
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced catalytic activity of solubilised species obtained by counter-cation exchange of K{CoII1.5[FeII(CN)6]} for water oxidation†
Yusuke
Seki
a,
Takashi
Nakazono
 b,
Hiroyasu
Tabe
c and
Yusuke
Yamada
*ab
b,
Hiroyasu
Tabe
c and
Yusuke
Yamada
*ab
aChemistry and Bioengineering, Graduate School of Engineering, Osaka Metropolitan University, Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: ymd@omu.ac.jp
bResearch Center for Artificial Photosynthesis, Osaka Metropolitan University, Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan
cInstitute for Integrated Cell-Material Sciences, Institute for Advanced Study, Kyoto University, Yoshida-Honmachi, Sakyo-ku, Kyoto 606-8501, Japan
First published on 23rd September 2024
Abstract
A cyano-bridged coordination polymer, K{CoII1.5[FeII(CN)6]} {(K)Co–Fe}, reported as a highly active heterogeneous catalyst for water oxidation was solubilised by a conventional counter-cation exchange of K+ with Me4N+ ions to provide the homogeneous catalyst of (Me4N){CoII1.5[FeII(CN)6]} {(Me4N)Co–Fe}. (Me4N)Co–Fe exhibited enhanced catalytic activity for photocatalytic water oxidation using [Ru(2,2′-bipyridine)3]2+ and S2O82− as a photosensitiser and a sacrificial electron acceptor, respectively, in terms of the initial reaction rate (1.26 μmol min−1), which is about twice that of (K)Co–Fe (0.61 μmol min−1). Powder X-ray diffraction, pair distribution function and electrospray ionization mass spectrometry measurements of (Me4N)Co–Fe manifested that small heptanuclear clusters of {Co4[Fe(CN)6]3}4− formed by depolymerisation are catalytically active species in solution.
Introduction
Water splitting utilising solar energy is a promising method to produce hydrogen as a clean fuel, contributing to realising sustainable society. Photocatalytic systems require at least three components, i.e., water reduction and oxidation catalysts and a photosensitiser for light harvesting and charge separation.1–8 Among the three components, development of water oxidation catalysts (WOCs) should be accelerated because water oxidation involving four-electrons and four-protons transfer is the most sluggish.9–15 So far, various types of heterogeneous and homogeneous catalysts have been examined as WOCs.16–26 In general, heterogeneous catalysts are advantageous for robustness compared with homogeneous ones; however, rational insight into the reaction mechanism at the atomic level is hardly achievable, especially identification of catalytically active species is almost unfruitful because of their insoluble nature. Solubilisation of heterogeneous catalysts without losing catalytic activity is beneficial to study reaction mechanisms at the atomic level.Recently, cyano-bridged coordination polymers (CPs) containing Co ions have been reported as highly active heterogeneous catalysts for photocatalytic and electrocatalytic water oxidation.27–37 Cyano-bridged CPs are insoluble solids; however, surface modification with hexacyanometallate enables achieving high dispersibility in water because of negatively charged surfaces.38–41 Also, the coordination of macrocyclic extra ligands to Co ions, such as Na4{[CoIII(Me3-Tacn)]4[FeII(CN)6]4} (Me3-Tacn = 1,4,7-trimethyl-1,4,7-triazacyclononane), succeeded in constructing discrete structures, resulting in high dispersion in water or an organic solvent.42–47 Such solubilised cyano-bridged CPs can be subjected to various analyses usually used for homogeneous systems. However, these methods have rarely been applied to cyano-bridged CPs acting as heterogeneous catalysts,48–51 because the modifications may change the surface conditions including active-site structures of heterogeneous catalysts.
In this context, counter-cation exchange would be a promising method to achieve high dispersibility without changing the surface conditions of cyano-bridged CPs. For example, the most typical cyano-bridged CP of FeIII4[FeII(CN)6]3 known as Prussian blue is insoluble in water; however, K{FeIII[FeII(CN)6]} occluding counter cations inside pores is highly soluble in water.52,53 However, K{CoII1.5[FeII(CN)6]} {(K)Co–Fe} showing high catalytic activity for water oxidation is insoluble in water although its structure is similar to that of K{FeIII[FeII(CN)6]}. In general, choice of counter cations highly affects the solubility of metal complexes so that cation exchange of K+ ions in (K)Co–Fe with larger ions can enhance the solubility.
We report herein the solubilisation of (K)Co–Fe with enhanced catalytic activity for water oxidation by the counter-cation exchange of K+ with Me4N+ ions to form (Me4N){CoII1.5[FeII(CN)6]} {(Me4N)Co–Fe}. Photocatalytic water oxidation conducted by photoirradiation of a phosphate buffer (pH 8.0) containing a cyano-bridged CP, [RuII(bpy)3]2+ ([RuII(2,2′-bipyridine)3]2+), and S2O82− indicated that the catalytic activity was enhanced after solubilisation in terms of the initial reaction rate and O2 yield. The solid structure of (Me4N)Co–Fe was compared with that of (K)Co–Fe based on powder X-ray diffraction (PXRD) and the pair distribution function (PDF). Additionally, electrospray ionization mass spectrometry (ESI-MS) was performed to identify the water-soluble species achieved after counter-cation exchange.
Experimental section
Materials
All chemicals were used as supplied without further purification. Aqueous solutions were prepared with ultrapure water provided using a Barnstead Smart2Pure water purification system (Thermo Scientific, US) where the electronic conductance was 18.2 MΩ cm. Cobalt(II) nitrate hexahydrate, silver(I) sulphate, potassium hexacyanoferrate(II) trihydrate, disodium hydrogen phosphate, sodium dihydrogen phosphate, sulphuric acid, and sodium persulphate were purchased from FUJIFILM-Wako Pure Chemical Industries Corporation. Tris(2,2′-bipyridine)ruthenium chloride ([Ru(bpy)3]Cl2) and tetramethylammonium chloride were purchased from Tokyo Chemical Industry Co., Ltd. Lead(IV) oxide was purchased from Kanto Chemical Co., Inc. Tris(2,2′-bipyridine)ruthenium sulphate ([Ru(bpy)3]SO4) was synthesised by adding one equivalent of Ag2SO4 to an aqueous solution of [Ru(bpy)3]Cl2 (0.13 mmol, 10 mL).54 The stock solution of [RuIII(bpy)3]3+ was prepared by oxidising [RuII(bpy)3]2+ with PbO2 in H2SO4 aq.21 The solution was kept in the dark under an Ar atmosphere. The molar concentrations of the solution containing a cyano-bridged CP were calculated based on the moles of Fe.Synthesis of K{CoII1.5[FeII(CN)6]} {(K)Co–Fe}
An aqueous solution of potassium hexacyanoferrate(II) (K4[FeII(CN)6], 100 mM, 1.5 mL) was slowly added to an aqueous solution of cobalt(II) nitrate (CoII(NO3)2, 300 mM, 1.5 mL). The formed precipitates were collected by centrifugation and washed with distilled water a couple of times. The precipitates were dried in vacuo for 12 h.Synthesis of water-dispersible (K)Co–Fe modified with [FeII(CN)6]4− (ref. 38)
(K)Co–Fe (0.4 mmol, 157 mg) was dispersed in an aqueous solution containing K4[FeII(CN)6] (60 mM, 2.0 mL). Stirring the dispersion for 3 days provided water-dispersible (K)Co–Fe. The water-dispersible (K)Co–Fe modified with [FeII(CN)6]4− was centrifugally collected for PXRD and TEM measurements.Synthesis of (Me4N){CoII1.5[FeII(CN)6]} {(Me4N)Co–Fe}
An aqueous dispersion of (K)Co–Fe (50 mg) containing Me4NCl (500 mM, 5 mL) was stirred at 50 °C for 20 h to achieve precipitates with a blight green supernatant. The precipitates collected by centrifugation were washed with a small amount of water once and then with methanol three times. The obtained precipitates were dried in vacuo for 12 h. Yield 25 mg (46%). Anal. calcd. for C10.8H18.4N7.2O2K0.1Cl0.1Co1.4Fe [(Me4N)1.2{Co1.4[Fe(CN)6]}·(H2O)2·(KCl)0.1]: C, 30.38: H, 4.34: N, 23.63. Found C, 30.21: H, 4.10: N, 23.81.Physical measurements
The atomic ratios of (K)Co–Fe and (Me4N)Co–Fe were determined by X-ray fluorescence (XRF) measurements using a Malvern PANalytical Epsilon 1. Powder X-ray diffraction (PXRD) patterns were recorded on a Rigaku MiniFlex 600. Incident X-ray radiation was produced using a Cu X-ray tube operating at 40 kV and 15 mA with Cu Kα radiation (λ = 1.54 Å). The scan rate was 5° min−1 from 2θ = 10–60°. Ultraviolet-visible (UV-vis) absorption spectra and diffused reflectance UV-vis spectra were recorded on a JASCO V-770 spectrometer. Infrared (IR) spectra were obtained on a Jasco FT/IR-6700 spectrometer with an attenuated total reflectance unit using a diamond window. ESI-TOF-MS measurements were performed with a JEOL JMS-T100LP mass spectrometer in the negative ion mode. The sample was dissolved in the mixed solvent of water and acetonitrile [1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :39 (v/v)]. Transmission electron microscope (TEM) images were taken on a JEM-2100 (Japan Electron Optics Laboratory Co., Ltd) operating at 200 kV. Dynamic laser scattering (DLS) measurements were conducted at room temperature using a Zetasizer Nano S90 particle size analyzer from Malvern Instruments. Nitrogen (N2) adsorption–desorption isotherms at −196 °C were obtained with a MicrotracBEL Belsorp-mini II. Weighed samples (∼50 mg) were used for adsorption analysis after pretreatment at 150 °C for 1 h in vacuo. The samples were exposed to N2 within a relative pressure range from 0.01 to 101.3 kPa. The adsorbed amounts of N2 were calculated from the pressure change in a cell after reaching equilibrium at −196 °C. X-ray photoelectron spectroscopy (XPS) analyses were performed using a Shimadzu ESCA-3400HSE. The incident radiation was a Mg Kα X-ray (1253.6 eV) at 200 W. The samples were mounted on a stage with double-sided carbon tape. The binding energy of each element was corrected using the C 1s peak (284.6 eV) from the carbon tape.
:39 (v/v)]. Transmission electron microscope (TEM) images were taken on a JEM-2100 (Japan Electron Optics Laboratory Co., Ltd) operating at 200 kV. Dynamic laser scattering (DLS) measurements were conducted at room temperature using a Zetasizer Nano S90 particle size analyzer from Malvern Instruments. Nitrogen (N2) adsorption–desorption isotherms at −196 °C were obtained with a MicrotracBEL Belsorp-mini II. Weighed samples (∼50 mg) were used for adsorption analysis after pretreatment at 150 °C for 1 h in vacuo. The samples were exposed to N2 within a relative pressure range from 0.01 to 101.3 kPa. The adsorbed amounts of N2 were calculated from the pressure change in a cell after reaching equilibrium at −196 °C. X-ray photoelectron spectroscopy (XPS) analyses were performed using a Shimadzu ESCA-3400HSE. The incident radiation was a Mg Kα X-ray (1253.6 eV) at 200 W. The samples were mounted on a stage with double-sided carbon tape. The binding energy of each element was corrected using the C 1s peak (284.6 eV) from the carbon tape.
X-ray total scattering and pair distribution function (PDF) analyses
The X-ray total scattering data of samples filled in quartz capillaries were collected with two CdTe detectors covering the Q range up to 25 Å at the BL04B2 beamline in SPring-8 (113.4 keV, λ = 0.11 Å). The collected scattering was normalised to obtain the Faber–Ziman total structure factor S(Q), being processed by absorption, background, and Compton scattering corrections.55 The PDF was calculated by the Fourier transformation of S(Q) with a Lorch modification function using Igor Pro software.56,57Electrocatalytic water oxidation
Electrocatalytic water oxidation was conducted in a phosphate buffer (pH 8.0, 50 mM) containing (Me4N)Co–Fe (1.0 mMFe) using glassy carbon as a working electrode, a Pt wire as a counter electrode, and a saturated calomel electrode (SCE) as a reference electrode. Currents and potentials were recorded with an EC stat 302 (EC Frontier Co. Ltd).General procedure for photocatalytic water oxidation
A typical procedure for photocatalytic water oxidation is as follows: a phosphate buffer (pH 8.0, 50 mM, 2.0 mL) containing a cyano-bridged CP, [Ru(bpy)3]SO4 (0.30 mM), and Na2S2O8 (5.0 mM) was flushed with Ar gas for 10 min in the dark. The solution was irradiated for a certain time with an LED lamp (a RELYON Twin LED Light) at 25 °C. Evolved oxygen gas in a headspace was quantified by using a BAS FireSting oxygen metre.General procedure of repetitive reactions for photocatalytic water oxidation
A typical procedure of repetitive reactions for photocatalytic water oxidation was as follows: an aliquot (100 μL) containing Na2S2O8 (10 μmol) with or without [Ru(bpy)3]SO4 (0.6 μmol) was added to the reaction solution after each run. The solution was flushed with Ar gas for 10 min in the dark. The oxygen gas evolved by photoirradiation of the solution with an LED lamp was quantified by using the oxygen metre.Thermal water oxidation
A typical procedure for thermal water oxidation is as follows: an aqueous solution (0.1 mL) of [RuIII(bpy)3]3+ (4.0 μmol) containing H2SO4 (50 mM) as a stabiliser flushed with Ar gas was added to a phosphate buffer (pH 8.0, 50 mM, 1.9 mL) containing (Me4N)Co–Fe (120 μMFe) flushed with Ar gas in the dark. The pH of the resultant solution decreased to 7.5. The oxygen gas evolved at 25 °C was quantified by using the oxygen metre.Determination of the quantum yield for photocatalytic water oxidation
A phosphate buffer (pH 8.0, 50 mM, 2.0 mL) containing Na2S2O8 (5.0 mM), [Ru(bpy)3]SO4 (0.30 mM) and (Me4N)Co–Fe (80 μMFe) was placed in a long neck quartz cell after being degassed with Ar gas for at least 10 min prior to the photocatalytic reaction. Photoirradiation was performed using a 450 nm laser (Kikoh Giken Co., Ltd). The photon flux was monitored using a power metre (Newport Corporation, 919P-003-10), while the oxygen gas was monitored using a BAS FireSting oxygen metre. Quantum yields (Q. Y.) were calculated using the following eqn (1), where the maximum quantum yield is 50%.| Q. Y. = (No. of O2 molecules)/(No. of absorbed photons) | (1) |
Results and discussion
Solid structure of (Me4N){CoII1.5[FeII(CN)6]} {(Me4N)Co–Fe}
(Me4N){CoII1.5[FeII(CN)6]} {(Me4N)Co–Fe} was obtained by immersing K{CoII1.5[FeII(CN)6]} {(K)Co–Fe} powder in water containing Me4NCl at 50 °C for 20 h. The formed precipitates were collected from blight green solution by centrifugation. X-ray photoelectron spectroscopy (XPS) measurements for Co 2p, Fe 2p, and N 1s of (Me4N)Co–Fe indicated no change in the oxidation states of the corresponding ions in (K)Co–Fe (Fig. S1†). Also, no significant difference was observed in diffuse reflectance UV-vis spectra of (Me4N)Co–Fe and (K)Co–Fe in terms of absorption maxima (Fig. S2†). The N2 adsorption and desorption isotherm of (Me4N)Co–Fe suggested that the BET surface area and micropore volume of (Me4N)Co–Fe were 164 m2 g−1 and 0.38 cm3 g−1, which were slightly smaller than those of (K)Co–Fe, 261 m2 g−1 and 0.44 cm3 g−1 (Fig. S3†). The smaller BET surface area and pore volumes resulted from the occlusion of Me4N+ ions in the pores after the cation exchange.The counter-cation exchange of K+ with Me4N+ ions was confirmed by X-ray fluorescence (XRF), elemental analysis, and infrared (IR) spectroscopy. The XRF measurements revealed that a negligible amount of K+ ions remained in (Me4N)Co–Fe with the molar ratio of CoII and FeII ions similar to that in (K)Co–Fe (Table S1†). The presence of Me4N+ ions in (Me4N)Co–Fe was evidenced by IR measurements. Two characteristic peaks were observed assignable to the symmetric deformation band of CH3 groups and the asymmetric stretching band of the C4N moiety of the Me4N+ ion at 1479 and 945 cm−1, respectively (Fig. S3a and c†). Additionally, the retention of the FeII–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N–CoII bridging structure was evidenced by the CN stretching band, νCN, of (Me4N)Co–Fe at 2073 cm−1, which was almost identical to that of (K)Co–Fe (Fig. S3b†), accompanied by shoulder peaks at 2081 and 2096 cm−1.58 The appearance of the shoulder peaks resulted from the elongation along one-axis in (Me4N)Co–Fe (vide infra). Also, an additional Fe–CN bending band appeared at 509 cm−1 in (Me4N)Co–Fe together with that in (K)Co–Fe at 478 cm−1.59 Thus, the bridging structure of FeII–CN–CoII is maintained in (Me4N)Co–Fe although the structure is slightly disordered, or the high crystallographic symmetry of (K)Co–Fe is lost.
N–CoII bridging structure was evidenced by the CN stretching band, νCN, of (Me4N)Co–Fe at 2073 cm−1, which was almost identical to that of (K)Co–Fe (Fig. S3b†), accompanied by shoulder peaks at 2081 and 2096 cm−1.58 The appearance of the shoulder peaks resulted from the elongation along one-axis in (Me4N)Co–Fe (vide infra). Also, an additional Fe–CN bending band appeared at 509 cm−1 in (Me4N)Co–Fe together with that in (K)Co–Fe at 478 cm−1.59 Thus, the bridging structure of FeII–CN–CoII is maintained in (Me4N)Co–Fe although the structure is slightly disordered, or the high crystallographic symmetry of (K)Co–Fe is lost.
The crystal structure of (Me4N)Co–Fe was investigated by powder X-ray diffraction (PXRD). The PXRD peaks obtained for (Me4N)Co–Fe were assigned to the tetragonal crystal system with the unit cell parameters of a, b = 16.30 Å, c = 11.58 Å, which were longer than those of (K)Co–Fe (cubic; a = 10.13 Å) (Fig. 1a and b). A tetragonal unit cell with a = ∼16 Å has been previously reported for cyano-bridged CPs involving the Me4N+ ion as a counter cation.61 The length is similar to the side length of the red square in Fig. 1c, which can be expressed as {(a/2)2 + (3a/2)2}1/2, where a = ∼10.3 Å. The unit-cell parameter of c in (Me4N)Co–Fe longer than that in (K)Co–Fe also evidenced that the penetration of the Me4N+ ion (∼4 Å) larger than the K+ ion (2.8 Å) into the lattice (Fig. 1d). Thus, (Me4N)Co–Fe maintains the jungle-gym like structure similar to (K)Co–Fe, with many vacancies forming pores due to the CoII/FeII ratio being close to 1.5.62
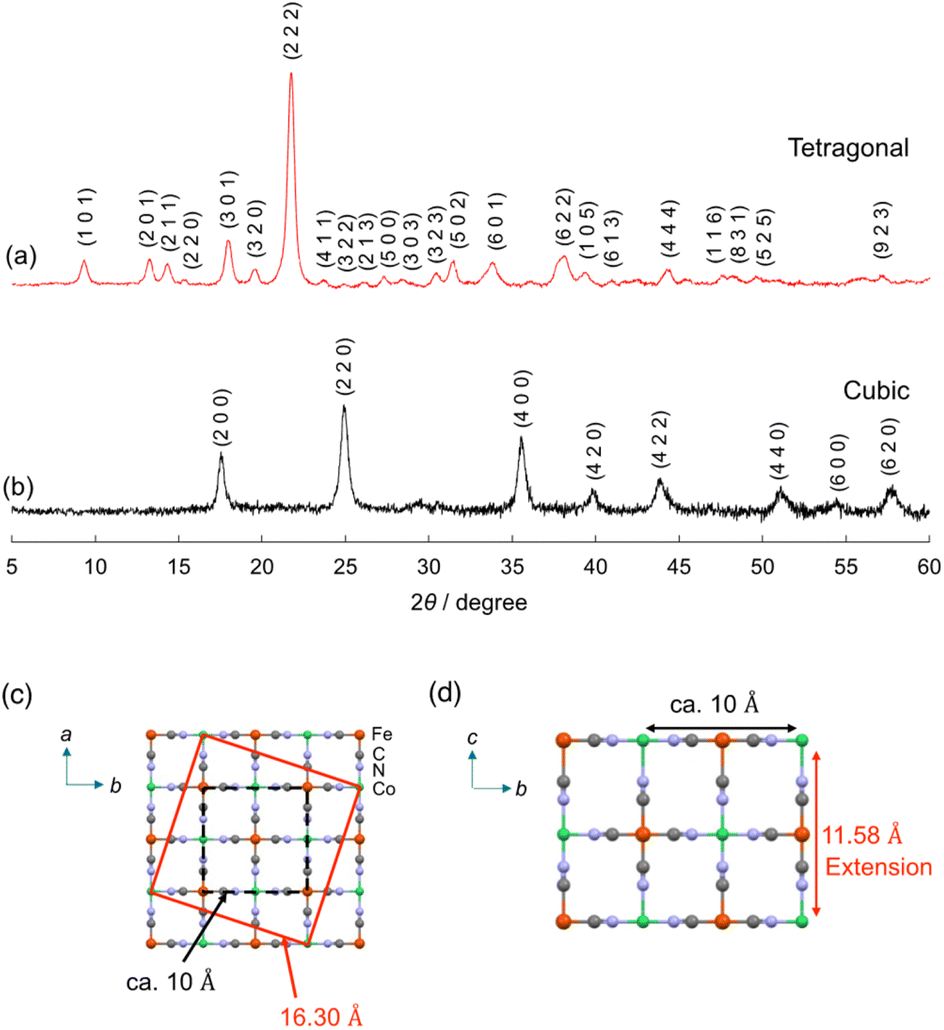 | ||
| Fig. 1 PXRD patterns with Miller indices (hkl) of (a) (Me4N){CoII1.5[FeII(CN)6]} {(Me4N)Co–Fe} and (b) K{CoII1.5[FeII(CN)6]} {(K)Co–Fe}. Miller indices were determined by Mcmaille.60 (c and d) A possible unit cell of (Me4N)Co–Fe. | ||
The solid structure of (Me4N)Co–Fe was also scrutinised by the pair distribution function (PDF) analyses in more detail (Fig. 2a). Atom-pair distance histograms were obtained by the PDF analysis of X-ray total scattering.57 The PDF peaks of (K)Co–Fe were almost identical to those of the reported cyano-bridged CP (Fig. 2b).63 The peaks observed in (Me4N)Co–Fe at 1.92, 3.11, 4.99, and 6.97 Å were attributed to FeII–C (or CoII–N), FeII–CN (CoII–NC), FeII–CN–CoII, and diagonal CoII⋯CoII (FeII⋯FeII), respectively. The pair distances derived from the extended FeII–C (or CoII–N), FeII–CN (CoII–NC), FeII–CN–CoII, and diagonal FeII⋯FeII (CoII⋯CoII) by the penetration of Me4N+ ions were observed at 2.53, 4.01, 5.81 and 7.73 Å, respectively. The pair distances were similar to those observed for (K)Co–Fe, 1.96, 3.12, 5.08, and 7.14 Å. On the other hand, peaks above 8 Å were hardly assignable for (Me4N)Co–Fe in contrast to those of (K)Co–Fe, indicating the lack of periodicity due to large structural disorder of (Me4N)Co–Fe, which may be caused by the positional disorder of Me4N+ ions in the lattice.
 | ||
| Fig. 2 Pair distribution function (PDF) of (a) (Me4N){CoII1.5[FeII(CN)6]} {(Me4N)Co–Fe} and (b) K{CoII1.5[FeII(CN)6]} {(K)Co–Fe}. | ||
The particle sizes of (Me4N)Co–Fe and (K)Co–Fe in the solid states were confirmed by transmission electron microscopy (TEM; Fig. S5a and b†). The TEM observation for powder specimens suggested that the sizes of primary particles were about 40 nm and the size of secondary particles were larger than 400 nm for both (Me4N)Co–Fe and (K)Co–Fe. The particle sizes of undissolved species in water were examined by dynamic light scattering (DLS) measurements (Fig. S5c†). The DLS measurements were performed on the solution of (Me4N)Co–Fe and the dispersion of (K)Co–Fe before the particles settled. The average particle sizes of (Me4N)Co–Fe and (K)Co–Fe in water were 32 and 370 nm, respectively. Most of the particles of (Me4N)Co–Fe were dissolved; however, a small number of particles remained undissolved as primary particles as evidenced by UV-vis measurements (vide infra). On the other hand, (K)Co–Fe is agglomerated as secondary particles maintaining heterogeneous nature. The disaggregation of (Me4N)Co–Fe results from the slightly increased size of open windows, which is beneficial for solvation.
(Me4N){CoII1.5[FeII(CN)6]} {(Me4N)Co–Fe} in water
The obtained (Me4N)Co–Fe was dissolved in water to provide transparent bright-green solution (Fig. 3a). The UV-vis adsorption spectrum measured for the aqueous solutions containing (Me4N)Co–Fe indicated two distinct absorption bands appearing at around 388 and 615 nm (Fig. 3b). The absorbance at 615 nm linearly increased in proportion to the concentrations of (Me4N)Co–Fe, following Beer's law in the concentration range 0–10 mMFe (Fig. 3c and d). The molar absorption coefficients calculated on the basis of the Fe amounts were 5900 and 1870 M−1 cm−1 at 388 and 615 nm bands, respectively. Assignments of UV-vis absorption bands have been reported for some cyano-bridged complexes containing Fe and Co with discrete structures.42,64 For example, the LMCT and IVCT bands of octanuclear {[CoII(pz)3CCH2OH]4[(pzTp)FeIII(CN)3]4}4+} (pzTp = tetra(pyrazolyl)borate) appeared at 440 nm as a broad intense absorption band (7077 M−1 cm−1) and 510 nm (4234 M−1 cm−1), respectively.42 The IVCT band of tetranuclear {[CoIII{Me2(μ-ET)cyclen}2{[(μ-NC)2FeII(CN)4]2}2−} (Me2(μ-ET)cyclen = 4,10-dimethyl-1,4,7,10-tetraazabicyclo[5.5.2]tetradecane) has been reported to appear at 515 nm (1650 M−1 cm−1).64 Thus, absorption bands at 388 and 615 nm observed for (Me4N)Co–Fe were assignable to LMCT and IVCT bands, respectively. The red shift of the IVCT band of (Me4N)Co–Fe could originate from the oxidation states of Fe and Co ions different from the reported complexes.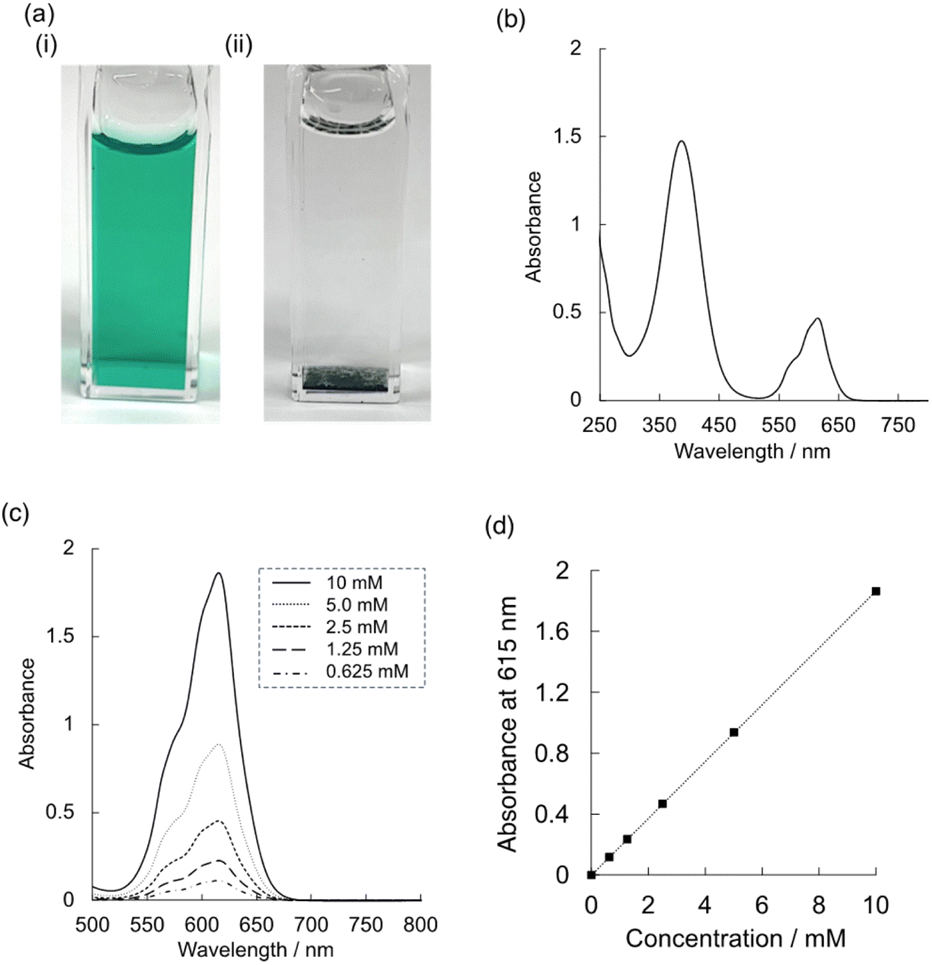 | ||
| Fig. 3 (a) (i) Photographs of (Me4N){CoII1.5[FeII(CN)6]} {(Me4N)Co–Fe} and (ii) K{CoII1.5[FeII(CN)6]} {(K)Co–Fe} in water. (b) UV-vis absorption spectrum of (Me4N)Co–Fe in water. (c) UV-vis spectra of (Me4N)Co–Fe at various concentrations ranging from 0.625 to 10 mMFe. (d) Absorbance at 615 nm as a function of the concentrations of (Me4N)Co–Fe in water. | ||
Water oxidation catalysis of (Me4N){CoII1.5[FeII(CN)6]} {(Me4N)Co–Fe}
Electrocatalytic water oxidation was examined by cyclic voltammetry (CV) of (Me4N)Co–Fe (1.0 mMFe) in a phosphate buffer (pH 7.0, 50 mM) in the potential range from 0 to 1.2 V vs. the saturated calomel electrode (SCE). The CV exhibited a large current attributed to water oxidation above 1.02 V, indicating that the overpotential was 0.42 V (Fig. S6†). The quasi-reversible redox couple observed at E1/2 = 0.92 V was assigned to the redox of CoII/III and/or FeII/III.65,66Photocatalytic water oxidation was performed under photoirradiation (LED lamp) of a phosphate buffer (pH 8.0, 50 mM, 2.0 mL) containing (CC+)Co–Fe (CC+ = K+ or Me4N+ ion), [Ru(bpy)3]SO4 (0.30 mM) and Na2S2O8 (5.0 mM) as a water oxidation catalyst, a photosensitiser and a sacrificial electron acceptor, respectively, at 25 °C. Time course of O2 evolution from the reaction solution with (Me4N)Co–Fe indicated that the initial O2-evolution rate (1.26 μmol min−1) and O2 yield based on Na2S2O8 (93%) were higher than those of (K)Co–Fe (0.61 μmol min−1, 56%) (Fig. 4). The saturation of O2 evolution mainly resulted from decomposition of the photosensitiser. The oxidized [Ru(bpy)3]3+ formed during the reaction is unstable under basic conditions, because of its high susceptibility to ˙OH attack leading to ligand oxidation.67 The difference in the O2 yield could be deeply related to the catalytic stability of each complex. Additionally, catalytic performance was examined for (K)Co–Fe modified with [FeII(CN)6]4− to increase water dispersibility. The PXRD patterns and particle sizes of the water-dispersible (K)Co–Fe modified with [FeII(CN)6]4− were almost identical to those of (K)Co–Fe (Fig. S8†). The O2 yield achieved for the photocatalytic system with (K)Co–Fe modified with [FeII(CN)6]4− was only 60%, which was comparable to that with the water-insoluble (K)Co–Fe (Fig. 4c). Thus, the high catalytic activity of (Me4N)Co–Fe results from its solution structure rather than the solubility.
 | ||
| Fig. 4 Time courses of O2 evolution under visible-light irradiation (LED light) of a phosphate buffer (pH 8.0, 50 mM, 2.0 mL) containing Na2S2O8 (5.0 mM), [Ru(bpy)3]SO4 (0.30 mM) and a water oxidation catalyst (80 mMFe): (a) (Me4N){CoII1.5[FeII(CN)6]} {(Me4N)Co–Fe; closed square}, (b) K{CoII1.5[FeII(CN)6]} {(K)Co–Fe; closed circle}, and (c) (K)Co–Fe modified with {[FeII(CN)6]4− (closed triangle)}. | ||
The catalytic activity of (Me4N)Fe–Co was examined in the repetitive experiments performed by the successive addition of an aliquot of Na2S2O8 with or without [Ru(bpy)3]SO4 (Fig. S7†). The O2 yields at the second and third runs using an aliquot containing Na2S2O8 with [Ru(bpy)3]SO4, 76 and 40%, were higher than those containing only Na2S2O8, 66 and 22%, respectively. Thus, the decomposition of [Ru(bpy)3]2+ contributed to the deactivation in the repetitive reaction, although contribution of decomposition of (Me4N)Co–Fe or the adsorption of SO42− cannot be ignored.
Photocatalytic water oxidation using only CoII ions or [FeII(CN)6]4− was also performed to confirm the necessity of the bridging structure of CoII–NC–FeII for high catalytic activity. The O2 yields obtained for the reaction systems using CoII ions and [FeII(CN)6]4− were 80% and almost 0%, respectively, which were inferior to that of (Me4N)Co–Fe (93%) (Fig. S9†). Thus, the bridging structure enhances the catalysis of Co ions.
Thermal water oxidation using [RuIII(bpy)3]3+ (2 mM) as an oxidant was performed in a phosphate buffer (pH 8.0) containing (Me4N)Co–Fe (120 μMFe) at 25 °C in the dark. No obvious temperature increase was observed during the reaction. The O2 yield based on the used amount of [RuIII(bpy)3]3+ exceeded 90% under the current reaction conditions although a decrease in the catalyst concentration resulted in lower O2 yield (Fig. S10†). This result ensures that photocatalytic water oxidation using (Me4N)Co–Fe is driven by [RuIII(bpy)3]3+ formed by the electron transfer to S2O82−.
The quantum yield of photocatalytic water oxidation was determined under photoirradiation (λ = 450 nm) of a phosphate buffer (pH 8.0, 50 mM, 2.0 mL) containing (Me4N)Co–Fe (80 μMFe), [Ru(bpy)3]SO4 (0.30 mM) and Na2S2O8 (5.0 mM) in a quartz cell (light path length: 1.0 cm). The quantum yield was calculated to be 14%, which was defined as the number of O2 molecules divided by the number of absorbed photons for 65 min (the maximum quantum yield: 50%) (Fig. S11†).
The concentration effect of (Me4N)Co–Fe on the O2-evolution rates was examined to determine the rate determining step. The photocatalytic water oxidation involves three reaction steps: (i) oxidation of photoexcited [Ru(bpy)3]2+ with S2O82−, (ii) electron transfer from (Me4N)Co–Fe to [RuIII(bpy)3]3+, and (iii) water oxidation on oxidised (Me4N)Co–Fe.68 The first-order dependence observed on the catalyst concentration ranging from 5 to 70 μMFe for (Me4N)Co–Fe and 80 to 160 μMFe for (K)Co–Fe indicates that step (ii) is the rate determining step for both catalysts (Fig. S12 and S13†). The highest O2 yields for (Me4N)Co–Fe and (K)Co–Fe reached 93% and 84%, respectively. The higher O2 yields with (Me4N)Co–Fe can result not only from less screening effect by dissolution but also from its solution structure.
