
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the antibacterial efficacy of vancomycin analogues: targeting metallo-β-lactamases and cell wall biosynthesis†
Paramita
Sarkar
a,
Weipan
Xu
b,
Melissa
Vázquez-Hernández
c,
Geetika
Dhanda
a,
Shubhandra
Tripathi
d,
Debajyoti
Basak
a,
Hexin
Xie
 b,
Lea
Schipp
c,
Pascal
Dietze
c,
Julia E.
Bandow
c,
Nishanth N.
Nair
d and
Jayanta
Haldar
*ae
b,
Lea
Schipp
c,
Pascal
Dietze
c,
Julia E.
Bandow
c,
Nishanth N.
Nair
d and
Jayanta
Haldar
*ae
aAntimicrobial Research Laboratory, New Chemistry Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bengaluru 560064, Karnataka, India. E-mail: jayanta@jncasr.ac.in; Tel: +91 802208 2565
bSchool of Pharmacy, East China University of Science and Technology, 130 Meilong Rd., Shanghai 200237, China
cApplied Microbiology, Faculty of Biology and Biotechnology, Ruhr University Bochum, Universitätsstraße 150, 44780 Bochum, Germany
dDepartment of Chemistry, Indian Institute of Technology Kanpur, Kanpur 20816, India
eSchool of Advanced Materials, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bengaluru 560064, Karnataka, India
First published on 9th September 2024
Abstract
Vancomycin is a crucial last-resort antibiotic for tackling Gram-positive bacterial infections. However, its potency fails against the more difficult-to-treat Gram-negative bacteria (GNB). Vancomycin derivatives have shown promise as broad-spectrum antibacterials, but are still underexplored. Toward this, we present a novel strategy wherein we substitute the sugar moiety of vancomycin with a dipicolyl amine group, yielding VanNHdipi. This novel glycopeptide enhances its efficacy against vancomycin-resistant bacteria by up to 100-fold. A comprehensive approach involving microbiological assays, biochemical analyses, proteomics, and computational studies unraveled the impact of this design on biological activity. Our investigations reveal that VanNHdipi, like vancomycin, disrupts membrane-bound steps of cell wall synthesis inducing envelope stress, while also interfering with the structural integrity of the cytoplasmic membrane, setting it apart from vancomycin. Most noteworthy is its potency against critical GNB producing metallo-β-lactamases (MBLs). VanNHdipi effectively inactivates various MBLs with IC50 in the range of 0.2–10 μM resulting in resensitization of MBL-producing bacteria to carbapenems. Molecular docking and molecular dynamics (MD) studies indicate that H-bonding interactions between the sugar moiety of the vancomycin derivative with the amino acids on the surface of NDM-1 facilitate enhanced binding affinity for the enzyme. This work expands the scope of vancomycin derivatives and offers a promising new avenue for combating antibiotic resistance.
Introduction
The emergence of antibiotic resistance in both Gram-positive and Gram-negative bacteria (GPB and GNB respectively) has become a major concern to global health. In particular, the rise of vancomycin-resistant Gram-positive and carbapenem-resistant Gram-negative superbugs has emerged as a dire threat, exacerbated by the scarcity of treatment options.1 Reports of carbapenem resistance are increasingly common among Gram-negative pathogens like Escherichia coli and Klebsiella pneumoniae.2 The prevalence of multidrug resistance necessitates the development of novel antibiotics with unique mechanisms of action. This concern is highlighted in the joint ECDC-EMEA report, which emphasizes the scarcity of new antibiotics with novel mechanisms, especially for combating GNB.3 One potential solution to this problem is the exploration of novel semi-synthetic antibiotics.Vancomycin, a glycopeptide antibiotic, is a crucial last-resort drug used to treat Gram-positive bacterial infections. It acts by binding to the D-Ala-D-Ala moieties of the cell wall precursors, thereby inhibiting the final stage of bacterial cell wall synthesis. However, in vancomycin-resistant bacteria, a 1000-fold reduction in binding affinity to the mutated target, D-Ala-D-Lac, results in the loss of its effectiveness.4 The second-generation glycopeptide antibiotics, dalbavancin, telavancin, and oritavancin use hydrophobic modifications on the vancosamine sugars to overcome resistance. Albeit, their efficacy varies depending on the strain and the type of vancomycin-resistance acquired by the corresponding bacteria.5–7 Given their success, the development of semi-synthetic glycopeptides has been a propitious approach toward antibacterial drug discovery.8 Some of the successful strategies that have been reported to overcome vancomycin resistance in GPB involve incorporating membrane disruptive moieties;9–13 the enhancement of binding affinity to the target peptide or other components of the bacterial membrane;14–16 and backbone modifications to substitute the lost binding affinity to the target.17–19 These glycopeptide antibiotics are however not active against GNB due to the outer membrane, which serves as a barrier to their entry. Although reports of glycopeptide derivatives with activity against GPB and GNB are limited, there remains significant potential for their further development against a broader spectrum of pathogens.20–24 The antibacterial activity and mechanisms of action of glycopeptide antibiotics pivot on the sites of functionalization of the glycopeptides.4,22 An understanding of how these modifications affect activity is still limited. This gap necessitates further research and refinement to better understand how chemical modifications impact antibacterial activity.
We have reported multifunctional vancomycin derivatives that exhibit activity against both multidrug-resistant GPB and GNB.22,25,26 We showed that the conjugation of cationic lipophilic moieties at the C-terminus of vancomycin results in activity against both types of bacteria. In contrast, the conjugation of cationic lipophilic moieties at the vancosamine sugar resulted in high antibacterial activity against the GPB but not against GNB.27 In another strategy, we showed that the conjugation of a Zn(II) chelating ligand, dipicolyl-1,6-hexadiamine to the C-terminus of vancomycin resulted in Dipi-Van (1, Fig. 1A), which demonstrated a 375-fold enhancement in activity against vancomycin-resistant Enterococci (VRE).14 The ability of the compound to form a Dipi-Van-Zn2+ complex with the pyrophosphate of lipid II on the outer surface of the bacterial membrane underpins its enhanced activity against VRE.14 Notably, it inhibits the metallo-β-lactamase NDM-1, re-sensitizing NDM-producing bacteria to meropenem, as demonstrated in both in vitro and in vivo experiments.28 The promising efficacy of Dipi-Van (1) called for further exploration and understanding of this design strategy to achieve even higher antibacterial activity against both GPB and GNB.
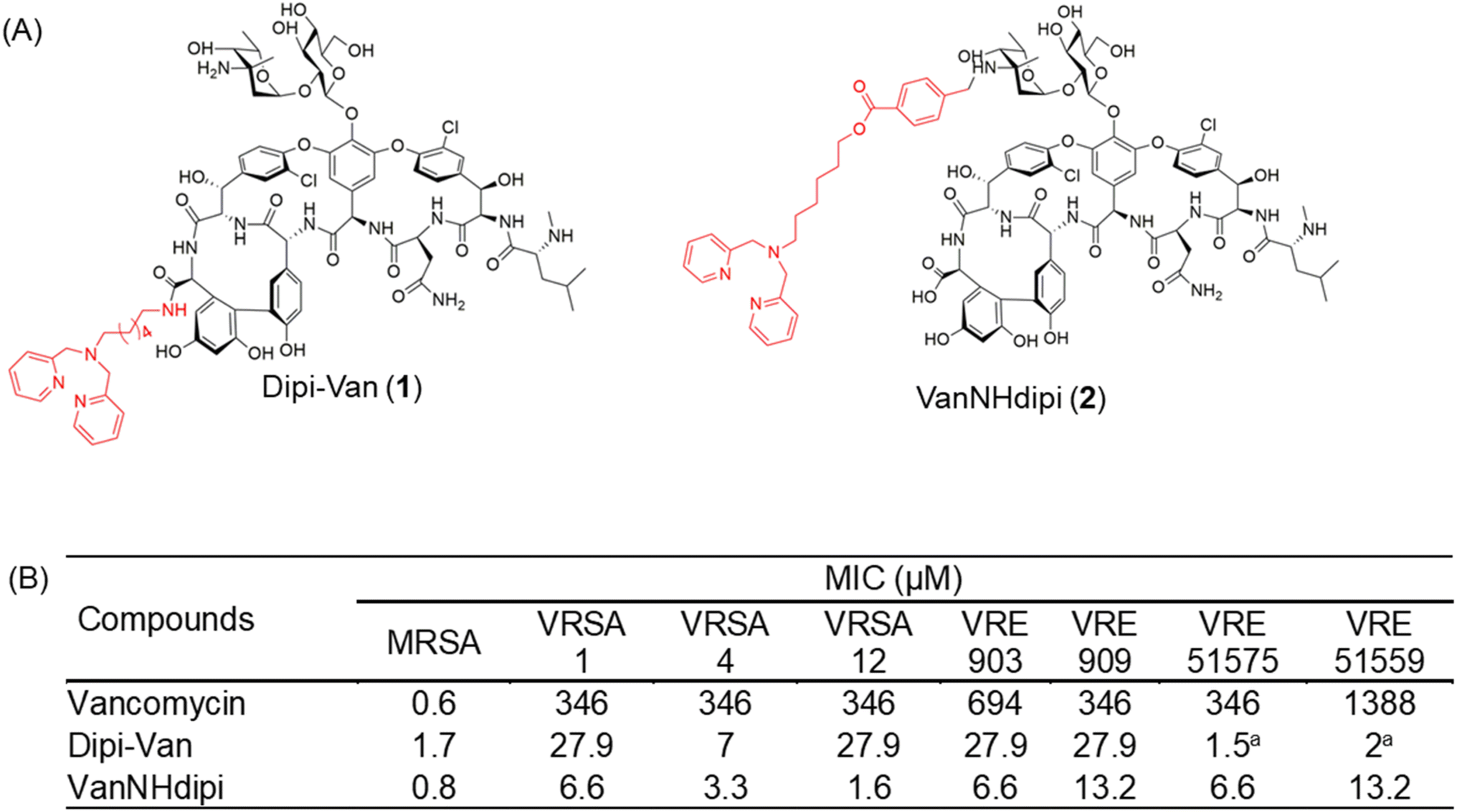 | ||
| Fig. 1 Structure and antibacterial activity of dipicolylamine functionalized vancomycin derivatives. (A) Vancomycin was functionalized at the C-terminus and the vancosamine nitrogen to yield Dipi-Van and VanNHdipi respectively, (B) MIC of vancomycin, Dipi-Van and VanNHdipi against various vancomycin-sensitive and resistant strains. MRSA, methicillin-resistant S. aureus (ATCC 33591); VRSA, vancomycin-resistant S. aureus; VRE vancomycin-resistant E. faecium (VanA phenotype, ATCC 51559); vancomycin-resistant E. faecalis (VanB phenotype, ATCC 51575); VRE 909 and 903 are vancomycin-resistant E. faecium. aActivity of Dipi-Van against VRE 51575 and VRE 51559 has been reported previously.14 | ||
In this study, we present the development of a next-generation vancomycin-dipicolyl amine conjugate, VanNHdipi (2, Fig. 1A). Unlike Dipi-Van, herein, the dipicolyl amine moiety was conjugated at the primary amine of the vancosamine sugar of vancomycin to yield, VanNHdipi (2) using the synthetic protocol shown in Scheme S1.† We explore the antibacterial attributes of VanNHdipi, elucidating its efficacy against multidrug-resistant GPB and their biofilms. Additionally, its efficacy against a wide range of MBLs, for which effective inhibitors are scarce, was established. We compare its activity with Dipi-Van (1) and demonstrate the enhanced antibacterial efficacy of VanNHdipi (2). Through a combination of biochemical assays, proteomics and computational analyses, we investigate the mechanisms of action that underlie its antibacterial activity. This study emphasizes the importance of positional modification to augment the antibacterial properties of vancomycin.
Results and discussion
Vancosamine-functionalized derivative displays potent activity against Gram-positive bacteria
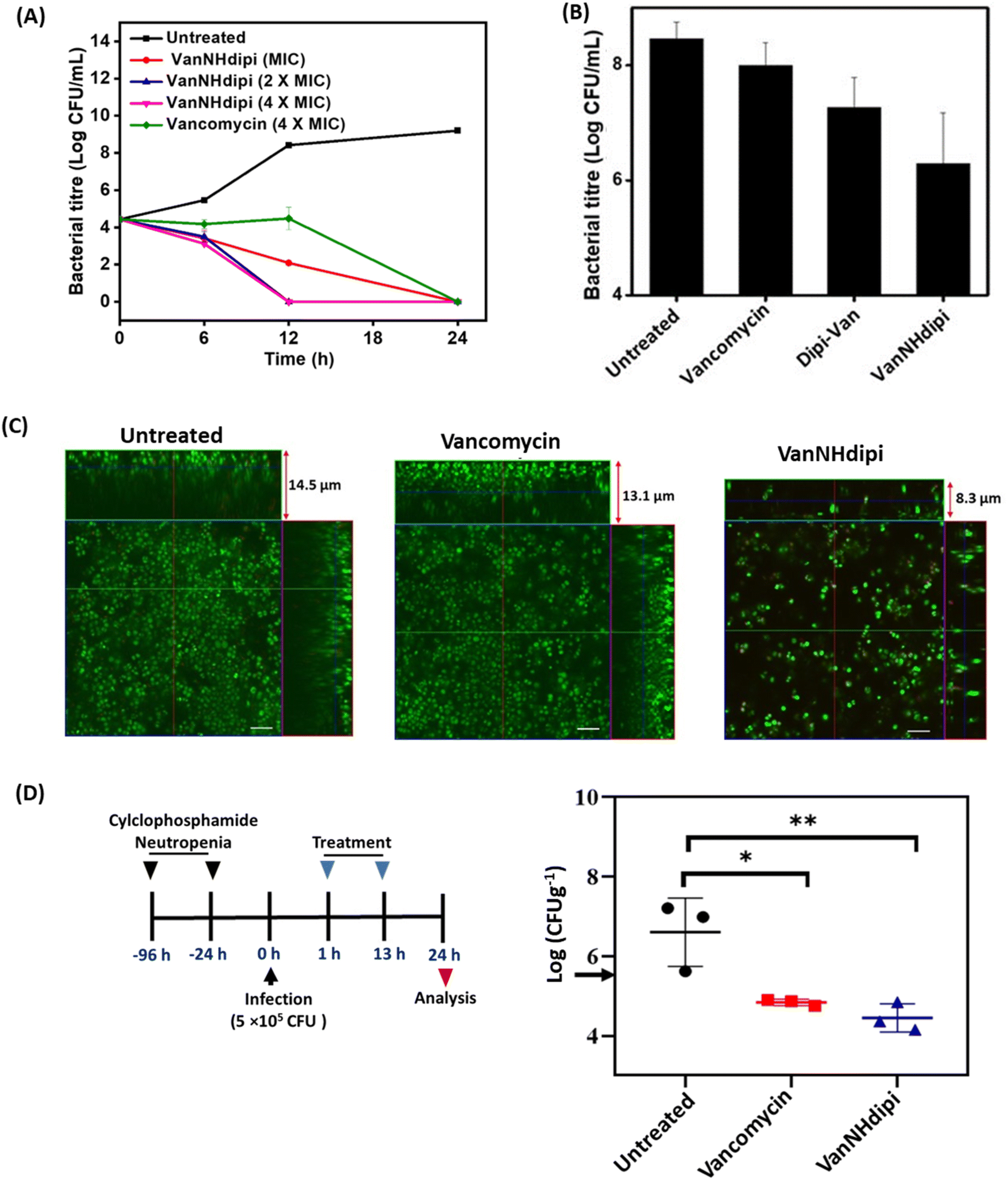 | ||
| Fig. 2 Efficacy of VanNHdipi (2) against methicillin-resistant S. aureus (MRSA) in exponential phase, biofilms and a mouse thigh infection model. (A) Kinetics of bactericidal activity of VanNHdipi (2) and vancomycin against exponential growth phase MRSA. ‘*’ indicates <50 CFU mL; (B) viability of cells within biofilms when treated with vancomycin and VanNHdipi at 20 μM; (C) confocal laser scanning microscopy when mature biofilms were left untreated, treated with vancomycin and VanNHdipi at 20 μM each; (scale bar = 5 μm); (D) in vivo efficacy of VanNHdipi and vancomycin against MRSA (n = 3/dose). Antibiotics were administered intraperitoneally twice at 12 h intervals at 12 mg kg−1 (left). Black arrow indicates pre-treatment bacterial load (right). Statistical analysis was done using One-way ANOVA. ‘*’ indicates p = 0.017, ‘**’ p = 0.006. | ||
VanNHdipi acts against Gram-positive bacteria through multiple modes of action
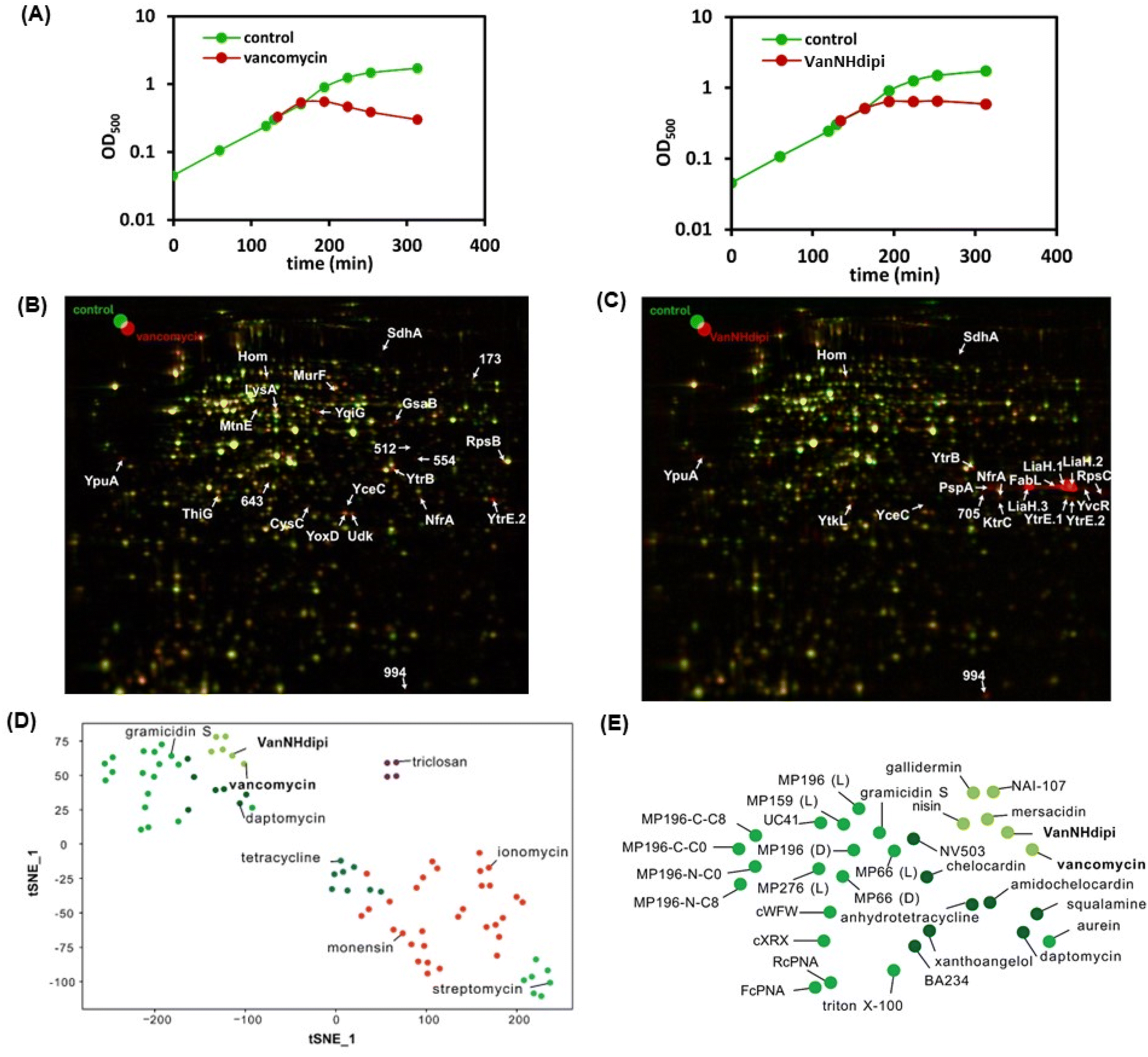 | ||
| Fig. 3 (A) Acute growth retardation upon treatment of cultures of B. subtilis 168 with vancomycin at 0.4 μM (2 × MIC) and VanNHdipi 0.16 μM (0.75 × MIC) upon treatment at the PEC; 2D gel-based proteome analysis of vancomycin-treated B. subtilis. Synthesis of cytosolic proteins of (B) vancomycin-treated (false-colored in red), (C) VanNHdipi-treated (false-colored in red), and untreated (false-colored in green) B. subtilis were compared based on [35S]-methionine labeling. In the overlaid autoradiographs, down-regulated proteins appear green, up-regulated proteins appear red, and proteins expressed at equal rates appear yellow. (D & E) The tSNE analysis serves to compare the similarity of proteome responses to a library of responses. (E) Details the upper left corner of (D). The response to VanNHdipi stress is most similar to responses to antibacterial agents inhibiting cell wall synthesis, such as vancomycin and lantibiotics. | ||
| Protein | Protein function | Biological process | Regulation factor | |
|---|---|---|---|---|
| Vancomycin/control | VanNHdipi/control | |||
| a Indicates that protein was identified by inference rather than LC-MS. | ||||
| CysC | Probable adenylyl-sulfate kinase | Sulfate assimilation | 2.3 ± 0.1 | |
| FabL | Enoyl-[acyl-carrier-protein] reductase [NADPH] | Fatty acid elongation | 41.9 ± 3.5a | |
| GsaB | Glutamate-1-semialdehyde 2,1-aminomutase 2 | Heme biosynthesis | 2.8 ± 0.4 | |
| Hom | Homoserine dehydrogenase | Amino acid metabolism | 5.0 ± 3.8* | 3.3 ± 1.0 |
| KtrC | Ktr system potassium uptake protein C | Ion transport | 5.4 ± 2.2 | |
| LiaH.1 | LiaH | Cell envelope stress | 67.3 ± 35.2 | |
| LiaH.2 | LiaH | Cell envelope stress | 131.3 ± 65.96 | |
| LiaH.3 | LiaH | Cell envelope stress | 45.7 ± 9.98 | |
| LysA | Diaminopimelate decarboxylase | Amino acid metabolism | 2.7 ± 0.3 | |
| MtnE | L-Glutamine-4-(methylsulfanyl)-2-oxobutanoate aminotransferase | Amino acid metabolism | 2.4 ± 0.3 | |
| MurF | UDP-N-acetylmuramoyl-tripeptide-D-alanyl-D-alanine ligase | Cell wall biosynthesis | 4.1 ± 1.6 | |
| NfrA | FMN reductase (NADPH) | Oxidative stress response | 3.3 ± 0.2 | 2.7 ± 0.2 |
| PspA | Phage shock protein A homolog | Cell envelope stress | 8.2 ± 2.5 | |
| RpsB | 30S ribosomal protein S2 | Translation | 2.7 ± 0.3 | |
| RpsC | 30S ribosomal protein S3 | Translation | 11.4 ± 12.7 | |
| SdhA | Succinate dehydrogenase flavoprotein | TCA cycle | 3.3 ± 1 | 3.1 ± 0.9a |
| ThiG | Thiazole synthase | Cofactor biosynthesis | 2.5 ± 0.1 | |
| Udk | Uridine kinase | Nucleotide biosynthesis | 3.1 ± 0.5 | |
| YceC | Stress response protein SCP2 | Cell envelope stress | 3.7 ± 0.9 | 3.4 ± 1 |
| YoxD | Uncharacterized oxidoreductase | Uncharacterized | 2.9 ± 0.1 | |
| YpuA | Uncharacterized protein | Cell envelope stress | 4.4 ± 0.3 | 6.7 ± 1.95a |
| YqiG | Probable NADH-dependent flavin oxidoreductase | Uncharacterized | 5.2 ± 1.3 | |
| YtkL | UPF0173 metal-dependent hydrolase | General stress | 5.7 ± 1.3 | |
| YtrB | ABC transporter ATP-binding protein | ABC transporter | 4.8 ± 0.5 | 3.17 ± 0.4 |
| YtrE.1 | ABC transporter ATP-binding protein | ABC transporter | 140.2 ± 40.5 | |
| YtrE.2 | ABC transporter ATP-binding protein | ABC transporter | 18.2 ± 8.98 | 13.55 ± 6.6a |
| YvcR | Uncharacterized ABC transporter ATP-binding protein | ABC transporter | ± 5.5 | |
Taken together, the proteome analysis indicates that the response to VanNHdipi shares hallmarks of the response to vancomycin, reflecting an interference with a membrane-bound step of cell wall biosynthesis. However, it also indicates that the mechanism of action is distinct from that of the parent drug, in that VanNHdipi interferes with the structural integrity of the membrane, like gallidermin, nisin and daptomycin (Fig. 3D and E).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mixture of acetic acid and methanol, bubbles appeared on the cell surface that show the membrane and cytoplasm blebbing out of these perforations. Bubble-like formations were observed on the bacterial cell surface upon treatment with both vancomycin and VanNHdipi (2) (Fig. 4A). These observations are consistent with both compounds inhibiting cell wall biosynthesis, compromising integrity of the cell wall, and resulting in structural vulnerabilities (Fig. 4A).
:3 mixture of acetic acid and methanol, bubbles appeared on the cell surface that show the membrane and cytoplasm blebbing out of these perforations. Bubble-like formations were observed on the bacterial cell surface upon treatment with both vancomycin and VanNHdipi (2) (Fig. 4A). These observations are consistent with both compounds inhibiting cell wall biosynthesis, compromising integrity of the cell wall, and resulting in structural vulnerabilities (Fig. 4A).
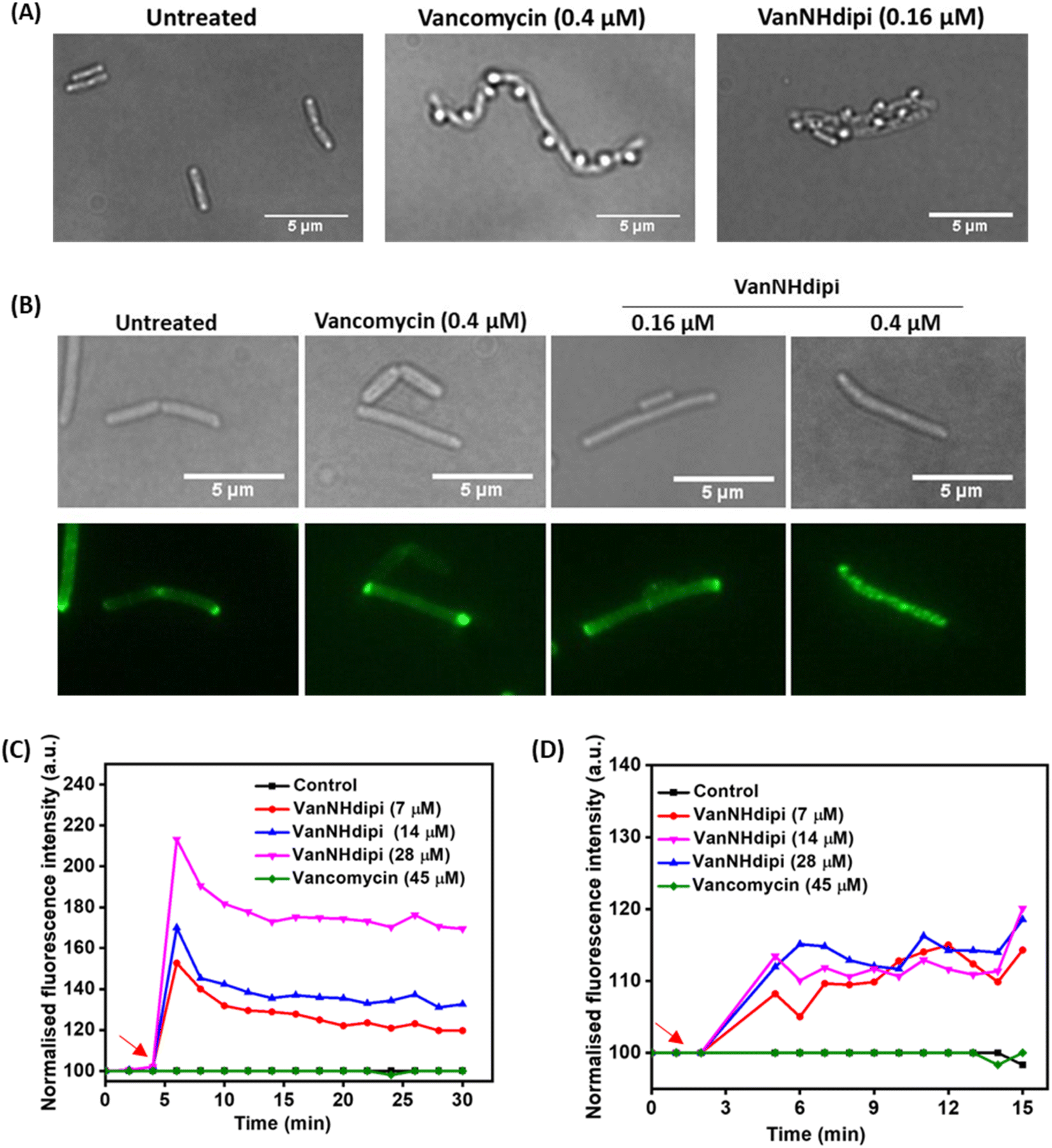 | ||
| Fig. 4 Investigating the effect of vancomycin and VanNHdipi on the cell envelope. (A) Inhibition of cell wall biosynthesis in B. subtilis by vancomycin (0.4 μM) and VanNHdipi (0.16 μM) upon treatment at respective PECs, are indicated by bubble like structures on the surface of bacteria. (B) Delocalisation of the GFP-tagged MinD protein in B. subtilis by vancomycin at 0.4 μM (PEC) and VanNHdipi (2) at 0.16 μM (PEC) and 0.4 μM (MIC). Membrane (C) depolarisation and (D) permeabilization of exponential phase MRSA upon treatment with VanNHdipi (2); compound addition is indicated with red arrows. For the measurement of: depolarization, λex 622 nm/λem 670 nm and for permeabilization, λex 535 nm/λem 617 nm. | ||
A second set of assays was directed at investigating the impact of VanNHdipi (2) treatment on the bacterial membrane potential and its propensity to induce permeabilization in MRSA. Membrane depolarization was assessed by monitoring the fluorescence of the membrane potential-sensitive dye DiSC3(5) (3,3′-dipropylthiadicarbocyanine iodide), while permeabilization was monitored by the uptake of the propidium iodide dye (Fig. 4C and D). While vancomycin exhibited no discernible influence on the bacterial membrane even at 45 μM (approximately 55 × MIC), VanNHdipi demonstrated the capability to depolarize it at 7 μM (approximately 11.5 × MIC). This depolarization phenomenon exhibited a concentration-dependent pattern, becoming more pronounced with concentrations increasing up to 28 μM (Fig. 4C). Unlike membrane depolarization, which intensified with increasing compound concentration, membrane permeabilization remained relatively low and constant across a range of concentrations (Fig. 4D). Overall, the membrane integrity assays show that VanNHdipi, which through the incorporation of the benzyl-hexan-dipicolyl amine moiety to vancomycin has an additional hydrophobic element, possesses the potential to interact with bacterial membranes. In conclusion, the improved antibacterial activity of VanNHdipi results from a combination of inhibiting a membrane-bound step of cell wall synthesis and damage of membrane integrity.
VanNHdipi synergizes with carbapenems against Gram-negative bacteria
We previously reported that Dipi-Van (1) inhibits the NDM-1 enzyme and resensitizes NDM-producing Gram-negative bacteria to carbapenems.28 Having validated the activity of VanNHdipi in Gram-positive bacteria, the applicability of the compound against Gram-negative bacteria was next investigated. The dipicolyl amine moiety is a known chelator of Zn(II) ion with a Kd of 160 nM.36 Therefore, it was imperative to investigate the influence of the positional variation of dipicolyl amine in VanNHdipi (2) on the inhibition of metallo-β-lactamases, but also other effects attributable to metal chelation (Fig. S4†), such as a potential destabilization of the outer membrane.| MIC (μg mL−1) | ||||||
|---|---|---|---|---|---|---|
| Meropenem + VanNHdipi (μg mL−1) | Doripenem + VanNHdipi (μg mL−1) | |||||
| Bacteria | 16 | 32 | 16 | 32 | ||
| K. pneumoniae EN5136 | 8 | 1 | 0.25 | 8 | 0.5 | 0.125 |
| K. pneumoniae BAA2146 | 8 | 4 | 0.5 | 16 | 2 | 0.5 |
| K. pneumoniae R3934 | 64 | 8 | 4 | 16 | 4 | 1 |
| E. coli EN5141 | 2 | 0.13 | 0.06 | 8 | 0.5 | 0.25 |
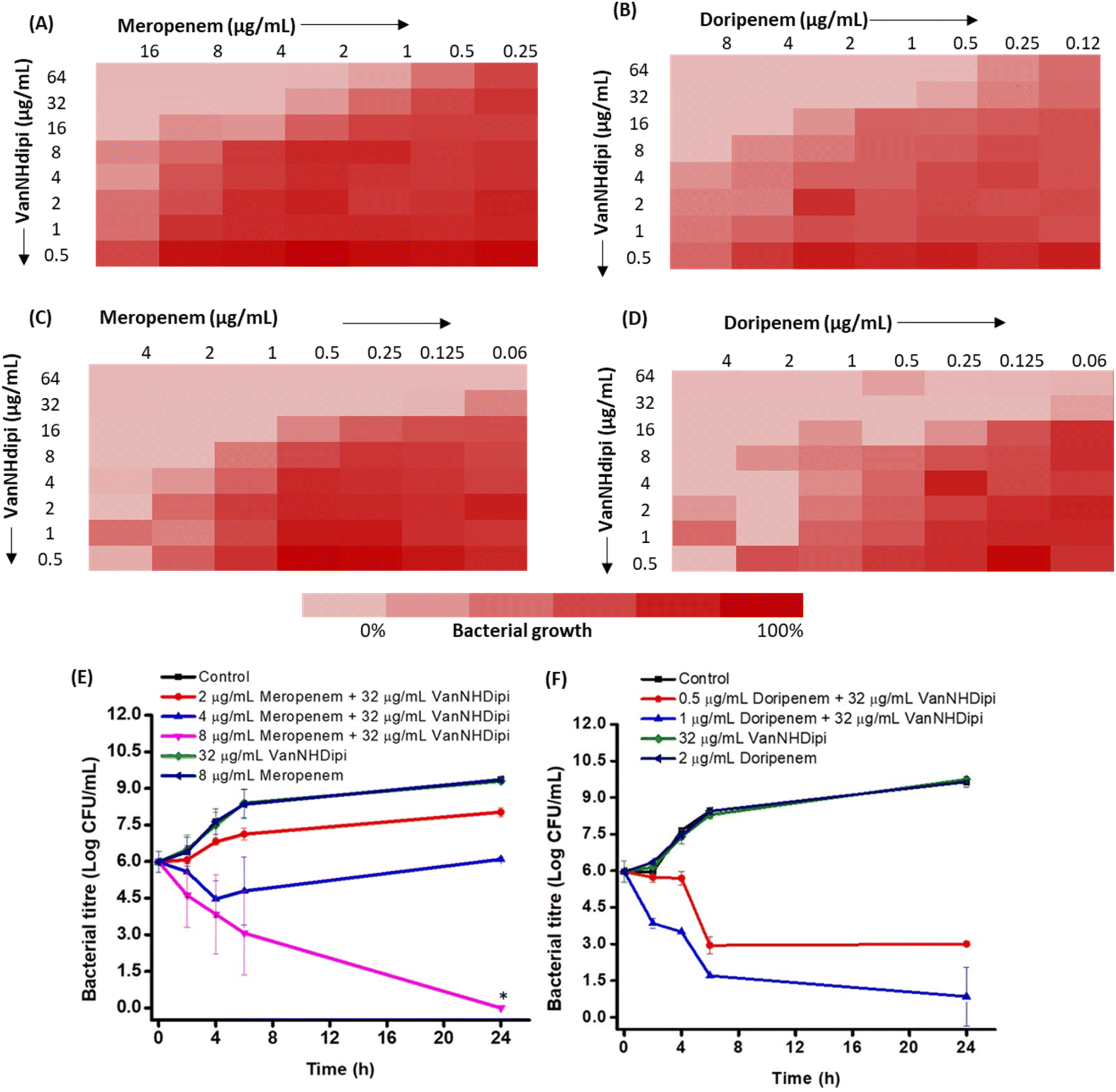 | ||
| Fig. 5 VanNHdipi resensitizes NDM-producing bacteria to carbapenems. (A–D) Checkerboard assay to assess the effect of the combination of VanNHdipi (2) with meropenem and doripenem against NDM-1 positive K. pneumoniae R3934 (A & B) and K. pneumoniae EN5136 (C & D). Time-kill kinetics of (E) meropenem (F) doripenem in combination with VanNHdipi (2) against NDM-1 positive K. pneumoniae R3934. ‘*’ indicates <50 CFU mL−1. The lines for control, 8 μg mL−1 meropenem and 2 μg mL−1 doripenem perfectly overlap in E and F. | ||
We evaluated two structurally similar carbapenems, doripenem and meropenem (Fig. 5A–D). Doripenem is more active and undergoes slower hydrolysis by carbapenemases.41 The clinical breakpoints of doripenem and meropenem against Gram-negative bacteria are 2 μg mL−1 and 1 μg mL−1, respectively.42 The MIC values of these carbapenems, against the clinical isolates tested ranged from 2 μg mL−1 to 64 μg mL−1 (Table 2). Susceptibility testing revealed that when exposed to 16 μg mL−1 of VanNHdipi (7 μM), NDM-1 expressing K. pneumoniae strains exhibited a 2-8-fold decrease in the MIC value of meropenem. Similarly, the MIC value of doripenem decreased by 4-16-fold for clinical isolates of NDM producing K. pneumoniae and E. coli in the presence of 16 μg mL−1 of VanNHdipi (7 μM) (Table 3). At a higher concentration of 32 μg mL−1 of VanNHdipi (14 μM), the MIC values of both meropenem and doripenem further reduced by 16–32-fold (MIC = 0.06–4 μg mL−1) against all strains. However, the addition of an equimolar amount of ZnSO4 resulted in the loss of sensitization of K. pneumoniae to carbapenem (meropenem). This indicates that the inhibitory activity of VanNHdipi is associated with Zn(II) chelation (Fig. S5†). Overall, VanNHdipi demonstrated the ability to resensitize K. pneumoniae and E. coli strains expressing multiple β-lactamase enzymes to carbapenems.
| MBLs | IC50 (μM) | |||
|---|---|---|---|---|
| VanNHdipi (Abs) | VanNHdipi (Fl) | Dipi-Van (Abs) | Dipi-Van (Fl) | |
| a The fluorescence (Fl)-based assay measured for different MBLs is based on cleavage of chromophores CDC-1 and CPC-1. | ||||
| Class B1 | ||||
| NDM-1 | 0.6 ± 0.1 | 0.2 ± 0.02 | 0.5 ± 0.1 | 0.4 ± 0.04 |
| NDM-12 | 2.3 ± 0.6 | 0.6 ± 0.2 | 0.3 ± 0.06 | 0.1 ± 0.005 |
| VIM-1 | 4.0 ± 0.4 | 3.6 ± 0.4 | 3.2 ± 0.6 | 1.9 ± 0.04 |
| VIM-27 | 2.7 ± 0.4 | 5.0 ± 0.2 | 1.4 ± 0.04 | 3.5 ± 0.6 |
| IMP-1 | 4.0 ± 1.7 | 1.2 ± 0.2 | >40 | >40 |
|
||||
| Class B2 | ||||
| CphA | — | 2.1 ± 1.1 | — | >40 |
|
||||
| Class B3 | ||||
| L1 | 11.0 ± 2.4 | 10.2 ± 2.8 | 7.2 ± 1.1 | 1.6 ± 0.3 |
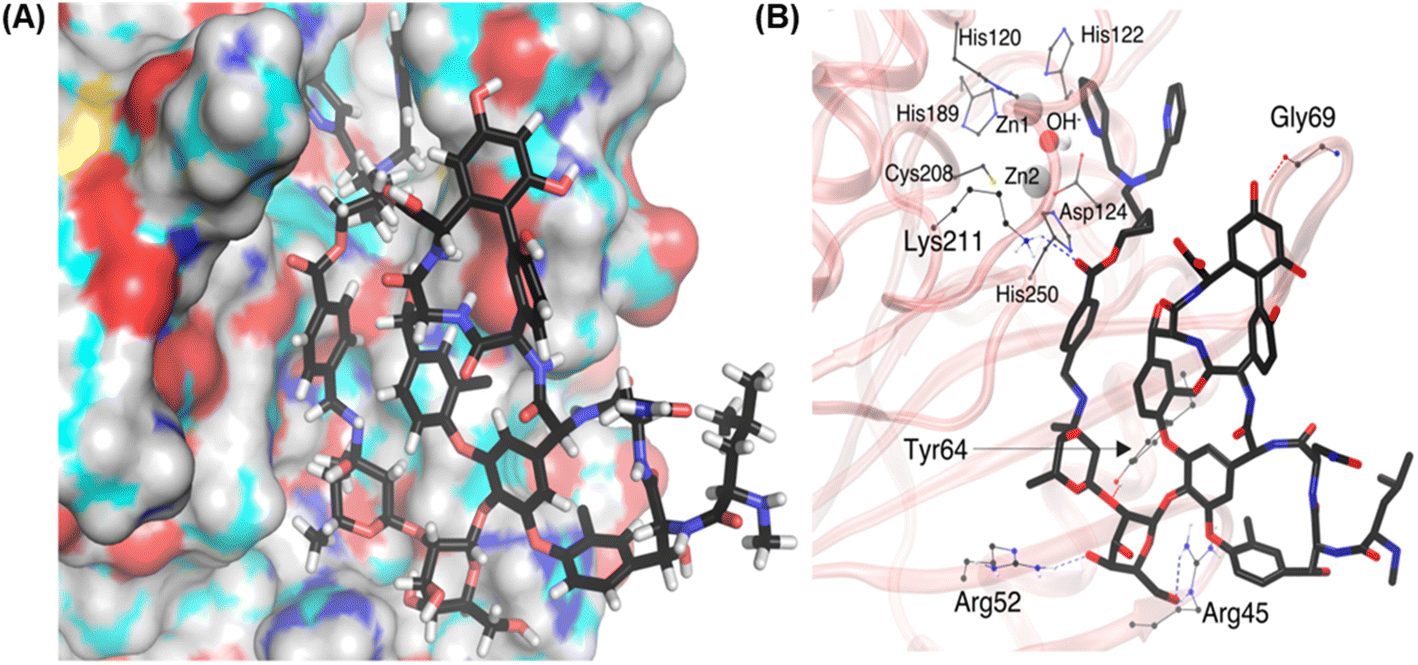 | ||
| Fig. 6 Molecular docking and MD simulation study of VanNHdipi-mediated inhibition of NDM enzyme. (A) VanNHdipi (thick-stick representation) bound to the NDM-1 (surface representation) is shown. (B) The residues Tyr64, Arg45, Arg52, Lys211, and Gly69 are forming H-bonding interactions with VanNHdipi. Color code: C (black), O (red), N (blue), Zn (silver). Protein surface in (A) is color-coded by atom colors except that C is in cyan for clarity. | ||
Importantly, the dipicolylamine fragment is in the vicinity of the active site of the NDM, which explains the deactivation of the NDM enzyme. In VanNHDipi, the stable vancomycin group stays on the surface of the protein, while the side chain compliments the contour of the protein surface and allows the dipicolylamine fragment to reach the active site (Fig. 6A). Dipicolylamine fragment likely forms a coordinated complex with Zn ions, thereby inhibiting NDM. However, as the MM force field we used is non-reactive, we could not see the formation of a covalent bond with Zn ions.
To explore the potential of Dipi-Van and VanNHdipi as broad-spectrum MBL inhibitors, we conducted in vitro tests on a panel of seven MBLs, representing each subclass. Two assays were employed: a nitrocefin-based absorbance assay and a fluorescence assay using chromophores CDC-1 and CPC-1.47,48 These chromophores serve as substrates for MBL enzymes.49 Hydrolysis of the β-lactam ring of nitrocefin by enzyme results in a shift in the ultraviolet absorption spectrum, allowing for visual detection of enzyme activity. On the other hand, CDC-1 and CPC-1 release a fluorescent probe, umbelliferone, upon hydrolysis, enabling quantification of enzyme activity.50
Class B1 MBLs are commonly associated with carbapenem resistance in human pathogens. These enzymes require one or two zinc ions at their active centers for full activity.51 Dipi-Van showed an IC50 of 0.1–0.5 μM against NDM-1 and NDM-12. It also inhibited VIMs (VIM-1 and VIM-27) with an IC50 of 1.4–3.5 μM, albeit with reduced efficacy compared to NDM enzymes (Table 3, ESI S4 & S5†). However, Dipi-Van was inactive against the IMP-1 enzyme. On the other hand, VanNHdipi effectively inhibited all 5 class B1 enzymes tested including IMP-1 (S4 & S5†), with IC50 values ranging from 0.2–5 μM in both the absorbance and fluorescence assays with a 2-4-fold difference in IC50 values between the two assays (Table 3).
Class B2 enzymes contain one Zn(II) ion at their active site and specifically hydrolyse carbapenems.52 Both compounds were tested against the CphA enzyme using the fluorescent probe CPC-1, which is a carbapenem-based probe.47 Dipi-Van (1) did not inhibit CphA activity even at concentrations up to 40 μM, whereas VanNHdipi (2) showed an IC50 of 2.1 μM against this enzyme. Class B3 MBL enzymes are di-zinc enzymes with active-site architectures similar to B1 MBLs.53 Against the Class B3 enzyme L1, both Dipi-Van and VanNHdipi demonstrated inhibitory activity, with IC50 values of 1.6–7.2 μM and 10.2–11 μM, respectively. It is important to note that the Zn(II)-chelating moiety, bis(pyridin-2-ylmethyl)amine (BPYA) alone, was found to inhibit enzyme activity with an IC50 of 1.2 μM against NDM-1 and 22.2 μM against VIM-27, while the IC50 against IMP-1 was greater than 40 μM (Fig. S8†). This indicates that there may be other factors contributing to the potency of VanNHdipi against IMP-1. The compounds were ineffective against the other β-lactamases, KPC-1 and AmpC, which are not MBLs (ESI S5†). Overall, we see that the two compounds differ in their spectrum of activity against MBLs. Although both derivatives share the same zinc binding moiety, they differ in their positioning on vancomycin and the linker. These differences in inhibitory activity suggest that the linker, orientation and steric factors imposed by the vancomycin core influence ligand access to the enzyme's active site. This is the first report of the use of glycopeptide analogues as inhibitors of a broad spectrum of MBLs. While this provides a promising starting point, the full potential of this application will require further systematic investigation in bacteria.
Conclusion
In this study, we introduced a dual-functional vancomycin derivative, VanNHdipi, that shows superior antibacterial activity over its predecessors. We not only demonstrate its enhanced activity against vancomycin-resistant bacteria, but also expand their application in resensitizing Gram-negative pathogens to last-line drugs, carbapenems. We emphasize that the position of modification of glycopeptide antibiotics significantly impact their biological activity and strategic modifications can be exploited to alter their efficacy. The vancosamine dipicolyl-amine vancomycin conjugate, VanNHdipi, shows superior activity against vancomycin-resistant Gram-positive bacteria as compared to the dipicolyl amine derivative functionalized at the C-terminus, Dipi-Van. VanNHdipi, also inhibits a broad spectrum of MBL enzymes while Dipi-Van, derivatized at the C-terminus, was limited in its activity. This leads the way for the design of molecules with better inhibitory activity against MBLs.We provide insights into the mechanisms of action of new glycopeptide analogues against Gram-positive pathogens by exploring the mechanisms of VanNHdipi. In addition to vancomycin-like inhibition of cell wall biosynthesis, VanNHdipi acts through other mechanisms of action including disruption of structural integrity of the cytoplasmic membrane and inhibition of the membrane-bound stage of cell wall synthesis. The multifaceted mechanisms of action is advantageous over drugs with a single target, in that, they could slow the development of resistance.
Most importantly, we investigated the inhibitory activity of glycopeptide analogs against the various classes of MBL enzymes, an application that has not been explored previously. While our docking and MD simulation studies provide insights into the potential binding modes of Dipi-Van and VanNHdipi, further experimental approaches will be needed to fully understand mechanistic differences in MBL inhibition. With limited options available against MBL-producing Gram-negative pathogens, VanNHdipi presents a versatile alternative. Since examples of metal chelators used in the clinics are limited, the therapeutic potential of these vancomycin derivatives will need further rigorous assessment in vivo models. Overall, our research expands the scope of vancomycin derivatives presenting a promising new avenue for addressing antibiotic resistance.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.† Data for the molecular dynamics simulation generated during the current study are not publicly available due to file size limitations but are available upon request.Author contributions
P. S. conceptualized, designed, executed, and coordinated project; synthesized and characterized compounds, performed biological assays, curated and analyzed data, prepared original draft and revised manuscript. W. X., H. X. designed and performed metallo-β-lactamase inhibition studies; S. T., N. N. N. performed molecular docking and molecular dynamics simulation studies; M. V. H., L. S., P. D. performed study of proteomic response of B. subtilis; J. E. B. supervised design and analysis of experiments for mechanistic studies in B. subtilis; D. B. synthesized and characterized compounds; G. D. performed in vivo activity against MRSA, checkerboard assays, and revised manuscript; J. H. conceptualized, supervised, coordinated research/manuscript writing, and acquired funding. All authors discussed the results and provided input to the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge DST-DAAD bilateral cooperation project (INT/FRG/DAAD/P-15/2018), DST-BRICS mutilateral cooperation project (DST/IMRD/BRICS/PilotCall2/MBLI/2018(G)) and JNCASR for funding. JEB acknowledges funding from the DAAD Program DST 2018 (57389759). We thank Dr Sidharth Chopra (Central Drug Research Institute, Lucknow) and Dr Sulagna Basu (National Institute of Cholera and Enteric Diseases, Kolkata) for the gift of drug-resistant clinical isolates.References
- H. M. Lee, J. Ren, K. M. Tran, B. M. Jeon, W. U. Park, H. Kim, K. E. Lee, Y. Oh, M. Choi, D. S. Kim and D. Na, Commun. Biol., 2021, 4, 205 CrossRef CAS PubMed.
- G. Meletis, Ther. Adv. Infect. Dis., 2016, 3, 15–21 CAS.
- https://www.ecdc.europa.eu, 2022.
- D. Zeng, D. Debabov, T. L. Hartsell, R. J. Cano, S. Adams, J. A. Schuyler, R. McMillan and J. L. Pace, Cold Spring Harbor Perspect. Med., 2016, 6, a026989 CrossRef PubMed.
- J. A. Karlowsky, K. Nichol and G. G. Zhanel, Clin. Infect. Dis., 2015, 61, S58–S68 CrossRef CAS PubMed.
- M. T. Guskey and B. T. Tsuji, Pharmacotherapy, 2010, 30, 80–94 CrossRef CAS PubMed.
- G. G. Zhanel, D. Calic, F. Schweizer, S. Zelenitsky, H. Adam, P. R. Lagace-Wiens, E. Rubinstein, A. S. Gin, D. J. Hoban and J. A. Karlowsky, Drugs, 2010, 70, 859–886 CrossRef CAS PubMed.
- G. Dhanda, P. Sarkar, S. Samaddar and J. Haldar, J. Med. Chem., 2019, 62, 3184–3205 CrossRef CAS PubMed.
- M. A. T. Blaskovich, K. A. Hansford, Y. Gong, M. S. Butler, C. Muldoon, J. X. Huang, S. Ramu, A. B. Silva, M. Cheng, A. M. Kavanagh, Z. Ziora, R. Premraj, F. Lindahl, T. A. Bradford, J. C. Lee, T. Karoli, R. Pelingon, D. J. Edwards, M. Amado, A. G. Elliott, W. Phetsang, N. H. Daud, J. E. Deecke, H. E. Sidjabat, S. Ramaologa, J. Zuegg, J. R. Betley, A. P. G. Beevers, R. A. G. Smith, J. A. Roberts, D. L. Paterson and M. A. Cooper, Nat. Commun., 2018, 9, 22 CrossRef PubMed.
- A. Antonoplis, X. Zang, M. A. Huttner, K. K. L. Chong, Y. B. Lee, J. Y. Co, M. R. Amieva, K. A. Kline, P. A. Wender and L. Cegelski, J. Am. Chem. Soc., 2018, 140, 16140–16151 CrossRef CAS.
- F. Umstatter, C. Domhan, T. Hertlein, K. Ohlsen, E. Muhlberg, C. Kleist, S. Zimmermann, B. Beijer, K. D. Klika, U. Haberkorn, W. Mier and P. Uhl, Angew Chem. Int. Ed. Engl., 2020, 59, 8823–8827 CrossRef PubMed.
- D. Guan, F. Chen, Y. Qiu, B. Jiang, L. Gong, L. Lan and W. Huang, Angew Chem. Int. Ed. Engl., 2019, 58, 6678–6682 CrossRef CAS PubMed.
- M. B. Chosy, J. Sun, H. P. Rahn, X. Liu, J. Brcic, P. A. Wender and L. Cegelski, ACS Infect. Dis., 2024, 10, 384–397 CrossRef CAS PubMed.
- V. Yarlagadda, P. Sarkar, S. Samaddar and J. Haldar, Angew Chem. Int. Ed. Engl., 2016, 55, 7836–7840 CrossRef CAS PubMed.
- Y. Jiang, M. Han, Y. Bo, Y. Feng, W. Li, J. R. Wu, Z. Song, Z. Zhao, Z. Tan, Y. Chen, T. Xue, Z. Fu, S. H. Kuo, G. W. Lau, E. Luijten and J. Cheng, ACS Cent. Sci., 2020, 6, 2267–2276 CrossRef CAS PubMed.
- D. Guan, F. Chen, Faridoon, J. Liu, J. Li, L. Lan and W. Huang, ChemMedChem, 2018, 13, 1644–1657 CrossRef CAS PubMed.
- A. Okano, N. A. Isley and D. L. Boger, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E5052–E5061 CrossRef CAS.
- Z. C. Wu, N. A. Isley and D. L. Boger, ACS Infect. Dis., 2018, 4, 1468–1474 CrossRef CAS PubMed.
- S. S. Printsevskaya, M. I. Reznikova, A. M. Korolev, G. B. Lapa, E. N. Olsufyeva, M. N. Preobrazhenskaya, J. J. Plattner and Y. K. Zhang, Future Med. Chem., 2013, 5, 641–652 CrossRef CAS PubMed.
- S. H. Werby, J. Brcic, M. B. Chosy, J. Sun, J. T. Rendell, L. F. Neville, P. A. Wender and L. Cegelski, RSC Med. Chem., 2023, 14, 1192–1198 RSC.
- A. Antonoplis, X. Zang, T. Wegner, P. A. Wender and L. Cegelski, ACS Chem. Biol., 2019, 14, 2065–2070 CrossRef CAS PubMed.
- P. Sarkar, K. De, M. Modi, G. Dhanda, R. Priyadarshini, J. E. Bandow and J. Haldar, Chem. Sci., 2023, 14, 2386–2398 RSC.
- Y. Acharya, S. Bhattacharyya, G. Dhanda and J. Haldar, ACS Infect. Dis., 2022, 8, 1–28 CrossRef CAS PubMed.
- E. van Groesen, P. Innocenti and N. I. Martin, ACS Infect. Dis., 2022, 8, 1381–1407 CrossRef CAS PubMed.
- V. Yarlagadda, G. B. Manjunath, P. Sarkar, P. Akkapeddi, K. Paramanandham, B. R. Shome, R. Ravikumar and J. Haldar, ACS Infect. Dis., 2016, 2, 132–139 CrossRef CAS PubMed.
- P. Sarkar, S. Samaddar, V. Ammanathan, V. Yarlagadda, C. Ghosh, M. Shukla, G. Kaul, R. Manjithaya, S. Chopra and J. Haldar, ACS Chem. Biol., 2020, 15, 884–889 CrossRef CAS PubMed.
- P. Sarkar, D. Basak, R. Mukherjee, J. E. Bandow and J. Haldar, J. Med. Chem., 2021, 64, 10185–10202 CrossRef CAS PubMed.
- V. Yarlagadda, P. Sarkar, S. Samaddar, G. B. Manjunath, S. D. Mitra, K. Paramanandham, B. R. Shome and J. Haldar, ACS Infect. Dis., 2018, 4, 1093–1101 CrossRef CAS PubMed.
- E. Banin, K. M. Brady and E. P. Greenberg, Appl. Environ. Microbiol., 2006, 72, 2064–2069 CrossRef CAS PubMed.
- D. G. Conrady, C. C. Brescia, K. Horii, A. A. Weiss, D. J. Hassett and A. B. Herr, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19456–19461 CrossRef CAS PubMed.
- M. Wenzel, B. Kohl, D. Munch, N. Raatschen, H. B. Albada, L. Hamoen, N. Metzler-Nolte, H. G. Sahl and J. E. Bandow, Antimicrob. Agents Chemother., 2012, 56, 5749–5757 CrossRef CAS PubMed.
- A. Muller, M. Wenzel, H. Strahl, F. Grein, T. N. V. Saaki, B. Kohl, T. Siersma, J. E. Bandow, H. G. Sahl, T. Schneider and L. W. Hamoen, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7077–E7086 CrossRef.
- C. H. R. Senges, J. J. Stepanek, M. Wenzel, N. Raatschen, U. Ay, Y. Martens, P. Prochnow, M. Vazquez Hernandez, A. Yayci, B. Schubert, N. B. M. Janzing, H. L. Warmuth, M. Kozik, J. Bongard, J. N. Alumasa, B. Albada, M. Penkova, T. Lukezic, N. A. Sorto, N. Lorenz, R. G. Miller, B. Zhu, M. Benda, J. Stulke, S. Schakermann, L. I. Leichert, K. Scheinpflug, H. Brotz-Oesterhelt, C. Hertweck, J. T. Shaw, H. Petkovic, J. M. Brunel, K. C. Keiler, N. Metzler-Nolte and J. E. Bandow, Antimicrob. Agents Chemother., 2020, 65, e01373 CrossRef PubMed.
- M. Wenzel, M. Patra, D. Albrecht, D. Y. Chen, K. C. Nicolaou, N. Metzler-Nolte and J. E. Bandow, Antimicrob. Agents Chemother., 2011, 55, 2590–2596 CrossRef CAS PubMed.
- H. Strahl and L. W. Hamoen, Proc. Natl. Acad. Sci. U.S.A., 2010, 107, 12281–12286 CrossRef CAS PubMed.
- B. A. Wong, S. Friedle and S. J. Lippard, J. Am. Chem. Soc., 2009, 131, 7142–7152 CrossRef CAS PubMed.
- K. L. May and M. Grabowicz, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8852–8854 CrossRef CAS PubMed.
- L. A. Clifton, M. W. Skoda, A. P. Le Brun, F. Ciesielski, I. Kuzmenko, S. A. Holt and J. H. Lakey, Langmuir, 2015, 31, 404–412 CrossRef CAS PubMed.
- E. R. Milaeva, D. B. Shpakovsky, Y. A. Gracheva, S. I. Orlova, V. V. Maduar, B. N. Tarasevich, N. N. Meleshonkova, L. G. Dubova and E. F. Shevtsova, Dalton Trans., 2013, 42, 6817–6828 RSC.
- N. T. Antunes, T. L. Lamoureaux, M. Toth, N. K. Stewart, H. Frase and S. B. Vakulenko, Antimicrob. Agents Chemother., 2014, 58, 2119–2125 CrossRef PubMed.
- A. M. Queenan, W. Shang, R. Flamm and K. Bush, Antimicrob. Agents Chemother., 2010, 54, 565–569 CrossRef CAS PubMed.
- https://www.eucast.org, 2021.
- M. F. Mojica, R. A. Bonomo and W. Fast, Curr. Drug Targets, 2016, 17, 1029–1050 CrossRef CAS.
- L. C. Ju, Z. Cheng, W. Fast, R. A. Bonomo and M. W. Crowder, Trends Pharmacol. Sci., 2018, 39, 635–647 CrossRef CAS PubMed.
- A. M. King, S. A. Reid-Yu, W. Wang, D. T. King, G. De Pascale, N. C. Strynadka, T. R. Walsh, B. K. Coombes and G. D. Wright, Nature, 2014, 510, 503–506 CrossRef CAS PubMed.
- M. Everett, N. Sprynski, A. Coelho, J. Castandet, M. Bayet, J. Bougnon, C. Lozano, D. T. Davies, S. Leiris, M. Zalacain, I. Morrissey, S. Magnet, K. Holden, P. Warn, F. De Luca, J. D. Docquier and M. Lemonnier, Antimicrob. Agents Chemother., 2018, 62, e00074 CrossRef CAS PubMed.
- H. Xie, J. Mire, Y. Kong, M. Chang, H. A. Hassounah, C. N. Thornton, J. C. Sacchettini, J. D. Cirillo and J. Rao, Nat. Chem., 2012, 4, 802–809 CrossRef CAS PubMed.
- S. S. van Berkel, J. Brem, A. M. Rydzik, R. Salimraj, R. Cain, A. Verma, R. J. Owens, C. W. Fishwick, J. Spencer and C. J. Schofield, J. Med. Chem., 2013, 56, 6945–6953 CrossRef CAS PubMed.
- X. Qian, S. Zhang, S. Xue, W. Mao, M. Xu, W. Xu and H. Xie, Bioorg. Med. Chem. Lett., 2019, 29, 322–325 CrossRef CAS.
- W. Mao, L. Xia and H. Xie, Angew Chem. Int. Ed. Engl., 2017, 56, 4468–4472 CrossRef CAS PubMed.
- M. N. Lisa, A. R. Palacios, M. Aitha, M. M. Gonzalez, D. M. Moreno, M. W. Crowder, R. A. Bonomo, J. Spencer, D. L. Tierney, L. I. Llarrull and A. J. Vila, Nat. Commun., 2017, 8, 538 CrossRef PubMed.
- M. Vanhove, M. Zakhem, B. Devreese, N. Franceschini, C. Anne, C. Bebrone, G. Amicosante, G. M. Rossolini, J. Van Beeumen, J. M. Frere and M. Galleni, Cell. Mol. Life Sci., 2003, 60, 2501–2509 CrossRef CAS PubMed.
- C. L. Tooke, P. Hinchliffe, E. C. Bragginton, C. K. Colenso, V. H. A. Hirvonen, Y. Takebayashi and J. Spencer, J. Mol. Biol., 2019, 431, 3472–3500 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: supplementary figures; materials; experimental methods for in vitro microbiological assays (antibacterial activity, biofilms, time-kill kinetics, proteomics and mechanistic studies) and toxicity, and in vivo activity and toxicity; characterization of intermediates and final compounds through (1HNMR, and HR-MS). See DOI: https://doi.org/10.1039/d4sc03577a |
| This journal is © The Royal Society of Chemistry 2024 |