mM. Encouragingly, after the formation of the disulfide bond, the intramolecular STL proceeded smoothly at low concentration (0.25 mM), reaching completion within 12 minutes (Table 1, entry 5). No adverse effect caused by excess 2a like the competing intermolecular STL was observed. In the case of the PD-STL of peptide 2b, the ligation rate remained faster (<78 min) compared to that of the control experiments without prior disulfide capture (Table 1, entry 6). The accelerating effect was less significant, because a larger and more flexible cyclic structure than that in the 1b/2a case was involved. This performance was in accordance with our former observations that STL generally showed good reaction kinetics in peptide cyclization.55–58
Subsequently, we explored how the performance of PD-STL was influenced by the locations of the two designated Cys residues, i.e., the size of the macrocycles formed in the ligation (Table 2). When the distance between Cys and the C-terminus of the peptide SAL ester was too small (Table 2, entries 2 and 3), the disulfide bond-assisted ligation was slow, which can be attributed to the strained reaction conformation. However, the ligation still proceeded more rapidly at 0.25 mM concentration than the STL without disulfide bond assistance. When the cysteine was located one amino acid further from the C-terminus of the peptide SAL ester (Table 2, entries 4–12), the ligation rate improved dramatically. The ligation was more sensitive to the location of the Cys residue in the peptide SAL ester, since 1d/2a ligation was much faster than that in the 1c/2c case, though the same 17-membered ring was formed in the ligation intermediate. Furthermore, the disulfide bond-assisted STL rate was the highest when the distance between the two designated cysteines was 3 or 4 amino acids (forming 17-membered and 20-membered rings, respectively) (Table 2, entries 4 and 5). All disulfide bond-assisted ligations exhibited much higher rates than the standard STL in control experiments. PD-STL can successfully enhance ligation performance, though the Cys location can influence the ligation rate.
| Entry | SAL esters | Thr peptides | No. of A.A. in between | Ligation time |
|---|---|---|---|---|
| a The SAL esters 1b–1d (1.0 equiv.) and peptides 2a–2h (1.5 equiv.) were dissolved in MeOH containing 0.1% TFA with a concentration of 10 mM (SAL ester) to form disulfide. After removing 2-thiopyridone, the crude peptides together with excess peptides 2a–2h were dissolved in pyridine/HOAc 1/1 v/v with a concentration of 0.25 mM for STL. The conversion was monitored at different time points by UPLC-MS. | ||||
| 1 | Fmoc-GPARGP-OSAL ester 1a | TCLVGSQKAPSEVPTAGF 2a | — | >3 days |
| 2 | Fmoc-GPRGC(SPyr)P-OSAL ester 1c | TCLVGSQKAPSEVPTAGF 2a | 2 | >12 h |
| 3 | Fmoc-GPRGC(SPyr)P-OSAL ester 1c | TACVGSQKAPSEVPTAGF 2c | 3 | >12 h |
| 4 | Fmoc-GPRC(SPyr)GP-OSAL ester 1d | TCLVGSQKAPSEVPTAGF 2a | 3 | ≤12 min |
| 5 | Fmoc-GPC(SPyr)RGP-OSAL ester 1b | TCLVGSQKAPSEVPTAGF 2a | 4 | ≤12 min |
| 6 | Fmoc-GPC(SPyr)RGP-OSAL ester 1b | TACVGSQKAPSEVPTAGF 2c | 5 | ≤63 min |
| 7 | Fmoc-GPC(SPyr)RGP-OSAL ester 1b | TALCGSQKAPSEVPTAGF 2d | 6 | ≤50 min |
| 8 | Fmoc-GPC(SPyr)RGP-OSAL ester 1b | TALVCSQKAPSEVPTAGF 2e | 7 | ∼40 min |
| 9 | Fmoc-GPC(SPyr)RGP-OSAL ester 1b | TALVGCQKAPSEVPTAGF 2f | 8 | ≤65 min |
| 10 | Fmoc-GPC(SPyr)RGP-OSAL ester 1b | TALVGSCKAPSEVPTAGF 2g | 9 | ≤52 min |
| 11 | Fmoc-GPC(SPyr)RGP-OSAL ester 1b | TALVGSQCAPSEVPTAGF 2h | 10 | ≤75 min |
| 12 | Fmoc-GPC(SPyr)RGP-OSAL ester 1b | TALVGSQKCPSEVPTAGF 2b | 11 | ≤78 min |
With the exciting results obtained, we applied the PD-STL to peptides with varied sequences to confirm its generality. As shown in Table 3, all PD-STL reactions were completed within 5 to 305 minutes at a low concentration of 0.25 mM. All those peptides resulted in rapid and high-yielding ligations (29–63% isolated yields) under high dilution conditions (0.25 mM), even for highly sterically hindered amino acids (e.g., Ile, Val, and Pro, Table 3, entries 3–5 and 7) at the C-terminus, which typically exhibited lower ligation rates and yields in both NCL and STL. These results demonstrated the capability of PD-STL.
