
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevisiting the activity origin of the PtAu24(SR)18 nanocluster for enhanced electrocatalytic hydrogen evolution by combining first-principles simulations with the experimental in situ FTIR technique†
Fang
Sun‡
a,
Lubing
Qin‡
b,
Zhenghua
Tang
 *b and
Qing
Tang
*a
*b and
Qing
Tang
*a
aSchool of Chemistry and Chemical Engineering, Chongqing Key Laboratory of Chemical Theory and Mechanism, Chongqing University, Chongqing 401331, China. E-mail: qingtang@cqu.edu.cn
bNew Energy Research Institute, School of Environment and Energy, South China University of Technology, Guangzhou Higher Education Mega Center, Guangzhou, 510006, China. E-mail: zhht@scut.edu.cn
First published on 6th September 2024
Abstract
Thiolate-protected metal nanoclusters (NCs) have been widely used in various electrocatalytic reactions, yet the dynamic evolution of metal NCs during electrocatalysis has been rarely explored and the activity origin remains largely ambiguous. Herein, using a PtAu24(SCH3)18 NC as a prototype model, we combined advanced first-principles calculations and attenuated total reflection surface-enhanced infrared spectroscopy (ATR-SEIRAS) to re-examine its active site and reaction dynamics in the hydrogen evolution reaction (HER). It has been previously assumed that the central Pt is the only catalytic center. However, differently, we observed the spontaneous desorption of thiolate ligands under moderate potential, and the dethiolated PtAu24 exhibits excellent HER activity, which is contributed not only by the central Pt atom but also by the exposed bridged Au sites. Particularly, the exposed Au exhibits high activity even comparable to Pt, and the synergistic effect between them makes dethiolated PtAu24 an extraordinary HER electrocatalyst, even surpassing the commercial Pt/C catalyst. Our predictions are further verified by electrochemical activation experiments and in situ FTIR (ATR-SEIRAS) characterization, where evident adsorption of Au–H* and Pt–H* bonds is monitored. This work detected, for the first time, the Au–S interfacial dynamics of the PtAu24 nanocluster in electrocatalytic processes, and quantitatively evaluated the essential catalytic role of the exposed Au sites that has been largely overlooked in previous studies.
Introduction
Electrocatalytic hydrogen evolution reaction (HER) is essential for efficiently producing H2 from water splitting, which provides a plausible alternative to decarbonization.1–4 To date, many types of catalysts have been developed for such energy conversion.2,5–7 Among them, some surface engineered solid catalysts exhibit high catalytic activity comparable to platinum.8 However, their inherent heterogeneity in the surface structure and composition seriously hinders the precise control at the molecular scale. Hence a highly homogeneous catalyst with an atomically precise structure is indispensable to achieve fine-tuning of the catalytic properties. Following the pioneering report by Kornberg et al.,9 a class of thiolate-protected gold nanoclusters (Aun(SR)m NCs), such as Au25(SR)18 and Au38(SR)24, have attracted considerable interest because of their unique properties and well-defined atomic structures confirmed by X-ray crystallography.5,10–13 Recently, doping foreign metals into the Au25(SR)18 NCs has been widely explored to tailor their electronic and catalytic properties, with PtAu24(SR)18 garnering the most interest.14–17 The existing experiments and theoretical calculations have confirmed the Pt dopant located at the centre of the core, replacing the central Au and ultimately resulting in a superatomic 6-electron configuration.17,18 This substitution leads to the splitting of the superatomic 1P orbitals accompanied by an Jahn–Teller-like distortion of the metal nucleus, making the electrochemical and optical properties significantly altered from the undoped Au25 NCs.18–21In 2017, Lee et al. reported the first electrocatalytic experiment of PtAu24(SC6H13)18 and found that this NC can efficiently produce hydrogen, even more than the benchmark platinum catalyst.18 The central Pt atom was suggested to be the key active site based on the thermodynamic static calculations of H adsorption in the intact PtAu24 NC. But is it really the case? Our recent constant potential calculations and experimental spectroscopic data have verified the spontaneous electrochemical dethiolation of surface SR ligands in the Au25(SCH3)18 NC, leading to the exposed under-coordinated Au atoms and formation of free HSCH3 molecule in solution.22 In other words, the metal–ligand interface does not remain intact, but is dynamically affected by the electrode potential and interface microenvironment, thus the traditional static calculations would be inaccurate in describing the reaction dynamics of thiolated metal NCs under real electrochemical conditions. Particularly, it can be anticipated that if the surface dethiolation (i.e., desorption of –SR) of PtAu24 can occur spontaneously, then the exposed new Au site, rather than the previously solely assumed Pt site, could also be accountable for the HER activity, which is yet to be explored.
To solve the puzzling question about the real active site of PtAu24, we performed advanced first-principles calculations and in situ experimental characterization techniques to make a clear understanding of its true active state and reaction dynamics during the electrocatalytic HER process. Our constant potential calculations combined with ab initio molecular dynamics (AIMD) simulations revealed that H+ from the water layer preferentially attacks the S atom, weakening the Au–S bond and ultimately stripping –SR from the PtAu24 surface. Importantly, the exposed under-coordinated surface Au atoms are efficient catalytic sites for hydrogen evolution. The bridged naked Au–Au atoms can adsorb the H* intermediate appropriately and exhibit extraordinary HER performance comparable to Pt, especially in the reduction potential range of approximately −0.3–0 V. Our theoretical prediction is further verified by the electrochemical activity experiments and in situ FTIR characterization. Our results showed that the surface Au sites originally passivated by thiolate ligands are reactivated via electrochemical induced ligand removal, which has been largely ignored in previous static calculations.
Results and discussion
Thus far, the prior thermodynamic evaluation of the HER activity of the PtAu24(SR)18 NC has solely relied on the computational hydrogen electrode (CHE) model calculations.18 Although the CHE model has been widely used in various electrochemical studies, it overlooks some important factors: (1) the grand free energy is usually approximated as a linear function of electrode potential, ignoring the capacitive contributions to the energy;7,22 (2) the electrochemical interactions between reaction intermediates and the surrounding solvent cannot be explicitly described;23 (3) the reaction energy is usually computed for the cluster's most stable charge state, which does not necessarily correspond to the charge state occurring under experimental electrode potential.21,24–26 In particular, the solvent effect and electrode potential are vital for accurately evaluating the electrochemical process.To address this issue, we firstly employed a constant-potential hybrid-solvent model to assess the potential-dependent reaction thermodynamics of the HER in the intact PtAu24(SCH3)18 NC. More details can be found in the methods section in the ESI.† A thick water slab containing 78H2O molecules in conjunction with Poisson–Boltzmann implicit solvation were constructed on the surface of PtAu24 (Fig. S1a†).27–29 The average density of water layers was calculated to be around 1 g cm−3 (Fig. S1b†).30 In intact PtAu24(SCH3)18, three adsorption sites for H were considered (Fig. 1a–c): the central Pt site, the S site from the exterior –SR ligand, and the staple Au site. For each case, six individual simulations were conducted at charges of 0e to 5e with decrements of 1e, and the electrode potential (URHE) for a given charge can be determined (Fig. S2†). The total free energy of each species can be described as a continuous quadratic function of URHE by further fitting the six potential-energy points that take into account the contributions of zero-point energy, enthalpy, and entropy (Fig. S3†). With these potential-energy curves, the free energy at a specific URHE can be obtained, thereby determining the corresponding free energy changes of the Volmer–Heyrovsky reaction in the HER (Volmer reaction, * + H+ + e− = H* (ΔG1) and Heyrovsky reaction, H* + H+ + e− = H2 + * (ΔG2)). Note that here we did not consider the Tafel process where two H* intermediates are firstly formed at contiguous active sites and then combine to generate H2, which is kinetically much more difficult on the surface of intact PtAu24 NCs. Moreover, the pH value is set as 3, which is consistent with the electrolyte acidity in the experiment (unless stated, all pH values are referred to as 3).
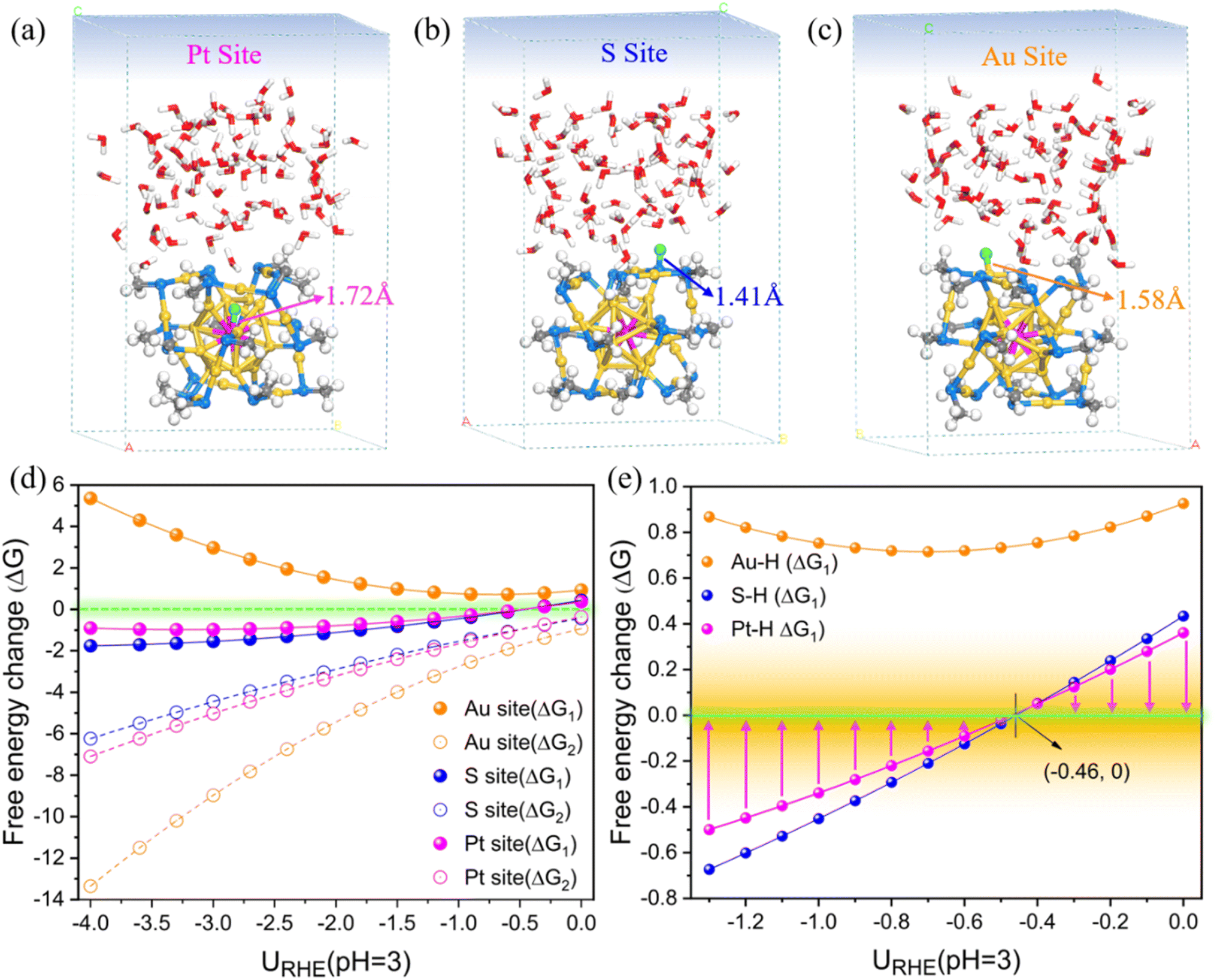 | ||
| Fig. 1 (a–c) The geometric structures of three adsorbed H* systems at different sites on intact PtAu24(SCH3)18 optimized under a zero charge (0e). Color codes: yellow, Au; purple, Pt; blue, S; grey, C; red, O; green, adsorbed H*; white, other H. (d) Free energy changes (ΔG) of the HER reaction steps as a function of electrode potential URHE in pH = 3 (ΔG1 represents the Volmer step for H* adsorption, while ΔG2 represents the Heyrovsky step for H2 generation), and (e) only shows the correlation between the Volmer step (ΔG1) and the potential from −1.30 to 0 V. | ||
From Fig. 1d, the H* formation step (Volmer step, ΔG1) at all three sites is identified as the potential-limiting step (PDS) of the entire HER process, while the subsequent Heyrovsky step to form H2 could be easily achieved with a very negative ΔG2 value. We further focused on the Volmer H* adsorption in a narrower URHE range from −1.30 to 0 V. As shown in Fig. 1e, the adsorption of H* at the Pt and S sites is energetically much more favorable than the Au site, and the overall activity follows the trend Pt > S > Au. We have to note that the Volmer H* adsorption on Pt and S site is exothermic below URHE = −0.46 V, whereas this reaction is endothermic above this threshold potential. In other words, at a potential of −0.46 V, the bonding of H on Pt and S reaches the most ideal state with ΔG1 close to 0 eV. As far as the current analysis is concerned, both Pt and S should be significant active sites for the HER, however, this thermodynamic evaluation is not convincing, and further kinetic calculations are required to detect the electrochemical structural evolution of the PtAu24(SCH3)18 NC.
Hence, we further evaluated the kinetic information of PtAu24 by combining the constant-potential hybrid-solvent model with the state-of-the-art ab initio molecular dynamics (AIMD) simulations at room temperature (300 K). Based on the model used in the above thermodynamics, we pre-executed 5 ps AIMD simulations to ensure the reasonable hydrogen bonding network in dynamic operation. The changes in energy and temperature during this simulation are shown in Fig. S4,† indicating that the structure is well-equilibrated. The radial distribution functions (RDFs) and the coordination number of O–O and O–H pairs (Fig. S5†) further validate the proper convergence of the water structure to bulk water behaviors, indicating that our model can correctly describe the behavior of liquid water.31 From Fig. S6a,† an extra H3O+ is placed on the local surface of PtAu24 NC to rationalize the presence of proton in an acidic system. Similarly, excess electrons were introduced to drive a determined potential during dynamic execution, known as the constant-potential AIMD simulation.
To better track the impact of potential, we initiated a series of dynamic potentiostatic simulations (Fig. S7†), and the results revealed the potential-dependent desorption of the –SCH3 ligand from the PtAu24 surface. In particular, only at a moderate reduction potential (URHE = −0.99 V), the S atom is able to spontaneously capture the surrounding H from H3O+ and weaken the Au–S bonds, ultimately causing the –SCH3 group to detach from the bonding of the surface Au and the staple Au and then form a free HSCH3 molecule in the solution (Fig. S7e†). Otherwise, the proton would either be transferred to water by hydrogen bonding to form a Zundel (H5O2+) cation (Fig. S7a,†URHE = −0.24 V) or the proton binds to the S atom in an adsorbed H* form, which can only break one surface Au–S bond and is not sufficient to completely peel off the –SCH3 ligand (Fig. S7b–d,†URHE = −0.29 V, −0.66 V, −0.87 V). Based on this, we propose that −0.3 VRHE could be the critical potential for cleaving the surface Au–S bond, and the entire thiolate group can be completely stripped at negative potentials ≤−1.0 VRHE (Fig. 2a). To trace a more detailed dynamic response of the –SR etching behavior, we focus on the local region around the broken bond during AIMD and label these key atoms (the detailed labeling can be found in Fig. S6b and the inset in Fig. 2e). Fig. 2b–e show the relative distance between representative atoms under different URHE. Obviously, the S13 atom is first disconnected from the surface Au (labeled as Au17) at potential ≤−0.3 V, and ultimately the distance of Au17⋯S13 reaches greater than or equal to 3.5 Å which is considered to be completely disconnected (Fig. 2b). However, only when the potential is sufficiently negative can the other Au18 (staple)–S13 bond be completely broken, otherwise they would still form a strong chemical bond with an equilibrium distance of 2.40 Å (Fig. 2c). In addition, we observed that the fracture of the Au–S bonds is accompanied by a significant increase in the distance between the Au17 (surface) and Au18 (staple) atoms (Fig. 2d), and then shortened to 2.53 Å after SR desorption. Note that the more negative the applied potential, faster the breaking kinetics of the Au–S bond. For example, at the lower potential of −1.0 V, the Au17(surface)/Au18(staple)–S13 bonds are the most easily broken, which can be completely detached at around 1.13 ps and 1.79 ps (marked by the red star in Fig. 2b and c), respectively. Overall, our constant potential AIMD simulations clearly tracked the preference of proton attack for S atom in the electrochemical process, which is stably attached to S13 (Fig. 2e) while simultaneously inducing the Au–S bond separation and Au–Au bond change. Therefore, the surface of PtAu24 NC should spontaneously promote dethiolation under moderate electrochemical potential, and then expose the under-coordinated Au sites.
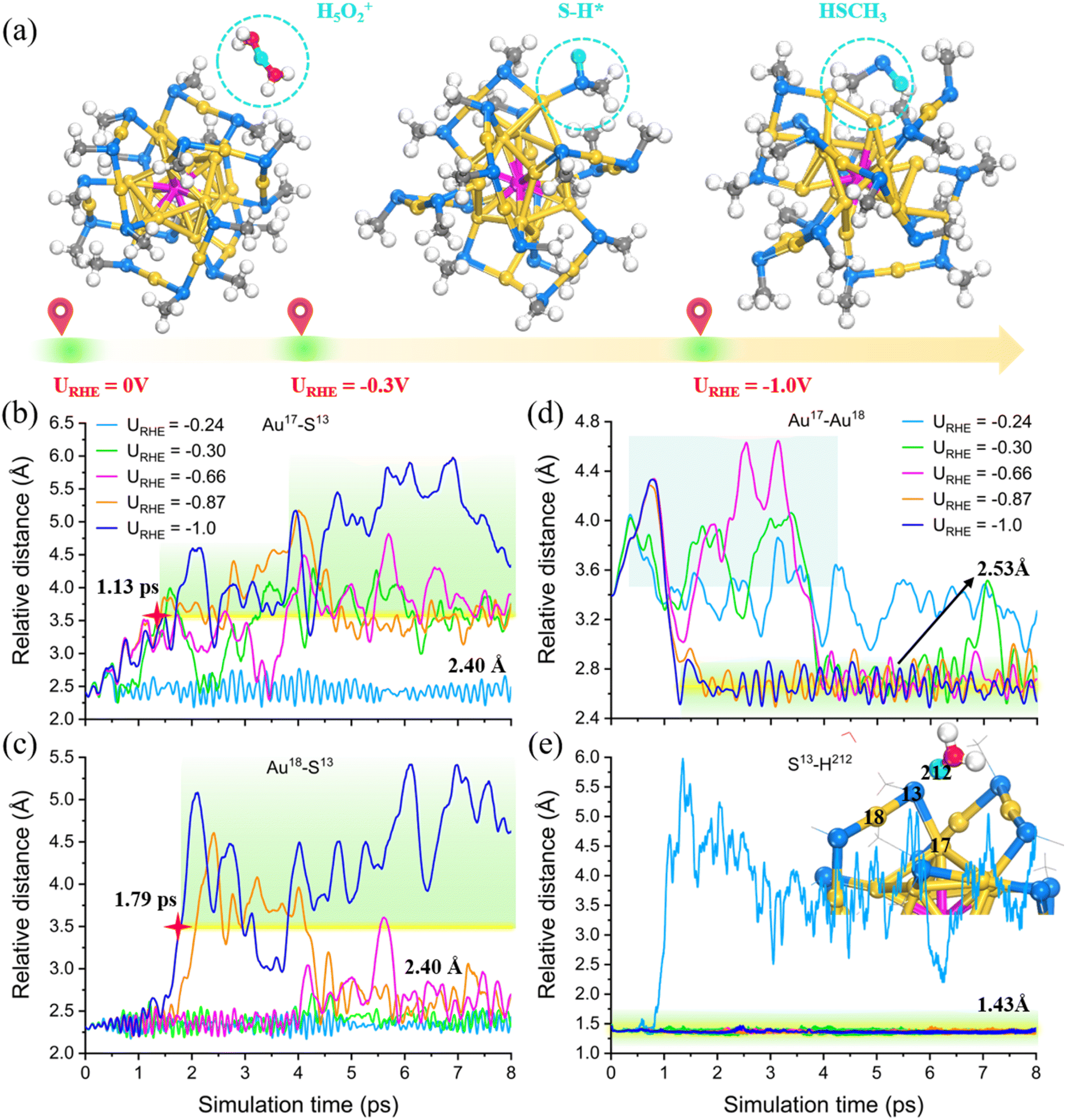 | ||
| Fig. 2 (a) Schematic illustration of the structural evolution of PtAu24 NC with the applied electrode potential. (b–e) Statistics of the relative distance between representative atoms during the equilibrated AIMD simulations under different potentials. The inset of (e) is the corresponding local structures. The transferred H atom from H3O+ to S atom is highlighted in turquoise. | ||
The dynamic origin of this unique SR etching behavior is attributed to the spatial accessibility around the S atom that can trigger the proton attack with the presence of its lone pair electron.22 What's more, the analysis of the radial distribution function (RDF) further confirmed that the average bond lengths of Au–Au, Au–Pt, Au–S and S–C bonds only fluctuated slightly before and after the ligand removal (Fig. S8†), suggesting that the PtAu24 NC could maintain the excellent structural stability under moderate electrochemical potential. However, it is critical to know whether the released –SR ligand can be recovered if the electrode potential is switched off to 0 V vs. RHE. This involves the evaluation of whether the active site can be stably retained after an electrochemical treatment. Fig. S9† verifies that the etching of the SR ligand is irreversible. Regardless of whether the Au–S bond is unilaterally or completely broken, the structural framework of the de-ligated PtAu24 is well maintained after converting the potential from −0.3 V and −1.0 V to 0 V. Obviously, the electrochemical treatment is an ideal strategy for creating stable active sites without destroying the structural framework of the metal NCs. However, in order to balance the electrocatalytic activity and the cluster stability, the applied potential should be moderate, otherwise the excessively negative potential can cause serious distortion of the metal framework (Fig. S7f,†URHE = −1.42 V).
According to the current kinetic results, it is undeniable that the –SCH3 ligand can spontaneously detach when subjected to a suitable reduction potential, yet the estimated threshold potential for the complete SR removal (around −1.0 V) needs to be further finalized. In the actual acidic electrolyte, since multiple protons are available on the catalyst surface, the exposed available surface Au site after the breaking of the first Au (surface)–S bond could be possibly passivated by the adsorbed proton (Fig. S10a†). To confirm this hypothesis, we constructed an adsorbed H on the exposed bridging Au sites for geometric relaxation at two different potentials. As shown in Fig. S10b and c,† as the potential becomes more negative, the distance of the other Au (staple)⋯S expands from 2.6 Å to 3.1 Å. Obviously, the hard-to-split Au (staple)–S bond is greatly weakened due to the binding of another H on the exposed surface Au site. This result emphasizes that the above threshold potential for ligand shedding (around −1.0 V) may be overestimated if the proton occupation at the bare Au site is ignored in the electrochemical process.
To this end, we further address this concern from a dynamic perspective. Based on the equilibrated AIMD snapshot of PtAu24(SCH3)18 with one broken Au (surface)–S bond at −0.3 V, we placed a H3O+ near the bridged Au atoms (Fig. S11a and b†) and similarly performed AIMD simulation lasting 8 ps under the same potential. As expected, the H from H3O+ eventually spontaneously capped the two exposed Au atoms in a bridging manner, and in the meantime, the adsorbed HSCH3 molecule was completely dissociated into the solution (Fig. S11c and d†). The corresponding locally magnified structures with key atomic markings from the initial and final AIMD snapshots can be seen in Fig. 3a and b. Specifically, the proton from H3O+ is stably attached to the two bare Au17 and Au18 atoms at a distance of ∼1.73 Å accompanied by the weakening of the Au (staple)–S bond, and the S13 atom remained disconnected from the surface Au17, and then completely separated from the other staple Au18 to quickly transform into a free HSCH3 molecule at ∼2.94 ps (Fig. 3c). The resultant two under-coordinated Au17 and Au18 atoms are then bonded closer with a shortened distance of 2.74 Å compared to that in the intact PtAu24 NC (3.4 Å). Obviously, the re-evaluated URHE = −0.3 V should be the real critical point for the complete dethiolation (Fig. 3d). Note that the adsorbed H* at this bridge site (marked by the green circle) should be an active state, and further desorption may occur under the reaction potential, which can then be replenished by adsorbing another proton from the solution. This process can be alternatively regarded as a Volmer reaction in the HER, therefore, it is reasonable to speculate that such exposed gold atoms could be the active HER sites.
 | ||
| Fig. 3 The corresponding local structures with some atomic markings from the initial (a) and final (b) AIMD snapshots of H adsorption to the bridging Au site in PtAu24(SCH3)18 after the breaking of the first Au (surface)–S bond under URHE = −0.3 V. (c) Statistics of the relative distance between representative atoms during the equilibrated AIMD simulation at URHE = −0.3 V. (d) Updated schematic illustration of the structural evolution of PtAu24 NC with electrode potential. | ||
Clearly, based on the above kinetic results, the thiolate-protected PtAu24 clusters would undergo facile dethiolation under a moderate electrochemical potential. Based on this, we applied a constant-potential method to revisit the HER activity of the singly dethiolated PtAu24(SCH3)17 with the consideration of a hybrid-solvent acid environment at pH = 3. After preliminary screening, the sites for H adsorption include the newly exposed Au–Au dual metals as well as the Pt and S atoms (Fig. S12†). Similarly, for each case, a set of calculations with a different system charge was conducted (Fig. S13†), which can establish the relationships between the free energy and the electrode potential (Fig. S14†). As shown in Fig. 4, the adsorbed H* is singly bound to the central Pt (Fig. 4a) and exterior S atom (Fig. 4b) with Pt–H and S–H bond length of 1.70 Å and 1.35 Å, but is readily adsorbed to the dethiolated Au–Au site in a bridging manner with a distance of 1.73 Å (Fig. 4c). Fig. S15† shows the corresponding free energy changes ΔG of the Volmer (ΔG1) and Heyrovsky (ΔG2) steps as a function of electrode potential URHE, wherein the results at a selected potential from −0.52 V to 0 V relevant for the HER experiments have been outlined separately in Fig. 4d for better clarity. For the S site, the Volmer H* adsorption has always been the PDS, while for the Au and Pt sites, the two elementary reactions both exhibit intersections around a potential of −0.07 V. In other words, the HER activity of the ligand S site is determined by the initial Volmer reaction, while the activity of Pt and the exposed bridging Au sites is determined by the Volmer reaction only when the potential is below −0.07 V, otherwise (−0.07 V < URHE < 0 V) it depends on the subsequent Heyrovsky reaction. Since the critical potential for the PDS crossover (−0.07 V) is almost close to 0 V, we can still assume that the HER performance at these three sites mainly relies on the Volmer step. A closer inspection of the Volmer H* adsorption (Fig. 4e) indicates that the central Pt site exhibits the most optimum H binding (|ΔG1| ≤ 0.15 eV) except for a better outperformance of the Au–Au bridge site in a narrow potential range of −0.08–−0.13 V. Note that the formation of H* on the dethiolated Au sites is thermodynamically more supported than the ligand S site above URHE = −0.27 V, which is reversed below this potential.
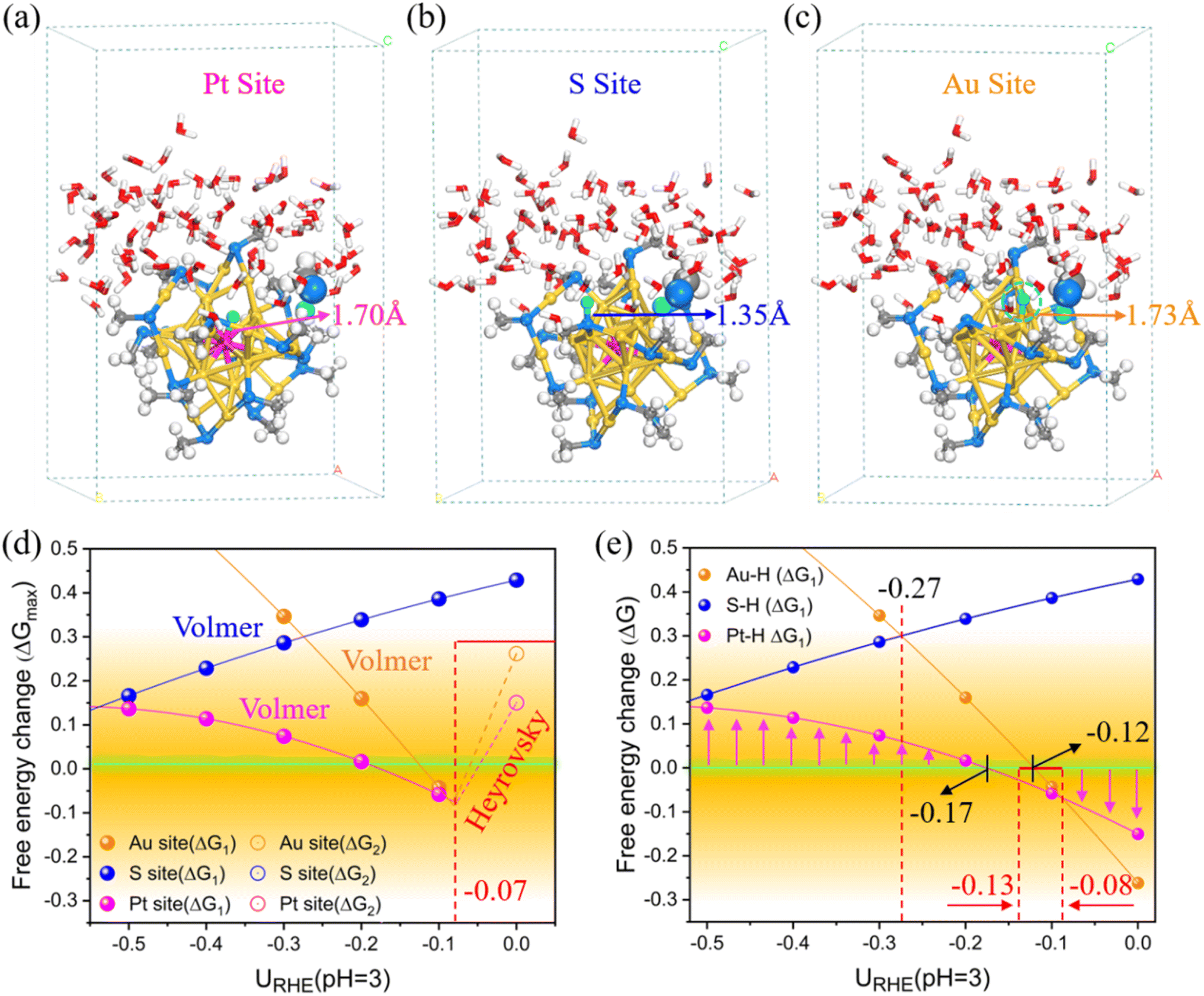 | ||
| Fig. 4 (a–c) Views of the geometric structures of three adsorbed H* systems at different sites on dethiolated PtAu24(SCH3)17 optimized under a zero charge (0e). Free energy changes of the (d) PDS (ΔGmax) and (e) Volmer reaction (ΔG1) as a function of the electrode potential URHE at pH = 3. | ||
Taking the Gibbs free energy of H adsorption |ΔG1| ≤ 0.3 eV as the reference criteria for an active HER catalyst, the comprehensive thermodynamic analysis suggests that both the Pt and the bare Au atoms are the favored active sites for the HER under a moderate reduction potential, however, when subjected to a more negative potential, the protons would prefer to bind at the Pt and exterior S atoms. This raises the question whether the similar Au–Au bridge sites, like the exposed Au sites in undoped Au25(SR)18 clusters,22 are also activated and possess high HER activity. To answer this, we also evaluated the HER performance of the activated Au sites in Au25 NCs at the same computational level. The constant-potential results are shown in Fig. S16,† and their energy–potential relationships are summarized in Fig. S17.† Apparently, a similar activity trend can be observed in the dethiolated Au25(SCH3)17 NC, where the exposed Au sites also exhibit excellent HER activity within a certain potential range (Fig. S16a†). The crossover potential of the two elementary reactions is at −0.2 V, indicating that the Volmer reaction is the PDS below −0.2 V, but above this potential the reaction is mainly limited by the Heyrovsky process. Compared with the doped PtAu24(SCH3)17 system, the performance of the exposed Au sites in both Au25(SCH3)17 and PtAu24(SCH3)17 are comparable, but the active potential range in Au25(SCH3)17 is wider (−0.6–−0.1 V, Fig. S16b†). However, given the ability to span a wider potential range (−1.3–−0.1 V) for an active HER (|ΔG1| ≤ 0.3 eV) and the extraordinarily active Pt site, the PtAu24(SCH3)17 shows overall superior HER performance than Au25(SCH3)17.
Noteworthily, since both the central Pt atom and the exposed Au sites are active for H adsorption, can these two adsorbed H* further combine to generate H2 through the Tafel step (*2H = H2 + *)? For this purpose, we conducted further calculations (Fig. S18 and S19†). If this mechanism is feasible, it is firstly necessary to ensure the binding of two H on the two active sites (Fig. S19a†). Our results show that when U ≤ −0.36 V, both the Pt and the bare bridging Au sites can each stably bond with an H (Fig. S19b†), and the adsorption of H on the exposed Au sites below −0.53 V (Volmer-2) is superior to the competitive Heyrovsky reaction on the Pt site (Fig. S19c†), which means that the subsequent Tafel step can be triggered only at potential lower than −0.53 V. However, further prediction of the reaction energy for the Tafel step indicates significant energy hindrance, with an endothermic ΔG up to 1 eV even at a very negative potential of −1.3 V (Fig. S19d†). Hence, the Volmer–Tafel mechanism is impossible, where the Tafel step is highly blocked, and our further constrained kinetic results also confirm this (Fig. S19e and S20,† the criterion for the end of this reaction is the formation of H2 (0.75 Å)), where a high barrier of 1.50 eV is required for the H2 formation at URHE = −0.60 V. Therefore, both the Pt and the bare Au sites would prefer the Volmer–Heyrovsky pathway. These observations indicate that the source of HER active sites in metal NCs is significantly influenced by the applied potential, and conducting detailed kinetic studies to track its structural evolution is of great significance in accurately revealing the underlying mechanism.
Overall, the investigated PtAu24 NCs undergo spontaneous electrochemical etching of the surface thiolate ligands under moderate potential, transforming from pre-synthesized fully ligand-protected catalysts into stable and truly catalytic dethiolated NCs. As shown in Fig. 5a, the originally inert Au site is activated due to dethiolation, and the adsorption of proton on the exposed bridge Au site becomes energetically favorable, greatly enhancing the Au–H interaction; the Pt–H bond is also enhanced under a higher potential, which is possibly due to the desorption of the electron-withdrawing –SR ligand that enhances the interaction between surface Au and staple Au while weakens the metal bond between Pt and the surface Au, thus strengthening the adhesion of H to Pt; however, the binding of the remaining exterior S atom to H is weakened after dethiolation. Our above analysis has identified that the HER activity of PtAu24 NCs mainly depends on the Volmer reaction (ΔG1), that is, the electrochemical adsorption of proton is the PDS. If the activity boundary is set as |ΔG1| ≤ 0.3 eV, then Pt and the bridged Au–Au atoms are both ideal HER active sites, while the S site tends to adsorb proton to assist in ligand stripping. As the number of detached ligands increases, the electrochemical etching of ligands would become increasingly difficult because of the gradually weakened affinity of the remaining S atom to H.
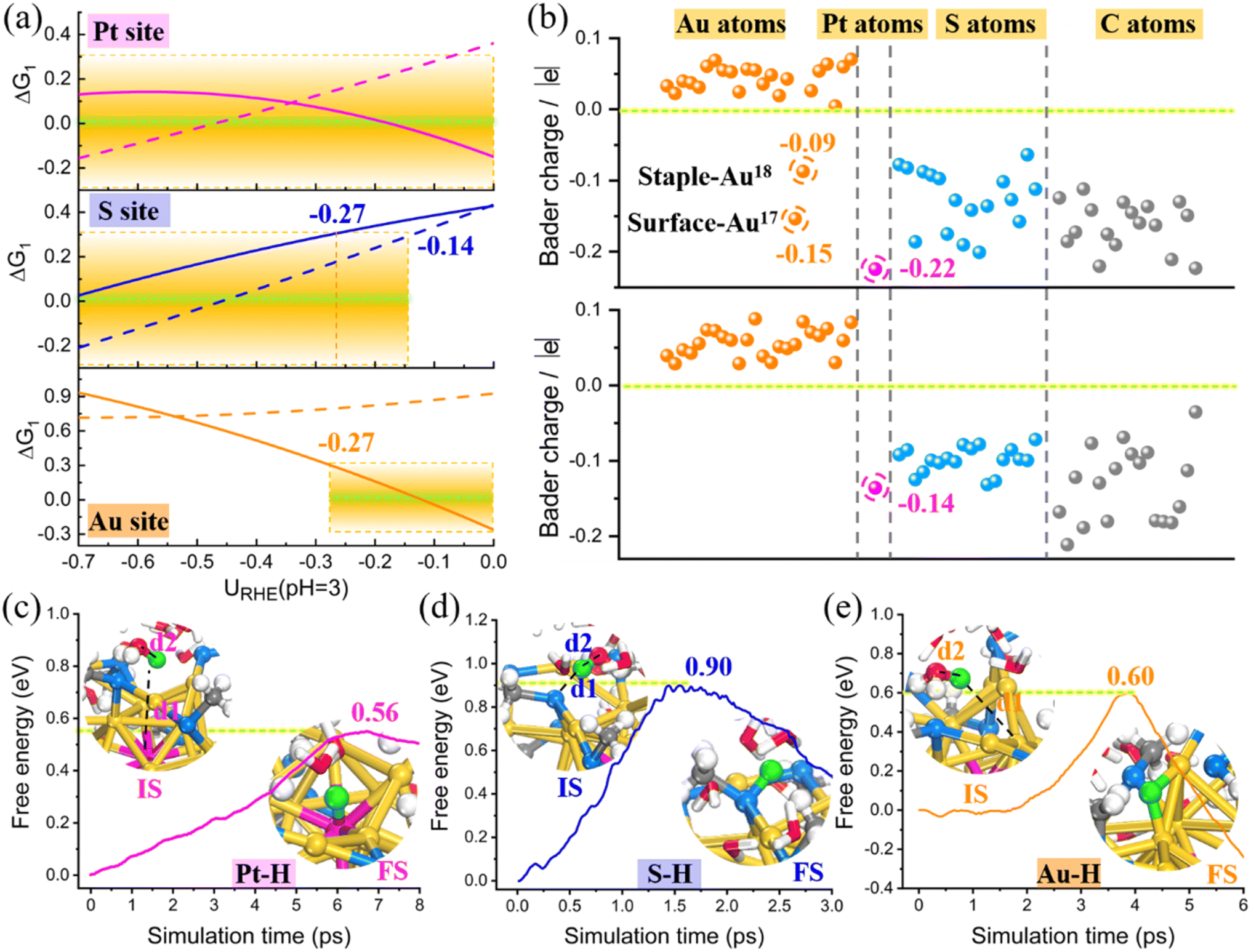 | ||
| Fig. 5 (a) Free energy changes of the Volmer reaction (ΔG1) at three different sites of intact PtAu24(SCH3)18 (dotted line) and dethiolated PtAu24(SCH3)17 (solid line) as a function of electrode potential URHE at pH = 3. The shaded area highlights the HER active region with |ΔG1| ≤ 0.3 eV. (b) Bader charge analysis of each atom in intact PtAu24(SCH3)18 (below) and dethiolated PtAu24(SCH3)17 (above). (c–e) Free energy profiles of solvated proton (H3O+) adsorption on three different sites in dethiolated PtAu24(SCH3)17 during the slow-growth simulation at Ucross = −0.1 V. The structures concerning the initial and final states are denoted as IS and FS, respectively. | ||
Furthermore, the dethiolation effect can also be reflected by the changes in Bader charge and projected density of states (PDOS) in PtAu24(SCH3)18 and PtAu24(SCH3)17 NCs. Fig. 5b clearly shows that only the exposed Au atoms have negative charges while the other thiolate-capped Au atoms show positive charges, indicating that the surface dethiolated Au is indeed activated for electrocatalysis. Although the central Pt atom is always negatively charged, its charge has significantly increased from 0.14 to 0.22 due to partial dethiolation, supporting the improvement in HER performance. In addition, the apparently negative charge of S atoms, combined with their spatial accessibility, can also explain their affinity for proton adsorption. The adsorption strength of H* is highly related to the electronic structure, and the d-states energy of active metal relative to the Fermi level is a good indicator of the bond strength. As shown in Fig. S21,† while the s-states change very little, the d-states of the active Pt and Au–Au atoms in PtAu24(SCH3)17 are located considerably higher in energy than those in intact PtAu24(SCH3)18, which explains its stronger H* adsorption. According to the quantified d-band center (εd), the εd of Pt (−1.64 eV) and bare Au (−2.43 eV) in dethiolated systems shift upward closer to the Fermi level compared with the fully ligand-protected NCs (−1.95 and −3.19 eV, respectively), also supporting the enhanced Pt–H/Au–H interactions. These results clearly explain the electronic origin of the extraordinary HER activity present in dethiolated PtAu24 NCs.
Combined with the thermodynamic calculations and the electronic structure analysis, we have successfully identified the possible HER active sites of PtAu24 and the activity trends. To better understand the concerned PDS Volmer reaction in dethiolated PtAu24 catalyst, the reaction kinetics are further examined. The “slow-growth” method was employed to monitor the free energy barrier for solvated proton (H3O+) adsorption on three different sites at −0.1 V. This potential is chosen because it is an approximate crossover potential (Ucross) between the two scaling lines in the ΔG1–U curve in Fig. 4e, where the Gibbs free energy of H adsorption at the Pt and Au site are equivalent, suggesting the comparable activity and similar Faraday efficiency for H2 production. The dynamic sampling results in Fig. 5c–e reveal that the H in H3O+ gradually separates with the elongation of the H⋯O bond, and is eventually donated to the active site. This step of proton transfer and adsorption has a lower free energy barrier for Pt and Au–Au bridge sites (0.56 and 0.60 eV), indicating the favorable kinetic promotion of H adsorption, while a relatively higher barrier of 0.9 eV is required at the S site. It has been known that the affinity of S atom for H from H3O+ should spontaneously facilitate the dethiolation and finally form a free HSCH3 molecule in solution. Therefore, it should be difficult to produce H2 through the subsequent Heyrovsky reaction after the adhesion of H to the S site. To verify this, we further performed constrained AIMD simulations to explore the protonation of H* by an explicit hydronium, i.e., H* + H3O+ + e− = H2 + * + H2O. Unsurprisingly, this process is greatly hampered at the S site, with a reaction free energy up to 2.08 eV (Fig. S22†). However, this Heyrovsky process is nearly barrierless at the Pt (0.25 eV) and dethiolated Au sites (0.27 eV) (Fig. S23†). Thus the HER activity of dethiolated PtAu24 should be mainly contributed by the Pt and Au atom, which have comparable reaction kinetics.
To verify our theoretical predictions, we then synthesized the Au24Pt(SCH2CH2Ph)18 (Au24Pt in short) and Au25(SCH2CH2Ph)18 (Au25 in short) nanoclusters and investigated their HER performances. Moreover, the attenuated total reflection surface-enhanced infrared spectroscopy (ATR-SEIRAS) technique was used to further study the reaction kinetics of Au25 and Au24Pt in the HER process, and the different roles of Au and Pt active sites in the HER process were also identified clearly.
Fig. 6a shows the UV-vis absorbance of Au24Pt and Au25 in CH2Cl2. The characteristic absorption peaks of Au24Pt are located at 596 nm and 369 nm, while the characteristic absorption peaks of Au25 are situated at 440 nm and 676 nm, and there are two weak absorption bands at 395 and 790 nm. The absorbance feature of these two clusters is in good agreement with the results reported previously.18,22 The molecular composition of Au24Pt NC was then determined by electrospray ionization mass spectrometry (ESI-MS). As shown in Fig. 6b, it only displays one main peak in the m/z ranging from 3000 to 6500 Da. Careful analysis of the high-resolution mass data reveals that the main peak at m/z = 3695.9547 Da is ascribed to [Au24Pt(SCH2CH2Ph)18]2+, whose isotopic pattern perfectly matches the simulated pattern (see the inset, Cal.: 3695.9505 Da, deviation: 0.0042 Da). Besides, the ESI-MS spectra also show a strong diffraction peak at m/z = 3942.9692, which belongs to [Au25Pt(SCH2CH2Ph)20]2+ (=[Au24Pt(SCH2CH2Ph)18]2+ + Au(SCH2CH2Ph)2− + Na+, Cal.: 3942.9855 Da, deviation: 0.0163 Da). The results of UV-vis absorption and ESI-MS concurrently confirm that high purity Au24Pt(SCH2CH2Ph)18 and Au25(SCH2CH2Ph)18 NCs were obtained.
 | ||
| Fig. 6 (a) The UV-vis absorbance spectra of Au24Pt and Au25 in DCM (a) and ESI-MS spectra of Au24Pt, the inset in (b) is the comparison figure of experimental and simulated isotope patterns. (c) LSV curves of commercial Pt/C, Au24Pt and Au25 after activation in 0.5 M Na2SO4 (pH = 3). (d) Corresponding Tafel plots of Au24Pt and Au25 after activation. | ||
We subsequently employed these two NCs as model catalysts for the HER. Fig. 6c presents the polarization curve of commercial Pt/C, Au24Pt and Au25 after electrochemical activation, and the required overpotentials are 301 mV, 242 mV and 288 mV, respectively, to afford a current density of 10 mA cm−2, indicating that the HER catalytic activity could be significantly enhanced by replacing the central Au of the Au25 cluster with Pt, which even surpasses the commercial Pt/C catalyst. It is worth noting that, without activation, the overpotentials of Au24Pt and Au25 under the same conditions are as high as 587 and 599 mV, respectively (Fig. S24†). The enhanced performance is caused by the effective exposure of the active site due to ligand shedding from the cluster surface after activation, which is more conducive to the adsorption and desorption of H* on the catalyst surface. Further characterization of electrochemical activation also confirmed that ligands on the surface of Au24Pt decreased as the applied potential decreased, as shown in Fig. S25,† which was consistent with Au25.22 However, under the same conditions, the number of remaining ligands on the surface of Au24Pt is significantly higher than that of Au25, indicating that Au24Pt has higher stability in an electrochemical environment. In addition, the XPS spectra of Au24Pt at different activation potentials revealed that the binding energy of the Au 4f orbitals is almost identical to the original Au24Pt (Fig. S25b†), further demonstrating that the Au active sites on the Au24Pt surface are not further reduced after ligand detachment, and the overall structure of Au24Pt remains intact during the electrochemical testing. The Tafel plots of the two tested samples are shown in Fig. 6d, and the Tafel slope of Au24Pt (113 mV dec−1) is lower than that of Au25 (127 mV dec−1), implying that the energy barrier for its rate-determining step (RDS) in the Volmer reaction is lower, and its activity is better.
We further performed the electrochemical impedance spectroscopy (EIS) and electrochemical active surface area tests, and the EIS fitted results are compiled in Table S1.† As shown in Fig. S26a and S27,† Au24Pt exhibits a much smaller semi-circle than Au25, indicating faster reaction kinetics. Based on the simulation results of the equivalent circuit diagram (insets in Fig. S27†), the Rct (charge transfer resistance) values for the activated Au24Pt and Au25 are 74.19 and 148.1 Ω, respectively, suggesting that Au24Pt exhibits markedly lower charge transfer resistance than Au25. To further understand the difference in their HER performances, the electrochemical active surface area (ECSA) values of the two samples were compared. The cyclic voltammetry curves of the two samples at different scanning rates are shown in Fig. S28,† and the double layer capacitance (Cdl) of the two samples can be obtained. The Au24Pt catalyst (1.06 mF cm−2) has a larger capacitance than the Au25 sample (0.87 mF cm−2) (Fig. S26b†). Note that the ECSA value is linearly proportional to the Cdl value, therefore, Au24Pt has a much larger ECSA value, which accounts for the superior HER activity. Similar to the results of the pre-activation polarization curve, the impedance and Cdl values of the two samples before activation are much lower than those after activation (Fig. S26†). It is noteworthy that Au24Pt exhibits a larger ECSA value than Au25. Therefore, determining whether the superior HER performance of Au24Pt compared to Au25 is due to the difference in intrinsic electrochemical activity or merely the effect of the area becomes necessary.20 The HER polarization curves of Au25 and Au24Pt normalized by ECSA are shown in Fig. S29.† At the same current density, the overpotential of Au24Pt is significantly lower than that of Au25, indicating that the superior HER performance of activated Au24Pt arises indeed from its higher intrinsic electrochemical activity compared to Au25.
