
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSnapshot of cyclooctyne ring-opening to a tethered alkylidene cyclic polymer catalyst†
Javier M.
Hurst
 a,
Rinku
Yadav
a,
Parker T.
Boeck
ab,
Ion
Ghiviriga
ab,
ChristiAnna L.
Brantley
ab,
Łukasz
Dobrzycki
ab and
Adam S.
Veige
*ab
a,
Rinku
Yadav
a,
Parker T.
Boeck
ab,
Ion
Ghiviriga
ab,
ChristiAnna L.
Brantley
ab,
Łukasz
Dobrzycki
ab and
Adam S.
Veige
*ab
aUniversity of Florida, Department of Chemistry, Center for Catalysis, P.O. Box 117200, Gainesville, FL 32611, USA. E-mail: veige@chem.ufl.edu
bUniversity of Florida, Department of Chemistry, George and Josephine Butler Polymer Research Laboratory, Center for Macromolecular Sciences and Engineering, Gainesville, FL 32611, USA
First published on 30th August 2024
Abstract
Cyclooctyne reacts with the trianionic pincer ligand supported alkylidyne [tBuOCO]WCC(CH3)3(THF)2 (1) to yield tungstacyclopropene (3) and tungstacyclopentadiene (4) complexes. The ratio of 3 and 4 in the reaction mixture depends on the stoichiometry of the reaction. The maximum concentration of 3 occurs with one equiv. of cyclooctyne and 4 is the exclusive product of the reaction above three equivalents. Both complexes 3 and 4 convert to the cyclooctyne ring-opened product 5 upon heating. While the conversion of 4 to 5 is accompanied by formation of polycyclooctyne as a white precipitate during the reaction, conversion of 3 to 5 is homogeneous. Exhibiting Ring Expansion Polymerization (REP), complexes 4 and 5 initiate the polymerization of phenylacetylene to generate cyclic poly(phenylacetylene) (c-PPA).
Cyclic polymers do not have chain ends1 and have notably different physical, biological, and chemical properties compared to their linear analogues with similar molar mass. Cyclic polymers exhibit higher glass transition temperature (Tg),2–4 smaller radii of gyration,5,6 lower intrinsic viscosity,7,8 and higher thermal stability.9,10 Changing polymer topology imparts a significant difference in biological properties. For instance, cyclic polymers exhibit higher tumor penetration ability,11–13 cytotoxicity,14 and nucleic acid delivery.12 When grafted on a surface, cyclic polymers have lower friction coefficients,15,16 smaller nanostructure domain spacing,17 and higher surface annealing rates.18 Increased nucleation density and crystallization rates were measured for cyclic polymers.19 Enhanced conjugation in cyclic polymers also affects photophysical properties including higher fluorescence emission and excited state-lifetime,20 and higher refractive index.21 Conventional synthetic routes to obtain cyclic polymers rely on coupling chain ends of linear polymers.22,23 Coupling chain ends requires multiple synthetic steps, is laborious, and often requires dilute conditions though improved approaches are emerging.24–26
In 2016, we reported the trianionic pincer27–30 alkylidyne complex 1 (ref. 31–33) transforms to the tethered tungsten alkylidene complex 2 (Scheme 1) when treated with 4,4-dimethyl-but-1-yne.34 Complex 2 rapidly initiates ring expansion polymerization (REP) of alkynes to generate cyclic polyenes in excellent yields with diverse functional groups.35
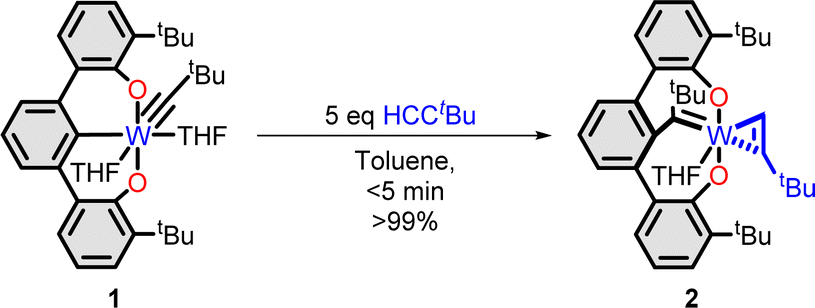 | ||
| Scheme 1 Reported synthesis of REP initiator 2. | ||
The REP mechanism, introduced by Lee and Kricheldorf using a dibutyltin catalyst in 1995,36 involves repeated monomer insertions into a growing ring.37–41 New REP initiators and an ever-expanding list of monomers are appearing in the literature.37,42 The initial proposal for ring formation with complex 2 was published in 2016 (Fig. 1). In that work, because complex 2 does not undergo metathesis with preformed cyclic polyphenylacetylene, the tethered alkylidene was not invoked as the site of ring expansion and only played the role of ancillary ligand. Instead, the mechanism implied sequential insertions to grow a metallacycle where an intermediate 2b (Fig. 1) forms and then eliminates the cyclic polymer in a reductive elimination step.34 Support at the time, and since, for 2b as an intermediate in the polymerization came from the isolation of several ring expanded metallacyclopentadiene species.43–45 The extreme rates (1.8 × 108 g mol−1 h−1)46 recorded for polymerizations initiated by 1 and 2 make it difficult to ascertain the mechanism of alkyne REP.
