
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective lignin depolymerization via transfer hydrogenolysis using Pd/hydrotalcite catalysts: model compounds to whole biomass†
Darren
Dolan
a,
Rebekah
Brucato
a,
Christopher
Reid
 a,
Adam F.
Lee
*b,
Karen
Wilson
*b and
Adelina M.
Voutchkova-Kostal
*a
a,
Adam F.
Lee
*b,
Karen
Wilson
*b and
Adelina M.
Voutchkova-Kostal
*a
aChemistry Department, The George Washington University, 800 22nd St NW, Washington D.C. 20910, USA. E-mail: avoutchkova@gwu.edu
bCentre for Catalysis and Clean Energy, Griffith University, Gold Coast QLD 4222, Australia. E-mail: adam.lee@griffith.edu.au
First published on 7th November 2024
Abstract
Cleavage of lignin ether bonds via transfer hydrogenolysis is a promising route to valorize lignin, thus processes that use mild reaction conditions and exploit renewable hydrogen donor solvents (rather than molecular hydrogen) are economically advantageous. Herein we demonstrate the efficient catalytic transfer hydrogenolysis and tandem decarbonylation of lignin model compounds possessing aromatic ether bonds (α-O-4, β-O-4 and 4-O-5 linkages), over transition metal-modified Pd hydrotalcite catalysts with ethanol as the hydrogen donor and solvent. Quantitative conversions and yields were attained for all model compounds, except for 4-O-5 models, which possess inherently strong sp2 C–O bonds. The latter demonstrates the utility of Pd hydrotalcite catalysts for transfer hydrogenolysis of model compounds. This process was employed to achieve whole pine biomass delignification with 97% yield and a 22% phenolic monomer yield, with 64% selectivity for 4-(3-hydroxypropyl)-2-methoxyphenol.
Introduction
The need to transition the manufacture of platform chemicals from fossil to non-food competitive, renewable resources is emphasized in Green Chemistry principles,1 the newly-defined term of “sustainable chemistry”,2 and is well-aligned with the UN Sustainable Development goals 7, 9 and 11.3 Lignin has received considerable interest as a non-food competitive, renewable resource that can yield high quality aromatics through selective depolymerization.4–6 Currently, ∼100 million tons per year of lignin are produced as waste from paper and ethanol manufacture, and the vast majority (>98%) is incinerated for energy recovery,7 which does not harness its potential as a source of chemical feedstocks. While there is a clear opportunity present, chemical manufacturing from lignin requires scalable, selective, energy-efficient, and clean depolymerization processes which are economically viable.The asymmetric and non-repeating nature of the lignin biopolymer presents a multitude of ether linkages through which to effect depolymerization, although the diversity of β-O-4, α-O-4, and 4-O-5 ether linkages and associated monomeric units is itself a challenge for selective depolymerization under mild conditions. Such C–O linkages have lower bond dissociation energies compared to C–C linkages (C–O: 210–290 kJ mol−1vs. C–C: 450–500 kJ mol−1)8 and hence are the chemical functionality targeted in the depolymerization of native lignin. Cleavage of C–C linkages often requires harsh conditions that in turn may result in undesired degradation and aromatic ring hydrogenation.
Most depolymerization strategies rely on hydrogenolysis with exogenous hydrogen derived from fossil sources.9–25 Alternative strategies, such as oxidation,26–31 hydrolysis,32–38 hydrothermal,39–42 photo-43,44 and electrochemical45 transformation of lignin have also been explored, with transfer hydrogenolysis (TH) being particularly promising for selective depolymerization under relatively mild conditions without molecular H2. TH is advantageous for minimizing energy input and avoiding undesired aromatic ring hydrogenation. Although some evidence suggests that hydrogen can be sourced directly from biomass during its valorization,46 the lack of a hydrogen donating solvent requires longer reaction times or is altogether insufficient for complete depolymerization. A homogenous Ru-Xantphos catalyst is reported to facilitate dehydrogenation of benzylic alcohol in β-O-4 compounds, but not γ-alcohols whose presence inhibits hydrogenolysis.47 Use of homogeneous catalysts for large-scale, continuous processing of whole biomass is problematic due to the challenges of product separation, catalyst reuse, waste minimization and expensive ligands, and is associated with poorer energy economy coefficients and environmental energy impact factor.48 The predominant hydrogen sources for TH of lignin model compounds are currently formic acid, formate salts and isopropyl alcohol – all derived from non-renewable sources.49,50 Shen and co-workers recently demonstrated an ethanol/isopropanol mix as solvent and hydrogen donor (H-donor) at very high temperature (270 °C) for the transfer hydrogenolysis of organosolv poplar lignin over high loading NiCu bimetallic catalysts,51 an approach that does not address the challenge of lignin fractionation from holocelluose pulp associated with processing whole biomass. In contrast, the use of ethanol as a hydrogen donor for TH is highly desirable due to its availability from fermentation of renewable sugars and starches, as well as its relatively low cost and low toxicity compared to alternative donors.52,53 Ethanol is reported as a hydrogen donor for the reductive catalytic fractionation (RCF) of birch, poplar, spruce, and pine under forcing conditions (e.g., 210 °C and 15 h).54 Aqueous ethanol was also used a solvent for the RCF of hybrid poplar clones in the presence of 0.36 M HCl (at 175 °C)55 over commercial 5 wt% Pd/C catalysts, albeit hemicellulose (not ethanol) was proposed as the internal H donor. Ethanol is not only an excellent external H-donor for TH and activation of cationic metal pre-catalysts, but also has good dissolution and solvolytic properties for lignin,55,56 providing multiple advantages for use in transfer hydrogenolysis.
Catalytic TH of lignin model compounds has been reported (Table 1 and Fig. S1†); however, tandem catalytic TH and decarbonylation over a single catalyst in a one-pot reaction is, to our knowledge, previously unreported. Optimizing the activity and selectivity of multifunctional catalysts capable of such tandem transformations is challenging, since the reaction steps can be interdependent. High selectivity requires control over the electronic and geometric properties of catalytically active species (and their spatial distribution), and mechanistic insight of underlying structure–activity relationships, including the role of catalyst supports.63 Efforts by our group and others to develop heterogeneous Pd catalysts on active, tunable supports for organic synthesis and biomass valorization have exploited cooperativity between strongly interacting support matrices and metals to regulate metal speciation, reactivity and stability.64–71 MgO and γ-Al2O3 supported metal catalysts exhibit a range of acid–base properties:72–75 for example, Saad et al. reported that Pt/MgO displays strong basicity, whereas Pt/Al2O3 displays moderate acidity;71 similar observations were made by Groppo et al. for Pd analogues.76 Layered double hydroxides (LDHs) possess tunable acidity and basicity and can support a number of transition metals, yielding supported catalysts that facilitate a multitude of catalytic transformations. LDHs comprise layers of metal hydroxides sandwiched between layers of weakly bound anions (and often water). Hydrotalcites (HTs) are a subset of LDHs, with the formula [M1−x2+Mx3+(OH)2]x+(An−)x/n·mH2O, wherein M2+ and M3+ are commonly Mg2+ and Al3+ (or interchangeable alkali earth and transition metal cations).77 Although HTs exhibit solid basicity approaching that of MgO,78 their variable cationic charge means that both O2− Lewis base and Al3+ Lewis acid centres are accessible to substrates.79,80 Catalytic applications of Pd/LDH (or HT) include aldehyde decarbonylation,66 aldol condensation64 and acceptorless alcohol and amine dehydrogenation,65,81 olefination of carbonyls via aldol-decarbonylative coupling64 and deoxygenative olefination of alcohols,82 among others.
| Substrate | Conditions | Products (yield/%) | Conversion/% | Whole lignin (monomer yield/%) | Renewable H-donor | Ref. |
|---|---|---|---|---|---|---|
| a Number of ether linkages between monolignols, and hence theoretical monomer yield (typically 30–40%), will depend on the wood type. See ESI for details on the quantification of ether linkages and associated theoretical monomer yield. | ||||||

|

|

|
100 | Yes (13)a | No | 57 |

|
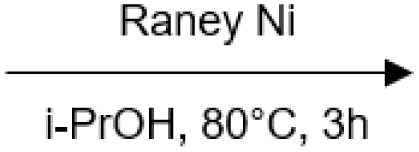
|

|
100 | No | No | 58 |

|
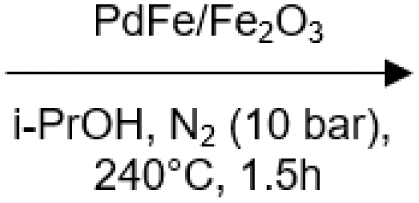
|

|
100 | No | No | 59 |

|
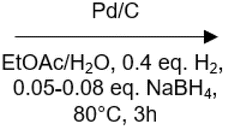
|

|
100 | No | No | 60 |
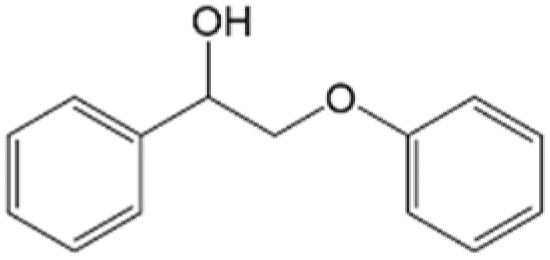
|

|

|
59 | No | No | 61 |

|

|

|
100 | Yes (20) | Yes | 62 |
|
|

|
98 | Yes (35)a | Yes | This work |
Here we demonstrate the feasibility of the one-pot, base-free cleavage of α-O-4, β-O-4 and 4-0-5 linkers of lignin with ethanol as H-donor over multifunctional Pd catalysts prepared by continuous flow synthesis (Fig. S2†), and featuring supports possessing different acid–base character:83,84 MgO, of primarily basic character; HT, exhibiting Lewis acidity and basicity; and γ-Al2O3, of primarily Lewis-acidic character. The same catalyst promotes TH, hydrogenolysis and decarbonylation. Novel aspects include: (i) efficient transfer hydrogenolysis of α-O-4, β-O-4 and 4-O-5 model compounds using ethanol as renewable hydrogen source; (ii) demonstration of a multifunctional catalyst for the tandem transfer hydrogenolysis and decarbonylation of β-O-4 motifs; and (iii) whole biomass valorization to phenolic derivatives using only ethanol as a hydrogen donor.
Results and discussion
Catalyst characterization
Pd/γ-Al2O3, Pd–MgO and pure and metal-modified Pd/HT catalysts were synthesized and characterized to: (i) assess their catalytic activity for the TH of lignin ether linkages to phenolic platform chemicals; and (ii) establish structure–reactivity relationships that identify physicochemical properties desirable for active and selective transformations. A Pd loading between 2–5 wt% was targeted for Pd/γ-Al2O3 and Pd–MgO, with that of (metal-modified) Pd/HT ∼5 wt%. Both oxide supported Pd catalysts were synthesized by wet impregnation of the supports with Pd(NO3)2·xH2O. MgO was prepared by a previously reported batch co-precipitation method.85 Pd/HT and transition metal-modified analogues (Table 3, A–G) were synthesized by a mesoscale, continuous flow precipitation method, adapted from our previous report.86 Compositions of A–G were highly reproducible (standard deviations <5% of molar metal content), with ICP-OES elemental analysis showing actual metal compositions was within 5% of targeted values (Table S1†). A decrease in the M2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M3+ ratio from the ideal value of 3.0 was observed for a few of the metal-modified Pd/HT samples, which is likely a consequence of the larger ionic radius of +2 dopant cations compared to that of Mg2+, or +3 dopant cations compared to the ionic radius of Al3+.
:M3+ ratio from the ideal value of 3.0 was observed for a few of the metal-modified Pd/HT samples, which is likely a consequence of the larger ionic radius of +2 dopant cations compared to that of Mg2+, or +3 dopant cations compared to the ionic radius of Al3+.
All Pd/HT samples exhibited a single crystalline phase from powder X-ray diffraction (XRD), associated with the hydrotalcite phase, characterized by (003), (006), (009), (015), (018), (110) and (113) reflections (Fig. S3†). No Pd or PdO crystalline phases (Pd: 2θ = 40.5° (111), 46.7° (200), 67.5° (220); PdO: 33.7° (101), 41.8° (110), 54.6° (112), 60.0° (103), 60.6° (200)), were observed in Pd/g-Al2O3 and the Pd/HT samples, even at 5 mol% Pd, suggesting that palladium is present either as low-nuclearity species of <2 nm particles, which are either dispersed over the HT support or incorporated as dopants within the HT cation layers. In contrast, Pd/MgO (Fig. S4†) exhibits weak reflections attributable to PdO. The (003), (006), (009) reflections of the HT are sensitive to the interlayer spacing (crystallographic parameter c), while the (110) and (113) reflections are related to the average metal–metal distance within the cation layers (crystallographic parameter a). The consistent values of a and c parameters for all doped HTs suggest that a common, crystalline HT structure was obtained in each case. Given that lattice expansion, reflected in parameter c (ESI Table S1†), does not vary significantly (range: 23.23–23.98 nm), there is no direct evidence that Pd was incorporated into the support structure to a significant extent. HT crystallite sizes, determined by applying the Scherrer equation, were insensitive to presence of Pd or other transition metal additives (10.8 ± 0.8 nm).
Fourier transform infrared (FTIR) spectra were consistent with a hydrotalcite phase, exhibiting characteristic bands for carbonate anions (1350–1370 cm−1) and interlayer water (∼1600–1700 cm−1) (Fig. S5†). Surface areas and pore size distributions for A–G were determined by N2 porosimetry (Fig. S6 and S7†) and subsequent application of the Brunauer–Emmett–Teller (BET) and Barrett, Joyner and Halenda (BJH) models (Fig. S8 and S9†), respectively (Table S2†). Surprisingly, the bimetal modified HTs all possessed higher surface areas that the parent (148 m2 g−1) or single metal modified (53–126 m2 g−1) HTs. We infer that the introduction of a second transition metal during the flow synthesis promotes the formation of highly dispersed Pd,M-containing clusters/nanoparticles with surface areas of 152–385 m2 g−1. We previously reported that pore volume decreases with increasing Pd loading,65 which is consistent with blockage of micropore entrances to interlayer spaces at higher loadings. However, the effects of transition metal doping on surface area and pore size are not fully elucidated. Thermogravimetric analysis (TGA) of all Pd/HTs revealed decomposition profiles consistent with a MgAl-HT.87 Three endothermic transitions were observed (Fig. S10†): at ∼100 °C due to the loss of physisorbed water; between 200–250 °C due to loss of interlayer water; and between 375–400 °C due to dehydroxylation and the loss of interlayer carbonate anions.77 These transitions occur within a relatively narrow temperature range for A–G, although a shift to lower temperatures is observed for metal-modified Pd/HTs relative to Pd-free HTs. This shift was most prominent for the high-temperature dehydroxylation/decarbonation, indicating that Pd likely destabilizes cation substitution and causes corresponding structural disorder.88
Analysis of the surface palladium by X-ray photoelectron spectroscopy (XPS) indicated the presence of multiple chemical environments in all samples. High-resolution Pd 3d XP spectra were dominated by a doublet with a 3d5/2 binding energy (BE) of 336.7 eV and spin–orbit splitting of 5.3 eV, assigned to Pd2+ in PdO (Fig. 2). A weaker doublet with a 3d5/2 BE of 335.4 eV attributable to metallic Pd was also apparent in all samples, with an additional, very weak doublet at 338.5 eV BE which we have previously assigned to Pd4+ substituted at low concentrations into the cation layers. Surprisingly, the diverse supports and metal modifiers exert little influence on the Pd surface speciation, which is dominated by PdO (70–80% of palladium present), alongside a modest amount of Pd metal (10–20%) and trace Pd4+ (<10%, likely in an octahedral coordination bound to 6 oxygen anions within the hydrotalcite layers). This observation was confirmed by complementary Pd K-edge X-ray absorption near edge spectroscopy (XANES), for which least-squares fitting identified Pd2+ in oxide clusters/nanoparticles as the dominant palladium phase (Fig. S11†). Mg 1s spectra of Pd/MgO, Pd/HT, and PdZn/HT shows that the presence of small amounts of surface magnesium carbonate species, which suggest presence of strongly basic sites (Fig. S12†). A high-energy XPS Mg 1s peak corresponding to surface carbonate was observed for Pd/MgO and PdZn/HT, suggesting presence of strong basic sites. In contrast, XPS Mg 1s of PdFe/HT and PdCu/HT were dominated by a peak characteristic of the pure HT, suggesting much lower basicity.
The morphology of samples B–G, as well as Pd/Al2O3 and Pd/MgO, were imaged by transmission electron microscopy (TEM). Uniformly dispersed metal-containing nanoparticles of ∼1–3 nm were visible for all materials (Fig. 3), consistent with the absence of crystalline phases (other than those of the supports) by XRD. Our previous studies of Pd/HT catalysts with nominal Pd loadings spanning 0.1 to 5 wt% revealed the presence of PdO and Pd metal clusters and nanoparticles for loadings >1 wt%,66 in accordance with Fig. 3.
These characterizations are consistent with a structure of the metal-modified 5 wt% PdHTs (B–G) that consists of atomically dispersed Pd4+ species incorporated into the HT cationic layers, low nuclearity Pd2+ species incorporated into the cationic layer or dispersed over the surface of HT layers, and Pd0 agglomerates on the surface. Unfortunately, we were not able to collect EDX mapping images of sufficient resolution to support this hypothesis. Furthermore, determination of Pd dispersion through common experimental means such as CO chemisorption could not be employed as this approach requires that CO titrates only Pd metal and not the support. However, weakly acidic CO molecules can bind to hydrotalcites as they are solid bases, thus hindering accurate dispersion measurements. Consequently, catalytic activity is quantitated based on the total Pd content, consistent with previous reports89–91 and hence reported TONs could underestimate the true catalyst performance.
Catalytic transfer hydrogenolysis of model compounds
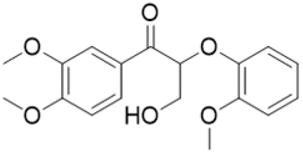
Catalyst activity for cleavage of different lignin motifs was subsequently explored for the synthetic model substrates in Fig. 1 (possessing α-O-4 (L-1), β-O-4 (L-2 to L-4) and 4-O-5 (L-5) linkages) and analogs lacking methoxy functions. Note that although we were unable to directly detect free H2 by GC-MS for any reactions below, hydrogen pop tests qualitatively evidenced in situ hydrogen formation from ethanol (as discussed later).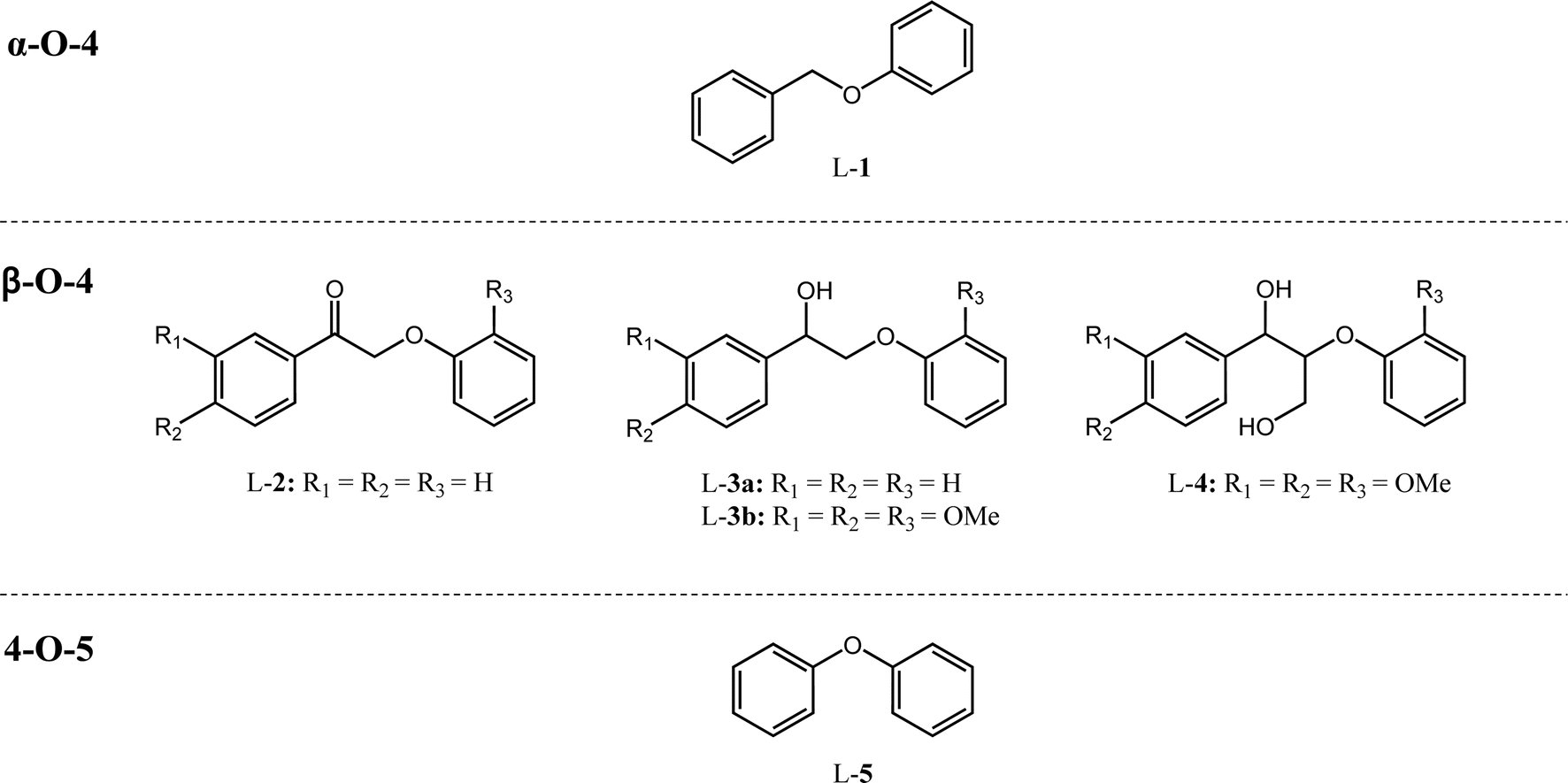 | ||
| Fig. 1 Model compounds L-1–L-5 with α-O-4, β-O-4 and 4-O-5 motifs. | ||
:1 with deionized water, but nevertheless afforded low yields (Table 2, entry 3). To probe whether dilution with water or the low viscosity of glycerol contributed to this low yield, additional reactions were performed with 1:1 isopropanol/water (Table 2, entry 4), which lowered the yields of a and b by ∼30% relative to the reaction with pure isopropanol. Similar modest yields were observed for reaction with a 1:1 isopropanol:glycerol solvent mixture (Table 2, entry 5), confirming that glycerol is not an effective hydrogen donor under these conditions. Note that the ability of glycerol to act as a hydrogen donor is strongly base-dependent due to the need to convert the initial dehydrogenation product, dihydroxyacetone, into the more thermodynamically stable lactate product.92–94 Consequently, addition of base could improve the viability of glycerol as a hydrogen donor. Although isopropanol delivered superior yields of hydrogenolysis products compared to ethanol, the latter offers a renewable source of hydrogen donor from fermentation of cellulosic waste, and hence was selected for further studies.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Conversion/% | Yield/% | |
| a | b | |||
| a Reaction conditions: 0.1 mmol benzyl phenyl ether (L-1), 3.0 mL solvent, 5 mol% Pd/HT catalyst (B), ∼50 W microwave heating at 150 °C, 2 h. | ||||
| 1 | Isopropanol | 80 | 80 | 78 |
| 2 | Ethanol | 62 | 62 | 61 |
| 3 | Glycerol:water (1:1) |
12 | 10 | 10 |
| 4 | Isopropanol:water (1:1) |
53 | 53 | 50 |
| 5 | Isopropanol:glycerol (1:1) |
50 | 50 | 50 |
The family of Pd catalysts were subsequently screened for TH of benzyl phenyl ether (L-1) with ethanol as the hydrogen-donor solvent under microwave heating at 150 °C for 2 h, with a Pd loading of 5 mol% relative to the substrate (Table 3). Transition metal-free HT (A, entry 1), 3 wt% Pd–Al2O3 (entry 2) and a homogeneous Pd(OAc)2 (entry 3) were inert under these conditions, while commercial 3 wt% Pd/MgO and 3 wt% Pd/C catalysts only achieved ∼30% conversion to toluene and phenol (Table 3, entries 4–5). In contrast, Pd-containing HT catalysts afforded high conversions (81–100%) with Pd-HT, Pd-FeHT, Pd-CuHT and Pd-CoHT (entries 6,7, 9,10) promoting quantitative conversion with full selectivity for toluene and phenol. To further differentiate the activity of the latter four, reaction time was decreased to 10 min, which allowed us to distinguish the following activity order: PdCu/HT (F) > PdFe/HT (C) > PdCo/HT (E) > Pd/HT (B) > PdZn/HT (G) > PdNi/HT (D). We note that PdFe/HT and PdCu/HT afforded >90% conversion in 10 min. We postulated that the superior activity of the Pd/HT catalysts relative to Pd/Al2O3, Pd/C and Pd/MgO is associated with their unique activity for alcohol dehydrogenation of ethanol. This was substantiated with substrate-free reactions in ethanol, where Pd/HT catalysts uniquely yielded dehydrogenation products (ethyl acetate and acetaldehyde). In conjunction with the similar Pd speciation observed for all catalysts (Fig. 2), these observations suggest that TH activity is primarily a function of the support acid–base properties, rather than electronic and geometric properties of the supported metals. The differences in activity between Pd/HTs modified with different transition metals can only be accounted for with EDX data that shows the metals are phase separated from Pd, alloyed with Pd, or in the HT cationic layers. Note that while α-O-4 linkages are not common in lignin, the preceding results demonstrate the broader potential application of mono- and bimetallic Pd/HTs for organic synthesis, and insight into structure–reactivity relationships pertinent to the transfer hydrogenolysis of 2-phenoxy-1-phenethanol (L-3a) described below, which involves ether cleavage and subsequent benzyl alcohol dehydrogenation.
| Entry | Catalyst | Conversion/% |
|---|---|---|
| a Reaction conditions: 0.1 mmol benzyl phenyl ether (L-1), 3.0 mL ethanol, 5 mol% catalyst, 50 W microwave heating at 150 °C, 2 h (or *10 min with resulting conversion in parentheses). | ||
| 1 | HT (A) | 0 |
| 2 | 3 wt% Pd/Al2O3 | 0 |
| 3 | Pd(OAc)2 | 0 |
| 4 | 3 wt% Pd/C | 30 |
| 5 | 3 wt% Pd/MgO | 34 |
| 6 | 5 wt% Pd/HT (B)* | 100 (47) |
| 7 | 5 wt% PdFe/HT (C)* | 100 (91) |
| 8 | 5 wt% PdNi/HT (D)* | 81 (37) |
| 9 | 5 wt% PdCo/HT (E)* | 100 (74) |
| 10 | 5 wt% PdCu/HT (F)* | 100 (93) |
| 11 | 5 wt% PdZn/HT (G)* | 88 (41) |
 | ||
| Fig. 2 Pd 3d XPs spectra of Pd/HTs (B–G), Pd–MgO, and Pd–Al2O3. | ||
 | ||
| Fig. 3 TEM images of metal modified PdM/HT catalysts (a) Pd/HT, (b) PdFe/HT, (c) PdCo/HT, (d) PdNi/HT, (e) PdCu/HT, and (f) PdZn/HT. Corresponding particle size distribution (bottom right corner of images a–f) histograms are shown in Fig. S13.† | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Conversion (%) | Yield (%)b | |||
| c | b | d | L-2 | |||
| a Reaction conditions: 0.1 mmol 2-phenoxy-1-phenethanol, 3.0 mL ethanol, 5 mol% catalyst, 50 W microwave heating at 150 °C, 2 h. Note that hydrogen may derive from ethanol dehydrogenation or intramolecular dehydrogenation of L-3a. b Unaccounted for yield is a result of aldol condensation between acetaldehyde and acetophenone forming ethyl benzoate. | ||||||
| 1 | HT (A) | 0 | 0 | 0 | 0 | 1 |
| 2 | 5% Pd/HT (B) | 68 | 66 | 66 | 0 | 2 |
| 3 | 5% PdFe/HT (C) | 97 | 79 | 86 | 4 | 11 |
| 4 | 5% PdNi/HT (D) | 6 | 5 | 5 | 0 | 1 |
| 5 | 5% PdCo/HT (E) | 59 | 51 | 52 | 0 | 7 |
| 6 | 5% PdCu/HT (F) | 98 | 93 | 96 | 0 | 2 |
| 7 | 5% PdZn/HT (G) | 16 | 8 | 11 | 0 | 5 |

The overall catalytic performance of transition metal modified Pd/HTs is summarized in Fig. 4, with PdCu/HT the most active for cleavage of all three linkages.
 | ||
| Fig. 4 Comparative yield of transfer hydrogenolysis products from L-1, L-2 and L-3a catalyzed by Pd/HTs. Reaction conditions: 0.1 mmol substrate, 3.0 mL ethanol, 5 mol% catalyst, 50 W microwave heating at 150 °C, 2 h (or *10 min for L-1). This data is a consolidation of Tables 3, S3†, and 4. †For L-1, cleavage yield includes toluene (a) and phenol (b); for L-2 and L-3a, cleavage yield includes acetophenone (c) and phenol (b). | ||
 | ||
| Fig. 5 (a) Reaction scheme for transfer hydrogenolysis of L-3b; and influence of (b) catalyst loading, (c) reaction temperature and (d) time on conversion and product selectivity. Reaction conditions: 0.1 mmol L-3b, 3.0 mL ethanol, PdCu/HT, 50 W microwave heating. aHydrogen may arise from ethanol dehydrogenation or dehydrogenation of the L-3 substrate. | ||
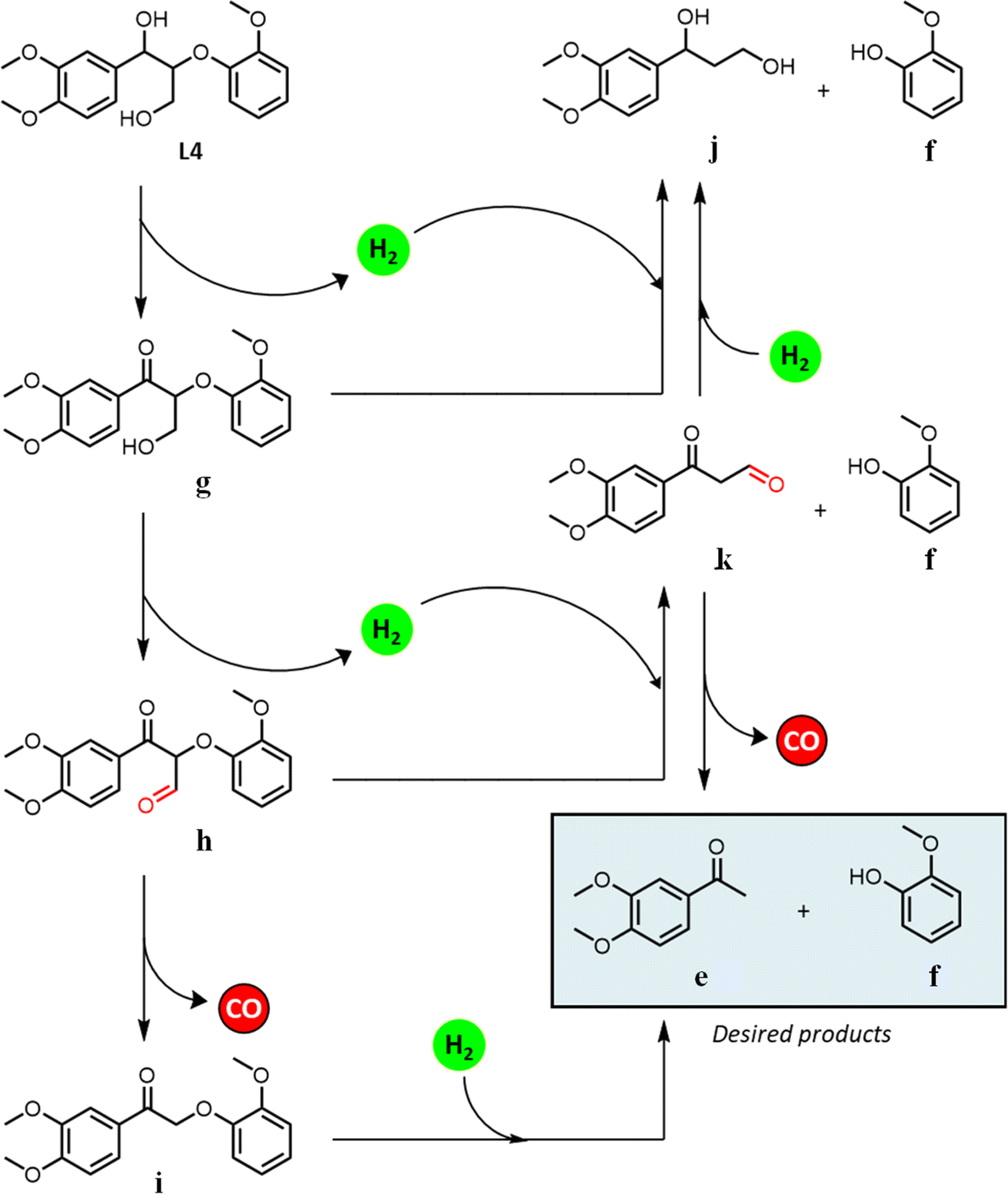 | ||
| Scheme 1 Proposed reaction pathway for the multistep transformation of L-4. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Conversion/% | Yield/% | ||
| i | e | f | |||
| a Reaction conditions: 0.1 mmol L-4, 3.0 mL solvent, 2.5 mol% PdCu/HT, 50 W microwave heating at 150 °C, 6 h. | |||||
| 1 | EtOH | 98 | 7 | 90 | 93 |
| 2 | p-Xylene | 88 | 75 | 5 | 4 |
| 3 | Dioxane | 13 | 11 | 2 | 1 |
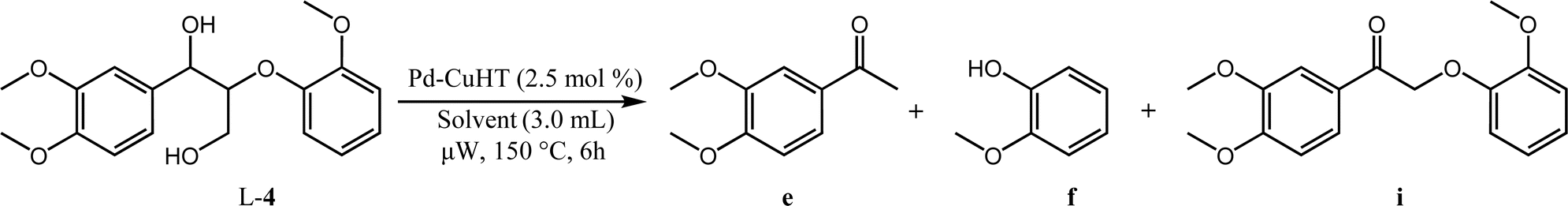
We hypothesize that the γ-hydroxy group undergoes dehydrogenation to an aldehyde, which is then eliminated via decarbonylation either before or after ether cleavage (dehydrogenation of α- and γ-hydroxy groups prior to ether cleavage is thermodynamically favorable).44 We have previously analyzed headspace gases of aldehyde decarbonylation reactions under similar conditions by GC-MS.66 Under air, such reactions evidenced CO2 formation, likely from the oxidation of CO with residual oxygen. Control reactions without aldehyde substrates afford no CO2, suggesting the source of CO2 is decarbonylation and oxidation, and exclude the possibility that aldehydes are first oxidized to carboxylic acids and undergo subsequent decarboxylation (by showing carboxylic acids are inert under our catalytic conditions).
Dehydrogenation of the benzylic alcohol is likely more thermodynamically favorable than that of the primary γ-hydroxy. The product distribution suggests that both hydroxy groups are dehydrogenated prior to decarbonylation and ether cleavage, as product i was observed as an intermediate, while j and k were not. Note that i is the dehydrogenated product of 1-(3,4-dimethoxyphenyl)-2-(2-methoxyphenoxy)-1,3-propanediol (L-3b), and thus likely follows the previously established pathways for its transfer hydrogenolysis (Table 4, starting from L-2). Dehydrogenation of benzylic alcohol produced from L-3a could also be followed by a retro-aldol cleavage98,99 to release formaldehyde and L-2, which in turn could undergo ether cleavage to form acetophenone; although the formaldehyde by-product was not observed with only decarbonylation followed by ether cleavage observed. In comparison to the benzylic alcohol, dehydrogenation of the primary γ-hydroxy is less kinetically and thermodynamically favored, but could likewise be followed by a retro-aldol cleavage (although we did not observed any products expected from such pathway either). The absence of a dehydrogenated γ-alcohol intermediate may thus reflect two possibilities: (i) once dehydrogenated, the γ-alcohol intermediate undergoes rapid decarbonylation and hence only attains a low steady state concentration; and/or (ii) ether cleavage occurs before decarbonylation, with subsequent rapid decarbonylation of the resulting intermediate k in Scheme 1.
To determine whether hydrogenolysis could be induced by intramolecular hydrogen from the α- and γ-hydroxy groups, additional reactions were performed in the aprotic solvents dioxane and p-xylene (Table 5). Although L-4 conversion in p-xylene was comparable to that in ethanol (88%, Table 5, entry 2), the carbonyl intermediate i (L-3b) was the dominant product, arising from α- and γ-dehydrogenation and decarbonylation. Very low conversion was observed in dioxane (13%, Table 5, entry 3), with i (L-3b) again the dominant product. Hydrogen donor solvents are thus critical for cleavage of ether linkages in these systems, and intramolecular hydrogen transfer is insufficient.
|
|
||||
|---|---|---|---|---|
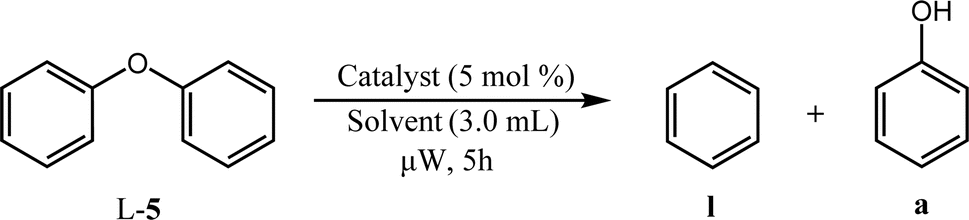
| Entry | Catalyst | Solvent | Temperature/°C | Conversionb/% |
| a Reaction conditions: 0.1 mmol L-5, 3.0 mL solvent, 5 mol% catalyst, 50 W microwave heating, 5 h. b Full selectivity is observed for each entry, thus conversion is equal to the yield of a and l. | ||||
| 1 | Pd/HT | EtOH | 150 | 2 |
| 2 | 180 | 2 | ||
| 3 | iPrOH | 150 | 22 | |
| 4 | 180 | 27 | ||
| 5 | PdCu/HT | EtOH | 150 | 1 |
| 6 | 180 | 3 | ||
| 7 | iPrOH | 150 | 0 | |
| 8 | 180 | 7 | ||
On the role of copper
The superior activity of PdCu/HT likely reflects a synergy between the metals that promotes ethanol dehydrogenation. Effective transfer hydrogenolysis requires a high rate of ethanol dehydrogenation, and a slow rate for the competing reaction between liberated H atoms to form (undesired) molecular H2. In the absence of substrate, Pd/HT is highly active for the ethanol dehydrogenation to ethyl acetate and acetaldehyde, the former product requiring an interplay between metal and acid sites. However, literature reports evidence only modest activity for ethanol dehydrogenation over Cu/HT even at higher temperature than in this work,100 and exclusively produces acetaldehyde. Single crystal studies reveal that alloying Pd with Cu enables the spillover of H adatoms from the former to the latter, while co-adsorbed CO inhibits H adatom combination and desorption over such single atom alloys that is otherwise rapid over pure Pd.101 Reactively-formed CO released by the decarbonylation of intermediates h and k (Scheme 1) and/or o (Scheme 2a) is thus expected to suppress molecular H2 formation over PdCu/HT versus Pd/HT. Future mechanistic studies will examine the sensitivity of ethanol dehydrogenation over Pd/HT and PdCu/HT to the presence of gas phase CO. | ||
| Scheme 2 Proposed pathways from hydroconiferyl alcohol (o) to (a) ethyl-, and (b) propyl guaiacols. | ||
Catalyst recycling
A catalyst recyclability study for PdCu/HT (F) was conducted to explore stability. L-3a was implemented as the model substrate for this purpose, utilizing the same reaction conditions as employed in the catalyst screen (Table 4). Postreaction, the mixture was centrifuged for 5 min at 5000 rpm, the supernatant was decanted off and sampled for product analysis. Fresh substrate and ethanol were added and heated at 150 °C for 2 h. This process was performed for five cycles. This method is beneficial as it does not necessitate filtration and washing, which can result in catalyst loss. Over five cycles, catalyst F retained >95% of its activity (Fig. 6), with a marginal decrease in selectivity for cleavage products due to the presence of unreacted dehydrogenated intermediate L-2. FTIR and PXRD of the used catalyst showed no change in catalyst phase or crystallite size (Fig. S19 and S20†), while TGA showed no evidence for retention of organic residues that might otherwise cause blocking of active-sites of micropores (Fig. S21†). Elemental analysis shows negligible loss of Cu or Pd (<5% of metal% by mass) post-reaction as compared to the as-prepared catalyst. XPS data of similar post-reaction catalysts for similar applications showed a reduction of Pdn+ to Pd0 after multiple cycles.65 Note that TEM revealed a small increase in Pd nanoparticle size after reaction (2.95 vs. 2.98 nm) (Fig. 7), with a doubling of the standard deviation (0.52 vs. 0.98), indicative of limited particle sintering and loss of active surface area that likely accounts for the minimal loss of initial activity (<5%) after five reaction cycles. Even if long-term (slow) leaching Pd or Cu is observed in future studies, such hydrotalcites can be readily regenerated by dissolution in nitric acid and reprecipitation using Na2CO3 and NaOH.65 | ||
| Fig. 6 Recyclability of PdCu/HT (F) for the transfer hydrogenolysis of 2-phenoxy-1-phenethanol (L-3a). Reaction conditions: substrate: 2-phenoxy-1-phenethanol (L-3a) (0.1 mmol), ethanol (3.0 mL), PdCu/HT (F) (2.5 mol%), 50 W microwave heating, 150 °C, 2 h. | ||
 | ||
| Fig. 7 TEM image of (a) fresh Pd-CuHT and (b) Pd nanoparticle size distribution of fresh 5 wt% PdCu/HT; (c) TEM image of used 5 wt% PdCu/HT after five reaction cycles for the TH of L-3a and (d) Pd nanoparticle size distribution of used 5 wt% PdCu/HT. | ||
Whole lignocellulose depolymerization
Application of Pd/HT catalysts for ether bond cleavage was extended to whole lignocellulose biomass (pine sawdust) through the RCF method, which circumvents the degradation of native lignin into a calcitrant structure observed during Kraft pulping.102,103 RCF is also referred to as a ‘lignin-first’ approach, and has been extensively reviewed.4,103–109 The RCF method was adapted for simultaneous:106,107 (i) lignin extraction from whole lignocellulose using a polar-protic solvent (ethanol in this case); and (ii) selective depolymerization using Pd/CuHT (F) with ethanol as hydrogen donor. Lignin extraction and depolymerization were thus combined in a novel single step process (Fig. 8), contrasting with the conventional two-step extraction of technical lignin and subsequent selective depolymerization. | ||
| Fig. 8 One-pot lignin extraction from whole biomass and integrated depolymerization in a sealed reactor. | ||
Pine wood sawdust was selected as the biomass source, with a ‘wet’ lignin content of 32 wt% (see ESI eqn (1)† for detailed procedure on lignin percent determination). The theoretical maximum monomer yield for a certain substrate can be calculated by considering a simplified, linear lignin polymer in which each building block is linked to two other units by either C–C or ether bonds. Lignin depolymerization occurs by transfer hydrogenolysis of the ether bonds. For building blocks possessing two ether bonds, cleavage of both will result in a monomer. To determine the efficiency of lignin conversion to monomers, the maximum theoretical monomer yield for the whole biomass sample was first calculated by nitrobenzene oxidation (NBO), a depolymerization method for subsequent quantitative analysis of the β-ether content by NMR.110–112 Note that NBO is not well-suited to determining guaiacyl/hydrophenyl monomer yields of poplar, aspen, willow and palms lignins.112 The NBO method afforded a theoretical maximum monomer yield of 23 wt% (see ESI† for further details of ether content and theoretical monomer yield determination). This value compares favorably to a theoretical yield of 25 wt% β-O-4 content in the lignin determined via the HSQC method.21,46,113
The one-pot reaction with pine sawdust was performed in a stainless-steel Parr 4560 stirred autoclave at 225 °C, which is sufficient for facilitating solubility and mass transfer of the whole pine biomass.114 Control reactions without catalyst (Table 7, entry 1) afford only delignification (62%, eqn (S2)†), i.e. lignin extraction from cellulose and hemicellulose, but without monomer and oligomer formation. This affirms the need for a catalyst to afford lignin depolymerization. Addition of 5 wt% Pd/CuHT (F) increased delignification to 97% (Table 7, entry 2), accompanied by a 22% monomer yield, approaching the theoretical maximum of 23% based on the NBO method discussed above (see ESI† for monomer yield determination, eqn (S3)†). The major monomer product was 4-(3-hydroxypropyl)-2-methoxyphenol (hydroconiferyl alcohol, HA, o), a monolignol derived from the dominant guaiacol components of pine lignin,46 which was obtained with 64% selectivity using PdCu/HT (14 wt% based on biomass). These values are greater than monophenolic yields of 10 wt% of initial wood mass obtained by RCF of polar clones in acidified aqueous ethanol, albeit over a different (Pd/C) catalyst.55 The balance of monolignols in the present work was accounted for by ethyl guaiacol and propyl guaiacol. Note that while the β-O-4 model compound L-4 afforded acetophenone and guaiacol, neither product was observed in one-pot reactions with pine sawdust, which instead favoured hydroconiferyl alcohol (HA, o, Scheme 1) and ethyl/propyl guaiacols. To explain this difference in selectivity of model compounds versus whole biomass, we suggest that in model compounds, dehydrogenation of β-O-4 moieties at the α-position forms g, which can undergo subsequent dehydrogenation and decarbonylation of the γ-OH to intermediate i, or transfer hydrogenolysis and further transfer hydrogenation to j (Scheme 1). In contrast, in whole biomass, the transfer hydrogenolysis is faster than γ-OH dehydrogenation, likely due to the more hindered γ-OH and stabilization of OH by H-bonding to other OH groups in lignin. The formation of ethyl guaiacol from biomass likely reflects the dehydrogenation of o to the corresponding aldehyde, followed by decarbonylation to ethyl guaiacol (Scheme 2a). The latter pathway is evidenced by a decrease in yield of coniferyl alcohol (HA, o in Scheme 2) with increasing reaction time and a concomitant increase in the yield of ethyl guaiacol (m, Scheme 2). Alternatively, propyl guaiacol could be produced through HA dehydration and hydrogenation (Scheme 2b). Further details on the mass balance and analysis of the lignin bio-oil are provided in Fig. S22, 23 and S24–S28† respectively.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Phenolic yieldb/wt% | Delignification/wt% | |||
| HAc | Other monomers | Total monomer | Dimers | |||
| a Reaction conditions: 5.0 g dry pine sawdust (32 wt% lignin), 50 mL ethanol, 0.6 g catalyst, 225 °C, 5 h. b Depolymerization to monomer units estimated from proportion of ether linkages in lignin content and monomer yield is a percentage of the maximum lignin present in the dried biomass (not just that present in extracted lignin). See ESI for detailed explanation for calculations of lignin percentage, delignification, maximum theoretical monomer yield, and experimentally obtained monomer yield. c 5-Hydroxy coniferyl alcohol. | ||||||
| 1 | N/A | 0.5 | <1 | <1 | <1 | 62 |
| 2 | 5 wt% Pd/CuHT | 14 | 8 | 22 | 21 | 97 |

In summary, the one-pot depolymerization of whole pine biomass in the presence of 5 wt% PdCu/HT promotes both delignification and lignin depolymerization, with a near-quantitative yield of hydroconiferyl alcohol, ethyl-, and propyl guaiacol monomers. Although these monomers differ from the products obtained from their representative model compounds, their formation involves the same transfer hydrogenolysis and decarbonylation pathways. Additional studies are underway to examine our catalyst performance towards organosolv lignin. Typical RCF processes use hydride donors as stoichiometric reducing agents. In our work, the only readily available sources of hydrogen are the ethanol solvent and alcohol groups in lignin. Evidence that ethanol is the primary hydrogen source is presented in Table 5, wherein replacing ethanol with nonhydrogen donating solvents, such as p-xylenes and dioxane, drastically lowered the yields of hydrogenation products in model compound studies (EtOH: 90%, p-xylene: 5%, dioxane: 2%). We thus conclude that hydrotalcite supported PdO clusters/nanoparticles are effective for RCF of lignin and the resulting one-pot synthesis of monolignols from woody biomass. The extent of delignification and attendant monomer yields from transfer hydrogenolysis over PdCu/HT are comparable to literature reports using molecular hydrogen.
Conclusions
Lignin model substrates of α-O-4 (L-1), β-O-4 (L-2 to L-4), and 4-O-5 (L-5) linkages (Fig. 1) were used to probe the catalytic activity of Pd-HTs. α-O-4 model compound benzyl phenylether (L-1) was used as substrate for an extensive screen of Pd/HT catalysts to determine the efficiency for transfer hydrogenolysis using ethanol as hydrogen donor. The initial catalyst screen identified PdFe/HT (C) and PdCu/HT (F) as the most active catalyst for benzyl phenylether, achieving full conversion using ethanol in under 30 min at 150 °C. This is the first system effective with ethanol as H-donor, furthermore, no base is required which is critical for making the system potentially economically viable. Similarly, a screen for the reactions that cleave the β-O-4 linker (L-2, L-3a, L-3b, and L-4) identified the same catalyst, PdCu/HT (F), as the most active and selective catalyst for both C2 and C3 type β-O-4 compounds. Extensive further optimization of reaction conditions has been carried out, as well as structure–activity studies to elucidate how catalytic activity for the multi-step process can be rationally optimized. For the 4-O-5 linkage, biphenyl ether (L-5), the best catalyst B only afforded 27% conversion. This is not surprising given the challenging nature of transfer hydrogenolysis of biphenyl ethers. Further catalyst optimization is ongoing for this linkage. The efficacy of these catalysts for the reductive catalytic fractionation of whole pine lignocellulose was also studied. PdCu/HT (F) provided the best activity, giving 97% delignification and 22% monomer yield, close to the theoretical maximum where one compound (4-(3-hydroxypropyl)-2-methoxyphenol) accounted for 64% of all monomers identified. Model compound studies demonstrated that Pd doped HTs can adequately cleave ether bonds via hydrogenolysis in the absence of exogenous hydrogen. Transition metal modified HTs are a viable option for RCF of whole lignocellulose using ethanol without external hydrogen or high pressures.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
A. M. V.-K. conceived the research concept. D. D., R. B. C. R. and A. M. V.-K. planned the research methods. A. M. V.-K. led the project supervision, A. F. L. and K. W. provided co-supervision. D. D. led the experimental work, aided by R. B. and C. R., and undertook the data analysis. D. D. prepared figures and drafted the manuscript and revisions. A. M. V.-K., A. F. L. and K. W. edited the manuscript draft and revisions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DD, RB, CR and AV sincerely thank NSF Chemistry (award #2154815 and #1805080) for financial support and Peter Deuss and group for insights on lignin depolymerization and organosolv process. AFL and KW thank the Australian Research Council (DP200100204, DP200100313 and LP190100849) and CRC-P (CRCPPEIGHT000194) for financial support.References
- P. T. Anastas and J. C. Warner, Green chemistry : theory and practice, Oxford University Press, Oxford England; New York, 1998 Search PubMed.
- United States, Joint Subcomitee on Environment, Innovation, and Public Health, Sustainable Chemistry Report: Framing the Federal Landscape, National Science and Technology Council, 2023, https://www.whitehouse.gov/wp-content/uploads/2023/08/NSTC-JCEIPH-SCST-Sustainable-Chemistry-Federal-Landscape-Report-to-Congress.pdf Search PubMed.
- P. Anastas, M. Nolasco, F. Kerton, M. Kirchhoff, P. Licence, T. Pradeep, B. Subramaniam and A. Moores, ACS Sustain. Chem. Eng., 2021, 9, 8015–8017 CrossRef CAS.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- Y. Song, J. K. Mobley, A. H. Motagamwala, M. Isaacs, J. A. Dumesic, J. Ralph, A. F. Lee, K. Wilson and M. Crocker, Chem. Sci., 2018, 9, 8127–8133 RSC.
- Z. Sun, B. Fridrich, A. d. Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS.
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, Ind. Crops Prod., 2019, 139, 111526 CrossRef CAS.
- R. Parthasarathi, R. A. Romero, A. Redondo and S. Gnanakaran, J. Phys. Chem. Lett., 2011, 2, 2660–2666 CrossRef CAS.
- K. Barta, G. R. Warner, E. S. Beach and P. T. Anastas, Green Chem., 2014, 16, 191–196 RSC.
- J. Chen, F. Lu, X. Si, X. Nie, J. Chen, R. Lu and J. Xu, ChemSusChem, 2016, 9, 3353–3360 CrossRef CAS PubMed.
- P. Ferrini and R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8634–8639 CrossRef CAS PubMed.
- Y. Hu, G. Jiang, G. Xu and X. Mu, Mol. Catal., 2018, 445, 316–326 CrossRef CAS.
- X. Huang, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ChemSusChem, 2014, 7, 2276–2288 CrossRef CAS PubMed.
- H. Konnerth, J. Zhang, D. Ma, M. H. G. Prechtl and N. Yan, Chem. Eng. Sci., 2015, 123, 155–163 CrossRef CAS.
- Z. Luo, Z. Zheng, L. Li, Y.-T. Cui and C. Zhao, ACS Catal., 2017, 7, 8304–8313 CrossRef CAS.
- B. Ma, H. Cui, D. Wang, P. Wu and C. Zhao, Nanoscale, 2017, 9, 5986–5995 RSC.
- R. Ma, W. Hao, X. Ma, Y. Tian and Y. Li, Angew. Chem., Int. Ed., 2014, 53, 7310–7315 CrossRef CAS PubMed.
- V. Molinari, C. Giordano, M. Antonietti and D. Esposito, J. Am. Chem. Soc., 2014, 136, 1758–1761 CrossRef CAS.
- Y. Shao, Q. Xia, L. Dong, X. Liu, X. Han, S. F. Parker, Y. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, S. Yang and Y. Wang, Nat. Commun., 2017, 8(1), 1–9 CrossRef CAS.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 RSC.
- Q. Xia, Z. Chen, Y. Shao, X. Gong, H. Wang, X. Liu, S. F. Parker, X. Han, S. Yang and Y. Wang, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- M. Zaheer and R. Kempe, ACS Catal., 2015, 5, 1675–1684 CrossRef CAS.
- J. Zhang, H. Asakura, J. Van Rijn, J. Yang, P. Duchesne, B. Zhang, X. Chen, P. Zhang, M. Saeys and N. Yan, Green Chem., 2014, 16, 2432–2437 RSC.
- J. Zhang, J. Teo, X. Chen, H. Asakura, T. Tanaka, K. Teramura and N. Yan, ACS Catal., 2014, 4, 1574–1583 CrossRef CAS.
- W. Deng, H. Zhang, X. Wu, R. Li, Q. Zhang and Y. Wang, Green Chem., 2015, 17, 5009–5018 RSC.
- Y. Gao, J. Zhang, X. Chen, D. Ma and N. Yan, ChemPlusChem, 2014, 79, 825–834 CrossRef CAS.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS PubMed.
- R. Lu, F. Lu, J. Chen, W. Yu, Q. Huang, J. Zhang and J. Xu, Angew. Chem., Int. Ed., 2016, 55, 249–253 CrossRef CAS PubMed.
- R. Ma, M. Guo and X. Zhang, ChemSusChem, 2014, 7, 412–415 CrossRef CAS PubMed.
- M. Wang, X. Zhang, H. Li, J. Lu, M. Liu and F. Wang, ACS Catal., 2018, 8, 1614–1620 CrossRef CAS.
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef CAS PubMed.
- J. Hu, D. Shen, S. Wu, H. Zhang and R. Xiao, Energy Fuels, 2014, 28, 4260–4266 CrossRef CAS.
- T. D. H. Nguyen, M. Maschietti, T. Belkheiri, L.-E. Åmand, H. Theliander, L. Vamling, L. Olausson and S.-I. Andersson, J. Supercrit. Fluids, 2014, 86, 67–75 CrossRef CAS.
- J. A. Onwudili and P. T. Williams, Green Chem., 2014, 16, 4740–4748 RSC.
- Y. Pan, R. Qiu, J. Li, L. Bi, B. Shang and J. Tschiersch, Qinghua Daxue Xuebao, 2013, 53, 1380–1384 CAS.
- A. Toledano, L. Serrano and J. Labidi, Fuel, 2014, 116, 617–624 CrossRef CAS.
- L. Yang, Y. Li and P. E. Savage, Ind. Eng. Chem. Res., 2014, 53, 2633–2639 CrossRef CAS.
- S. Adhikari, V. Srinivasan and O. Fasina, Energy Fuels, 2014, 28, 4532–4538 CrossRef CAS.
- G. Jiang, Y. Hu, G. Xu, X. Mu and H. Liu, ACS Sustain. Chem. Eng., 2018, 6, 5772–5783 CrossRef CAS.
- G. T. Neumann, B. R. Pimentel, D. J. Rensel and J. C. Hicks, Catal. Sci. Technol., 2014, 4, 3953–3963 RSC.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef CAS.
- H. Liu, H. Li, J. Lu, S. Zeng, M. Wang, N. Luo, S. Xu and F. Wang, ACS Catal., 2018, 8, 4761–4771 CrossRef CAS.
- J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed.
- D. Shao, J. Liang, X. Cui, H. Xu and W. Yan, Chem. Eng. J., 2014, 244, 288–295 CrossRef CAS.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2014, 7, 2154–2158 CrossRef CAS PubMed.
- A. Wu, B. O. Patrick, E. Chung and B. R. James, Dalton Trans., 2012, 41, 11093–11106 RSC.
- K. Wang, J. Horlyck, N. An and A. Voutchkova-Kostal, Green Chem., 2024, 26, 3546–3564 RSC.
- M. J. Gilkey and B. Xu, ACS Catal., 2016, 6, 1420–1436 CrossRef CAS.
- A. Shivhare, D. Jampaiah, S. K. Bhargava, A. F. Lee, R. Srivastava and K. Wilson, ACS Sustain. Chem. Eng., 2021, 9, 3379–3407 CrossRef CAS.
- C. Cheng, P. Li, W. Yu, D. Shen and S. Gu, Bioresour. Technol., 2021, 319, 124238 CrossRef CAS PubMed.
- C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC.
- K. Tekin, N. Hao, S. Karagoz and A. J. Ragauskas, ChemSusChem, 2018, 11, 3559–3575 CrossRef CAS PubMed.
- M. V. Galkin, A. T. Smit, E. Subbotina, K. A. Artemenko, J. Bergquist, W. J. J. Huijgen and J. S. M. Samec, ChemSusChem, 2016, 9, 3280–3287 CrossRef CAS PubMed.
- A. Adler, I. Kumaniaev, A. Karacic, K. R. Baddigam, R. J. Hanes, E. Subbotina, A. W. Bartling, A. J. Huertas-Alonso, A. Moreno, H. Håkansson, A. P. Mathew, G. T. Beckham and J. S. M. Samec, Joule, 2022, 6, 1845–1858 CrossRef CAS.
- X. Wang and R. Rinaldi, ChemSusChem, 2012, 5, 1455–1466 CrossRef CAS PubMed.
- L. Jiang, H. Guo, C. Li, P. Zhou and Z. Zhang, Chem. Sci., 2019, 10, 4458–4468 RSC.
- X. Wang and R. Rinaldi, Energy Environ. Sci., 2012, 5, 8244–8260 RSC.
- E. Paone, C. Espro, R. Pietropaolo and F. Mauriello, Catal. Sci. Technol., 2016, 6, 7937–7941 RSC.
- M. V. Galkin, C. Dahlstrand and J. S. M. Samec, ChemSusChem, 2015, 8, 2187–2192 CrossRef CAS PubMed.
- H.-T. Wang, Z.-K. Li, H.-L. Yan, Z.-P. Lei, J.-C. Yan, S.-B. Ren, Z.-C. Wang, S.-G. Kang and H.-F. Shui, Fuel, 2022, 326, 125027 CrossRef CAS.
- J. Huang, C. Zhao and F. Lu, Polymers, 2018, 10, 1077 CrossRef PubMed.
- M. Crespo-Quesada, A. Yarulin, M. S. Jin, Y. N. Xia and L. Kiwi-Minsker, J. Am. Chem. Soc., 2011, 133, 12787–12794 CrossRef CAS PubMed.
- D. Ainembabazi, C. Reid, A. Chen, N. An, J. Kostal and A. Voutchkova-Kostal, J. Am. Chem. Soc., 2020, 142, 696–699 CrossRef CAS PubMed.
- D. Ainembabazi, N. An, J. C. Manayil, K. Wilson, A. F. Lee and A. M. Voutchkova-Kostal, ACS Catal., 2019, 9, 1055–1065 CrossRef CAS.
- N. An, D. Ainembabazi, C. Reid, K. Samudrala, K. Wilson, A. F. Lee and A. Voutchkova-Kostal, ChemSusChem, 2020, 13, 312–320 CrossRef CAS PubMed.
- M. A. Stranick, M. Houalla and D. M. Hercules, J. Catal., 1990, 125, 214–226 CrossRef CAS.
- A. Davantès, C. Schlaup, X. Carrier, M. Rivallan and G. Lefèvre, J. Phys. Chem. C, 2017, 121, 21461–21471 CrossRef.
- O. Borg, P. Dietzel, A. Spjelkavik, E. Tveten, J. Walmsley, S. Diplas, S. Eri, A. Holmen and E. Rytter, J. Catal., 2008, 259, 161–164 CrossRef CAS.
- B. N. Shelimov, J. F. Lambert, M. Che and B. Didillon, J. Mol. Catal. A: Chem., 2000, 158, 91–99 CrossRef CAS.
- F. Saad, J. D. Comparot, R. Brahmi, M. Bensitel and L. Pirault-Roy, Appl. Catal., A, 2017, 544, 1–9 CrossRef CAS.
- B.-Q. Xu, T. Yamaguchi and K. Tanabe, Chem. Lett., 1988, 17, 1663–1666 CrossRef.
- H. P. Aytam, V. Akula, K. Janmanchi, S. R. R. Kamaraju, K. R. Panja, K. Gurram and J. W. Niemantsverdriet, J. Phys. Chem. B, 2002, 106, 1024–1031 CrossRef CAS.
- W. Gac, Appl. Surf. Sci., 2011, 257, 2875–2880 CrossRef CAS.
- C. Poupin, R. Maache, L. Pirault-Roy, R. Brahmi and C. T. Williams, Appl. Catal., A, 2014, 475, 363–370 CrossRef CAS.
- E. Groppo, S. Bertarione, F. Rotunno, G. Agostini, D. Scarano, R. Pellegrini, G. Leofanti, A. Zecchina and C. Lamberti, J. Phys. Chem. C, 2007, 111, 7021–7028 CrossRef CAS.
- F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- Y. He, P. Yang, J. Fan, Y. Liu, Y. Du, J. Feng, F. Fan and D. Li, RSC Adv., 2015, 5, 74907–74915 RSC.
- M. G. Álvarez, R. J. Chimentão, F. Figueras and F. Medina, Appl. Clay Sci., 2012, 58, 16–24 CrossRef.
- O. D. Pavel, D. Tichit and I.-C. Marcu, Appl. Clay Sci., 2012, 61, 52–58 CrossRef CAS.
- J. Bain, P. Cho and A. Voutchkova-Kostal, Green Chem., 2015, 17, 2271–2280 RSC.
- D. Ainembabazi, J. Horlyck, D. Dolan, M. Finn, A. F. Lee, K. Wilson and A. Voutchkova-Kostal, ACS Sustainable Chem. Eng., 2021, 9, 14657–14662 CrossRef CAS.
- G. J. S. Dawes, E. L. Scott, J. Le Nôtre, J. P. M. Sanders and J. H. Bitter, Green Chem., 2015, 17, 3231–3250 RSC.
- J. A. Lopez-Ruiz and R. J. Davis, Green Chem., 2014, 16, 683–694 RSC.
- S. R. Akuri, C. Dhoke, K. Rakesh, S. Hegde, S. A. Nair, R. Deshpande and P. Manikandan, Catal. Lett., 2017, 147, 1285–1293 CrossRef CAS.
- P. Yaseneva, N. An, M. Finn, N. Tidemann, N. Jose, A. Voutchkova-Kostal and A. Lapkin, Chem. Eng. J., 2019, 360, 190–199 CrossRef CAS.
- C. A. A. R. Jules, J. A. Bokhoven, J. A. van Dillen, J. W. Geus and K. P. de Jong, Chem. - Eur. J., 2002, 8, 5571–5579 CrossRef.
- J. Pérez-Ramırez, G. Mul and J. A. Moulijn, Vib. Spectrosc., 2001, 27, 75–88 CrossRef.
- N. Agarwal, S. J. Freakley, R. U. McVicker, S. M. Althahban, N. Dimitratos, Q. He, D. J. Morgan, R. L. Jenkins, D. J. Willock, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Science, 2017, 358, 223–227 CrossRef CAS PubMed.
- A. Corma, P. Concepción, M. Boronat, M. J. Sabater, J. Navas, M. J. Yacaman, E. Larios, A. Posadas, M. A. López-Quintela, D. Buceta, E. Mendoza, G. Guilera and A. Mayoral, Nat. Chem., 2013, 5, 775–781 CrossRef CAS PubMed.
- J. K. Edwards and G. J. Hutchings, Angew. Chem., Int. Ed., 2008, 47, 9192–9198 CrossRef CAS PubMed.
- A. Azua, M. Finn, H. N. Yi, A. B. Dantas and A. Voutchkova-Kostal, ACS Sustainable Chem. Eng., 2017, 5, 3963–3972 CrossRef CAS.
- M. Finn, J. A. Ridenour, J. Heltzel, C. Cahill and A. Voutchkova-Kostal, Organometallics, 2018, 37, 1400–1409 CrossRef CAS.
- J. M. Heltzel, M. Finn, D. Ainembabazi, K. Wang and A. M. Voutchkova-Kostal, Chem. Commun., 2018, 54, 6184–6187 RSC.
- Z. Zhang, D. S. Zijlstra, C. W. Lahive and P. J. Deuss, Green Chem., 2020, 22, 3791–3801 RSC.
- S. Kim, S. C. Chmely, M. R. Nimlos, Y. J. Bomble, T. D. Foust, R. S. Paton and G. T. Beckham, J. Phys. Chem. Lett., 2011, 2, 2846–2852 CrossRef CAS.
- Z. Zhang, C. W. Lahive, D. S. Zijlstra, Z. Wang and P. J. Deuss, ACS Sustain. Chem. Eng., 2019, 7, 12105–12116 CAS.
- C. S. Lancefield, H. L. J. Wienk, R. Boelens, B. M. Weckhuysen and P. C. A. Bruijnincx, Chem. Sci., 2018, 9, 6348–6360 RSC.
- C. S. Lancefield, L. W. Teunissen, B. M. Weckhuysen and P. C. A. Bruijnincx, Green Chem., 2018, 20, 3214–3221 RSC.
- A. Ausavasukhi, N. Krukrathok and P. Singthaisong, J. Ind. Eng. Chem., 2023, 117, 371–385 CrossRef CAS.
- M. D. Marcinkowski, A. D. Jewell, M. Stamatakis, M. B. Boucher, E. A. Lewis, C. J. Murphy, G. Kyriakou and E. C. H. Sykes, Nat. Mater., 2013, 12, 523–528 CrossRef CAS PubMed.
- S. Van Den Bosch, T. Renders, S. Kennis, S. F. Koelewijn, G. Van Den Bossche, T. Vangeel, A. Deneyer, D. Depuydt, C. M. Courtin, J. M. Thevelein, W. Schutyser and B. F. Sels, Green Chem., 2017, 19, 3313–3326 RSC.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2016, 9, 1544–1558 CrossRef CAS PubMed.
- J.-M. Ha, K.-R. Hwang, Y.-M. Kim, J. Jae, K. H. Kim, H. W. Lee, J.-Y. Kim and Y.-K. Park, Renewable Sustainable Energy Rev., 2019, 111, 422–441 CrossRef CAS.
- E. Paone, T. Tabanelli and F. Mauriello, Curr. Opin. Green Sustainable Chem., 2020, 24, 1–6 CrossRef.
- T. Renders, S. Van den Bosch, S. F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- Y. Song, in Chemical Catalysts for Biomass Upgrading, 2020, DOI:10.1002/9783527814794.ch9, pp. 395–437.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- V. E. Tarabanko and N. Tarabanko, Int. J. Mol. Sci., 2017, 18, 2421 CrossRef PubMed.
- R. Katahira and F. Nakatsubo, J. Wood Sci., 2001, 47, 378–382 CrossRef CAS.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- D. S. Zijlstra, C. W. Lahive, C. A. Analbers, M. B. Figueirêdo, Z. Wang, C. S. Lancefield and P. J. Deuss, ACS Sustain. Chem. Eng., 2020, 8, 5119–5131 CrossRef CAS.
- T. Renders, G. Van den Bossche, T. Vangeel, K. Van Aelst and B. Sels, Curr. Opin. Biotechnol., 2019, 56, 193–201 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03942d |
| This journal is © The Royal Society of Chemistry 2024 |