| Entry | N-terminal peptide | C-terminal peptide | Ligation time | Isolatedb yield |
|---|---|---|---|---|
| a The SAL esters 1e–1k (1.0 equiv.) and peptides 2i–2o (1.3–2.0 equiv.) were dissolved in MeCN/H2O 1/1 v/v containing 0.1% TFA with a concentration of 10 mM (SAL ester) to form disulfide. After lyophilization and 2-thiopyridone removal via ether wash, the crude peptides together with excess peptides 2i–2o were dissolved in pyridine/HOAc 1/1 v/v with a concentration of 0.25 mM for STL. The conversion was monitored at different time points by UPLC-MS. b The ligation intermediate was treated with TFA/H2O/TIPS 95/2.5/2.5 v/v/v for acidolysis and 20 mM TCEP in MeCN/H2O 1/1 v/v for disulfide reduction, followed by HPLC purification. | ||||
| 1 | LEQLKC(SPyr)GF-OSAL ester 1e | SGGCGLFDVVKG 2i | 5 min | 30% |
| 2 | FRKSGFC(SPyr)GT-OSAL ester 1f | TCRSYFPGSTYG 2j | 23 min | 50% |
| 3 | GMTHGLIC(SPyr)GI-OSAL ester 1g | TGKCQRM 2k | 50 min | 29% |
| 4 | PTIPC(SPyr)KARGI-OSAL ester 1h | TCHYIPRPKPR 2l | 44 min | 57% |
| 5 | IPAC(SPyr)IAGV-OSAL ester 1i | TKKCFLGGLMKA 2m | 88 min | 52% |
| 6 | GRKSDC(SPyr)FPA-OSAL ester 1j | SRKCFLI 2n | 23 min | 63% |
| 7 | GSKKPVPIIYC(SPyr)NRP-OSAL ester 1k | SGFCAFLKSPS 2o | 305 min | 49% |
Synthesis of CAV-1 (2-104)
To showcase the effectiveness of this disulfide bond-assisted Ser/Thr ligation for chemical protein synthesis, we synthesized the N-terminal cytoplasmic domain (2-104) of caveolin-1 (Cav-1). Cav-1 is a V-shaped membrane protein that plays multiple roles in cellular processes including signal transduction, metabolism, endocytosis (via caveola formation), and differentiation.59,60 Cav-1 is also involved in several pathogenic processes like kidney diseases and cancer.61–63 Cav-1 bears various post-translational modifications (acetylation, phosphorylation, palmitoylation, etc.),59,60 and the Tyr14 phosphorylation located in the N-terminal cytoplasmic domain has been implicated in numerous biological functions, including focal adhesion, cancer biology,64–66 endothelial cell signalling in sepsis-induced lung injury,67 regulation of mechanotransduction,68–70 and insulin signaling.71 Chemical synthesis of Tyr14 phosphorylated or non-phosphorylated molecular probes will be important for biological studies. Full length Cav-1 bearing triple S-palmitoylations has been successfully synthesized by Hojo, via sequential thioester ligations in DMSO assisted by iNoc (4-pyridylmethoxycarbonyl) protected O-isopeptides and Lys residues.72 The chemical synthesis of a phosphorylated Cav-1 N-terminal cytoplasmic domain has not been reported.Our synthetic plan is shown in Fig. 2. Since Cav-1 (2-104) does not contain a native Cys residue, we mutated Ala31 and Ala44 to Cys to facilitate disulfide linkage formation. Ala87 was also mutated to Cys to install a reducible solubilizing tag (RST).73 An extra Lys residue was added to the C-terminus to install a biotin, which would be used for probe enrichment by streptavidin beads in future chemical biology study. For protein assembly, we devised an N-to-C sequential ligation process, which involved the Leu36–Ser37 juncture as the first ligation site (PD-STL) and the Gly77–Thr78 juncture as the second ligation site (conventional STL).