So far, the experimental results are consistent with our theoretical predictions, that is, Au24Pt shows better HER performance than Au25. However, the underlying mechanism predicted by theory at the atomic level also needs to be further verified by the experimental data, especially the specific roles of Au and Pt in the HER. To that end, the ATR-SEIRAS technology was employed to investigate the reaction kinetics and key adsorption species of Au24Pt and Au25 in the HER. As shown in Fig. 7a, the ATR-SEIRAS test is carried out in a special H-cell where the catalyst was cast on the surface of a monocrystalline Si coated with an Au film. The Au film is able to express a surface-enhanced infrared absorption effect for the adsorbed species on the catalyst surface.32
 | ||
| Fig. 7 (a) Schematic of in situ ATR-SEIRAS configuration; a carbon rod electrode, Au film coated Si crystal and Ag/AgCl were used as the counter electrode, working electrode and reference electrode, respectively. The in situ ATR-SEIRAS spectra of Au24Pt (b) and Au25 (d) for the HER at different potentials in 0.5 M Na2SO4 (pH = 3). The 2D ATR-SEIRAS contour map of Au24Pt (c) and Au25 (e) for the HER. | ||
As shown in Fig. 7b and c, for Au24Pt, with the decrease of the applied potential, the intensity of the absorption peaks at 3374 cm−1and 1650 cm−1 from the adsorbed *H2O and *OH increases.33,34 From the in situ ATR-SEIRAS spectra in Fig. 7b and d, the absorption peaks for water in Au24Pt are higher and sharper compared to Au25. The difference in the position of the water peaks was caused by the different adsorption capacity of the catalyst to water. Au24Pt has stronger adsorption capacity for water, resulting in the shift of the water peak of Au24Pt to a lower wavenumber. Moreover, the two-dimensional (2D) spectrum indicates the absorbance through the contrast of colors and sets the highest peak as red. According to Fig. 7b and d, as the potential increases, the absorption peak for water in Au25 is comparatively lower, consequently, when plotting the 2D contour map (Fig. 7c and e), the water peak of Au24Pt is green and that of the Au25 is yellow. Meanwhile, two weak absorption bands are also observed at ∼1370 cm−1 and ∼2100 cm−1, and the absorption peak at 1370 cm−1 corresponds to the bridged Au–H* bonding.35 To obtain more in-depth understanding, we also carried out in situ infrared (ATR-SEIRAS) tests on Au25 and commercial Pt/C (20 wt%) catalyst under the same conditions. The strong absorption peaks of *H2O and *OH can be easily identified in the in situ infrared spectrum of the Pt/C catalyst (Fig. S30†). One may notice that there is one peak location slightly different from that of the cluster catalyst, specifically, Pt/C has an obvious absorption peak at 2024 cm−1, which is a typical absorption peak of the Pt–H* bonding.36 In the case of Au25, in addition to *H2O and *OH, the adsorption of bridging Au–H* bonding was observed at 1370 cm−1 (Fig. 7d and e) and its intensity was higher than that in Au24Pt, indicating that the Au active sites in Au25 have a better adsorption capacity for bridging H* than that in Au24Pt. Such a finding is highly consistent with the activity of the exposed Au sites of PtAu24vs. Au25 as we predicted earlier (Fig. S16b†). More importantly, there is an obvious absorption peak at 2087 cm−1, while no peak can be identified at this position in Pt/C, indicating that the adsorption species is independent of the carbon substrate. Moreover, the infrared absorption peak of the S–H bond (∼2500 cm−1) is also distinguished from this peak. Note that the shape and position of this peak are very close to Pt–H* (∼2024 cm−1). Since Au and Pt are adjacent elements in the periodic table, it can be concluded that the absorption peak at 2087 cm−1 corresponds to the adsorption state of the staple Au–H* bonding. In addition, we also performed the cyclic voltametric measurements for Au25 and Au24Pt before and after activation to understand their surface features at −0.2 V to 1.45 V (vs. Ag/AgCl). As illustrated in Fig. S31,† the voltammetric response of Au25 and Au24Pt before activation displayed only the capacitive features, indicating that the surface is passivated with thiol ligands. The activated clusters exhibit a higher current density, and the voltammetry of Au24Pt shows the oxidation peak of Au and the cathodic gold oxidation–reduction peak, but the reduction peak of oxidized Pt is not observed. The voltammetries of Au24Pt and Au25 are very similar overall, indicating that Au24Pt exposes Au sites during the electrochemical reaction process.
Based on the above analysis and the corresponding adsorption states of different catalysts, we believe that the absorption peak at ∼2100 cm−1 in the in situ infrared spectrum of Au24Pt should correspond to staple Au–H* and Pt–H*. Although Pt has a better adsorption capacity for H* than Au, the Pt atom in Au24Pt is located in the center of the cluster and there is only one single Pt atom, thus the Pt–H* infrared absorption peak generated by the active Pt site is much weaker than the Pt/C. Meanwhile, the doping of Pt would weaken the adsorption of H* by the peripheral Au active sites in Au24Pt, resulting in the weakening of the absorption peaks of both staple Au–H* and bridged Au–H* bonds, consequently leading to the formation of a weak and wide absorption band at ∼2100 cm−1. These results indicate that the excellent HER performance of Au24Pt is not only due to the adsorption capacity of the central Pt for H*, but also ascribed to the strong adsorption capacity of the Au active sites for *H. The synergistic effect of the Pt and Au makes Au24Pt an excellent electrocatalyst for the HER.
Our current experiments and calculations have confirmed that thiolate-protected metal clusters in electrochemical processes can spontaneously promote dethiolation and expose available metal sites, thus significantly improving their catalytic activity. From this, we can infer that metal clusters protected by other ligands may have similar structural evolution behavior during electrochemical processes, such as halogen ligands and alkynyl ligands. Some recent studies have shown that ligands on these surfaces should also partially shed during electrocatalysis,37,38 indicating that the spontaneous desorption of ligands on metal clusters under electrochemical treatment may be a relatively common behavior, which further stimulates the far-reaching significance of our study.
Conclusion
In summary, using the prototype PtAu24(SCH3)18 NC as a model catalyst, we tracked its dynamic structural evolution under electrochemical potential via advanced constant potential AIMD simulations, and observed the spontaneous desorption of peripheral thiolate ligands. The activity prediction indicates that the dethiolated PtAu24 exhibits excellent HER activity, which is contributed not only by the suitable adsorption capacity of the central Pt atom for H*, but also by the bonding affinity of exposed Au active sites for H*, where the two exhibit comparable reaction kinetics. The synergistic effect between Pt and Au makes PtAu24 an extraordinary HER electrocatalyst, which even surpasses the state-of-the-art platinum catalyst. Electrochemical measurements and in situ FTIR (ATR-SEIRAS) characterization further validated our predictions. Our research indicates that the surface passivated Au sites can be reactivated under moderate electrochemical potential, which is sensitive to external potential and can even exhibit high electrocatalytic HER activity comparable to Pt, thus needs to be given special attention when unraveling the reaction mechanisms of ligand-protected metal nanoclusters.Data availability
The data supporting this article, including the computational and experimental details, the simulation results, and the electrochemical characterization data, have been included as part of the ESI.†Author contributions
Q. T. conceived the idea. F. S. performed the theoretical calculation. L. Q. performed the experiment and analyzed the data under the guidance of Z. T. F. S. wrote the manuscript with inputs from all authors. Q. T. and Z. T. finalized the manuscript, all the authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21903008), the Chongqing Science and Technology Commission (cstc2020jcyj-msxmX0382), and the financial support from Guangdong Natural Science Funds (No. 2023A0505050107).References
- J. D. Holladay, J. Hu, D. L. King and Y. Wang, An overview of hydrogen production technologies, Catal. Today, 2009, 139, 244–260 CrossRef CAS.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Advancing the Electrochemistry of the Hydrogen-Evolution Reaction through Combining Experiment and Theory, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- R. M. Bullock, A. M. Appel and M. L. Helm, Production of hydrogen by electrocatalysis: making the H-H bond by combining protons and hydrides, Chem. Commun., 2014, 50, 3125–3143 RSC.
- V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Complexes of earth-abundant metals for catalytic electrochemical hydrogen generation under aqueous conditions, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- X. Tian, P. Zhao and W. Sheng, Hydrogen Evolution and Oxidation: Mechanistic Studies and Material Advances, Adv. Mater., 2019, 31, 1808066 CrossRef PubMed.
- Y. Zheng, Y. Jiao, A. Vasileff and S.-Z. Qiao, The Hydrogen Evolution Reaction in Alkaline Solution: From Theory, Single Crystal Models, to Practical Electrocatalysts, Angew. Chem., Int. Ed., 2018, 57, 7568–7579 CrossRef CAS PubMed.
- F. Sun, Q. Tang and D.-E. Jiang, Theoretical Advances in Understanding and Designing the Active Sites for Hydrogen Evolution Reaction, ACS Catal., 2022, 12, 8404–8433 CrossRef CAS.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. B. Chorkendorff and J. K. Norskov, Computational high-throughput screening of electrocatalytic materials for hydrogen evolution, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. J. S. Kornberg, Structure of a Thiol Monolayer-Protected Gold Nanoparticle at 1.1 Å Resolution, Science, 2007, 318, 430–432 CrossRef CAS PubMed.
- M. Zhou, T. Higaki, G. Hu, M. Y. Sfeir, Y. Chen, D.-e. Jiang and R. Jin, Three-orders-of-magnitude variation of carrier lifetimes with crystal phase of gold nanoclusters, Science, 2019, 364, 279–282 CrossRef CAS PubMed.
- Y. Lu and W. Chen, Sub-nanometre sized metal clusters: from synthetic challenges to the unique property discoveries, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC.
- K. Kwak and D. Lee, Electrochemistry of Atomically Precise Metal Nanoclusters, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS PubMed.
- X. Ma, Z. Tang, L. Qin, J. Peng, L. Li and S. Chen, Unravelling the formation mechanism of alkynyl protected gold clusters: a case study of phenylacetylene stabilized Au144 molecules, Nanoscale, 2020, 12, 2980–2986 RSC.
- R. Jin, R. Jin, S. Zhao, C. Liu, M. Zhou, G. Panapitiya, Y. Xing, N. L. Rosi and J. P. Lewis, Controlling Ag-doping in [AgxAu25-x(SC6H11)18] nanoclusters: cryogenic optical, electronic and electrocatalytic properties, Nanoscale, 2017, 9, 19183–19190 RSC.
- C. A. Fields-Zinna, M. C. Crowe, A. Dass, J. E. F. Weaver and R. W. Murray and Colloids, Mass Spectrometry of Small Bimetal Monolayer-Protected Clusters, Langmuir, 2009, 25, 7704–7710 CrossRef CAS PubMed.
- S. Wang, Y. Song, S. Jin, X. Liu, J. Zhang, Y. Pei, X. Meng, M. Chen, P. Li and M. Zhu, Metal Exchange Method Using Au25 Nanoclusters as Templates for Alloy Nanoclusters with Atomic Precision, J. Am. Chem. Soc., 2015, 137, 4018–4021 CrossRef CAS PubMed.
- H. Qian, D.-E. Jiang, G. Li, C. Gayathri, A. Das, R. R. Gil and R. Jin, Monoplatinum Doping of Gold Nanoclusters and Catalytic Application, J. Am. Chem. Soc., 2012, 134, 16159–16162 CrossRef CAS PubMed.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D. E. Jiang and D. Lee, A molecule-like PtAu24(SC6H13)18 nanocluster as an electrocatalyst for hydrogen production, Nat. Chem., 2017, 8, 14723 Search PubMed.
- K. Kwak, Q. Tang, M. Kim, D. E. Jiang and D. Lee, Interconversion between Superatomic 6-Electron and 8-Electron Configurations of M@Au24(SR)18 Clusters (M = Pd, Pt), J. Am. Chem. Soc., 2015, 137, 10833–10840 CrossRef CAS PubMed.
- P. Mymoona, J. V. Rival, Nonappa, E. S. Shibu and C. Jeyabharathi, Platinum-Grafted Twenty-Five Atom Gold Nanoclusters for Robust Hydrogen Evolution, Small, 2024, 20, 2308610 CrossRef CAS PubMed.
- W. Choi, G. Hu, K. Kwak, M. Kim, D.-E. Jiang, J.-P. Choi and D. Lee, Effects of metal-doping on hydrogen evolution reaction catalyzed by MAu24 and M2Au36 nanoclusters (M= Pt, Pd), ACS Appl. Mater. Interfaces, 2018, 10, 44645–44653 CrossRef CAS PubMed.
- F. Sun, L. Qin, Z. Tang, G. Deng, M. S. Bootharaju, Z. Wei, Q. Tang and T. Hyeon, SR removal or –R removal? A mechanistic revisit on the puzzle of ligand etching of Au25(SR)18 nanoclusters during electrocatalysis, Chem. Sci., 2023, 14, 10532–10546 RSC.
- X. Zhao and Y. Liu, Unveiling the Active Structure of Single Nickel Atom Catalysis: Critical Roles of Charge Capacity and Hydrogen Bonding, J. Am. Chem. Soc., 2020, 142, 5773–5777 CrossRef CAS PubMed.
- O. López-Estrada, N. Mammen, L. Laverdure, M. M. Melander, H. Häkkinen and K. Honkala, Computational Criteria for Hydrogen Evolution Activity on Ligand-Protected Au25-Based Nanoclusters, ACS Catal., 2023, 13, 8997–9006 CrossRef.
- G. Hu, Q. Tang, D. Lee, W. Zili and D.-E. Jian, Metallic Hydrogen in Atomically Precise Gold Nanoclusters, Chem. Mater., 2017, 29, 4840–4847 CrossRef CAS.
- D. R. Kauffman, D. Alfonso, C. Matranga, P. Ohodnicki, X. Deng, R. C. Siva, C. Zeng and R. Jin, Probing active site chemistry with differently charged Au25q nanoclusters (q = -1, 0, +1), Chem. Sci., 2014, 5, 3151–3157 RSC.
- T. Cheng, L. Wang, B. V. Merinov and W. A. Goddard III, Explanation of dramatic pH-dependence of hydrogen binding on noble metal electrode: Greatly weakened water adsorption at high pH, J. Am. Chem. Soc., 2018, 140, 7787–7790 CrossRef CAS PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. Arias and R. G. Hennig, Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- K. Mathew, V. Kolluru, S. Mula, S. N. Steinmann and R. G. Hennig, Implicit self-consistent electrolyte model in plane-wave density-functional theory, J. Chem. Phys., 2019, 151, 234101 CrossRef PubMed.
- T. Giorgino, Computing 1-D atomic densities in macromolecular simulations: the Density Profile Tool for VMD, Comput. Phys. Commun., 2014, 185, 317–322 CrossRef CAS.
- K. T. Wikfeldt, M. Leetmaa, M. P. Ljungberg, A. Nilsson and L. G. M. Pettersson, On the Range of Water Structure Models Compatible with X-ray and Neutron Diffraction Data, J. Phys. Chem. B, 2009, 113, 6246–6255 CrossRef CAS PubMed.
- X.-Y. Ma, W.-Y. Zhang, K. Ye, K. Jiang and W.-B. Cai, Electrolyte-Layer-Tunable ATR-SEIRAS for Simultaneous Detection of Adsorbed and Dissolved Species in Electrochemistry, Anal. Chem., 2022, 94, 11337–11344 CrossRef CAS PubMed.
- S. Liu, T. Qian, M. Wang, H. Ji, X. Shen, C. Wang and C. Yan, Proton-filtering covalent organic frameworks with superior nitrogen penetration flux promote ambient ammonia synthesis, Nat. Catal., 2021, 4, 322–331 CrossRef CAS.
- S. Zhu, X. Qin, F. Xiao, S. Yang, Y. Xu, Z. Tan, J. Li, J. Yan, Q. Chen, M. Chen and M. Shao, The role of ruthenium in improving the kinetics of hydrogen oxidation and evolution reactions of platinum, Nat. Catal., 2021, 4, 711–718 CrossRef CAS.
- J. Dong, Z.-H. Gao, Q.-F. Zhang and L.-S. Wang, The Synthesis, Bonding, and Transformation of a Ligand-Protected Gold Nanohydride Cluster, Angew. Chem., Int. Ed., 2021, 60, 2424–2430 CrossRef CAS PubMed.
- H. Shi, T. Wang, J. Liu, W. Chen, S. Li, J. Liang, S. Liu, X. Liu, Z. Cai, C. Wang, D. Su, Y. Huang, L. Elbaz and Q. Li, A sodium-ion-conducted asymmetric electrolyzer to lower the operation voltage for direct seawater electrolysis, Nat. Commun., 2023, 14, 3934 CrossRef CAS PubMed.
- H. Seong, K. Chang, F. Sun, S. Lee, S. M. Han, Y. Kim, C. H. Choi, Q. Tang and D. Lee, ClAg14(C≡CtBu)12 Nanoclusters as Efficient and Selective Electrocatalysts Toward Industrially Relevant CO2 Conversion, Adv. Sci., 2024, 11, 2306089 CrossRef CAS PubMed.
- Z. Chen, F. Sun and Q. Tang, Thermal Stability and Electronic Properties of N-Heterocyclic Carbene-Protected Au13 Nanocluster and Phosphine-Protected Analogues, J. Phys. Chem. Lett., 2023, 14, 10648–10656 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04212c |
| ‡ F. S. and L. Q. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |