
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of bismuthanyl-substituted monomeric triel hydrides†‡
Robert
Szlosek
a,
Christian
Marquardt
a,
Oliver
Hegen
a,
Gábor
Balázs
a,
Christoph
Riesinger
a,
Alexey Y.
Timoshkin
 b and
Manfred
Scheer
*a
b and
Manfred
Scheer
*a
aInstitute of Inorganic Chemistry, University of Regensburg, 93053 Regensburg, Germany. E-mail: manfred.scheer@ur.de
bInstitute of Chemistry, St. Petersburg State University, Universitetskaya nab. 7/9, 199034, St. Petersburg, Russia
First published on 12th August 2024
Abstract
The syntheses and characterizations of the first bismuthanylborane monomers stabilized only by a donor in D·BH2Bi(SiMe3)2 (D = DMAP 1a, IDipp 1b, IMe41c; DMAP = 4-dimethylaminopyridine, IDipp = 1,3-bis(2,6-diisopropylphenyl)-imidazolin-2-ylidene, IMe4 = 1,3,4,5-tetramethylimidazol-2-ylidene) are presented. All compounds were synthesized by salt metathesis reactions between D·BH2I and KBi(SiMe3)2(THF)0.3 and represent some of the extremely rare compounds featuring a 2c–2e B–Bi bond in a molecular compound. The products display high sensitivity towards air and light and slowly decompose in solution even at −80 °C. By the reaction of IDipp·GaH2(SO3CF3) with KBi(SiMe3)2(THF)0.3, the synthesis of the first bismuthanylgallane IDipp·GaH2Bi(SiMe3)2 (2) stabilized only by a 2-electron donor was possible, as evident from single crystal X-ray structure determination, NMR spectroscopy and mass spectrometry. Computational studies shed light on the stability of the products and the electronic nature of the compounds.
The attention paid to the chemistry of bismuth has increased tremendously within the last decade.1–4 As bismuth can adopt all oxidation states from +V to −III and can often feature a stereoactive 6s atomic orbital, it is capable of extraordinary structural diversity. What is more, strong spin–orbit coupling and relativistic effects influence the chemical and physical properties of bismuth compounds.5 Prominent representatives for this flexibility are e.g. highly charged bismuth cluster anions.2,6 The flexibility of binding modes makes organic and inorganic bismuth compounds very useful reagents. E.g. inorganic salts BiX3 (X = Hal, OTf, NO3; OTf = SO3CF3) have been commonly employed in organic transformations either stoichiometrically or catalytically.7
In addition, more and more new bismuth-based catalyst systems have recently been developed, capable of impressive organic transformations.3,8 For instance, the Cornella group has pushed the boundaries of bismuth-catalysed organic transformations ranging from transfer hydrogenations and amide reductions to oxidative coupling reactions and many other applications.4,9–12 Significantly, it could be demonstrated that bismuth-based catalysis not only relies on the Lewis acidic properties of Bi(III), but that the latter also displays reaction behaviour similar to regular transition metals since important steps such as single-electron transfer can also be achieved.10–12
Important contributions to the chemistry of trivalent bismuth were also attained by Chitnis and co-workers who exploited the geometric perturbation of planar trivalent bismuthanes to achieve an unusual reactivity thanks to the steric and electronic properties of these compounds.13,14 Furthermore, planar Bi(III) complexes proved to act as tuneable platforms for catalytic ring opening polymerizations of cyclic esters with good dispersity values and high molecular weights, with only low quantities of the corresponding bismuth triamide catalyst being needed.14 The success of Bi in catalysis is based on rather strong Bi–C bonds, whereas Bi–triel bonds (triel = group 13 element) often suffer weak bonding because of rather low bond dissociation enthalpies. Thus, isolated examples of such compounds are quite rare, especially when heavy triels come into play. Therefore, such compounds are often unstable and difficult to isolate without prior decomposition.
To the best of our knowledge, the only examples of isolable compounds featuring covalent B–Bi compounds are found in a boryldiphenylbismuthane (Fig. 1, I)15 as well as in a boryl-dibismuthene (Fig. 1, II).16 Here, the B atom is incorporated in a stabilizing cycle. The von Hänisch group made important contributions to the field of covalent bismuth compounds and could obtain several highly sensitive interpnictogen chain compounds that include bismuth.17 This group was recently also able to report on donor-free bismuth cations capable of intriguing reactivity.18 The group of S. Schulz further advanced this field by the activation of bismuthanes with monovalent group 13 compounds of (Dipp2Nacnac)M (dipp = 2,6 diisopropylphenyl, M = Al, Ga, In; Fig. 1, IIIa–c, IVa).19 R. A. Fischer and co-workers reported similar dibismuthenes supported by (Dipp2Nacnac)Ga as well as (IVb/c).20 Von Hänisch and co-workers were able to synthesize similar compounds by a related strategy (Fig. 1, V, VI, VIIa/b).21 Again, also in these compounds, the group 13 element is embedded in a cyclic system. Very recently, the groups of Coles and McMullin synthesized a unique bicyclic-like compound featuring a covalent 2c–2e In–Bi bond (Fig. 1, VIII).22 However, in all reports about Bi–triel bonded compounds, none of the mentioned examples contained parent H-substituted triel moieties and they were all embedded in chain compounds.