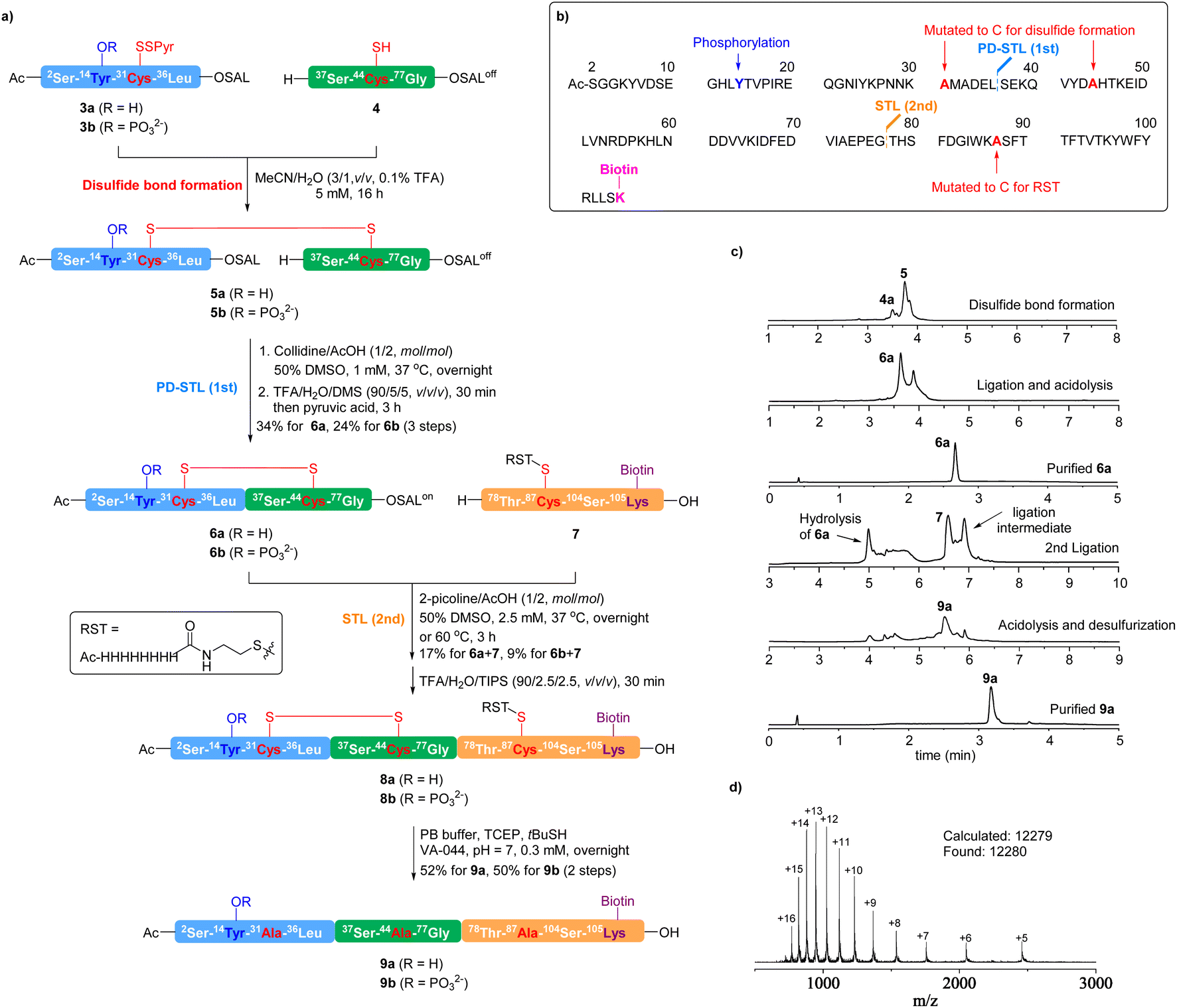 | ||
| Fig. 2 Total chemical synthesis of caveolin-1 (2-104) and the Tyr14 phosphorylated form facilitated by PD-STL. (a) Synthesis of caveolin-1 (2-104) and the Y14 phosphorylated variant. (b) Sequence of Cav-1 (2-104) and the synthetic plan. (c) LC traces of steps in the synthesis of Cav-1 (2-104) 9a. (d) ESI-MS of 9a. | ||
The peptide fragments 3a, 4 and 7 were readily synthesized via the standard Fmoc-SPPS. The C-terminus of 4 was modified to the peptide SALoff ester state74 to avoid head-to-tail cyclization. As shown in Fig. 2a, fragments 3a and 4 were dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in MeCN/H2O (3/1, v/v, containing 0.1% TFA) at a concentration of 5 mM to form the disulfide linkage. During the disulfide formation, peptide fragments 3a and 4 and product 5 showed good solubility in the solvent system we used. After achieving full conversion (16 h), the solvent was removed through freeze-drying to yield the crude disulfide-linked intermediate 5. As the precursor of PD-STL, 5a demonstrated good solubility in the ligation buffer at a concentration of 1 mM (collidine/AcOH 1/2 mol/mol containing 50% DMSO), where hydrophilic fragment c3a acted as a solubilizing tag to improve the solubility of 4. After stirring at 37 °C overnight, the PD-STL achieved near quantitative conversion, with negligible hydrolysis of the SAL ester observed (Fig. S84†). The ligation intermediate (N-peptidyl N,O-benzylidene acetal) was precipitated using diethyl ether and directly subjected to the acidolysis cocktail (TFA/H2O/dimethylsulfide 95/5/5 v/v/v). After 30 minutes, pyruvic acid was added to facilitate the SALoff-to-SALon switch, and the product 6a was isolated in 34% yield by reversed-phase preparative HPLC. As a comparison, we also tested the conventional STL between peptides 3′ and 4′ without Ala-to-Cys mutation and prior disulfide capture (refer to the ESI for details†). After overnight reaction at a concentration of 5 mM, only a minimal amount of the ligation intermediate was detected (Fig. S80†), largely due to the poor solubility of peptide 4′. When the concentration