
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriplet dynamics reveal loss pathways in multi-resonance thermally activated delayed fluorescence emitters†
Alexandra N.
Stuart
 *ab,
Katrina
Bergmann
c,
Inseong
Cho
ab,
William J.
Kendrick
bd,
Zachary M.
Hudson
c,
Wallace W. H.
Wong
bd and
Girish
Lakhwani
*ab
*ab,
Katrina
Bergmann
c,
Inseong
Cho
ab,
William J.
Kendrick
bd,
Zachary M.
Hudson
c,
Wallace W. H.
Wong
bd and
Girish
Lakhwani
*ab
aDepartment of Chemistry, The University of Sydney, Camperdown, New South Wales 2000, Australia. E-mail: a.stuart@sydney.edu.a; girish.lakhwani@sydney.edu.au
bAustralian Research Council Centre of Excellence in Exciton Science, Parkville 3010, Australia
cDepartment of Chemistry, The University of British Columbia, Vancouver, British Columbia V6T 1Z1, Canada
dSchool of Chemistry, The University of Melbourne, Parkville 3010, VIC, Australia
First published on 2nd August 2024
Abstract
Multi-resonance thermally activated delayed fluorescence (MR-TADF) materials are of interest for light-emitting applications due to their narrow emission bandwidths and high photoluminescence quantum yields. Whilst there have been numerous examples of multi-resonance molecules exhibiting efficient TADF, the photophysics and mechanism of TADF in multi-resonance emitters have not been investigated to the same extent as the more conventional spatially separated donor–acceptor TADF materials, limiting the development of MR-TADF devices. Here we study the photophysics of a multi-resonance TADF material, OQAO(mes)2, using transient absorption spectroscopy to spectrally resolve the triplet population(s). We identify multiple triplet populations with distinct spectral contributions, and resolve the dynamics between them. Unlike conventional donor–acceptor TADF materials that have previously been studied, we find these triplet states are not formed in equilibrium, instead exhibiting a slow evolution from a high-energy triplet to a low-energy triplet. Delayed fluorescence predominantly reflects the lifetime of the high-energy triplet state, indicating that the formation of the low-energy triplet is a loss pathway for TADF. We also find that greater amounts of the low-energy triplet are formed in a higher dielectric environment, which leads to less delayed fluorescence. These triplet dynamics have significant implications for TADF in devices, as depending on the identity of the triplet formed by electrical excitation, there will either be a significant barrier to TADF, or a competing nonradiative decay pathway.
1 Introduction
Thermally activated delayed fluorescence (TADF) is a desirable process for organic light-emitting applications, as it can convert non-emissive triplet excited states generated from electrical excitation to emissive singlet excited states, increasing the efficiency of light emission. This is done through the conversion of triplet excitons to singlet excitons through reverse intersystem crossing (RISC). For TADF to be achievable at room temperature, the difference in energy between singlet and triplet excited states should be small enough to be thermally accessible (≲0.26 eV, or 1 kT). A common design strategy to satisfy this requirement is to use donor–acceptor (D–A) type molecules, with spatially separated electron-donating and electron-withdrawing groups (Fig. 1).1–3 The separation between donor and acceptor moieties decreases the overlap between HOMO and LUMO wavefunctions, which decreases the electronic exchange energy and in turn the singlet–triplet energy gap.1,4–6 This design strategy has yielded a plethora of spatially separated D–A molecules that exhibit high TADF efficiencies.3,7,8 However, this D–A architecture also comes with some disadvantages. The strong charge transfer (CT) character of the excited states of these materials generally results in broad emission bandwidths, which are undesirable for light-emitting applications as they reduce color purity.9–12 Additionally, the HOMO–LUMO arrangements of spatially separated D–A molecules lead to low oscillator strengths for electronic transitions to and from the ground state, and consequently lower photoluminescence quantum yields (PLQYs) and large losses through thermal relaxation.3,11 | ||