 | ||
| Fig. 1 Selected examples of stabilization motifs of molecular 13/15 compounds featuring E–Bi bonds in (a) cyclic systems, (b) stabilized by β-diketiminates and (c) in bicyclic systems (Mes = 2,4,6-trimethylphenyl, dipp = 2,6-diisopropylphenyl, E = triel, Nacnac = bis(2,6-diaryl)-2,2'dimethyl-β-diketiminate); (d) bismuthanyl-substituted triel hydrides presented in this work. | ||
Our group is interested in the stabilization of parent group 13/15 element compounds stabilized only by Lewis acids or bases, which can be used e.g. as building blocks for selective oligo- and polymerization reactions.23 These monomers are usually synthesized by salt metathesis or hydrogen elimination reactions and show interesting reactivities towards electrophiles and nucleophiles.24 The group 15 element within such compounds also enables a broad range of coordination possibilities,25,26 even leading to promising luminescent materials.26 By varying the group 15 element, the parent phosphanyl-, arsanyl- as well as stibanylboranes stabilized only by one donor molecule D·BH2PnH2 (Pn = P, As, Sb) could be accessed.27,28 The corresponding phosphanyl- and arsanylgallanes and -alanes IDipp·EH2PnH2 (E = Ga, Al; Pn = P, As) were also recently obtained, but only with the support of a bulky N-heterocyclic carbene (NHC) as donor.29,30 Higher degrees of substitution at the group 13 entities could also be achieved by multiple salt metatheses, leading to the compounds IDipp·EH3−x(PnH2)x (x = 2, 3) and even the triphosphanylindiumane IDipp·In(PH2)3.30,31
Whereas phosphanyl-, arsanyl- and stibanylboranes have already been synthesized, the synthesis of the homologous bismuthanylboranes has so far not been realized. So, the question is whether such compounds are accessible and, if so, whether it is possible to synthesize even the parent compounds. Herein, we report on the synthesis of the first chain-like, donor-stabilized bismuthanylboranes featuring covalent 2c–2e electron B–Bi bonds which contain unique BH2-moieties as well as on the isolation of an unprecedented bismuthanylgallane, making each of them the first representatives of their particular kinds.
 | (1) |
The first step was to target the synthesis of a suitable bismuth source to use it to prepare the donor-stabilized bismuthanylboranes. As salt metathesis presents a reliable pathway towards this type of 13/15 compounds, we started off by investigating the salt metathesis of D·BH2I with an alkali metal bismuthanide. Unfortunately, MBiH2 (M = alkali metal) remains inaccessible so that we decided to introduce SiMe3 groups by using MBi(SiMe3)2,32 which offers the possibility towards future Bi–Si bond cleavage. KBi(SiMe3)2(THF)0.3 was prepared by the reaction of Bi(SiMe3)3 with KOtBu in THF (cf. ESI‡).
Subsequently, this reagent was reacted with iodinated donor-stabilized boranes D·BH2I (D = IDipp, IMe4 (=1,3,4,5-tetramethylimidazol-2-ylidene), DMAP) by adding neat KBi(SiMe3)2(THF)0.3 to a cold solution (−80 °C) of the corresponding boranes in THF under exclusion of light (eqn (1)). Upon reaching room temperature, full conversion of the starting materials is achieved according to NMR spectroscopy. After extraction with toluene, the donor-stabilized bismuthanylboranes D·BH2Bi(SiMe3)2 (1a: D = IDipp, 1b: D = IMe4, 1c D = DMAP) were obtained in moderate to good yields. The compounds are obtained as grey powders, which display very high sensitivity towards air and light and decompose at room temperature under visible deposition of elemental bismuth. A very slow decomposition occurs even upon prolonged storage at −30 °C, complicating the storage of the solid products for longer than a month.
Decomposition in solution happens even faster and at temperatures as low as −80 °C, with 1c displaying the highest tendency towards decomposition. Presumably, the donor strength of NHCs significantly contributes to the overall better kinetic and thermodynamic stability of the compounds which keeps the molecules intact. This is further supported by quantum chemical calculations (vide infra), which show a higher dissociation energy for the NHC adducts 1a and 1b. Furthermore, this is in line with the isolated yields from the reactions (cf.eqn (1)). The NMR spectra of solutions of 1a–c in C6D6 show characteristic triplet resonances in the 11B NMR spectra and singlets (Si(CH3)3) as well as broadened quartets (BH2) in the 1H NMR spectra (cf.Table 1). Moreover, 1a and 1b could also be characterized by their molecular ion peaks at m/z = 756.44 (1a) and m/z = 492.20 (1b) in the LIFDI mass spectra. It was also possible to crystallize all bismuthanylboranes 1a–c by storing saturated solutions in n-hexane (1b) or toluene (1c) at −30 °C overnight or slow evaporation (1a) of n-pentane at −30 °C. By single-crystal X-ray structure determination, the structures in the solid state could be elucidated (Fig. 2).