was reduced to 1 mM, even worse ligation performance was observed, with the formation of a tiny amount of ligation intermediate and numerous side products after overnight reaction (Fig. S81†). This result further demonstrates the rate-accelerating capability of the PD-STL strategy in the synthesis of poorly soluble peptides.
:1 ratio in MeCN/H2O (3/1, v/v, containing 0.1% TFA) at a concentration of 5 mM to form the disulfide linkage. During the disulfide formation, peptide fragments 3a and 4 and product 5 showed good solubility in the solvent system we used. After achieving full conversion (16 h), the solvent was removed through freeze-drying to yield the crude disulfide-linked intermediate 5. As the precursor of PD-STL, 5a demonstrated good solubility in the ligation buffer at a concentration of 1 mM (collidine/AcOH 1/2 mol/mol containing 50% DMSO), where hydrophilic fragment c3a acted as a solubilizing tag to improve the solubility of 4. After stirring at 37 °C overnight, the PD-STL achieved near quantitative conversion, with negligible hydrolysis of the SAL ester observed (Fig. S84†). The ligation intermediate (N-peptidyl N,O-benzylidene acetal) was precipitated using diethyl ether and directly subjected to the acidolysis cocktail (TFA/H2O/dimethylsulfide 95/5/5 v/v/v). After 30 minutes, pyruvic acid was added to facilitate the SALoff-to-SALon switch, and the product 6a was isolated in 34% yield by reversed-phase preparative HPLC. As a comparison, we also tested the conventional STL between peptides 3′ and 4′ without Ala-to-Cys mutation and prior disulfide capture (refer to the ESI for details†). After overnight reaction at a concentration of 5 mM, only a minimal amount of the ligation intermediate was detected (Fig. S80†), largely due to the poor solubility of peptide 4′. When the concentration was reduced to 1 mM, even worse ligation performance was observed, with the formation of a tiny amount of ligation intermediate and numerous side products after overnight reaction (Fig. S81†). This result further demonstrates the rate-accelerating capability of the PD-STL strategy in the synthesis of poorly soluble peptides.