 | ||
| Fig. 1 Original proposed REP pathway for the synthesis of cyclic polyenes. | ||
In this work, a snapshot of cyclooctyne (CyO) ring-opening across the tethered alkylidene of 2 provides new insight into the possible mechanism and suggests the tethered alkylidene may participate in the REP mechanism after all. Complex 2 rapidly polymerizes small non-coordinating monosubstituted alkynes but does not react with bulky or disubstituted alkynes at room temperature.35 A simple strategy to trap an intermediate in such a case is to hinder propagation with an appropriately bulky substrate. Metallacycopentadiene trapping events employ a similar strategy; however, those reactions require elevated temperatures (75 °C) to induce insertion.43 It is possible to miss reactive intermediates at high temperatures. Ideally, the reaction probe uses relatively lower temperatures to observe elusive intermediates. CyO is disubstituted, thus bulky enough to inhibit propagation but also significantly strained, making initial insertions favorable. Therefore, CyO was chosen for this study resulting in the first isolation of a complex featuring a ring opened CyO.
Treating a dark red benzene solution of 1 with CyO results in an immediate color change to amber and finally to olive green. The reaction between 1 and CyO simultaneously yields tungstacyclopropene (3) and tungstacyclopentadiene (4) tethered alkylidene complexes (Scheme 2). Complex 4 forms exclusively when combining 2.5 (or above) equivalents of CyO and 1. However, 3 always coexists with 4 in solution even at sub-stoichiometric additions. Table 1 and Fig. 2 depict the relationship between the relative amounts of complexes 1, 3, and 4 that form in benzene solution vs. equiv. of CyO added; the maximum concentration of 3 occurs with 1 equiv. It is important to notice that insertion of CyO in 1 is rapid leading to a mixture of either 3 and 4, or solely 4. Therefore, it is impossible to exclusively obtain 3 from 1. Exposing 1 to CyO in the presence of excess THF does favor the formation of 3. For example, addition of one equiv. of CyO in the presence of 10 equiv. of THF in benzene results in a relative ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8:2 of complexes 1:3:4, respectively. Performing the same reaction with THF as solvent does not improve the outcome in favor of 3.
:8:2 of complexes 1:3:4, respectively. Performing the same reaction with THF as solvent does not improve the outcome in favor of 3.