| NMR spectrum | 1a | 1b | 1c | 2 |
|---|---|---|---|---|
| 1H Si(CH3)3 signal | 0.53, s | 0.81, s | 0.95, s | 0.70, s |
| 1H EH2 signal (E = B, Ga) | 2.24, q, br 1JBH = 100 Hz | 2.43, q, br 1JBH = 107 Hz | 4.74, q, br 1JBH = 120 Hz | 4.97, s, br |
| 11B{1H} | −42.1, s, br | −39.5, s, br | −15.4, s, br | — |
| 11B | −42.1, t, br 1JBH = 100 Hz | −39.5, t, br 1JBH = 107 Hz | −15.2, t, br 1JBH = 120 Hz | — |
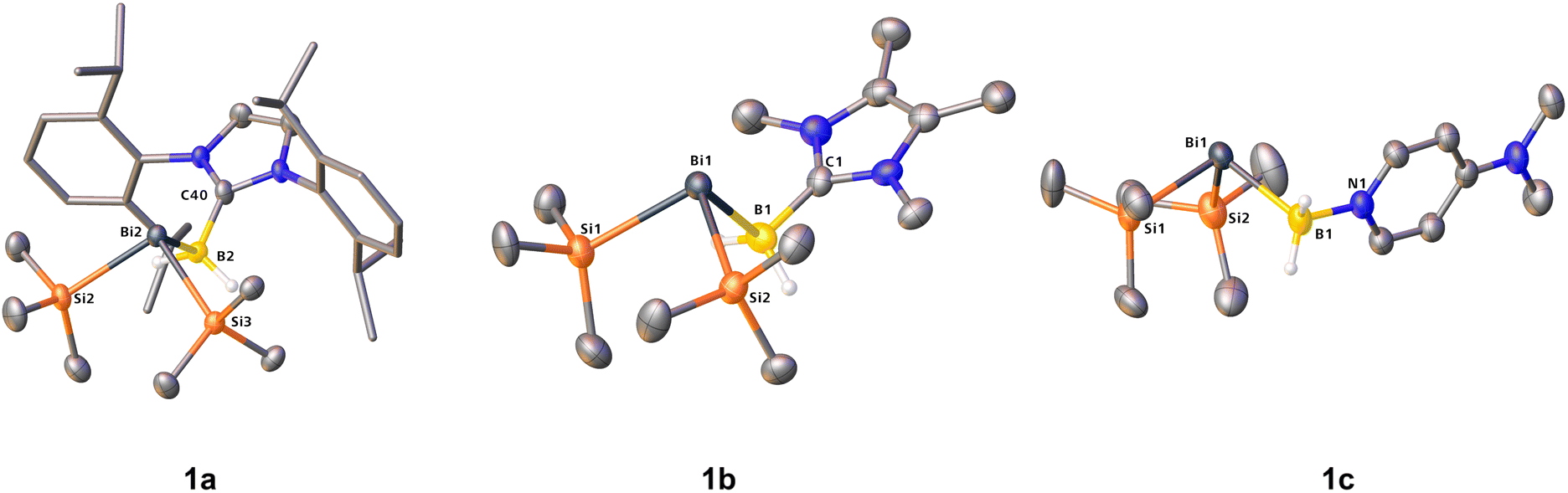 | ||
| Fig. 2 Molecular structures of 1a, 1b and 1c in the solid state. Anisotropic displacement ellipsoids are shown at 50% probability level. Hydrogen atoms bound to carbon are omitted for clarity. Dipp groups are displayed as stick models for improved clarity. Selected bond lengths and angles: 1a C40–B2 1.587(7), B2–Bi2 2.424(5), Bi2–Si2 2.639(4), Bi2–Si3 2.6494(15), C40–B2–Bi2 110.9(3), B2–Bi2–Si2 90.86(15), B2–Bi2–Si3 97.09(14). 1b C1–B1 1.569(9), B1–Bi1 2.442(7), Bi1–Si1 2.6278(17), Bi1–Si2 2.6452(18), C1–B1–Bi1 107.2(4), B1–Bi1–Si1 89.12(18), B1–Bi1–Si2 97.05(19). 1c N1–B1 1.578(5), B1–Bi1 2.424(5), Bi1–Si1 2.6331(9), Bi1–Si2 2.6322(11), N1–B1–Bi1 106.9(2), B1–Bi1–Si1 94.53(10), B1–Bi1–Si2 100.10(12). | ||
1a and 1b show eclipsed conformations at the chain positions, whereas 1c is arranged in a gauche conformation (cf.Fig. 2 and 3). All atoms within the core structure motifs are bound in a (pseudo)tetrahedral manner, with the B–Bi–Si bond angles bent at almost 90° as a result of very poor s–p orbital mixing (1a: 90.83(13)° & 97.10(12)°; 1b: 89.12(18)° & 97.05(19)°, 1c: 94.53(10)° & 100.10(12)°), which is significantly lower than for the lighter homologs (cf. Me3N·BH2P(SiMe3)2: Si–P–B 102.30(8)°).28
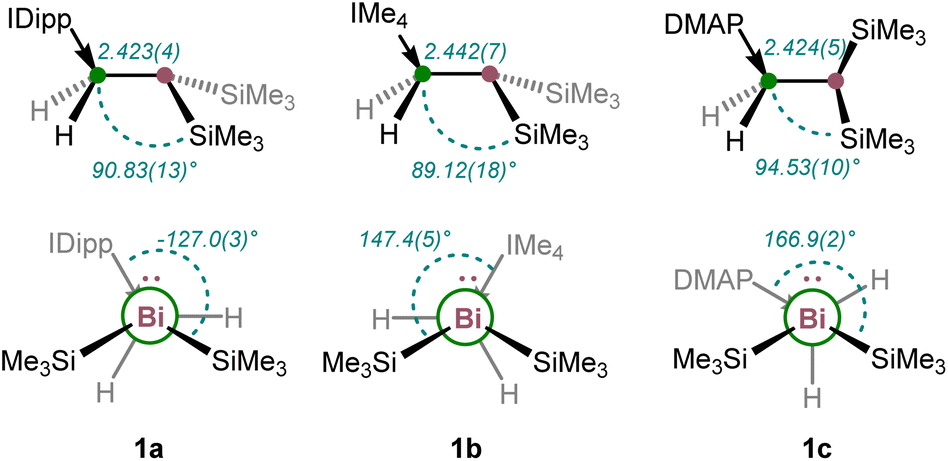 | ||
| Fig. 3 Structural features derived by X-ray crystallographic structure determination of 1a–c including key bond distances [Å] and angles (top). Bottom: Newman projections along the B–Bi σ-bonds alongside the corresponding dihedral angles. | ||
The B–Bi bond distances of 1a–c are between 2.423(4) Å and 2.442(7) Å and represent the first known examples of B–Bi 2c–2e bonds with tetracoordinate boron atoms within chain compounds. Compared to the B–Bi bonds in boryldiphenylbismuthane I (B–Bi: 2.343(6) Å)15 and boryldibismuthene II (B–Bi: 2.326(7)/2.317(9) Å),16 the bonds are significantly elongated due to the different electronic and steric environment of, especially, the boron atom.
The SiMe3 groups on the Bi atom of 1a–c provided a possible opportunity to generate the only hydrogen-substituted parent compounds of 1a–c D·BH2BiH2 by cleavage of the SiMe3 residues. Attempts at the cleavage of the SiMe3 groups were performed by the addition of MeOH, mixtures of MeOH/MeONa, hexanol or KF·HF. In all cases, however, only decomposition products or unreacted starting materials could be detected. The most promising attempt was the reaction of 1a with 1,4-benzoquinone as a {SiMe3} scavenger in the presence of MeOH-d4 in THF (cf. ESI‡), which showed the cleavage of at least one {SiMe3} group at the bismuth atom in the 1H NMR spectrum of the reaction mixture. Simultaneously, besides unreacted 1a, IDipp·BH2(OMe) and IDipp·BH3, a new, broad signal at −31.4 ppm was detected in the 11B NMR spectrum. The 2H NMR spectrum shows a broad signal at 2.96 ppm but, unfortunately, a clear identification of the product was not possible. Conceivably, IDipp·BH2Bi(SiMe3)2−xDx (x = 1–2) is formed as a transient species, but unequivocal evidence of this species could not be obtained. Hence, despite numerous efforts the parent compounds containing H2Bi–BH2 units have so far remained inaccessible.
After the successful synthesis of donor-stabilized bismuthanylboranes, the question arose as to whether it is also possible to synthesize the heavier gallium homolog. Thus, a reaction between IDipp·GaH2(OTf) and KBi(SiMe3)2(THF)0.3 was attempted. After cooling a solution of IDipp·GaH2(OTf) in tetrahydrofuran to −80 °C, the solid bismuthide was added and the mixture was slowly warmed to room temperature over the course of 18 hours (eqn (2)). The 1H NMR spectrum of the reaction mixture showed full conversion of the starting material (0.63 ppm, CD3CN) with a characteristic singlet including 29Si satellites at 0.71 ppm for the {SiMe3} residues (18H, 2JHSi = 7.15 Hz).
 | (2) |
2 is extremely air-, light- and temperature-sensitive and decomposes very slowly in solution already at −80 °C under deposition to elemental bismuth and other unidentified compounds. The 1H NMR spectra of solutions of 2 in C6D6 show characteristic signals at 0.70 ppm (Si(CH3)3) and 4.97 ppm (GaH2; cf.Table 1). The sensitivity of 2 greatly complicates the handling of the product, but, fortunately, by slow evaporation of a solution of 2 in n-pentane under reduced pressure at −80 °C, it was possible to grow a few single crystals of 2, which were of suitable quality for single crystal X-ray structure determination (Fig. 4). 2 crystallizes in the orthorhombic space group Pbca as clear colourless plate-shaped crystals. The structure in the solid state shows (pseudo)tetrahedral geometries at the [GaH2Bi(SiMe3)2] unit with a Ga–Bi bond length of 2.7234(4) Å, which is only slightly elongated compared to the Ga–Bi bond distance of 2.7171(8) Å of {(Dipp2NacNac)GaHBiMe}2.21 The C1–Ga1–Bi1 angle (110.10(8)°) indicates a tetrahedral geometry at the Ga atom while the Ga1–Bi1–Si1 (95.26(2)°) and Ga1–Bi1–Si2 (87.85(2)°) angles are significantly distorted from a regular tetrahedral binding situation. Similar to 1a and 1b, 2 is arranged in an eclipsed conformation (C1–Ga1–Bi1–Si1: 113.33(9)°). The presence of 2 could also be confirmed by mass spectrometry, as the LIFDI-MS spectrum of a solution of 2 in toluene shows a molecular ion peak at m/z = 814.34 besides the decomposition products [IDipp-H]+ (m/z = 389.31) and [IDipp-GaH2]+ (m/z = 459.24).
 | ||
| Fig. 4 Molecular structure of 2 in the solid state. Anisotropic displacement ellipsoids are shown at 50% probability level. Hydrogen atoms bound to carbon are omitted for clarity. Dipp residues are depicted as stick models for better clarity. Selected bond lengths [Å] and angles [°]: N1–C1 1.352(4), N2–C1 1.357(4), C1–Ga1 2.061(3), Ga1–Bi1 2.7234(4), Bi1–Si1 2.6487(9), Bi1–Si2 2.6403(9), N1–B1–Bi1 106.9(2), N1–C1–N2 104.2(3), N1–C1–Ga1 129.2(2), C1–Ga1–Bi1 110.10(8), Ga1–Bi1–Si1 95.26(2), B1–Bi1–Si2 87.85(2). | ||
To analyse the stability and electronic structure of all obtained products, quantum chemical calculations were performed on 1a–2 (B3LYP/def2-TZVP, ECP on Bi; cf. ESI‡). The stability with respect to the dissociation of the Lewis acid/base adducts decreases in the order of IMe4 > IDipp > DMAP and B > Ga, which is in line with the isolated yields and the observed stabilities in solution. The standard Gibbs energy for the dissociation of 2 into IDipp and GaH2Bi(SiMe3)2 is endergonic only by +27 kJ mol−1, which makes this compound the least stable one within the synthesized series with respect to dissociation. Analyses of structural characteristics, NBO and Wiberg bond indices (WBIs) indicate that upon complex formation, the E–Bi bond distances increase by 0.1–0.2 Å and the WBIs of the E–Bi bonds decrease by 0.1–0.37, in line with the above-mentioned order of thermodynamic stability (cf. ESI‡). The B–Bi bond in free BH2Bi(SiMe3)2 has a partial double bond character (WBI 1.21) due to the π-interaction between the lone pair of Bi (75.5% s character) and the empty orbital of boron (99.4% p-character) with an E(2) value of 3.74 kcal mol−1 (Table S6 in ESI‡). In case of the gallium analogue GaH2Bi(SiMe3)2, the similar π-interaction is much less pronounced (E(2) value 1.59 kcal mol−1) and the WBI value is close to one. The occupancies of vacant p-orbitals on boron (0.17ē) and gallium (0.08ē) also indicate smaller involvement of the Ga atom in π-interactions with Bi. After complexation with IDipp, π-interactions between Bi–B and Bi–Ga are absent in both 1a and 2.
Regarding the methanolysis reactions of 1a–1c, the formation of IDipp·BH2BiH2, IMe4·BH2BiH2 and DMAP·BH2BiH2, respectively, is predicted to be highly exothermic and exergonic by 187–206 kJ mol−1, while the formed hydride compounds are predicted to be thermodynamically unstable with respect to the decomposition to LB·BH3, H2 and solid Bi (processes are exergonic by 120–150 kJ mol−1) (cf. ESI‡).
In summary, it could be shown that the synthesis of donor-stabilized bismuthanylboranes and gallanes can be achieved by salt metathesis with KBi(SiMe3)2(THF)0.3, yielding IDipp·BH2Bi(SiMe3)2 (1a), IMe4·BH2Bi(SiMe3)2 (1b), DMAP·BH2Bi(SiMe3)2 (1c), and IDipp·GaH2Bi(SiMe3)2 (2). All compounds could be characterized by multinuclear NMR spectroscopy and LIFDI-MS. Moreover, the crystal structures of all products 1a–2 could be determined, representing the first solid-state structures of bismuthanyltrielanes that contain parent {EH2} triel moieties and possess E–Bi σ-bonds in chain-like compounds. Specifically, 1a–c represent the first examples of structures containing covalent B–Bi σ-bonds where the boron atom is not embedded in a cyclic system. All compounds display a very high sensitivity towards air and light. Desilylation attempts have been unsuccessful, which is likely to be also due to the extreme instability of the potentially formed {BH2BiH2} moiety. Future investigations will focus on the reactivity of these compounds regarding catenation, coordination as well as deposition experiments.
Data availability
The data supporting this article have been included as part of the ESI.‡ CCDC-2347535 (1a), CCDC-2340840 (1b), CCDC-2340841 (1c), and CCDC-2340842 (2), contain the supplementary crystallographic data for this paper.Author contributions
R. Szlosek performed the experimental work (incl. reproductions and analytical data of 1b and 1c), wrote the original draft, acquisition (1a, 2) and refinement (2) of X-ray data. C. Marquardt prepared 1c and acquisition/refinement of X-ray data for 1c. O. Hegen prepared 1b and acquisition/refinement of X-ray data for 1b. G. Balázs conceptualization and project management. C. Riesinger performed the refinement of X-ray data for 1a. A. Y. Timoshkin performed quantum chemical calculations, conceptualization, project management. M. Scheer wrote the original draft, supervised the project and conceptualization, funding and project management.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a joint DFG-RSF project (DFG Sche 384/41-1, Russian Science Foundation (RSF) grant 21-43-04404). The use of computational resources of the research centre “Computing Center” of the St. Petersburg State University is acknowledged. R. S. is grateful to the Fonds der Chemischen Industrie (FCI) for a PhD fellowship. Matthias Hautmann is acknowledged for the preparation of KBi(SiMe3)2(THF)0.3. Lisa Zimmermann and Dr Martin Weber are acknowledged for fruitful discussions.Notes and references
- (a) M. Kanatzidis, H. Sun and S. Dehnen, Inorg. Chem., 2020, 59, 3341 CrossRef CAS PubMed; (b) P. Ondet, G. Lemière and E. Duñach, Eur. J. Org Chem., 2017, 2017, 761 CrossRef CAS; (c) E. Lopez, S. C. Thorp and R. S. Mohan, Polyhedron, 2022, 222, 115765 CrossRef CAS.
- F. Pan, B. Peerless and S. Dehnen, Acc. Chem. Res., 2023, 56, 1018 CrossRef CAS PubMed.
- T. Ollevier, Org. Biomol. Chem., 2013, 11, 2740 RSC.
- H. W. Moon and J. Cornella, ACS Catal., 2022, 12, 1382 CrossRef CAS PubMed.
- (a) X. Gonze, J.-P. Michenaud and J.-P. Vigneron, Phys. Scr., 1988, 37, 785 CrossRef CAS; (b) A. Isaeva and M. Ruck, Inorg. Chem., 2020, 59, 3437 CrossRef CAS PubMed.
- (a) J. Heine, B. Peerless, S. Dehnen and C. Lichtenberg, Angew. Chem., Int. Ed., 2023, 62, e202218771 CrossRef CAS PubMed; (b) F. Pan, S. Wei, L. Guggolz, A. R. Eulenstein, F. Tambornino and S. Dehnen, J. Am. Chem. Soc., 2021, 143, 7176 CrossRef CAS PubMed; (c) A. R. Eulenstein, Y. J. Franzke, P. Bügel, W. Massa, F. Weigend and S. Dehnen, Nat. Commun., 2020, 11, 5122 CrossRef CAS PubMed; (d) A. R. Eulenstein, Y. J. Franzke, N. Lichtenberger, R. J. Wilson, H. L. Deubner, F. Kraus, R. Clérac, F. Weigend and S. Dehnen, Nat. Chem., 2021, 13, 149 CrossRef CAS PubMed; (e) B. Peerless, A. Schmidt, Y. J. Franzke and S. Dehnen, Nat. Chem., 2023, 15, 347 CrossRef CAS PubMed.
- (a) J. M. Bothwell, S. W. Krabbe and R. S. Mohan, Chem. Soc. Rev., 2011, 40, 4649 RSC; (b) R. Hua, Curr. Org. Synth., 2008, 5, 1 CrossRef CAS.
- (a) D. Janssen-Müller and M. Oestreich, Angew. Chem., Int. Ed., 2020, 59, 8328 CrossRef PubMed; (b) B. Jousseaume, C. Laporte, T. Toupance and J.-M. Bernard, Tetrahedron Lett., 2002, 43, 6305 CrossRef CAS; (c) T. Kitanosono, T. Ollevier and S. Kobayashi, Chem.–Asian J., 2013, 8, 3051 CrossRef CAS PubMed; (d) C. Lichtenberg, Chem. Commun., 2021, 57, 4483 RSC; (e) H. Qin, N. Yamagiwa, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2006, 128, 1611 CrossRef CAS PubMed; (f) H. Qin, N. Yamagiwa, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2007, 46, 409 CrossRef CAS PubMed; (g) J. Ramler, I. Krummenacher and C. Lichtenberg, Angew. Chem., Int. Ed., 2019, 58, 12924 CrossRef CAS PubMed; (h) M. Rueping, B. J. Nachtsheim and W. Ieawsuwan, Adv. Synth. Catal., 2006, 348, 1033 CrossRef CAS; (i) J. A. Salvador and S. M. Silvestre, Tetrahedron Lett., 2005, 46, 2581 CrossRef CAS; (j) S. Vidal, Synlett, 2001, 2001, 1194 CrossRef; (k) N. Worz, A. Brandner and P. Claus, J. Phys. Chem. C, 2010, 114, 1164 CrossRef; (l) Z.-P. Zhang, N. Dong and X. Li, Chem. Commun., 2017, 53, 1301 RSC; (m) K. Zhou, W. Wang, Z. Zhao, G. Luo, J. T. Miller, M. S. Wong and F. Wei, ACS Catal., 2014, 4, 3112 CrossRef CAS; (n) M. Pramanik, M. G. Guerzoni, E. Richards and R. L. Melen, Angew Chem. Int. Ed. Engl., 2024, 63, e202316461 CrossRef CAS PubMed; (o) M. Mato and J. Cornella, Angew. Chem., Int. Ed., 2024, 63, e202315046 CrossRef CAS PubMed.
- (a) M. Magre and J. Cornella, J. Am. Chem. Soc., 2021, 143, 21497 CrossRef CAS PubMed; (b) M. Magre, J. Kuziola, N. Nöthling and J. Cornella, Org. Biomol. Chem., 2021, 19, 4922 RSC; (c) Y. Pang, M. Leutzsch, N. Nöthling and J. Cornella, J. Am. Chem. Soc., 2020, 142, 19473 CrossRef CAS PubMed; (d) Y. Pang, M. Leutzsch, N. Nöthling, F. Katzenburg and J. Cornella, J. Am. Chem. Soc., 2021, 143, 12487 CrossRef CAS PubMed; (e) F. Wang, O. Planas and J. Cornella, J. Am. Chem. Soc., 2019, 141, 4235 CrossRef CAS PubMed; (f) O. Planas, F. Wang, M. Leutzsch and J. Cornella, Science, 2020, 367, 313 CrossRef CAS PubMed; (g) X. Yang, J. Kuziola, V. A. Béland, J. Busch, M. Leutzsch, J. Burés and J. Cornella, Angew. Chem., Int. Ed., 2023, e202306447 CAS; (h) X. Yang, E. J. Reijerse, K. Bhattacharyya, M. Leutzsch, M. Kochius, N. Nöthling, J. Busch, A. Schnegg, A. A. Auer and J. Cornella, J. Am. Chem. Soc., 2022, 144, 16535 CrossRef CAS PubMed; (i) X. Yang, E. J. Reijerse, N. Nöthling, D. J. SantaLucia, M. Leutzsch, A. Schnegg and J. Cornella, J. Am. Chem. Soc., 2023, 145, 5618 CrossRef CAS PubMed; (j) H. W. Moon, F. Wang, K. Bhattacharyya, O. Planas, M. Leutzsch, N. Nöthling, A. A. Auer and J. Cornella, Angew. Chem., Int. Ed., 2023, e202313578 CAS; (k) T. Tsuruta, D. Spinnato, H. W. Moon, M. Leutzsch and J. Cornella, J. Am. Chem. Soc., 2023, 145, 25538 CrossRef CAS PubMed.
- M. Mato, D. Spinnato, M. Leutzsch, H. W. Moon, E. J. Reijerse and J. Cornella, Nat. Chem., 2023, 15, 1138–1145 CrossRef CAS PubMed.
- O. Planas, V. Peciukenas and J. Cornella, J. Am. Chem. Soc., 2020, 142, 11382 CrossRef CAS PubMed.
- O. Planas, V. Peciukenas, M. Leutzsch, N. Nöthling, D. A. Pantazis and J. Cornella, J. Am. Chem. Soc., 2022, 144, 14489 CrossRef CAS PubMed.
- (a) S. S. Chitnis, N. Burford, A. Decken and M. J. Ferguson, Inorg. Chem., 2013, 52, 7242 CrossRef CAS PubMed; (b) S. S. Chitnis, A. P. M. Robertson, N. Burford, B. O. Patrick, R. McDonald and M. J. Ferguson, Chem. Sci., 2015, 6, 6545 RSC; (c) S. S. Chitnis, K. A. Vos, N. Burford, R. McDonald and M. J. Ferguson, Chem. Commun., 2016, 52, 685 RSC; (d) T. Hynes, J. D. Masuda and S. S. Chitnis, ChemPlusChem, 2022, 87, e202200244 CrossRef CAS PubMed; (e) M. B. Kindervater, T. Hynes, K. M. Marczenko and S. S. Chitnis, Dalton Trans., 2020, 49, 16072 RSC; (f) M. B. Kindervater, K. M. Marczenko, U. Werner-Zwanziger and S. S. Chitnis, Angew. Chem., Int. Ed., 2019, 58, 7850 CrossRef CAS PubMed; (g) K. M. Marczenko and S. S. Chitnis, Chem. Commun., 2020, 56, 8015 RSC; (h) K. M. Marczenko, J. A. Zurakowski, M. B. Kindervater, S. Jee, T. Hynes, N. Roberts, S. Park, U. Werner-Zwanziger, M. Lumsden, D. N. Langelaan and S. S. Chitnis, Chem. - Eur. J., 2019, 25, 16414 CrossRef CAS PubMed; (i) K. M. Marczenko, S. Jee and S. S. Chitnis, Organometallics, 2020, 39, 4287 CrossRef CAS.
- T. J. Hannah, W. M. McCarvell, T. Kirsch, J. Bedard, T. Hynes, J. Mayho, K. L. Bamford, C. W. Vos, C. M. Kozak, T. George, J. D. Masuda and S. S. Chitnis, Chem. Sci., 2023, 14, 4549 RSC.
- W. Lu, H. Hu, Y. Li, R. Ganguly and R. Kinjo, J. Am. Chem. Soc., 2016, 138, 6650 CrossRef CAS PubMed.
- D. Dange, A. Davey, J. A. B. Abdalla, S. Aldridge and C. Jones, Chem. Commun., 2015, 51, 7128 RSC.
- (a) T. Dunaj, K. Dollberg and C. von Hänisch, Dalton Trans., 2022, 51, 7551 RSC; (b) T. Dunaj, K. Dollberg, C. Ritter, F. Dankert and C. Hänisch, Eur. J. Inorg. Chem., 2021, 2021, 870 CrossRef CAS; (c) T. Dunaj, M. Egorycheva, A. Arebi, K. Dollberg and C. von Hänisch, Z. Anorg. Allg. Chem., 2023, 649 Search PubMed; (d) T. Dunaj and C. von Hänisch, Chem.–Eur. J., 2022, 28, e202202932 CrossRef CAS PubMed; (e) C. Ritter, N. Michel, A. Rinow, B. Ringler and C. Hänisch, Eur. J. Inorg. Chem., 2021, 2021, 2514 CrossRef CAS; (f) C. Ritter, F. Weigend and C. von Hänisch, Chem.–Eur. J., 2020, 26, 8536 CrossRef CAS PubMed.
- T. Dunaj, J. Schwarzmann, J. Ramler, A. Stoy, S. Reith, J. Nitzsche, L. Völlinger, C. von Hänisch and C. Lichtenberg, Chem.–Eur. J., 2023, 29, e202204012 CrossRef CAS PubMed.
- (a) C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Chem. Commun., 2014, 50, 12382 RSC; (b) L. Tuscher, C. Helling, C. Wölper, W. Frank, A. S. Nizovtsev and S. Schulz, Chem.–Eur. J., 2018, 24, 3241 CrossRef CAS PubMed.
- G. Prabusankar, C. Gemel, P. Parameswaran, C. Flener, G. Frenking and R. A. Fischer, Angew. Chem., Int. Ed., 2009, 48, 5526 CrossRef CAS PubMed.
- S. Schneider and C. von Hänisch, Chem. Commun., 2022, 58, 1522 RSC.
- M. D. Anker, A. O'Reilly, C. L. McMullin and M. P. Coles, Polyhedron, 2023, 246, 116695 CrossRef CAS.
- (a) C. Marquardt, O. Hegen, A. Vogel, A. Stauber, M. Bodensteiner, A. Y. Timoshkin and M. Scheer, Chem. - Eur. J., 2018, 24, 360 CrossRef CAS PubMed; (b) M. Bodensteiner, A. Y. Timoshkin, E. V. Peresypkina, U. Vogel and M. Scheer, Chem.–Eur. J., 2013, 19, 957 CrossRef CAS PubMed; (c) M. Bodensteiner, U. Vogel, A. Y. Timoshkin and M. Scheer, Angew. Chem., Int. Ed., 2009, 48, 4629 CrossRef CAS PubMed; (d) C. Marquardt, T. Jurca, K.-C. Schwan, A. Stauber, A. V. Virovets, G. R. Whittell, I. Manners and M. Scheer, Angew. Chem., Int. Ed., 2015, 54, 13782 CrossRef CAS PubMed; (e) C. Thoms, C. Marquardt, A. Y. Timoshkin, M. Bodensteiner and M. Scheer, Angew. Chem., Int. Ed., 2013, 52, 5150 CrossRef CAS PubMed; (f) J. Braese, F. Lehnfeld, V. T. Annibale, T. Oswald, R. Beckhaus, I. Manners and M. Scheer, Chem. Eur. J., 2023, 29, e202301741 CrossRef CAS PubMed.
- (a) R. Szlosek, M. Seidl, G. Balázs and M. Scheer, Chem.–Eur. J., 2023, 29, e202301752 CrossRef CAS PubMed; (b) O. Hegen, A. V. Virovets, A. Y. Timoshkin and M. Scheer, Eur. J. Inorg. Chem., 2018, 24, 16521–16525 CAS; (c) F. Lehnfeld, M. Seidl, A. Y. Timoshkin and M. Scheer, Eur. J. Inorg. Chem., 2022, 3, e202100930 CrossRef; (d) C. Marquardt, T. Kahoun, J. Baumann, A. Y. Timoshkin and M. Scheer, Z. Anorg. Allg. Chem., 2017, 643, 1326 CrossRef CAS; (e) M. T. Ackermann, M. Seidl, F. Wen, M. J. Ferguson, A. Y. Timoshkin, E. Rivard and M. Scheer, Chem. - Eur. J., 2022, 28, e20210370 CrossRef PubMed; (f) C. Marquardt, G. Balázs, J. Baumann, A. V. Virovets and M. Scheer, Chem.–Eur. J., 2017, 23, 11423 CrossRef CAS PubMed; (g) C. Marquardt, C. Thoms, A. Stauber, G. Balázs, M. Bodensteiner and M. Scheer, Angew. Chem., Int. Ed., 2014, 53, 3727 CrossRef CAS PubMed; (h) M. Elsayed Moussa, T. Kahoun, M. T. Ackermann, M. Seidl, M. Bodensteiner, A. Y. Timoshkin and M. Scheer, Organometallics, 2022, 41, 1572 CrossRef CAS; (i) C. Marquardt, T. Kahoun, A. Stauber, G. Balázs, M. Bodensteiner, A. Y. Timoshkin and M. Scheer, Angew. Chem., Int. Ed., 2016, 55, 14828 CrossRef CAS PubMed; (j) C. Marquardt, O. Hegen, T. Kahoun and M. Scheer, Chem.–Eur. J., 2017, 23, 4397 CrossRef CAS PubMed; (k) U. Vogel, A. Y. Timoshkin and M. Scheer, Angew. Chem., Int. Ed., 2001, 40, 4409 CrossRef CAS; (l) U. Vogel, P. Hoemensch, K.-C. Schwan, A. Y. Timoshkin and M. Scheer, Chem.–Eur. J., 2003, 9, 515 CrossRef CAS PubMed; (m) F. Lehnfeld, M. Seidl, A. Y. Timoshkin and M. Scheer, Eur. J. Inorg. Chem., 2022, 3, e202100930 CrossRef; (n) A. Adolf, M. Zabel and M. Scheer, Eur. J. Inorg. Chem., 2007, 2007, 2136 CrossRef; (o) F. Lehnfeld, M. Seidl, A. Timoshkin and M. Scheer, Eur. J. Inorg. Chem., 2023, 26, e202300338 CrossRef CAS; (p) R. Szlosek, A. S. Niefanger, G. Balázs, M. Seidl, A. Y. Timoshkin and M. Scheer, Chem.–Eur. J., 2024, 30, e202303603 CrossRef CAS PubMed.
- (a) A. Besson, C. R., 1890, 516 Search PubMed; (b) M. Elsayed Moussa, J. Braese, C. Marquardt, M. Seidl and M. Scheer, Eur. J. Inorg. Chem., 2020, 26, 2501 CrossRef; (c) M. Elsayed Moussa, C. Marquardt, O. Hegen, M. Seidl and M. Scheer, New J. Chem., 2021, 45, 14916 RSC; (d) K.-C. Schwan, A. Adolf, M. Bodensteiner, M. Zabel and M. Scheer, Z. Anorg. Allg. Chem., 2008, 634, 1383 CrossRef CAS; (e) R. Szlosek, M. T. Ackermann, C. Marquardt, M. Seidl, A. Y. Timoshkin and M. Scheer, Chem.–Eur. J., 2023, 29, e202202911 CrossRef CAS PubMed.
- J. Braese, A. Schinabeck, M. Bodensteiner, H. Yersin, A. Y. Timoshkin and M. Scheer, Chem.–Eur. J., 2018, 24, 10073 CrossRef CAS PubMed.
- (a) K.-C. Schwan, A. Y. Timoshkin, M. Zabel and M. Scheer, Chem.–Eur. J., 2006, 12, 4900 CrossRef CAS PubMed; (b) C. Marquardt, O. Hegen, M. Hautmann, G. Balázs, M. Bodensteiner, A. V. Virovets, A. Y. Timoshkin and M. Scheer, Angew. Chem., Int. Ed., 2015, 54, 13122 CrossRef CAS PubMed.
- C. Marquardt, A. Adolf, A. Stauber, M. Bodensteiner, A. V. Virovets, A. Y. Timoshkin and M. Scheer, Chem. - Eur. J., 2013, 19, 11887 CrossRef CAS PubMed.
- M. A. K. Weinhart, A. S. Lisovenko, A. Y. Timoshkin and M. Scheer, Angew. Chem., Int. Ed., 2020, 59, 5541 CrossRef CAS PubMed.
- M. A. K. Weinhart, M. Seidl, A. Y. Timoshkin and M. Scheer, Angew. Chem., 2021, 133, 3850 CrossRef.
- R. Szlosek, M. A. K. Weinhart, G. Balázs, M. Seidl, L. Zimmermann and M. Scheer, Chem.–Eur. J., 2023, 29, e202300340 CrossRef CAS PubMed.
- K. Dollberg, A. Marx, R.-M. Richter, L. Erlemeier, T. Dunaj, F. Weigend and C. von Hänisch, Chem.–Eur. J., 2024, 30, e202303734 CrossRef CAS PubMed.
Footnotes |
| † In memoriam on Ian Manners. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2347535 and 2340840–2340842. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03926b |
| This journal is © The Royal Society of Chemistry 2024 |