In Hojo's synthesis, two O-isopeptides were used to overcome the hydrophobicity of fragment Thr78–Ser104.72 In our case, since STL is conducted under non-reducing conditions, we applied our reducible solubilizing tag (RST) strategy73 to incorporate a His8 tag into this sequence at the Cys87 mutated from Ala via disulfide linkage. The RST modified fragment 7 was synthesized in 30% yield and subjected to the final STL with 6a. In this step, through condition screening, we found that using 2-picoline instead of collidine in the ligation buffer gave much better results by suppressing the hydrolysis of 6a. After overnight reaction at a concentration of 2.5 mM at 37 °C, the ligation intermediate was precipitated using diethyl ether. After acidolysis by TFA/H2O/TIPS treatment, product 8a overlapped with 7 as monitored by UPLC-MS (Fig. S87†), which hampered efficient separation. To tackle this problem, the ligation intermediate was purified by RP-HPLC with an isolated yield of 17% (Fig. 2B and S88†). After acidolysis for 30 minutes, the peptide 8a was directly subjected to disulfide reduction and desulfurization in a phosphate buffer at pH 7 containing TCEP, tBuSH, and VA-044. After overnight reaction, the Cav-1 (2-104) product 9a was isolated in 52% yield over two steps.
Following the same protocols, Tyr14 phosphorylated Cav-1 (2-104) was also synthesized, where the phosphorylated fragment 3b was used instead of 3a. The PD-STL between 3b and 4 showed comparable performance to the former case, giving rise to 6b in 24% yield. In contrast to the former case, the STL between 6b and 7 was less effective in picoline/HOAc/DMSO buffer, leading to serious hydrolysis of the SAL ester and only a tiny amount of the ligation intermediate. To address this issue, a higher reaction temperature (60 °C) was employed, allowing 6b to be consumed within 3 hours with an acceptable level of hydrolysis, affording the ligation intermediate in 9% isolated yield. After acidolysis, disulfide reduction and desulfurization, the product 9b was isolated in 50% yield.
Conclusions
Prior capture-mediated ligation, in other words, converting intermolecular reactions into intramolecular reactions, can effectively proceed regardless of the concentration, providing a promising strategy for large molecule synthesis. The key to this strategy is identifying an efficient capture reaction that can proceed at low concentrations and under a wide range of conditions. In this study, we integrated disulfide bond-forming chemistry with Ser/Thr ligation to develop prior disulfide bond-mediated Ser/Thr ligation (PD-STL). To this end, a peptide salicylaldehyde ester with a Cys-S-pyridine residue first reacts with another peptide featuring an N-terminal Ser/Thr and an internal Cys residue. A hetero-disulfide bond is readily formed, and the disulfide linkage is stable under non-reducing STL conditions. Subsequently, an intramolecular STL takes place. We demonstrated its effectiveness at low concentrations (e.g., 0.25 mM) even for the C-terminal proline at the ligation site. Furthermore, we applied PD-STL for the syntheses of the N-terminal cytoplasmic domain (2-104) of caveolin-1 and its Tyr14 phosphorylated form. Compared with the reported templated native chemical ligation strategies which required peptide sidechain or backbone modification, PD-STL uses the native or mutated cysteine residues, thus simplifying the operation. We anticipate that this strategy will inspire the development of prior capture-based peptide ligation for challenging protein synthesis.Data availability
The data supporting this article (“Prior disulfide bond-mediated Ser/Thr ligation”) have been included as part of the ESI.†Author contributions
X. L. conceived and supervised the project. H. L. and H. Y. C. designed the methodology and model study. J. L. and P. S. conducted the peptide synthesis, peptide ligation and protein synthesis. X. L. and P. S. wrote the manuscript. All the authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22177097) and Research Grants Council of Hong Kong (17312022, AoE/P-705/16), X. L. is a recipient of the Research Grants Council-Senior Research Fellow Scheme (SPFS2324-7S01). We thank Dr Han Liu for his editorial inputs.Notes and references
- Y. Tan, H. Wu, T. Wei and X. Li, J. Am. Chem. Soc., 2020, 142, 20288–20298 CrossRef CAS.
- O. Harel and M. Jbara, Angew. Chem., Int. Ed., 2023, 62, e202217716 CrossRef CAS PubMed.