Solution structure of (Me4N){CoII1.5[FeII(CN)6]} {(Me4N)Co–Fe}
The solution structure of (Me4N)Co–Fe was investigated by electrospray mass spectrometry (ESI-MS; negative) in a mixed solution of water and acetonitrile [1:39 (v/v)]. The absorbance at 615 nm of the mixed solution containing (Me4N)Co–Fe obeyed Beer's law in the concentration range of 0–1.0 mMFe (Fig. S14†). No peak appearance was confirmed under the measurement conditions (Fig. S15†). The ESI-MS spectrum for the solution of (Me4N)Co–Fe showed 17 anion peaks, which appeared equally spaced with the width of m/z = 74 derived from Me4N+ ions (Fig. 5 and S16†). Each peak was composed of {Co4[Fe(CN)6]3}n−, Me4N+, and extra-ligands, such as H2O, OH−, or Cl−. For example, the observed pattern containing five Me4N+ ions around m/z = 1312.17 agreed with the simulated pattern calculated under the assumption that {Co4[Fe(CN)6]3}n− with [4 × OH−], [H2O + 3 × OH−], [2 × Cl−], and [2 × H2O + Cl−] is concomitantly generated (Fig. S16†). Chemical species composed of polynuclear metal complexes accompanying many counter cations or solvent molecules have been occasionally detected in the ESI-MS measurements. For example, a negatively charged polyoxometalate accompanying 0–9 tetrabutylammonium cations has been detected by ESI-MS.69,70 Also, a positively charged cubane-type metal complex (C5Me5Mo3S4FeCl) together with 0–7 tetrahydrofuran molecules was observed in the ESI-MS spectrum.71 The increase in positive charge by increasing the number of Me4N+ ions can be compensated for by the reduction of metal ions in the mass chamber (Fig. S17†). These results indicate that (Me4N)Co–Fe is an anionic polynuclear metal complex, such as {Co4[Fe(CN)6]3}4−, formed by depolymerisation in solution.
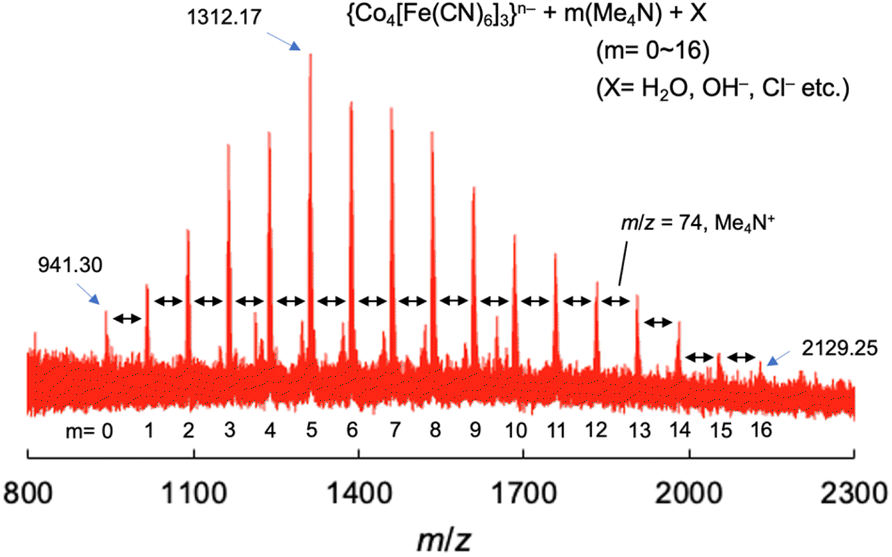 | ||
| Fig. 5 Electrospray ionization mass spectrum (negative) of (Me4N){CoII1.5[FeII(CN)6]} {(Me4N)Co–Fe} in a mixed solvent of water and acetonitrile [1:39 (v/v)]. | ||
The PDF measurements of (Me4N)Co–Fe in water indicated that the water-soluble species maintained multinuclear structures, because the peaks assignable to the atomic distances at around 1.92, 4.01, 4.99, 5.88, and 6.97 Å were observed similar to those in the solid state (Fig. S18†). The disappearance of peaks above 8 Å suggested that (Me4N)Co–Fe was depolymerised in water. (K)Co–Fe is thought to have a porous structure, in which the vacancies of [FeII(CN)6]4− units are connected to form larger pores.62 The penetration of Me4N+ ions into the larger pores resulting from extrusion of K+ ions allowed the invasion of water molecules for depolymerisation with hydration. As a result, the depolymerisation proceeds to provide heptanuclear clusters of {Co4[Fe(CN)6]3}4− with extra-open sites on Co ions, resulting in the enhancement of water oxidation catalysis (Fig. 6).
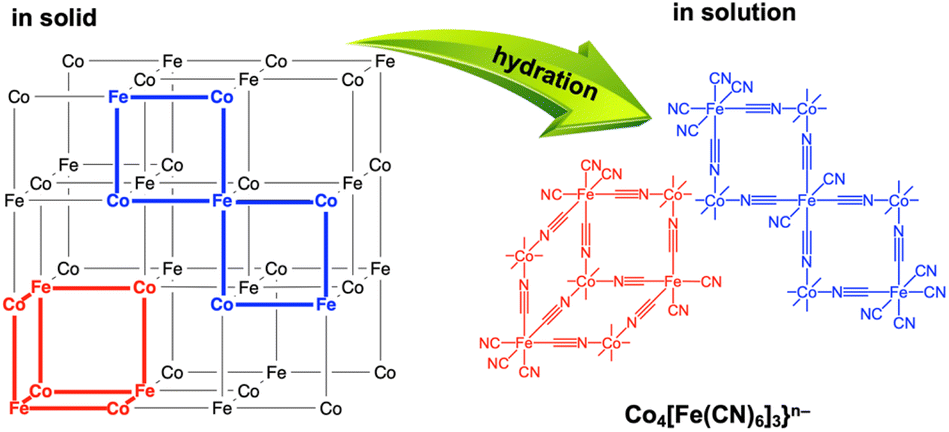 | ||
| Fig. 6 Depolymerisation by hydration of frameworks of (Me4N){CoII1.5[FeII(CN)6]} {(Me4N)Co–Fe} with many vacancies into {Co4[Fe(CN)6]3}n−. | ||
Conclusions
A cyano-bridged coordination polymer (CP), K{CoII1.5[FeII(CN)6]} {(K)Co–Fe}, acting as a heterogeneous catalyst for water oxidation, was depolymerised by the counter-cation exchange of K+ with Me4N+ ions. The water-soluble (Me4N){CoII1.5[FeII(CN)6]} (Me4N)Co–Fe catalyst exhibited enhanced catalysis compared with (K)Co–Fe in visible-light-driven water oxidation. The surface modification of (K)Co–Fe with {[FeII(CN)6]}4− provided dispersibility in the water; however, no catalytic enhancement was observed. Thus, a reason for the enhanced catalysis would be the increased number of open sites on Co ions to capture water molecules, because the catalytically active species of (Me4N)Co–Fe in water is {Co4[Fe(CN)6]3}4− formed by Co–N bond cleavages. The solubilisation of catalytically active CPs would be promising for rational elucidation of the reaction mechanism, which is difficult for conventional heterogeneous catalysts. Also, the solubilisation of heterogeneous water oxidation catalysts is beneficial to be combined with other photocatalytic semiconductors and/or photosensitisers to construct artificial photosynthesis systems.Data availability
Supporting data of this study are available within the paper. Further details regarding the data are available from the corresponding author upon reasonable request.Author contributions
Y. S.: conceptualization, investigation, methodology, writing – original draft, writing – review and editing. T. N.: conceptualization, funding acquisition, methodology, writing – original draft, writing – review and editing. H. T.: investigation, methodology, writing – review and editing. Y. Y.: conceptualization, funding acquisition, methodology, writing – original draft, writing – review and editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (No. JP19KK0144; JP22H01871; JP23K04770) and JST, the establishment of university fellowships towards the creation of science technology innovation, Grant Number JPMJFS2138, Osaka Metropolitan University Strategic Research Promotion Project (Priority Research), and MEXT the Joint Usage/Research Center for Catalysis (24AY0577 and 23CY0314). The synchrotron radiation experiments were performed using BL04B2 at SPring-8 with the approval of JASRI (Proposal No. 2023B1721 and 2023B2001). The authors thank Dr Hiroki Yamada (JASRI) and Mr Taichi Nishiguchi (Kyoto University) for their support throughout the synchrotron experiments.Notes and references
- J. Xiao, T. Hisatomi and K. Domen, Acc. Chem. Res., 2023, 56, 878 CrossRef CAS PubMed.
- S. Nishioka, F. E. Osterloh, X. Wang, T. E. Mallouk and K. Maeda, Nat. Rev. Methods Primers, 2023, 3, 42 CrossRef CAS.
- K. Sun, Y. Qian and H.-L. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202217565 CrossRef CAS.
- M. Sohail, S. Rauf, M. Irfan, A. Hayat, M. M. Alghamdi, A. A. El-Zahhar, D. Ghernaout, Y. Al-Hadeethi and W. Lv, Nanoscale Adv., 2024, 6, 1286 RSC.
- L. Lin, Y. Ma, J. J. M. Vequizo, M. Nakabayashi, C. Gu, X. Tao, H. Yoshida, Y. Pihosh, Y. Nishina, A. Yamakata, N. Shibata, T. Hisatomi, T. Takata and K. Domen, Nat. Commun., 2024, 15, 397 CrossRef CAS.
- J. Guan, K. Koizumi, N. Fukui, H. Suzuki, K. Murayama, R. Toyoda, H. Maeda, K. Kamiya, K. Ohashi, S. Takaishi, O. Tomita, A. Saeki, H. Nishihara, H. Kageyama, R. Abe and R. Sakamoto, ACS Catal., 2024, 14, 1146 CrossRef CAS.
- Y. Tomita, N. Taira, K. Sakai and H. Ozawa, ACS Catal., 2024, 14, 5788 CrossRef CAS.
- G. Fu, D. Yang, S. Xu, S. Li, Y. Zhao, H. Yang, D. Wu, P. S. Petkov, Z.-A. Lan, X. Wang and T. Zhang, J. Am. Chem. Soc., 2024, 146, 1318 CrossRef CAS.
- M. A. Khan, S. Khan, S. Sengupta, B. N. Mongal and S. Naskar, Coord. Chem. Rev., 2024, 504, 215679 CrossRef.
- Y. Zhao, D. P. Adiyeri Saseendran, C. Huang, C. A. Triana, W. R. Marks, H. Chen, H. Zhao and G. R. Patzke, Chem. Soc. Rev., 2023, 123, 6257 CrossRef CAS.
- N. Vereshchuk, M. Gil-Sepulcre, A. Ghaderian, J. Holub, C. Gimbert-Suriñach and A. Llobet, Chem. Soc. Rev., 2023, 52, 196 RSC.
- M. Kondo, H. Tatewaki and S. Masaoka, Chem. Soc. Rev., 2021, 50, 6790 RSC.
- G. Gao, X. Chen, L. Han, G. Zhu, J. Jia, A. Cabot and Z. Sun, Coord. Chem. Rev., 2024, 503, 215639 CrossRef CAS.
- Y. Mu, R. Ma, S. Xue, H. Shang, W. Lu and L. Jiao, Carbon Neutralization, 2024, 3, 4 CrossRef CAS.
- C. Wang, M. Humayun, D. P. Debecker and Y. Wu, Chem. Rev., 2023, 478, 214973 CAS.
- Q. Hu, H. Zhou, Y. Ding, T. Wågberg and X. Han, ACS Catal., 2024, 14, 5898 CrossRef CAS.
- H.-T. Zhang, Y.-H. Guo, Y. Xiao, H.-Y. Du and M.-T. Zhang, Angew. Chem., Int. Ed., 2023, 135, e202218859 CrossRef.
- X.-B. Han, D.-X. Wang, E. Gracia-Espino, Y.-H. Luo, Y.-Z. Tan, D.-F. Lu, Y.-G. Li, T. Wågberg, E.-B. Wang and L.-S. Zheng, Chin. J. Catal., 2020, 41, 853 CrossRef CAS.
- T. Ishizuka, A. Watanabe, H. Kotani, D. Hong, K. Satonaka, T. Wada, Y. Shiota, K. Yoshizawa, K. Ohara, K. Yamaguchi, S. Kato, S. Fukuzumi and T. Kojima, Inorg. Chem., 2016, 55, 1154 CrossRef CAS PubMed.
- T. Nakazono, N. Amino, R. Matsuda, D. Sugawara and T. Wada, Chem. Commun., 2022, 58, 7674 RSC.
- T. Nakazono and T. Wada, Inorg. Chem., 2021, 60, 1284 CrossRef CAS PubMed.
- E. Zhou, X. Zhang, L. Zhu, E. Chai, J. Chen, J. Li, D. Yuan, L. Kang, Q. Sun and Y. Wang, Sci. Adv., 2024, 10, eadk8564 CrossRef CAS PubMed.
- X. Li, Q. Yang, Y. Yuan, Y. shama and H. Yan, Small, 2024, 2401168 CrossRef CAS.
- S. Li, W. Jiang, Q. Sui, Y. Gao and Y. Jiang, Dalton Trans., 2024, 53, 3975 RSC.
- W. Kang, R. Wei, H. Yin, D. Li, Z. Chen, Q. Huang, P. Zhang, H. Jing, X. Wang and C. Li, J. Am. Chem. Soc., 2023, 145, 3470 CrossRef CAS.
- L. Reith, C. A. Triana, F. Pazoki, M. Amiri, M. Nyman and G. R. Patzke, J. Am. Chem. Soc., 2021, 143, 15022 CrossRef CAS PubMed.
- N. S. Peighambardoust, S. S. Akbari, R. Lomlu, U. Aydemir and F. Karadas, ACS Mater. Au, 2024, 4, 214 CrossRef CAS PubMed.
- A. A. Ahmad, T. G. U. Ghobadi, M. Buyuktemiz, E. Ozbay, Y. Dede and F. Karadas, Inorg. Chem., 2022, 61, 3931 CrossRef CAS PubMed.
- G. Gundogdu, T. G. U. Ghobadi, S. S. Akbari, E. Ozbay and F. Karadas, Chem. Commun., 2021, 57, 508 RSC.
- S. S. Akbari, U. Unal and F. Karadas, ACS Appl. Energy Mater., 2021, 4, 12383 CrossRef CAS.
- S. Goberna-Ferrón, W. Y. Hernández, B. Rodríguez-García and J. R. Galán-Mascarós, ACS Catal., 2014, 4, 1637 CrossRef.
- H. Matsuoka, O. Tomita, H. Tabe, H. Suzuki, Y. Yamada and R. Abe, J. Photochem. Photobiol., A, 2022, 426, 113753 CrossRef CAS.
- H. Tabe, A. Kitase and Y. Yamada, Appl. Catal., B, 2020, 262, 118101 CrossRef CAS.
- Y. Yamada, K. Oyama, R. Gates and S. Fukuzumi, Angew. Chem., Int. Ed., 2015, 54, 5613 CrossRef CAS.
- R. C. Oglou, T. G. U. Ghobadi, F. S. Hegner, J. R. Galán-Mascarós, N. López, E. Ozbay and F. Karadas, Angew. Chem., Int. Ed., 2023, 62, e202308647 CrossRef.
- F. S. Hegner, F. A. Garcés-Pineda, J. González-Cobos, B. Rodríguez-García, M. Torréns, E. Palomares, N. López and J. R. Galán-Mascarós, ACS Catal., 2021, 11, 13140 CrossRef CAS.
- S. Pintado, S. Goberna-Ferrón, E. C. Escudero-Adán and J. R. Galán-Mascarós, J. Am. Chem. Soc., 2013, 135, 13270 CrossRef CAS.
- M. Ishizaki, M. Sakamoto, H. Tanaka, T. Kawamoto and M. Kurihara, Mol. Cryst. Liq. Cryst., 2011, 539, 18 CAS.
- M. Ishizaki, K. Kanaizuka, M. Abe, Y. Hoshi, M. Sakamoto, T. Kawamoto, H. Tanaka and M. Kurihara, Green Chem., 2012, 14, 1537 RSC.
- A. Gotoh, H. Uchida, M. Ishizaki, T. Satoh, S. Kaga, S. Okamoto, M. Ohta, M. Sakamoto, T. Kawamoto and H. Tanaka, Nanotechnology, 2007, 18, 345609 CrossRef.
- E. Chelebaeva, Y. Guari, J. Larionova, A. Trifonov and C. Guérin, Chem. Mater., 2008, 20, 1367 CrossRef CAS.
- D. Li, R. Clérac, O. Roubeau, E. Harté, C. Mathonière, R. Le Bris and S. M. Holmes, J. Am. Chem. Soc., 2008, 130, 252 CrossRef CAS PubMed.
- Y.-Z. Zhang, P. Ferko, D. Siretanu, R. Ababei, N. P. Rath, M. J. Shaw, R. Clérac, C. Mathonière and S. M. Holmes, J. Am. Chem. Soc., 2014, 136, 16854 CrossRef CAS PubMed.
- A. Gallen, J. Jover, M. Ferrer and M. Martínez, Inorg. Chim. Acta, 2023, 545, 121282 CrossRef CAS.
- M. A. Gonzálvez, P. V. Bernhardt, M. Font-Bardia, A. Gallen, J. Jover, M. Ferrer and M. Martínez, Inorg. Chem., 2021, 60, 18407 CrossRef.
- M. A. Gonzálvez, A. Gallen, M. Ferrer and M. Martínez, Inorg. Chem., 2020, 59, 1582 CrossRef PubMed.
- E. Hruska, Q. Zhu, S. Biswas, M. T. Fortunato, D. R. Broderick, C. M. Morales, J. M. Herbert, C. Turro and L. Robert Baker, J. Am. Chem. Soc., 2024, 146, 8031 CrossRef CAS PubMed.
- X. Meng, J. Yang, C. Zhang, Y. Fu, K. Li, M. Sun, X. Wang, C. Dong, B. Ma and Y. Ding, ACS Catal., 2022, 12, 89 CrossRef CAS.
- A. A. Ahmad, T. G. U. Ghobadi, E. Ozbay and F. Karadas, Chem. Commun., 2022, 58, 9341 RSC.
- H. Tabe, Y. Seki, M. Yamane, T. Nakazono and Y. Yamada, J. Phys. Chem. Lett., 2023, 14, 158 CrossRef CAS PubMed.
- Y. Seki, H. Tabe and Y. Yamada, J. Phys. Chem. C, 2022, 126, 5564 CrossRef CAS.
- M. Ware, J. Chem. Educ., 2008, 85, 612 CrossRef CAS.
- L. Samain, F. Grandjean, G. L. Long, P. Martinetto, P. Bordet and D. Strivay, J. Phys. Chem. C, 2013, 117, 9693 CrossRef CAS.
- D. Hong, Y. Yamada, T. Nagatomi, Y. Takai and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 19572 CrossRef CAS.
- T. E. Faber and J. M. Ziman, Philos. Mag., 1965, 11, 153 CrossRef CAS.
- E. Lorch, J. Phys. C: Solid State Phys., 1969, 2, 229 CrossRef.
- S. Kohara, M. Itou, K. Suzuya, Y. Inamura, Y. Sakurai, Y. Ohishi and M. Takata, J. Phys.: Condens.Matter, 2007, 19, 506101 CrossRef.
- R. G. Romero, A. D. Morales, J. D. Rodríguez and J. F. Bertrán, Transition Met. Chem., 1992, 17, 573 CrossRef.
- S. N. Ghosh, J. Inorg. Nucl. Chem., 1974, 36, 2465 CrossRef CAS.
- A. Le Bail, Powder Diffr., 2004, 19, 249 CrossRef CAS.
- M. Witzel, B. Ziegler and D. Babel, Z. Anorg. Allg. Chem., 2000, 626, 471 CrossRef CAS.
- A. Simonov, T. De Baerdemaeker, H. L. Boström, M. L. Gomez, H. J. Gray, D. Chernyshov, A. Bosak, H. B. Bürgi and A. L. Goodwin, Nature, 2020, 578, 256 CrossRef CAS PubMed.
- N. Ma, R. Ohtani, H. M. Le, S. S. Sørensen, R. Ishikawa, S. Kawata, S. Bureekaew, S. Kosasang, Y. Kawazoe, K. Ohara, M. M. Smedskjaer and S. Horike, Nat. Commun., 2022, 13, 4023 CrossRef CAS PubMed.
- L. Alcázar, G. Aullón, M. Ferrer and M. Martínez, Chem.–Eur. J., 2016, 22, 15227 CrossRef PubMed.
- E. P. Alsaç, E. Ülker, S. V. K. Nune, Y. Dede and F. Karadas, Chem.–Eur. J., 2018, 24, 4856 CrossRef.
- W. Deng, Z. Li, Y. Ye, Z. Zhou, Y. Li, M. Zhang, X. Yuan, J. Hu, W. Zhao, Z. Huang, C. Li, H. Chen, J. Zheng and R. Li, Adv. Energy Mater., 2021, 11, 2003639 CrossRef CAS.
- P. K. Ghosh, B. S. Brunschwig, M. Chou, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1984, 106, 4772 CrossRef CAS.
- B. Limburg, E. Bouwman and S. Bonnet, ACS Catal., 2016, 6, 5273 CrossRef CAS.
- C. R. Mayer, M. Hervé, H. Lavanant, J.-C. Blais and F. Sécheresse, Eur. J. Inorg. Chem., 2004, 973 CrossRef CAS.
- C. R. Mayer, C. Roch-Marchal, H. Lavanant, R. Thouvenot, N. Sellier, J.-C. Blais and F. Sécheresse, Chem.–Eur. J., 2004, 10, 5517 CrossRef CAS PubMed.
- Y. Ohki, K. Munakata, Y. Matsuoka, R. Hara, M. Kachi, K. Uchida, M. Tada, R. E. Cramer, W. M. C. Sameera, T. Takayama, Y. Sakai, S. Kuriyama, Y. Nishibayashi and K. Tanifuji, Nature, 2022, 607, 86 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRF spectra, IR spectra, DRS, XPS, N2 adsorption–desorption isotherms, TEM, DLS, CV, time courses of O2 evolution, ESI-MS, and PDF. See DOI: https://doi.org/10.1039/d4sc04390a |
| This journal is © The Royal Society of Chemistry 2024 |