| Fig. 1 Schematic illustrating the design and photophysics of conventional D–A emitters, and the design and photophysics of the multi-resonance TADF emitter in this study. Energy levels are not necessarily to scale. IC is internal conversion. | ||
Multi-resonance TADF (MR-TADF) molecules have emerged as an alternative to the conventional D–A emitters.9,11–13 Rather than separated donor and acceptor groups, MR-TADF emitters have electron-rich and electron-deficient atoms together in a fused polycyclic aromatic hydrocarbon structure. By arranging the electron-rich and electron-deficient atoms such that their resonances are opposite, alternating HOMO and LUMO orbitals are generated that are separated enough to allow for small singlet–triplet energy gaps, whilst also retaining the oscillator strength necessary for high PLQYs (Fig. 1). The orbital arrangement and the rigidity of the MR-TADF structure reduces vibronic coupling and structural rearrangements, which reduces losses from thermal relaxation, and, importantly, results in narrow emission bandwidths. Aside from the narrow emission bandwidths making MR-TADF beneficial for the color purity of organic light emitting diodes (OLEDs), they are also attractive systems for modifying the photophysics through strong light-matter coupling.14
Whilst many MR-TADF emitters have now been designed and tested,12 the mechanistic details of TADF in these systems have not been well studied. As mentioned above, a consequence of the D–A structure is the strong CT character of the singlet and triplet excited states. Direct conversion between singlets and triplets with substantial CT character and the same molecular orbital spatial occupation (i.e.1CT and 3CT) is forbidden,15–17 and conversion must be instead mediated by mixing with triplet states with less CT character, namely, local exciton triplets (3LE).16,18–20 The 3LE states can be the triplet state corresponding to either the donor or the acceptor, and conversion or mixing between 3LE and 3CT has been shown to occur through vibronic coupling.3,18,20 The local exciton triplet involvement has significant consequences on the TADF photophysics. Many studies have shown that the energy gap between the 1CT and 3LE state governs the efficiency of TADF, rather than 1CT and 3CT.2,19,21,22 Additionally, the efficiency of RISC is highly dependent on the degree of vibronic coupling.3,20 Calculations have shown that vibrational modes are responsible for driving systems from CT to LE states,18 as well as decreasing singlet–triplet energy barriers.23 Experiments have also shown that increasing rigidity or steric hindrance in D–A molecules, and therefore decreasing vibronic coupling, is able to switch RISC off entirely.19,24 Given that the CT character in MR-TADF materials is generally weaker than D–A emitters, and the rigidity of MR structures reduces vibronic coupling, the differences in the TADF mechanism for these molecules remains uncertain. In particular, the relevance of local exciton triplet states and vibronic coupling in MR-TADF molecules, and the implications for MR-TADF OLEDs, is poorly understood.
Here, we investigate the photophysics and mechanism of TADF in the MR-TADF emitter (2,6-dimesitylbenzo)[9,1]quinolizino[3,4,5,6,7-klmn]phenoxazine-8,12-dione, or OQAO(mes)2 (Fig. 2a). This was derived from a previously synthesized MR-TADF emitter, OQAO, reported by Zou et al., with bulky mesityl groups added to increase solution processibility.25 Mesityl groups have also been shown to reduce aggregation induced quenching when added to the QAO moiety.26 We have previously shown that OQAO(mes)2 undergoes TADF, with a singlet–triplet energy gap of ∼0.15 eV,14 similar to the 0.16 eV gap in the unsubstituted OQAO.25 Here we investigate the photophysics of OQAO(mes)2 in detail through time-resolved emission and transient absorption spectroscopy. By changing the dielectric environment, we systematically alter the dynamics of the triplet excited states, revealing that multiple triplet states are formed, with an early-time high-energy triplet (Tearly) slowly converting to a late-time low-energy triplet (Tlate). This is distinct from previous studies on 3CT and 3LE in spatially-separated D–A emitters, which are reported to form in equilibrium.2 Secondly, we show that delayed emission predominantly corresponds to the lifetime of Tearly, indicating that forming Tlate is a competing pathway for RISC. Third, we show Tlate formation increases with solvent polarity whilst the amount of delayed emission decreases, further indicating that formation of Tlate is a loss pathway for TADF. Finally, time-dependent density functional theory suggests the identity of the Tearly state is T2, which undergoes internal conversion to T1 (Tlate).
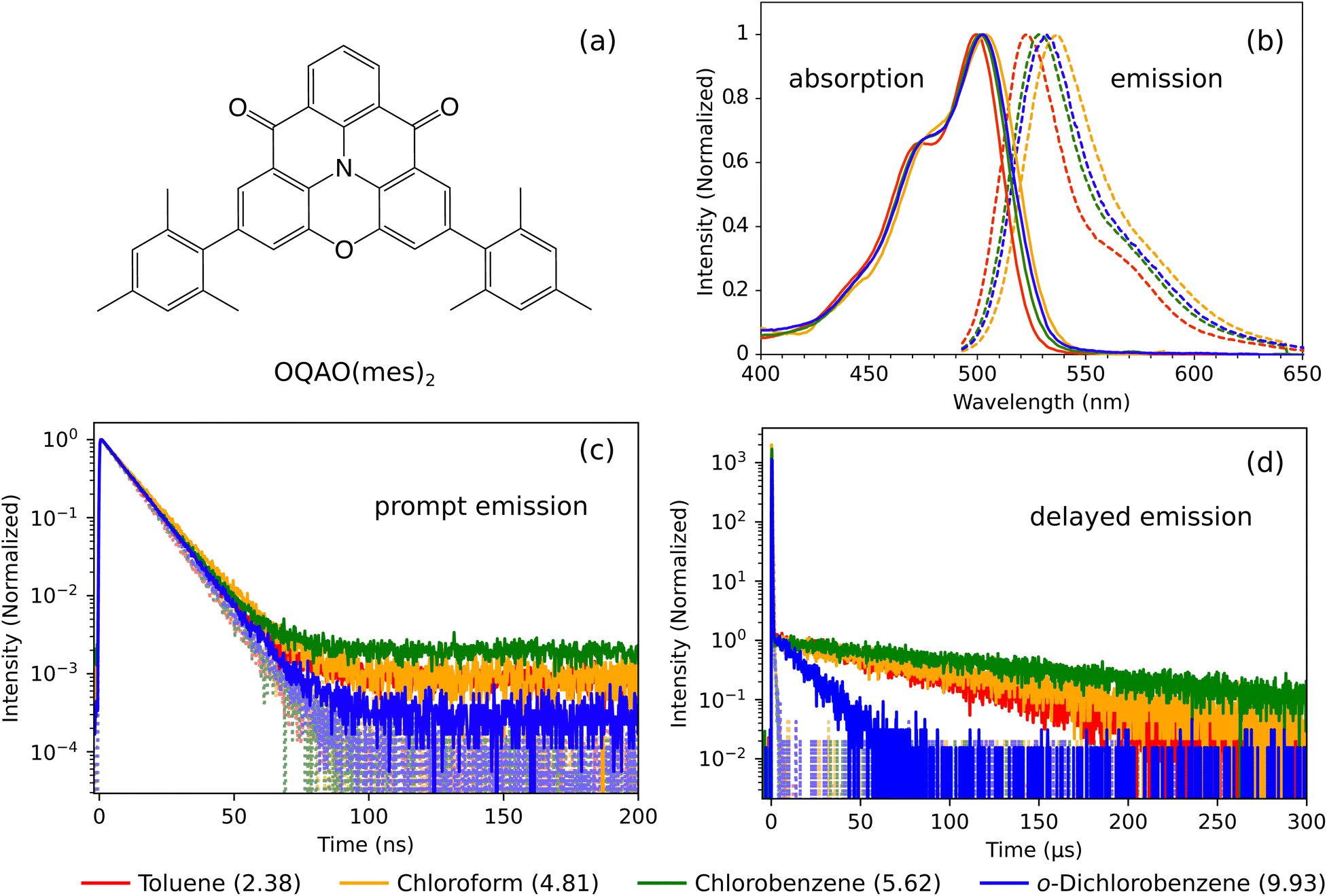 | ||
| Fig. 2 (a) The molecular structure of OQAO(mes)2. (b) Steady-state absorption (full lines) and emission exciting at 480 nm (dashed lines) of OQAO(mes)2 in various solvents at 298 K. Both absorption and emission are normalized to the maximum value. (c) Prompt emission and (d) delayed emission of OQAO(mes)2 in various solvents at 298 K, exciting at 480 nm and monitoring at the emission maximum. Full lines show solutions under N2, and dotted lines under O2. Prompt emission is normalized to the maximum intensity, and delayed emission is normalized to the intensity at 3 μs. The dielectric constant of the solvent is indicated in brackets in the legend. Additional plots of time-resolved emission data can be found in the ESI Section S2.† | ||
The presence of this low-energy triplet has significant implications for the application of this molecule in light-emitting devices. If T2 is formed through electrical injection, some RISC to bright singlets can occur, but the formation of T1 will be a significant competing loss pathway. If T2 is formed through electrical injection, then very little RISC will occur, particularly in low polarity environments. Our results hence demonstrate that the TADF efficiency in an optically excited system may significantly differ from that in an electrically injected system, or in other words, optical excitation may not be a good model for actual devices. Additionally, this work highlights the benefit of exploring the optically dark states in TADF systems with methods such as transient absorption, as these loss pathways are not evident from emission spectroscopy alone.
2 Results and discussion
OQAO(mes)2 was synthesized as reported previously,14 and solutions prepared as described in the ESI.† The steady-state (ground-state) absorption and emission of OQAO(mes)2 in various solvents are shown in Fig. 2b. The absorption and emission red-shift to lower energies with increasing dielectric constant of the solvent environment, indicating the stabilization of the singlet excited state with increasing polarity of the solvent. The exception to this is chloroform, which has a lower dielectric constant than chlorobenzene and o-dichlorobenzene, but stabilizes the singlet state further. This is likely due to specific solute–solvent interactions which are not captured by solely comparing dielectric constants.16,27 For example, hydrogen bond donating ability, which, although weak, is larger for chloroform than the other solvents.28–30 The trends here are consistent with empirical polarity parameters that account for multiple solute–solvent interactions, such as Reichardt's normalized solvent polarity parameter, ENT, which is shown in Table 1.31 The emission spectra also lose some vibronic features as polarity is increased, which is indicative of an increase in CT character. The time-resolved emission of OQAO(mes)2 in various solvents at 298 K is shown in Fig. 2c and d, both for solutions under N2 (full lines) and after exposure to air (dotted lines). The OQAO(mes)2 solutions exhibit both prompt emission (Fig. 2c, measured using time correlated single photon counting (TCSPC)) and delayed emission (Fig. 2d, measured using multi-channel scaling (MCS)). Details of these measurements are given in the ESI (Section S1†). Upon exposure to air, the delayed emission is almost entirely quenched, due to deactivation of triplet states by oxygen. Additionally, the background level of the prompt emission decays are reduced, and the prompt emission becomes near identical for each solvent. The time-resolved emission of each solvent is also shown individually in the ESI,† where these trends are more evident (Fig. S1†). Given the repetition rate required to collect the prompt emission decay is 1.22 MHz, the background (or pre-time zero) emission is recorded before the sample has completely relaxed to the ground state, resulting in delayed emission being present both before and after time zero. When delayed emission is quenched by oxygen, the background drops to zero, as in Fig. 2c. The background of the prompt emission is thus indicative of the magnitude and lifetime of delayed fluorescence, and follows the trend in delayed fluorescence lifetime. o-Dichlorobenzene shows the fastest delayed emission decay and the lowest prompt background level, at only 0.02% of the maximum prompt emission intensity. A shorter delayed emission lifetime can imply a greater rate of RISC, however the low background in the prompt emission suggests this is not the case here. It is more likely that the more rapid o-dichlorobenzene decay is due to the triplet state being depleted faster through some other, competing pathway, which increases with the polarity of the solvent. The delayed emission lifetime also appears to increase with dielectric constant from toluene to chlorobenzene, before rapidly decreasing for o-dichlorobenzene, the solvent with the highest dielectric constant. However, the delayed emission lifetime is extremely sensitive to quenching by even small amounts of oxygen, and the differing oxygen contents in each solution cannot be ruled out. To further complicate the emission data, the prompt fluorescence decay also becomes slightly faster on exposure to air, indicating that the singlet excited state is quenched by oxygen in addition to triplet states (see ESI Section S2†). Because of this, we cannot reliably use the ratio of emission intensity under N2 and O2 to calculate the rate of RISC, as has been occasionally done in other studies.3| Dielectric constant/ENTa | τ RISC , (μs) | τ 2 , (μs) | τ R2 , (μs) | k ISC/kRISC | k 2/kR2 | η Tearly (%) | η Tlate (%) | |
|---|---|---|---|---|---|---|---|---|
| a Reichardt's normalized polarity parameter.31 b Time constant of reverse intersystem crossing. c Time constant of conversion from Tearly to Tlate. d Time constant of conversion from Tlate to Tearly. e Errors are one standard deviation of the fitted parameters. f Photons out/excitons formed from charge recombination to Tearly and S1. g Photons out/excitons formed from charge recombination to Tlate and S1. | ||||||||
| Toluene | 2.38/0.099 | 156 ± 10 | 55 ± 2 | 240 ± 20 | 4.4 ± 0.4 | 8400 ± 500 | 24.3 | 19.7 |
| Chloroform | 4.81/0.259 | 53 ± 4 | 5.20 ± 0.08 | 51 ± 2 | 10.0 ± 0.4 | 1800 ± 100 | ||
| Chlorobenzene | 5.62/0.188 | 78 ± 24 | 1.70 ± 0.07 | 10.6 ± 0.5 | 6.2 ± 0.4 | 2700 ± 800 | ||
| o-Dichlorobenzene | 9.93/0.225 | 20 ± 2 | 0.59 ± 0.01 | 5.2 ± 0.5 | 8.7 ± 0.8 | 1000 ± 100 | 31.4 | 31.0 |
To understand the differences in delayed emission for each solvent, we next turn to transient absorption (TA) measurements of OQAO(mes)2 in solution. Fig. 3a shows the TA of OQAO(mes)2 in o-dichlorobenzene under N2. At early times, there is a sharp excited-state absorption (ESA) at 460 nm, which decays by around 50 ns. A combination of ground-state bleach (GSB) and stimulated emission (SE) is evident around 500 nm, with the SE component (around 530 nm) decaying on the same timescale as the 460 nm ESA and the prompt emission decay, leading us to assign the 460 nm ESA as singlet exciton absorption. The signals that persist after the singlet excited state has completely decayed (i.e. after 50 ns) we assign to triplet ESAs. ESAs from 600–800 nm are evident at all times, but change spectral shape significantly. Fig. 3b shows the spectra normalized to the GSB, demonstrating the clear changes in shape. The sharper feature at 630 nm increases relative to the GSB as the singlet exciton features decay, indicating a conversion of singlet to triplet through intersystem crossing (ISC). However, after this feature reaches a maximum at 50 ns, it then decreases, and the ESA becomes much broader and flatter. The spectrum continues to evolve until approximately 10 μs. This spectral evolution indicates at least three spectrally distinct components are present in the sample. This late-time spectral evolution is distinct from the TA of more conventional spatially separated D–A emitters, for which no spectral evolution is observed after the initial triplet formation.2,21 The TA spectra of all solvents studied here show similar features to varying extents (shown in ESI Section S3†). As a general trend, the late-time component becomes smaller and evolves slower with decreasing dielectric constant.
 | ||
| Fig. 3 TA spectra of OQAO(mes)2 in o-dichlorobenzene under N2 at 298 K, excited at 355 nm. (a) The raw TA spectra. (b) The TA spectra normalized to the GSB. Data is omitted from 705–715 nm due to scattering of the excitation pulse. (c) The deconvoluted spectral components of the TA spectra, alongside the (inverted) steady-state (SS) absorption and emission spectra. (d) The proportions of each spectral component, proportional to the concentration of the corresponding species, over time. The vertical dashed line indicates a change from linear to log scale. | ||
To identify and examine the dynamics of the different excited states present in the TA spectra, we deconvoluted the spectra into their base components. The methodology is described in detail in the ESI Section S4.† Briefly, we take the early-time spectrum (1 ns) as a singlet basis spectrum, which we label S1. Two spectra assigned to triplet states are extracted: one from very late times after the spectra have stopped evolving (>300 us), which we label Tlate, and one from intermediate times (55 ns), after the S1 state has predominantly decayed (which can be confirmed both by the prompt emission decay and the decay of the 460 nm ESA), which we label Tearly. Our assignment of these late time species as triplets is confirmed by triplet sensitization experiments (ESI Section S5†). The resulting basis spectra can be seen for o-dichlorobenzene in Fig. 3c, with the remaining solvents shown in the ESI Section S4.2.† A linear combination of these basis spectra was fit at each delay time to determine the relative concentration of each component, as shown in Fig. 3d. Note that we did not use global target analysis for this deconvolution, rather, each delay time was deconvoluted independently, and no model was assumed. A 2-component spectral deconvolution was also performed (ESI Section S4.1†), which results in poor fits, verifying that a minimum of 3 components are required. The amount of each basis spectrum required for the deconvolution, which is proportional to the concentration of that species, is plotted over time in Fig. 3d. It can be seen that the rise of Tearly is correlated with the decay of S1, which in turn is in good agreement with the prompt emission decay, as shown in the ESI (Fig. S19†). At later times, Tearly decays and the Tlate state concurrently rises. There is a small tail in the decay of the Tearly state for o-dichlorobenzene, indicating a small degree of reverse reaction from Tlate to Tearly.
The spectral deconvolution was performed for the TA data of each solvent, resulting in the traces in Fig. 4. The decay of S1 (Fig. 4a) is relatively independent of environment, which is consistent with the lack of trend in the prompt emission decay. This suggests that the decay pathways for S1 are largely insensitive to stabilization of the S1 state with solvent. The red-shift of the steady-state emission (Fig. 2b) indicates that between toluene and chloroform the S1 state is stabilized by 0.06 eV. If the energy gap between S1 and Tearly is much larger than this, the effect of polarity on the rate constant of ISC may be minimal. Alternatively, the size of the ISC rate constant may be dominated by the magnitude of spin–orbit coupling, rather than the energy difference between the states, again resulting in minimal change with solvent. Contrastingly, there is a significant trend in the decay of the Tearly population and concurrent rise in the Tlate population (Fig. 4b and c). As the dielectric constant is increased, Tearly decays faster and Tlate rises faster (and more of it is formed), suggesting the rate of conversion between these states is maximized as solvent polarity is increased.
 | ||
| Fig. 4 Deconvoluted concentrations from the TA data of OQAO(mes)2 in various solvents. (a) The S1 component. (b) The Tearly component. (c) The Tlate component. All states are scaled by the maximum 1CT concentration. The vertical dashed line indicates a change from linear to logarithmic scale. | ||
OQAO(mes)2 in o-dichlorobenzene exhibits the fastest Tearly to Tlate conversion, which is consistent with the trends in delayed emission. This confirms that the fast delayed emission decay and small prompt emission background observed in o-dichlorobenzene (Fig. 2c and d) is due to the enhancement of a pathway that competes with RISC, i.e. the formation of Tlate. Fig. 5 shows the delayed emission data together with the Tearly and Tlate kinetics from TA for each solvent. For toluene and chlorobenzene the delayed emission clearly mirrors the decay of Tearly, with Tlate lasting substantially longer than the emission. This confirms that in these solvents Tlate contributes negligibly to the delayed emission, and instead Tlate formation is a competing pathway to RISC. For the chloroform and o-dichlorobenzene, the correlation between delayed emission and triplet decay becomes less clear. On initial inspection, the delayed emission of chloroform almost corresponds with the Tlate decay, but is slightly shorter lived. However, upon closer inspection, there is a small long-lived tail in the Tearly population, and when magnified (dashed lines) it can be seen that the delayed emission can feasibly correspond to this decay. For o-dichlorobenzene the agreement between the delayed emission and either Tlate or Tearly decay is poor, with the delayed emission seemingly decaying too fast for either population. This may be due to an overestimation of the contribution of Tearly at late times. Note that there is some uncertainty in determining the basis spectrum of Tearly, but this does not alter the decay of either Tearly or Tlate, and therefore does not change the agreement with the delayed emission, as discussed in the ESI (Section S4.3†). We also deconvoluted the TA data of solutions exposed to oxygen (ESI Section S7†), and can see that the lifetime of both triplet populations is reduced, consistent with the trends in the delayed emission data.
 | ||
| Fig. 5 Deconvoluted concentrations of Tearly and Tlate from the TA data of OQAO(mes)2 in various solvents with the delayed emission data. Delayed emission is scaled to best match either Tearly or Tlate, and plot only for times >1 μs to avoid the instrument response/prompt emission. The dashed line in (b) is the Tearly multiplied by 13. | ||
To understand the kinetics determined from TA further, we fit each excited state population to a simple 3-state kinetic model, described by the system of differential equations
 | (1) |
 | (2) |
 | (3) |
To fully determine the extent of the effect of Tlate, we used the rate constants determined from fitting to the TA kinetics to evaluate the model under “electrical excitation” rather than optical, and calculated a theoretical OLED efficiency. In other words, under optical excitation, the initially excited states are singlet excitons only, but in an OLED (electrical excitation), 75% of excitons generated from the recombination of charges are triplets. We therefore evaluated the model using an initial concentration of 75% triplet excitons, and 25% singlet excitons. The resulting kinetics are shown in the ESI Section S8.1,† and efficiencies (η = maximum photons emitted over initial concentration of injected excitons) are given in Table 1. We note that real devices will have several additional loss pathways not considered by this model, and the photophysics in the solid state are likely to differ from solution. Despite this, the efficiencies predicted by this model are comparable to the quantum efficiencies reported for OQAO devices by Zou et al. (20.3%).25 The efficiencies are dependent on whether Tearly or Tlate is initially formed by the recombination of charges (ηTearly or ηTlate, respectively). We see that for both solvents considered (toluene and o-dichlorobenzene), the amount of photons produced is less when Tlate is generated (ηTlate). Comparing only the emission resulting from the 75% triplet population (Table S4†), for o-dichlorobenzene, the difference is minimal, with recombination of charges to Tlate resulting in only 3% less photons than recombination to Tearly. However, for toluene, charge recombination to Tlate results in less than half the amount of photons as exciting Tearly. We additionally considered the performance in the absence of Tlate formation (i.e. with k2 = 0), which was 43% higher for both solvents, further demonstrating that, although some conversion of Tlate back to Tearly does occur, Tlate ultimately acts as a loss pathway. The calculated efficiencies of o-dichlorobenzene are higher than that of toluene, but this predominantly arises from differences in rate constants associated with the singlet state (kS1, kISC, and kRISC). When these rate constants were fixed to be the same as toluene, the o-dichlorobenzene efficiencies become lower than for toluene (ESI Table S4†).
To summarize, we see that for higher polarity environments, there is less difference between whether Tlate or Tearly is initially formed in a device, but the presence of Tlate is more detrimental. For low polarity environments, there are less losses due to Tlate when Tearly is initially formed, and the performance is similar to high polarity when Tlate is initially formed, all other processes being equal. An ideal situation in an OLED (ignoring differences in kS1, kISC, and kRISC) would thus be charge recombination to Tearly in a low polarity environment.
To determine the identity of the Tearly and Tlate states, we performed time dependent density functional theory calculations (TDDFT) under the Tamm–Dancoff approximation (TDA) at the TPSSh/6-31G(d)/IEFPCM(toluene) level of theory (benchmarking details in the ESI Section S9†). The electron–hole distributions at the ground state geometry are shown in Fig. 6. To determine the CT character of each excited state, the lambda index (Λ) was used to indicate the extent of overlap between the hole and the electron, as shown in Fig. 6a.33Λ = 0.0 represents full separation of electron and hole (i.e. a CT state) and Λ = 1.0 represents full overlap (LE). For the excited states in MR-TADF emitters, the degree of CT character is relatively low, hence we avoid assigning states as 1CT or 3CT as is done for spatially-separated D–A emitters, and do not distinguish between LE and CT states. The orbital arrangement between S1 and T1 states is near identical, indicating that conversion between these states should not be significant.15 However, the T2 state has different orbital character to S1, increasing the probability of conversion between these states. Spin–orbit coupling (SOC) constants were calculated to evaluate the likelihood of ISC to either T1 or T2. The S1 to T1 SOC constant was calculated to be zero as expected from their nearly identical character, whilst the S1 to T2 SOC constant was found to be non-zero at 0.02 cm−1, which is consistent with SOC constants of emitters with similarly slow ISC/rISC rate constants.34 This confirms that ISC and rISC are more likely to occur between S1 and T2 than S1 and T1. We therefore assign the Tearly state as T2, and Tlate as T1. TA data thus shows ISC occurring from S1 to T2, followed by internal conversion (IC) of T2 to T1 (described by k2). Interestingly, this is similar to theoretical predictions of Shizu and Kaji on the mechanism of TADF in the multi-resonance emitter DABNA,35 though we note they did not explicitly identify the low energy triplet state as a loss pathway, and multiple spectrally distinct triplet states have not yet been observed for DABNA experimentally.36
 | ||
| Fig. 6 (a) Electron–hole distributions for OQAO(mes)2 calculated at the TPSSh/6-31G(d)/IEFPCM(toluene) level of theory. Λ indicates the extent of overlap between the hole and electron. (b) Proposed model describing TADF in OQAO(mes)2. Optically excited S1 excitons undergo ISC to a higher energy triplet state, T2, which can then undergo internal conversion to a lower energy triplet state, T1. The formation of T1 competes with RISC back to S1. The T2 state has larger CT character, so the stabilization by higher polarity solvents is more significant than in S1 and T1. Note that energy levels are not necessarily to scale, and differences with solvent are exaggerated for clarity. | ||
The difference in orbital character between the T2 and T1 states can be used to rationalize some of the observations in the TA and PL data. In Fig. 6b we propose a model describing the photophysical processes of OQAO(mes)2 in various solvents. Steady-state emission suggests that the S1 energy is stabilized in the order indicated, with chloroform resulting in the lowest energy S1 state. Since T1 has an identical orbital arrangement to S1, we tentatively postulate that the different solvents will interact with and stabilize this state in the same way, i.e. following the trend in Reichardt's polarity parameter (Table 1). Because of the different electron distribution of the T2 state, the T2-solvent interactions may differ from that of T1, such that the stabilization of T2 with solvent better follows the trend in dielectric constant. Additionally, we note that the T2 state has a larger degree of CT character than S1 and T1, and so the effects of different solvents are likely to be larger. This can result in the energy of T2 becoming substantially stabilized in o-dichlorobenzene, whilst the energy of T1 changes little. Thus, changing the solvent has the effect of bringing the energies of the T2 and T1 states into resonance, analogous to what has been previously reported for CT and LE states in spatially-separated D–A emitters,32 such that the forward and reverse rates of conversion between T1 and T2 are fastest in o-dichlorobenzene, and slowest in toluene (see Fig. 6b). Internal conversion from T2 to T1 may also be influenced by solvent viscosity, with higher viscosities leading to slower rates. But, in this case, the highest viscosity solvent (o-dichlorobenzene) exhibits the fastest conversion, and the lowest viscosities (toluene or chloroform) the slowest, so the solvent polarity is likely the more dominant factor. As mentioned in the above discussion, the rates of ISC from S1 to T2 are largely unaffected by solvent, indicating that either this process is sufficiently downhill such that the change in energy of T2 is inconsequential, or that the rate is dominated by the magnitude of SOC, rather than the difference in energies.
Finally, we consider the implications of the results of this study for the application in solid-state OLED devices. As discussed above, this is dependent on whether the Tlate/T1 or Tearly/T2 states are generated from the recombination of charges in a device. If T2 is formed, T1 will act as a loss pathway that competes with RISC. This was more significant for high polarity solutions, suggesting a low polarity solid state matrix would be more beneficial for devices in this case. On the other hand, if T1 is formed from the recombination of charges, few triplets will be converted back to T2, and even fewer will be converted to S1via RISC, so the efficiency of TADF will be low. This was particularly the case for lower-polarity solvents, bringing the performance down to similar levels as the high-polarity. Thus, in this case, it may make little difference whether a high or low polarity environment is used (assuming all other parameters are the same). The polarity of the matrix will also effect the charge mobility of the device, so compromises between charge mobility and RISC efficiency may have to be considered. We have performed preliminary studies on OQAO(mes)2 in solid states matrices of different dielectric constants (ESI Section S10†), however the results are dominated by aggregation and excimer formation, and work of resolving the TADF photophysics in these systems is ongoing. Tri-mesityl substitution has previously been shown to reduce excimer formation on QAO,26 so future work will focus on using a tri-mesityl substituted OQAO to resolve differences between solution and solid-state photophysics. We also note that there has been some suggestion that in MR-TADF systems much of the TADF is facilitated by exciplex formation between the chromophore and the host matrix.36 Investigating how the solid-state interactions and exciplex formation affect the dynamics between the two triplet states will be an interesting avenue of further study.
3 Conclusions
TA measurements reveal triplet dynamics of the MR-TADF emitter, OQAO(mes)2, that are not detectable through time-resolved emission. Specifically, we show the formation of an additional species at late times after delayed emission has decayed, which increases with dielectric environment, and ultimately acts as a competing pathway for RISC. TD-DFT calculations suggest this is a low-energy triplet state (T1), formed from the internal conversion of high-energy triplets (T2) that are initially formed from ISC. RISC from T1 to S1 is negligible, and conversion of the low-energy triplets back to T2 is relatively slow, so if the T1 state is formed through electrical excitation in devices, then RISC back to bright singlets will be a significant challenge. Regardless, this study shows that time-resolved emission alone does not reveal the full picture of photophysics in TADF systems, and that the TADF efficiency in optically excited systems will not necessarily be realized under electrical excitation.Data availability
Raw transient absorption data, time-resolved emission data, and code for the deconvolution and modeling of transient absorption data can be found at https://hdl.handle.net/2123/32599. Additional data is available upon request from the corresponding author.Author contributions
A. N. S. and G. L. conceived the project. A. N. S. prepared solutions, performed spectroscopic experiments, analyzed data, and wrote the original manuscript. K. B. performed computational studies and wrote the computational details sections of the ESI.† I. C. prepared thin films of OQAO(mes)2. W. J. K. synthesized the OQAO(mes)2 molecule. W. W. H. W. and Z. M. H. supervised the work of W. J. K. and K. B., respectively. G. L. supervised the work of A. N. S. and I. C. and the overall project. All authors revised and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. N. S., I. C., W. J. K., W. W. H. W., and G. L. acknowledge support from the Australian Research Council Centre of Excellence in Exciton Science (CE170100026). Z. M. H. and K. B. thank the Natural Sciences and Engineering Research Council of Canada (NSERC) for support, and the Digital Research Alliance of Canada for computational resources. K. B. thanks NSERC for a Vanier Canada Graduate Scholarship and Z. M. H. is grateful for a Canada Research Chair and a Killam Accelerator Research Fellowship.Notes and references
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- T. Hosokai, H. Matsuzaki, H. Nakanotani, K. Tokumaru, T. Tsutsui, A. Furube, K. Nasu, H. Nomura, M. Yahiro and C. Adachi, Sci. Adv., 2017, 3, e1603282 CrossRef PubMed.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef.
- T. J. Penfold, J. Phys. Chem. C, 2015, 119, 13535–13544 CrossRef CAS.
- T. J. Penfold, F. B. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 RSC.
- X. Li, S. Fu, Y. Xie and Z. Li, Rep. Prog. Phys., 2023, 86, 096501 CrossRef PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- K. R. Naveen, P. Palanisamy, M. Y. Chae and J. H. Kwon, Chem. Commun., 2023, 59, 3685–3702 RSC.
- M. Sugawara, S.-Y. Choi and D. Wood, IEEE Signal Process. Mag., 2014, 31, 170–174 Search PubMed.
- S. Madayanad Suresh, D. Hall, D. Beljonne, Y. Olivier and E. Zysman-Colman, Adv. Funct. Mater., 2020, 30, 1908677 CrossRef CAS.
- H. J. Kim and T. Yasuda, Adv. Opt. Mater., 2022, 10, 2201714 CrossRef CAS.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- I. Cho, W. J. Kendrick, A. N. Stuart, P. Ramkissoon, K. P. Ghiggino, W. W. H. Wong and G. Lakhwani, J. Mater. Chem. C, 2023, 11, 14448–14455 RSC.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- C. M. Marian, Ann. Phys. Chem., 2021, 72, 617–640 CrossRef CAS PubMed.
- B. T. Lim, S. Okajima, A. Chandra and E. Lim, Chem. Phys. Lett., 1981, 79, 22–27 CrossRef CAS.
- C. M. Marian, J. Phys. Chem. C, 2016, 120, 3715–3721 CrossRef CAS.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- T. Hosokai, H. Noda, H. Nakanotani, T. Nawata, Y. Nakayama, H. Matsuzaki and C. Adachi, J. Photonics Energy, 2018, 8, 032102 Search PubMed.
- J. Lu, B. Pattengale, Q. Liu, S. Yang, W. Shi, S. Li, J. Huang and J. Zhang, J. Am. Chem. Soc., 2018, 140, 13719–13725 CrossRef CAS PubMed.
- K. Bergmann, R. Hojo and Z. M. Hudson, J. Phys. Chem. Lett., 2023, 14, 310–317 CrossRef CAS PubMed.
- J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Dias and M. R. Bryce, Chem. Commun., 2016, 52, 2612–2615 RSC.
- S.-N. Zou, C.-C. Peng, S.-Y. Yang, Y.-K. Qu, Y.-J. Yu, X. Chen, Z.-Q. Jiang and L.-S. Liao, Org. Lett., 2021, 23, 958–962 CrossRef CAS PubMed.
- D. Hall, S. M. Suresh, P. L. dos Santos, E. Duda, S. Bagnich, A. Pershin, P. Rajamalli, D. B. Cordes, A. M. Z. Slawin, D. Beljonne, A. Köhler, I. D. W. Samuel, Y. Olivier and E. Zysman-Colman, Adv. Opt. Mater., 2020, 8, 1901627 CrossRef CAS.
- Y. Marcus, Chem. Soc. Rev., 1993, 22, 409–416 RSC.
- M. J. Kamlet, J. L. Abboud and R. W. Taft, J. Am. Chem. Soc., 1977, 99, 6027–6038 CrossRef CAS.
- V. Ravi Kumar, C. Verma and S. Umapathy, J. Chem. Phys., 2016, 144, 064302 CrossRef PubMed.
- J. E. Elenewski and J. C. Hackett, J. Chem. Phys., 2013, 138, 224308 CrossRef PubMed.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- M. J. G. Peach, P. Benfield, T. Helgaker and D. J. Tozer, J. Chem. Phys., 2008, 128, 044118 CrossRef PubMed.
- H. Miranda-Salinas, J. Wang, A. Danos, T. Matulaitis, K. Stavrou, A. P. Monkman and E. Zysman-Colman, J. Mater. Chem. C, 2024, 12, 1996–2006 RSC.
- K. Shizu and H. Kaji, Commun. Chem., 2022, 5, 53 CrossRef CAS PubMed.
- X. Wu, B.-K. Su, D.-G. Chen, D. Liu, C.-C. Wu, Z.-X. Huang, T.-C. Lin, C.-H. Wu, M. Zhu, E. Y. Li, W.-Y. Hung, W. Zhu and P.-T. Chou, Nat. Photonics, 2021, 15, 780–786 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further detail on experimental and computational methods, data analysis and modelling, additional transient absorption data, experimental data sets and modelling code. See DOI: https://doi.org/10.1039/d4sc03649b |
| This journal is © The Royal Society of Chemistry 2024 |