- Z. Sun, H. Liu and X. Li, Chem, 2024, 10, 767–799 CAS.
- S. Bondalapati, M. Jbara and A. Brik, Nat. Chem., 2016, 8, 407–418 CrossRef CAS PubMed.
- L. Liu, Isr. J. Chem., 2019, 59, 64–70 CrossRef CAS.
- C. C. Hanna, J. Kriegesmann, L. J. Dowman, C. F. W. Becker and R. J. Payne, Angew. Chem., Int. Ed., 2022, 61, e202111266 CrossRef CAS PubMed.
- H. Zhuang, H. Hao, Y. Qiu, J. Gan and T. Li, Chin. J. Chem., 2023, 41, 2010–2024 CrossRef CAS.
- J. Zhao, F. Ye, P. Huang and P. Wang, Curr. Opin. Chem. Biol., 2023, 77, 102405 CrossRef CAS PubMed.
- Y.-K. Qi, J.-S. Zheng and L. Liu, Chem, 2024, 10, 2390–2407 CAS.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed.
- P. A. Cistrone, M. J. Bird, D. T. Flood, A. P. Silvestri, J. C. J. Hintzen, D. A. Thompson and P. E. Dawson, Curr. Protoc. Chem. Biol., 2019, 11, e61 CrossRef.
- V. R. Pattabiraman, A. O. Ogunkoya and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 5114–5118 CrossRef CAS PubMed.
- I. Pusterla and J. W. Bode, Nat. Chem., 2015, 7, 668–672 CrossRef CAS PubMed.
- J. W. Bode, Acc. Chem. Res., 2017, 50, 2104–2115 CrossRef CAS PubMed.
- X. Li, H. Y. Lam, Y. Zhang and C. K. Chan, Org. Lett., 2010, 12, 1724–1727 CrossRef CAS.
- Y. Zhang, C. Xu, H. Y. Lam, C. L. Lee and X. Li, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6657–6662 CrossRef CAS.
- H. Liu and X. Li, Acc. Chem. Res., 2018, 51, 1643–1655 CrossRef CAS PubMed.
- S. S. Kulkarni, J. Sayers, B. Premdjee and R. J. Payne, Nat. Rev. Chem, 2018, 2, 0122 CrossRef CAS.
- J.-S. Zheng, S. Tang, Y.-C. Huang and L. Liu, Acc. Chem. Res., 2013, 46, 2475–2484 CrossRef CAS PubMed.
- B. Zhang, Y. Li, W. Shi, T. Wang, F. Zhang and L. Liu, Chem. Res. Chin. Univ., 2020, 36, 733–747 CrossRef CAS.
- K. S. A. Kumar, S. N. Bavikar, L. Spasser, T. Moyal, S. Ohayon and A. Brik, Angew. Chem., Int. Ed., 2011, 50, 6137–6141 CrossRef CAS.
- S. Tang, L.-J. Liang, Y.-Y. Si, S. Gao, J.-X. Wang, J. Liang, Z. Mei, J.-S. Zheng and L. Liu, Angew. Chem., Int. Ed., 2017, 56, 13333–13337 CrossRef CAS.
- H. Sun, S. M. Mali, S. K. Singh, R. Meledin, A. Brik, Y. T. Kwon, Y. Kravtsova-Ivantsiv, B. Bercovich and A. Ciechanover, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7805–7812 CrossRef CAS PubMed.
- M. T. Weinstock, M. T. Jacobsen and M. S. Kay, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11679–11684 CrossRef CAS.
- A. Pech, J. Achenbach, M. Jahnz, S. Schülzchen, F. Jarosch, F. Bordusa and S. Klussmann, Nucleic Acids Res., 2017, 45, 3997–4005 CrossRef CAS PubMed.
- W. Xu, W. Jiang, J. Wang, L. Yu, J. Chen, X. Liu, L. Liu and T. F. Zhu, Cell Discovery, 2017, 3, 17008 CrossRef PubMed.
- W. Jiang, B. Zhang, C. Fan, M. Wang, J. Wang, Q. Deng, X. Liu, J. Chen, J. Zheng, L. Liu and T. F. Zhu, Cell Discovery, 2017, 3, 17037 CrossRef CAS.