 | ||
| Scheme 2 Synthesis of complexes 3 and 4. | ||
| CyO | 1 | 3 | 4 |
|---|---|---|---|
| 0.25 | 83 | 11 | 6 |
| 0.58 | 60 | 22 | 18 |
| 1.11 | 27 | 35 | 38 |
| 1.52 | 13 | 34 | 53 |
| 1.97 | 0 | 3 | 97 |
| 2.50 | 0 | 0 | 100 |
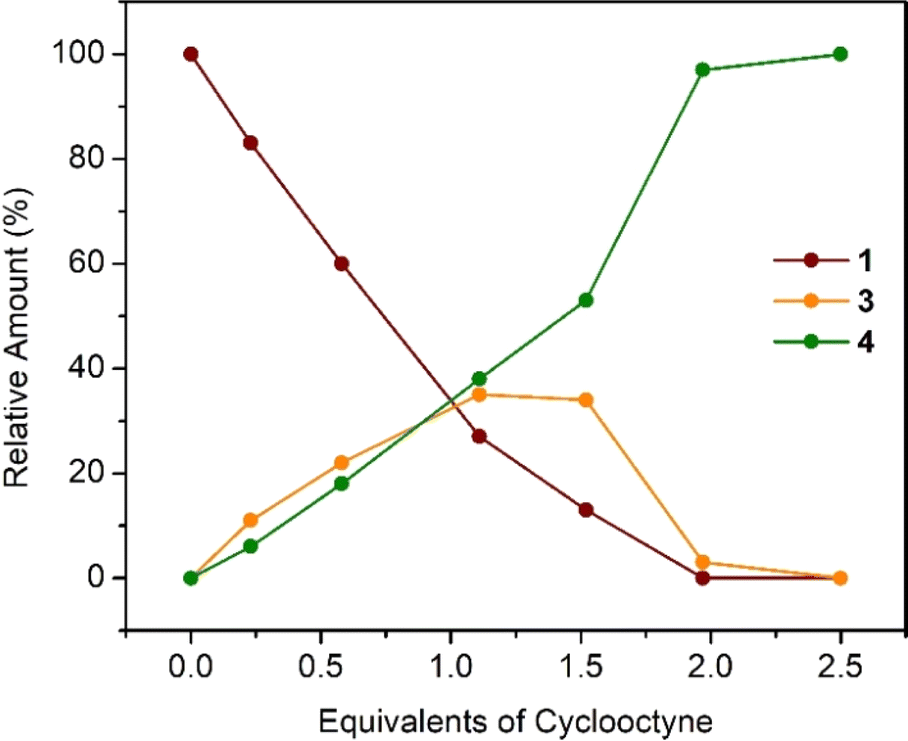 | ||
| Fig. 2 Yield (%) of 1, 3, and 4vs. CyO equivalents. | ||
Complex 3 co-crystallizes as light orange crystals with dark olive green crystals of 4 from a concentrated Et2O solution of 1:3:4 in ratio 7:8:2, respectively, within several days at −30 °C. Consequently, it is not possible to obtain pure 3 in large quantities. Single crystals of 4 deposit exclusively from a concentrated Et2O solution of 4 at −30 °C overnight. Single crystal X-ray diffraction experiments provide unambiguous assignment of 3 as a tungstacyclopropene and 4 as a tungstacyclopentadiene (Fig. 3); Table 2 lists selected bond lengths and angles. Both 3 and 4 are pseudo-Cs symmetric with the plane of symmetry bisecting the OCO pincer ligand and W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond.
C bond.
 | ||
| Fig. 3 Synthesis of complex 5 and crystal structures of 3, 4, and 5. | ||
| 3 | 4 | 5 | |
|---|---|---|---|
| W1–C27 | 1.904(2) | 1.908(6) | 1.895(2) |
| W1–C32 | 2.043(2) | 2.071(9) | — |
| W1–C38 | — | — | 2.022(2) |
| W1–C39 | 2.027(2) | — | 2.037(2) |
| C32–C39 | 1.309(4) | 1.381(6) | — |
| C38–C39 | — | — | 1.316(3) |
Complex 3 consists of a formally W(VI) ion that diverges from a conventional polyhedral geometry. The W1–C27 bond length of 1.904(2) Å resembles previously reported W(VI) alkylidene complexes; for example, 2 has a W1–C27 bond length of 1.9021(19) Å.34 The W1–C32 (2.043(2) Å), W1–C39 (2.027(2) Å) and C32–C39 (1.309(4) Å) bond lengths are statistically similar to reported η2-bound W-alkyne complexes (Table S4†).34,47–50 An elongated W1–O3 bond length of 2.3003(17) Å implies that the alkylidene exerts a strong trans influence on bound THF. Solution state NMR studies indicate 3 retains Cs symmetry in solution. The 1H NMR spectrum of 3 in benzene-d6 exhibits two singlets at 0.89 and 1.26 ppm in a relative integration ratio of 1:2 for the alkylidene-tBu and aryl-tBu protons, respectively. A 13C{1H} spectrum of 3 reveals a resonance at 263.4 ppm for the alkylidene carbon.
For complex 4, an Addison parameter value τ of 0.53 suggests that the ligand geometry around the formally W(VI) ion is between square pyramidal and trigonal bipyramidal.51 The W1–C32 (2.071(9) Å), W1–C47 (2.080(7) Å), C32–C39 (1.381(6) Å) and C40–C47 (1.370(6) Å) bond lengths in 4 are all similar to reported tungstacyclopentadiene complexes.43,52–54 Differences in the bond lengths and angles of the metallacyclopentadiene ring are likely attributable to the ring strain of the CyO substituents; however, the alkylidene is statistically the same bond length as that of a previously reported metallacyclopentadiene complex (Table S5†). Complex 4 also retains Cs symmetry in solution. The 1H NMR spectrum of 4 in benzene-d6 exhibits two distinct singlets at 0.98 ppm and 1.55 ppm with a relative integration of 1:2 for the alkylidene-tBu and pincer-tBu protons, respectively. A resonance for the alkylidene carbon appears at 311.3 ppm in the 13C NMR spectrum. Although, it is unusual for an alkylidene carbon to resonate so far downfield, a similar signal was observed for other tungstacyclopentadienes.43
Complexes 3 and 4 form as the result of a migratory insertion of the alkylidyne carbon into the tungsten arene bond of the pincer ligand. The migratory insertion event is a distinctive property of 1 and occurs with various substrates with high rates.34,43,50,55–59 The ring strain of cyclooctyne (∼18 kcal mol−1)60–63 must play a role in the swift insertion to give 4 since disubstituted alkynes require heating to induce metallacylopentadiene formation.
According to the originally proposed REP mechanism (Fig. 1), successive addition of alkyne monomers in the metallacyclopropene ring generates a cyclic polymer. Perhaps signifying an alternative mechanism, isolating a metallacycloheptatriene by adding additional alkyne equivalents was not successful. However, 4 initiates the polymerization of phenylacetylene even at −30 °C. Monitoring a 1:1 (4:phenylacetylene) reaction by 1H NMR spectroscopy reveals the alkyne directly converts into polymer without showing any detectable signals of an intermediate species (Fig. S12†). As is typical for these polymerizations a significant amount of unreacted 4 remains during the polymerization, indicative of slow initiation and rapid propagation kinetics. Exposing 4 to excess CyO leads to an insoluble white cyclooctyne polymer. Consistent with reported literature, polycyclooctyne is insoluble in THF, toluene, o-dichlorobenzene, and other common organic solvents consequently preventing its characterization.64,65 The IR spectrum of the white precipitate does not exhibit a C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretch (Fig. S39†). Instead, it exhibits a CC stretch at 1440 cm−1 indicating the conversion of CyO to poly-CyO.
C stretch (Fig. S39†). Instead, it exhibits a CC stretch at 1440 cm−1 indicating the conversion of CyO to poly-CyO.
Providing insight into the role of the tethered alkylidene and its ability to engage in metathesis with alkynes, heating 3 at 80 °C for 30 h provides the cyclooctyne ring opened product 5 (Fig. 3). A similar transformation occurs in the case of 4 as well, but it is slower and requires 120 °C to complete in the same time frame. The color of the solution changes from orange (3) or olive green (4) to red orange (5) during these transformations. The formation of 5 from 3 and 4 at elevated temperatures indicates that 5 is the thermodynamic product from the overall reaction between 1 and CyO. Conversion of 3 to 5 occurs homogeneously, whereas conversion of 4 to 5 is accompanied with the formation of a white precipitate. The white precipitate is insoluble in common organic solvents leading to the conclusion that it is also polycyclooctyne, that results from shedding one equivalent of cyclooctyne.
Single crystals suitable for X-ray data collection accumulate from a concentrated Et2O solution of 5 at −30 °C within several days. Fig. 3 depicts the solid-state structure of 5. Similar to 3, 5 also contains a tungstacyclopropene, belongs to Cs symmetry in the solid-state, and exhibits analogous structural features (Table 2). A noticeable and unexpected structural difference among 3, 4, and 5 is a contracted alkylidene bond in 5 (1.895(2) Å), presumably due to the tether between the alkylidene and η2-bound alkyne. Additional evidence suggesting the tether alters the alkylidene is the bond angle between the alkylidene and the adjacent carbon of the bound alkyne is considerably smaller for 5 by 5.64(14)°.
The coordinated Et2O, from solvent, is weakly bound in 5 (2.300 (17) Å) akin to THF in 3 (2.390 (17) Å). Key to the structure is the unambiguous presence of a tether between the alkylidene and the η2-bound alkyne. Evidence of the tether in solution comes from 1H, 13C, HSQC, HMBC, and COSY NMR data (see ESI† for spectra).
Akin to their parent complexes 1, 4 and 5 polymerize phenylacetylene. In a typical polymerization reaction, the required amount of a stock solution of pure initiator 4 or 5 was added to a stirring solution of freshly dried and distilled phenylacetylene in toluene. As the reaction stirs, the color of the solution changes from green to deep red. Reactions using 4 as the catalyst were stirred for 12 h, while reactions with 5 were stirred for 3 h (Fig. 4, Table 3). Adding the reaction mixture dropwise into oxygen free methanol precipitates the polymer. Precipitation of polyenes requires oxygen free methanol as conjugated polyenes degrade via oxidation.35 The polymerization experiments were conducted in triplicate for 4 and 5, (see ESI†).
 | ||
| Fig. 4 Polymerization of phenylacetylene to generate cyclic polyphenylacetylene (c-PPA) with 4 and 5 as catalysts. | ||
For comparison, linear polyphenylacetylene (l-PPA) was prepared according to literature procedures using (acetylacetonato)(1,5-cyclooctadiene)rhodium(I) [acac(Rh(I)cod)]. Importantly, PPA samples prepared with 4, 5, and [acac(Rh(I)cod)]66,67 contain >90% cis double bonds, as determined by NMR spectroscopy (Fig. S30–S35†).
Cyclic polymers exhibit distinct physical properties compared to linear polymers of comparable molecular weight, enabling the determination of their topology by size exclusion chromatography (SEC) coupled to viscometer measurements and static light scattering techniques.68–71Fig. 5 depicts a plot of the log of molar mass versus retention time for l-PPA and c-PPA synthesized by 4. It is clear from the graph that PPA synthesized by 4 elutes later than l-PPA at a given molar mass, consistent with a cyclic topology. Fig. 6 depicts a plot of root mean square radii of gyration vs. log of molar mass. Again, it is apparent from the plot that the radius of gyration of PPA produced from complex 4 is smaller than a known linear analogue at a given molar mass.9 PPA synthesized using 5 also has the same relationship to the linear version indicating a cyclic topology (Fig. S37 and S38†).
 | ||
| Fig. 5 Log (molar mass(g mol−1)) versus retention time graph for c-PPA (red) synthesized by 4 and linear polyphenylacetylene (l-PPA, black). | ||
 | ||
| Fig. 6 Root mean square radii of gyration versus Log (molar mass(g mol−1)) graph for c-PPA (red) synthesized by 4 and linear poly-phenylacetylene (l-PPA, black). | ||
Discussion
Cyclooctyne instantaneously reacts with 1 at room temperature. The outcome of the reaction is heavily dependent on the stoichiometry. Surprisingly, the expected tungstacyclopropene 3, is not a major product of the reaction especially at higher equivalents of cyclooctyne. Unlike previously reported tungstacyclopropene complexes,433 does not establish a detectable equilibrium with its metathesis isomer 5 at ambient temperature. For example, alkylidyne 1 reacts with 1 equiv. of 1-phenyl-1-propyne to yield two metathesis isomers, 6a and 6b in a 1:2 ratio respectively (Scheme 3).43 Presumably, isomerization proceeds through a traditional metallacylcobutadiene intermediate by cleavage of the C–C bond to the pincer backbone. Published examples indicate the C–C bond cleavage is surprisingly facile in this pincer framework and is a hallmark of the high reactivity. For 3, due to the cyclic nature of CyO, forming a metallacyclobutadiene intermediate followed by metathesis yields the ring opened product as the tethered alkylidene 5.
 | ||
| Scheme 3 Proposed role of metallacyclobutadiene in tethered alkylidene formation. | ||
In an alternative route, heating 4 also produces the tethered alkylidene 5 presumably by first expelling an equiv. of cyclooctyne to revert to 3 followed by metathesis as in Scheme 3. Monitoring the conversion of 4 to 5via1H NMR spectroscopy does not reveal any detectable signals or buildup of 3.
Conclusions
Alkylidyne 1 is an initiator for the polymerization of alkynes to give cyclic polymers by first transforming into tethered alkylidene with a bound alkyne according to Scheme 1. A previously proposed mechanism34 neglected the potential role of the tethered alkylidene in the propagation of the growing ring. The main conclusion from this work is the isolation of and existence of 5 demonstrates that the tether is a reasonable composition for possible active species in both ring expansion metathesis polymerization (REMP) of alkynes and cyclic alkenes such as norbornene. Complexes 4 and 5 polymerize phenylacetylene to give cyclic polymers. Though much work remains to elucidate other intermediates in the polymerization, isolation of a tethered alkylidene within 5, at least from a compositional standpoint, offers new insight. Complex 5 is also a snapshot of a ring-opened cyclic alkyne via metathesis, an elusive intermediate until now.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
JMH carried out all experiments. CLB, RY, and LD collected and refined the X-ray data. IG assisted in NMR characterizations. PTB characterized the polymer samples. RY and JMH drafted the manuscript. RY, JMH, ASV, and PTB revised the manuscript. ASV and RY supervised the work. All authors have given approval to the final version of the manuscript.Conflicts of interest
ASV has an equity position in Oboro Labs Inc.Acknowledgements
This material is based upon work supported by the National Science Foundation CHE-2154377 (A. S. V.). The GC/EI-MS instrument is funded by NIH S10 OD021758-01A1. LD acknowledges the NSF (CHE-1828064) and UF for the purchase of X-ray equipment.Notes and references
- Z. Jia and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2085–2097 CrossRef CAS.
- D. J. Bannister and J. A. Semlyen, Polymer, 1981, 22, 377–381 CrossRef CAS.
- S. J. Clarson, K. Dodgson and J. A. Semlyen, Polymer, 1985, 26, 930–934 CrossRef CAS.
- L. Gao, J. Oh, Y. Tu, T. Chang and C. Y. Li, Polymer, 2019, 170, 198–203 CrossRef CAS.
- J. Roovers, J. Polym. Sci., 1985, 23, 1117–1126 CAS.
- T. E. Gartner, F. M. Haque, A. M. Gomi, S. M. Grayson, M. J. A. Hore and A. Jayaraman, Macromolecules, 2019, 52, 4579–4589 CrossRef CAS.
- Y. Jeong, Y. Jin, T. Chang, F. Uhlik and J. Roovers, Macromolecules, 2017, 50, 7770–7776 CrossRef CAS.
- R. Liénard, J. De Winter and O. Coulembier, J. Polym. Sci., 2020, 58, 1481–1502 CrossRef.
- B. H. Zimm and W. H. Stockmayer, J. Chem. Phys., 1949, 17, 1301–1314 CrossRef CAS.
- R. Zhang, M. M. Billingsley and M. J. Mitchell, J. Controlled Release, 2018, 292, 256–276 CrossRef CAS PubMed.
- C. E. Wang, H. Wei, N. Tan, A. J. Boydston and S. H. Pun, Biomacromolecules, 2016, 17, 69–75 CrossRef CAS PubMed.
- B. Chen, K. Jerger, J. M. J. Fréchet and F. C. Szoka, J. Controlled Release, 2009, 140, 203–209 CrossRef CAS PubMed.
- B. Golba, E. M. Benetti and B. G. De Geest, Biomaterials, 2021, 267, 120468 CrossRef CAS PubMed.
- N. Nasongkia, B. Chen, N. Macaraeg, M. E. Fox, J. M. J. Fréchet and F. C. Szoka, J. Am. Chem. Soc., 2009, 131, 3842–3843 CrossRef PubMed.
- G. Morgese, L. Trachsel, M. Romio, M. Divandari, S. N. Ramakrishna and E. M. Benetti, Angew. Chem., Int. Ed., 2016, 55, 15583–15588 CrossRef CAS PubMed.
- A. Patel, T. Cosgrove and J. A. Semlyen, Polymer, 1991, 32, 1313–1317 CrossRef CAS.
- A. D. Goodson, J. E. Troxler, M. S. Rick, H. S. Ashbaugh and J. N. L. Albert, Macromolecules, 2019, 52, 9389–9397 CrossRef CAS.
- L. Wang, L. Xu, B. Liu, T. Shi, S. Jiang and L. An, Soft Matter, 2017, 13, 3091–3098 RSC.
- N. Zaldua, R. Liénard, T. Josse, M. Zubitur, A. Mugica, A. Iturrospe, A. Arbe, J. De Winter, O. Coulembier and A. J. Müller, Macromolecules, 2018, 51, 1718–1732 CrossRef CAS.
- X. Zhu, N. Zhou, Z. Zhang, B. Sun, Y. Yang, J. Zhu and X. Zhu, Angew. Chem., Int. Ed., 2011, 50, 6615–6618 CrossRef CAS PubMed.
- D. J. Orrah, J. A. Semlyen and S. B. Ross-Murphy, Polymer, 1988, 29, 1455–1458 CrossRef CAS.
- D. Pasini, Molecules, 2013, 18, 9512–9530 CrossRef CAS PubMed.
- K. Pangilinan and R. Advincula, Polym. Int., 2014, 63, 803–813 CrossRef CAS.
- H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 251–284 CrossRef CAS.
- M. Kapnistos, M. Lang, D. Vlassopoulos, W. Pyckhout-Hintzen, D. Richter, D. Cho, T. Chang and M. Rubinstein, Nat. Mater., 2008, 7, 997–1002 CrossRef CAS PubMed.
- W. Lee, H. Lee, H. C. Lee, D. Cho, T. Chang, A. A. Gorbunov and J. Roovers, Macromolecules, 2002, 35, 529–538 CrossRef CAS.
- J. Koller, S. Sarkar, K. A. Abboud and A. S. Veige, Organometallics, 2007, 26, 5438–5441 CrossRef CAS.
- S. Sarkar, K. P. McGowan, J. A. Culver, A. R. Carlson, J. Koller, A. J. Peloquin, M. K. Veige, K. A. Abboud and A. S. Veige, Inorg. Chem., 2010, 49, 5143–5156 CrossRef CAS PubMed.
- S. S. Nadif, M. E. O'Reilly, I. Ghiviriga, K. A. Abboud and A. S. Veige, Angew. Chem., Int. Ed., 2015, 54, 15138–15142 CrossRef CAS PubMed.
- M. E. O'Reilly and A. S. Veige, Chem. Soc. Rev., 2014, 43, 6325–6369 RSC.
- S. Sarkar, K. P. McGowan, S. Kuppuswamy, I. Ghiviriga, K. A. Abboud and A. S. Veige, J. Am. Chem. Soc., 2012, 134, 4509–4512 CrossRef CAS.
- S. Kuppuswamy, A. J. Peloquin, I. Ghiviriga, K. A. Abboud and A. S. Veige, Organometallics, 2010, 29, 4227–4233 CrossRef CAS.
- S. Sarkar, A. R. Carlson, M. K. Veige, J. M. Falkowski, K. A. Abboud and A. S. Veige, J. Am. Chem. Soc., 2008, 130, 1116–1117 CrossRef CAS PubMed.
- C. D. Roland, H. Li, K. A. Abboud, K. B. Wagener and A. S. Veige, Nat. Chem., 2016, 8, 791–796 CrossRef CAS PubMed.
- P. T. Boeck, R. Yadav, B. S. Sumerlin and A. S. Veige, Macromolecules, 2024, 57, 71–77 CrossRef.
- H. R. Kricheldorf and S. R. Lee, Macromolecules, 1995, 28, 6718–6725 CrossRef CAS.
- Y. A. Chang and R. M. Waymouth, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2892–2902 CrossRef CAS.
- M. Ouchi, H. Kammiyada and M. Sawamoto, Polym. Chem., 2017, 8, 4970–4977 RSC.
- B. A. Laurent and S. M. Grayson, Chem. Soc. Rev., 2009, 38, 2202–2213 RSC.
- R. Tuba, Pure Appl. Chem., 2014, 86, 1685–1693 CAS.
- M. Ahmad and S. M. Grayson, Mass Spectrom. Rev., 2024, 1–29 Search PubMed.
- T. Kubo, R. Yadav and A. S. Veige, Topological Polymer Chemistry: Concepts and Practices, Springer Nature, 2022, pp. 261–275 Search PubMed.
- K. P. Mcgowan, M. E. O'Reilly, I. Ghiviriga, K. A. Abboud and A. S. Veige, Chem. Sci., 2013, 4, 1145–1155 RSC.
- A. M. Esper, PhD Thesis, University of Florida, 2023 Search PubMed.
- C. D. Roland, T. Zhang, S. Venkatramani, I. Ghiviriga and A. S. Veige, Chem. Commun., 2019, 55, 13697–13700 RSC.
- Z. Miao, D. Pal, W. Niu, T. Kubo, B. S. Sumerlin and A. S. Veige, Macromolecules, 2020, 53, 7774–7782 CrossRef CAS.
- S. M. Holmes, D. F. Schafer, P. T. Wolczanski and E. B. Lobkovsky, J. Am. Chem. Soc., 2001, 123, 10571–10583 CrossRef CAS PubMed.
- T. W. Hayton, J. M. Boncella, B. L. Scott, K. A. Abboud and R. C. Mills, Inorg. Chem., 2005, 44, 9506–9517 CrossRef CAS PubMed.
- R. J. Beattie, P. S. White and J. L. Templeton, Organometallics, 2016, 35, 32–38 CrossRef CAS.
- R. Yadav, I. Ghiviriga, K. K. Abboud and A. S. Veige, Chem. Commun., 2023, 59, 12899–12902 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- M. H. Chisholm, K. Folting, D. M. Hoffman, J. C. Huffman and J. Leonelli, J. Chem. Soc., Chem. Commun., 1983, 589–591 RSC.
- I. M. Bartlett, N. G. Connelly, M. S. Legge, A. J. Martin, B. Metz and A. G. Orpen, Chem. Commun., 1996, 2, 1877–1878 RSC.
- L. J. Canoira, J. L. Davidson, G. Douglas and K. W. Muir, J. Organomet. Chem., 1989, 362, 135–146 CrossRef CAS.
- S. A. Gonsales, T. Kubo, M. K. Flint, K. A. Abboud, B. S. Sumerlin and A. S. Veige, J. Am. Chem. Soc., 2016, 138, 4996–4999 CrossRef CAS PubMed.
- S. S. Nadif, T. Kubo, S. A. Gonsales, S. Venkatramani, I. Ghiviriga, B. S. Sumerlin and A. S. Veige, J. Am. Chem. Soc., 2016, 138, 6408–6411 CrossRef CAS PubMed.
- V. K. Jakhar, A. M. Esper, I. Ghiviriga, K. A. Abboud, C. Ehm and A. S. Veige, Angew. Chem., Int. Ed., 2022, 61, e202203073 CrossRef CAS PubMed.
- V. Jakhar, D. Pal, I. Ghiviriga, K. A. Abboud, D. W. Lester, B. S. Sumerlin and A. S. Veige, J. Am. Chem. Soc., 2021, 143, 1235–1246 CrossRef CAS PubMed.
- R. Yadav, A. M. Esper, I. Ghiviriga, K. A. Abboud, W. Lester, C. Ehm and A. S. Veige, ChemCatChem, 2024, e202301678 CrossRef CAS.
- R. D. Bach, J. Am. Chem. Soc., 2009, 131, 5233–5243 CrossRef CAS PubMed.
- T. Harris and I. V. Alabugin, Mendeleev Commun., 2019, 29, 237–248 CrossRef CAS.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- G. De Almeida, E. M. Sletten, H. Nakamura, K. K. Palaniappan and C. R. Bertozzi, Angew. Chem., Int. Ed., 2012, 51, 2443–2447 CrossRef CAS PubMed.
- T. J. Katz and S. J. Lee, J. Am. Chem. Soc., 1980, 102, 422–424 CrossRef CAS.
- K. Yamada, R. Nomura and T. Masuda, Macromolecules, 2000, 33, 9179–9181 CrossRef CAS.
- P. Mastrorilli, F. Nobile, A. Rizzuti, G. P. Suranna, D. Acierno and E. Amendola, J. Mol. Catal. A: Chem., 2002, 178, 35–42 CrossRef CAS.
- O. Trhlíková, J. Zedník, H. Balcar, J. Brus and J. Sedláček, J. Mol. Catal. A: Chem., 2013, 378, 57–66 CrossRef.
- C. W. Bielawski, D. Benitez and R. H. Grubbs, Science, 2002, 297, 2041–2044 CrossRef CAS PubMed.
- W. Niu, S. A. Gonsales, T. Kubo, K. C. Bentz, D. Pal, D. A. Savin, B. S. Sumerlin and A. S. Veige, Chem, 2019, 5, 237–244 CAS.
- Z. Miao, T. Kubo, D. Pal, B. S. Sumerlin and A. S. Veige, Macromolecules, 2019, 52, 6260–6265 CrossRef CAS.
- J. N. Hoskins and S. M. Grayson, Polym. Chem., 2011, 2, 289–299 RSC.
Footnote |
| † CCDC 2360874–2360876. For crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04411h |
| This journal is © The Royal Society of Chemistry 2024 |