- C. Fan, Q. Deng and T. F. Zhu, Nat. Biotechnol., 2021, 39, 1548–1555 CrossRef CAS.
- Y. Xu and T. F. Zhu, Science, 2022, 378, 405–412 CrossRef CAS PubMed.
- M. Paradís-Bas, J. Tulla-Puche and F. Albericio, Chem. Soc. Rev., 2016, 45, 631–654 RSC.
- L. K. Mueller, A. C. Baumruck, H. Zhdanova and A. A. Tietze, Front. Bioeng. Biotechnol., 2020, 8, 162 CrossRef PubMed.
- D. S. Kemp, Biopolymers, 1981, 20, 1793–1804 CrossRef CAS.
- M. Fotouhi, N. G. Galakatos and D. S. Kemp, J. Org. Chem., 1989, 54, 2803–2817 CrossRef.
- G. S. Beligere and P. E. Dawson, J. Am. Chem. Soc., 1999, 121, 6332–6333 CrossRef CAS.
- O. Vázquez and L. Seitz, J. Pept. Sci., 2014, 20, 78–86 CrossRef.
- R. J. Giesler, P. W. Erickson and M. S. Kay, Curr. Opin. Chem. Biol., 2020, 58, 37–44 CrossRef CAS.
- D. H. Lee, J. R. Granja, J. A. Martinez, K. Severin and M. R. Ghadiri, Nature, 1996, 382, 525–528 CrossRef CAS PubMed.
- N. Kashiwagi, H. Furuta and Y. Ikawa, Nucleic Acids Res., 2009, 37, 2574–2583 CrossRef CAS PubMed.
- U. Reinhardt, J. Lotze, S. Zernia, K. Morl, A. G. Beck-Sickinger and O. Seitz, Angew. Chem., Int. Ed., 2014, 53, 10237–10241 CrossRef CAS PubMed.
- U. Reinhardt, J. Lotze, K. Mörl, A. G. Beck-Sickinger and O. Seitz, Bioconjugate Chem., 2015, 26, 2106–2117 CrossRef CAS.
- N. Brauckhoff, L. Fang, A. Haim and T. N. Grossmann, Chem. Commun., 2023, 59, 5241–5244 RSC.
- S. Ficht, A. Mattes and O. Seitz, J. Am. Chem. Soc., 2004, 126, 9970–9981 CrossRef CAS PubMed.
- A. Roloff and O. Seitz, Chem. Sci., 2013, 4, 432–436 RSC.
- S. Middel, C. H. Panse, S. Nawratil and U. Diederichsen, ChemBioChem, 2017, 18, 2328–2332 CrossRef CAS.
- P. W. Erickson, J. M. Fulcher, P. Spaltenstein and M. S. Kay, Bioconjugate Chem., 2021, 32, 2233–2244 CrossRef CAS.
- B. Zhang, Y. Zheng, G. Chu, X. Deng, T. Wang, W. Shi, Y. Zhou, S. Tang, J.-S. Zheng and L. Liu, Angew. Chem., Int. Ed., 2023, 62, e202306270 CrossRef CAS PubMed.
- T. M. Postma and F. Albericio, Eur. J. Org Chem., 2014, 3519–3530 CrossRef CAS.
- L. Z. Yan and P. E. Dawson, J. Am. Chem. Soc., 2001, 123, 526–533 CrossRef CAS PubMed.
- W. Qian and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef.
- Z. Sun, W. Ma, Y. Cao, T. Wei, X. Mo, H. Y. Chow, Y. Tan, C. H. P. Cheung, J. Liu, H. K. Lee, E. C. M. Tse, H. Liu and X. Li, Chem, 2022, 8, 2542–2557 CAS.
- S. B. Pollock and S. B. H. Kent, Chem. Commun., 2011, 47, 2342–2344 RSC.
- C. T. T. Wong, T. Li, H. Y. Lam, Y. Zhang and X. Li, Front. Chem., 2014, 2, 28 Search PubMed.
- D. Andreu, F. Albericio, N. A. Solé, M. C. Munson, M. Ferrer and G. Barany, Peptide synthesis protocols, 1995, pp. 91–169 Search PubMed.
- H. Y. Lam, Y. Zhang, H. Liu, J. Xu, C. T. T. Wong, C. Xu and X. Li, J. Am. Chem. Soc., 2013, 135, 6272–6279 CrossRef CAS PubMed.
- C. T. T. Wong, H. Y. Lam, T. Song, G. Chen and X. Li, Angew. Chem., Int. Ed., 2013, 52, 10212–10215 CrossRef CAS PubMed.
- Z. Sun, Z. Shang, N. Forelli, K. H. L. Po, S. Chen, S. F. Brady and X. Li, Angew. Chem., Int. Ed., 2020, 59, 19868–19872 CrossRef CAS.
- J. Wang, D. Lin, M. Liu, H. Liu, P. Blasco, Y. C. Cheung, S. Chen and X. Li, J. Am. Chem. Soc., 2021, 143, 12784–12790 CrossRef CAS.
- W. M. Krajewska and I. Maslowska, Cell. Mol. Biol. Lett., 2004, 9, 195–220 CAS.
- A. F. Quest, J. L. Gutierrez-Pajares and V. A. Torres, J. Cell. Mol. Med., 2008, 12, 1130–1150 CrossRef CAS PubMed.
- S. Luo, M. Yang, H. Zhao, Y. Han, N. Jiang, J. Yang, W. Chen, C. Li, Y. Liu, C. Zhao and L. Sun, Front. Pharmacol, 2021, 12, 768100 CrossRef CAS.
- Z. C. Nwosu, M. P. Ebert, S. Dooley and C. Meyer, Mol. Cancer, 2016, 15, 71 CrossRef.
- L. Simon, A. Campos, L. Leyton and A. F. G. Quest, Cancer Metastasis Rev., 2020, 39, 435–453 CrossRef CAS.
- M. Nethe, E. C. Anthony, M. Fernandez-Borja, R. Dee, D. Geerts, P. J. Hensbergen, A. M. Deelder, G. Schmidt and P. L. Hordijk, J. Cell Sci., 2010, 123, 1948–1958 CrossRef CAS PubMed.
- F. Meng, S. Saxena, Y. Liu, B. Joshi, T. H. Wong, J. Shankar, L. J. Foster, P. Bernatchez and I. R. Nabi, Mol. Biol. Cell, 2017, 28, 2190–2201 CrossRef CAS PubMed.
- R. Ortiz, J. Díaz, N. Díaz, L. Lobos-Gonzalez, A. Cárdenas, P. Contreras, M. I. Díaz, E. Otte, J. Cooper-White, V. Torres, L. Leyton and A. F. G. Quest, Oncotarget, 2016, 7, 40571–40593 CrossRef PubMed.
- H. Jiao, Y. Zhang, Z. Yan, Z.-G. Wang, G. Liu, R. D. Minshall, A. B. Malik and G. Hu, J. Immunol., 2013, 191, 6191–6199 CrossRef CAS PubMed.
- C. Radel and V. Rizzo, Am. J. Physiol.: Heart Circ. Physiol., 2005, 288, H936–H945 CrossRef CAS PubMed.
- B. Zhang, F. Peng, D. Wu, A. J. Ingram, B. Gao and J. C. Krepinsky, Cell. Signal., 2007, 19, 1690–1700 CrossRef CAS PubMed.
- B. Joshi, M. Bastiani, S. S. Strugnell, C. Boscher, R. G. Parton and I. R. Nabi, J. Cell Biol., 2012, 199, 425–435 CrossRef CAS PubMed.
- J. Chen, F. Capozza, A. Wu, T. deAngelis, H. Sun, M. Lisanti and R. Baserga, J. Cell. Physiol., 2008, 217, 281–289 CrossRef CAS PubMed.
- H. Hojo, T. Takei, Y. Asahina, N. Okumura, T. Takao, M. So, I. Suetake, T. Sato, A. Kawamoto and Y. Hirabayashi, Angew. Chem., Int. Ed., 2021, 60, 13900–13905 CrossRef CAS PubMed.
- J. Liu, T. Wei, Y. Tan, H. Liu and X. Li, Chem. Sci., 2022, 13, 1367–1374 RSC.
- C. L. Lee, H. Liu, C. T. T. Wong, H. Y. Chow and X. Li, J. Am. Chem. Soc., 2016, 138, 10477–10484 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04825c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |