
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper catalyzed benzylic sp3 C–H alkenylation†
Ting-An
Chen
ab,
Richard J.
Staples
 b and
Timothy H.
Warren
*b
b and
Timothy H.
Warren
*b
aDepartment of Chemistry, Georgetown University, Washington, D.C. 20057, USA
bDepartment of Chemistry, Michigan State University, East Lansing, Michigan 48824, USA. E-mail: warre155@msu.edu
First published on 19th September 2024
Abstract
The prenyl group is present in numerous biologically active small molecule drugs and natural products. We introduce benzylic C–H alkenylation of substrates Ar-CH3 with alkenylboronic esters (CH2)3O2B–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CMe2 as a pathway to form prenyl functionalized arenes Ar-CH2CHCMe2. Mechanistic studies of this radical relay catalytic protocol reveal diverse reactivity pathways exhibited by the copper(II) alkenyl intermediate [CuII]-CHCMe2 that involve radical capture, bimolecular C–C bond formation, and hydrogen atom transfer (HAT).
CMe2 as a pathway to form prenyl functionalized arenes Ar-CH2CHCMe2. Mechanistic studies of this radical relay catalytic protocol reveal diverse reactivity pathways exhibited by the copper(II) alkenyl intermediate [CuII]-CHCMe2 that involve radical capture, bimolecular C–C bond formation, and hydrogen atom transfer (HAT).
Introduction
The prenyl group (–CH2CHCMe2) is a prevalent functionality found in natural products and biologically active small molecules.1–6 Prenyltransferase, an essential enzyme involved in prenylation, enhances protein stability and anchors proteins to cell membranes due to the hydrophobic nature of the prenyl group.7–10 The addition of a prenyl group to molecules can influence their biological activities.11–14 For instance, the prenyl group is the primary inhibitory component of the HIV inhibitor Osthol (Fig. 1a).15 Additionally, both experimental and computational methods have been used to determine that prenylated chrysin functions as a more potent inhibitor for P-glycoprotein, a determinant of drug accumulation in leukemia cells.16,17
 | ||
| Fig. 1 (a) Prenylated natural products and drug molecules. (b) Prenylation via cross-coupling. (c) sp3 C–H styrenylation. (d) Net prenylation via benzylic C–H alkenylation. | ||
Methods for installing prenyl groups onto aromatic rings typically involve C–C coupling through allylation of an aryl halide (or pseudohalide) (Fig. 1b). For instance, Pd catalyzed Suzuki or Negishi coupling reactions produce prenylated aryl derivatives.18–20 Alternatively, sp2 C–H prenylation of arene C–H bonds with 1,1-dimethylallene also leads to aryl prenyl derivatives.21–24
This report presents an alternative strategy to prenyl-functionalized molecules through C–H alkenylation of benzylic C–H bonds via the alkenylboronic ester (CH2)3O2B–CHCMe2 (Fig. 1d). Previous examples of direct sp3 C–H alkenylation with alkenylboronic esters required highly acidic C–H bonds in aryl difluoromethyl derivatives Ar-CF2H.25 Alternatively, benzylic sp3 C–H styrenylation occurs with styrenyl carboxylates and nitrites in the presence of tBuOOtBu (Fig. 1c).26–30
This report utilizes the dimethylethenyl (–CHCMe2) group as the alkenyl source for sp3 C–H functionalization. Our research team and other groups have employed radical relay approaches for C–H functionalization (Fig. 2a).31–35 Based on sp3 C–H alkynylation,36 arylation,37–39 and methylation40 that proceed via [CuII]-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CAr, [CuII]-Ar, and [CuII]-Me intermediates, respectively (Fig. 2b), we anticipated that [CuII]-CHCMe2 intermediates (Fig. 2c) could lead to benzylic sp3 C–H alkenylation. This would convert an Ar–CH2–H group to Ar–CH2–CHCMe2, enabling net prenylation (Fig. 1d).
CAr, [CuII]-Ar, and [CuII]-Me intermediates, respectively (Fig. 2b), we anticipated that [CuII]-CHCMe2 intermediates (Fig. 2c) could lead to benzylic sp3 C–H alkenylation. This would convert an Ar–CH2–H group to Ar–CH2–CHCMe2, enabling net prenylation (Fig. 1d).
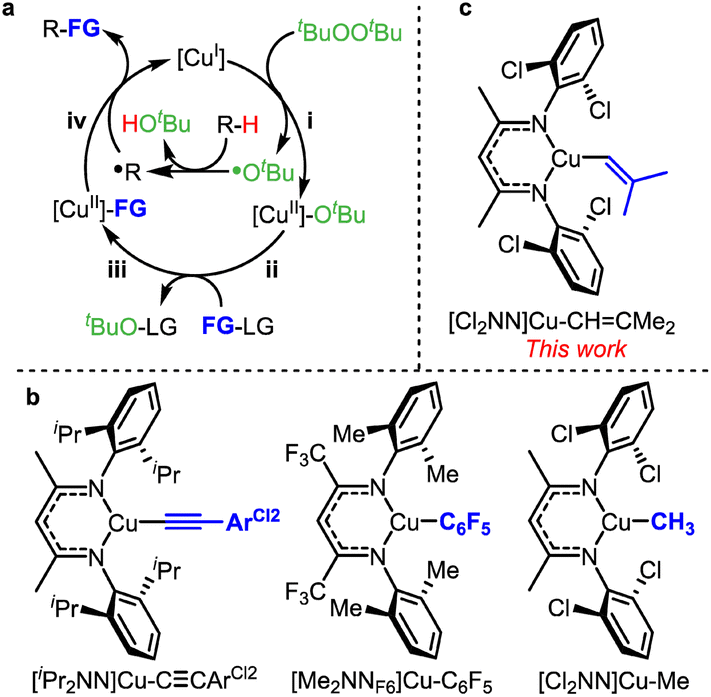 | ||
| Fig. 2 (a) Radical relay mechanism for C–H functionalization. (b) β-Diketiminato copper(II) intermediates in C–H alkynylation, arylation and methylation. (c) Proposed copper(II) alkenyl intermediate for C–H alkenylation. | ||
Results and discussion
Reaction discovery and optimization
We sought to enable benzylic C–H alkenylation, employing toluene as a representative benzylic substrate using Cu(I) β-diketiminato complexes as catalysts along with di-tert-butyl peroxide (tBuOOtBu) as the oxidant. We chose alkenylboronic esters as the alkenyl source but recognized that the nature of the boronic ester backbone could play an important role (Scheme 1). For example, 4,4,6-trimethyl-2-phenyl-1,3,2-dioxaborinane exhibited a higher yield in sp3 C–H arylation compared to phenylboronic acid pinacol ester.39 Therefore, we investigated 2-alkenyl-1,3,2-dioxaborinane (2a) and 4,4,6-trimethyl-2-alkenyl-1,3,2-dioxaborinane (2b) as possible alkenyl group transfer reagents for C–H alkenylation of toluene (Scheme 1). Use of the less sterically hindered boronic ester 2a produced a higher C–H alkenylation yield of product 3a than with the more sterically hindered boronic ester 2b (63% vs. 15%, respectively). Importantly, the more hindered boronic ester 2b led to toluene C–H etherification to give PhCH2-OtBu that signals capture of the benzyl radical PhCH2˙ by the [CuII]-OtBu intermediate (Scheme S1†).41 We hypothesize that a higher rate of transmetalation between the [CuII]-OtBu intermediate and the less sterically hindered alkenylboronic ester 2a to form a [CuII]–CCMe2 intermediate inhibits the formation of PhCH2OtBu (Scheme S1†).
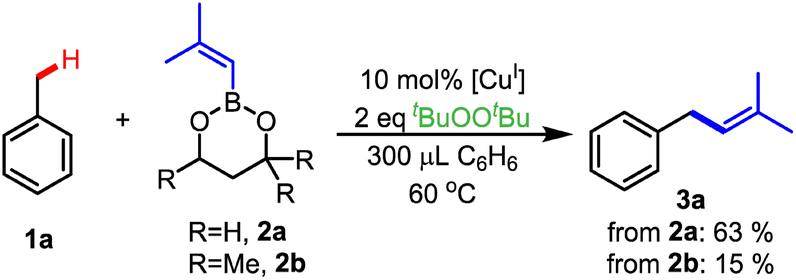 | ||
| Scheme 1 Size of alkenylboronic ester impacts alkenylation yield. | ||
Employing boronic ester 2a, we explored various parameters including the solvent, catalyst loading (Table S1†), oxidant (Table S2†), temperature (Table S4†). Using optimized conditions (10 mol% [CuI], 2 equiv. tBuOOtBu, 20 equiv. R–H, 300 μL benzene, and 60 °C), we also examined different copper β-diketiminate catalysts with diverse electronic and steric properties (Table 1). Among the tested catalysts, [Cl2NN]Cu (Table 1, entry 1) gave the highest product yield. Interestingly, a very electron-poor catalyst (Table 1, entry 2) gave a very low yield, yet catalysts with electron-donating groups on the β-diketiminato N-aryl rings also decreased the product yield.
| a Conditions: 20 equiv. R–H, 2 equiv. tBuOOtBu, 60 °C, 1 h. Yield determined by GCMS anaiysis. |
|---|

|
Benzylic C–H alkenylation leading to net prenylation
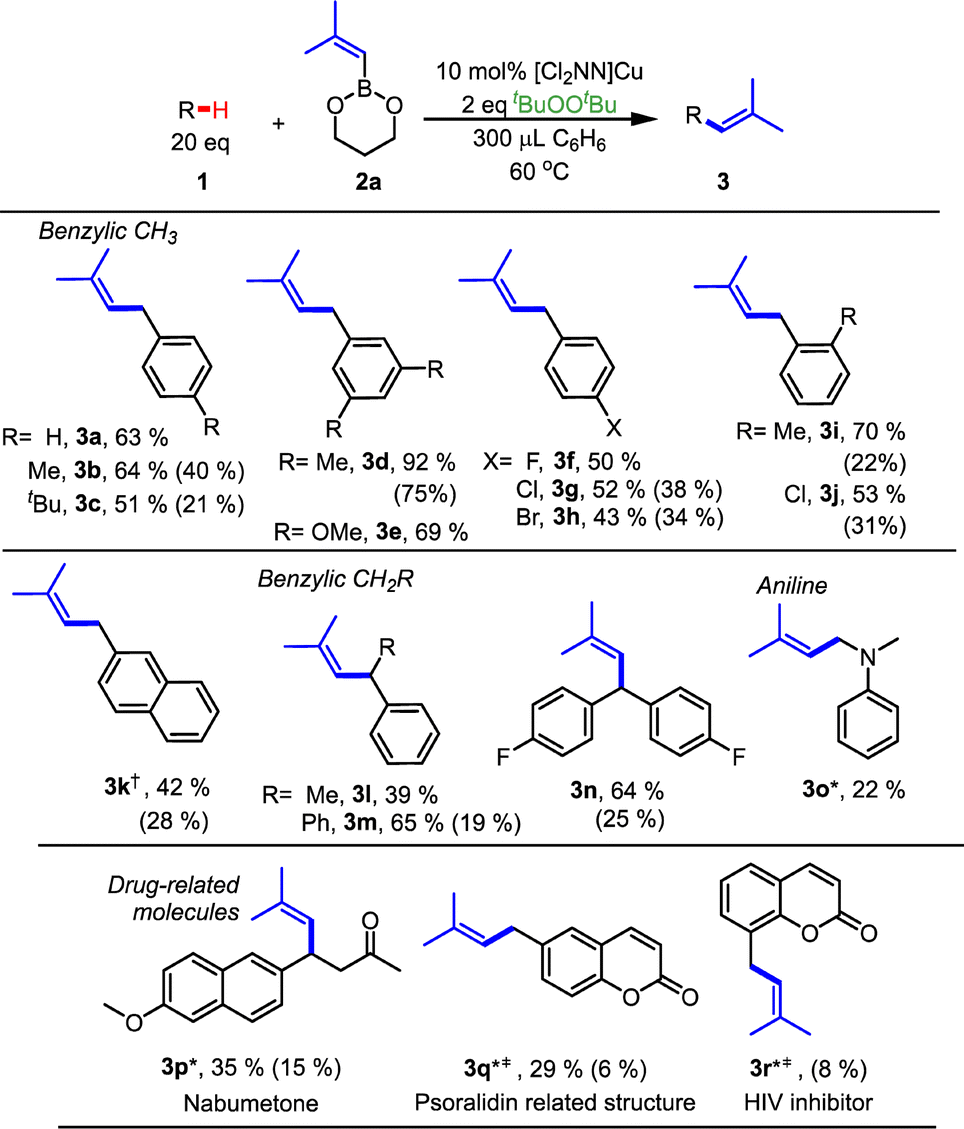
We systematically examined a range of benzylic R–H substrates, assessing reaction yields via GCMS analysis (Table 2). We initially focused on sp3 C–H alkenylation of commercially available 1° benzylic substrates Ar-CH3 that lead to prenyl derivatives ArCH2–CHCMe2 (3a–3i) in 42–92% yield. Slightly higher yields result in more electron-rich substrates Ar-CH3 (3b, 3d, 3e; 64–92%). This method tolerates aryl halides Ar-X (3f–3h) that typically serve as substrates in more traditional cross-coupling reactions. The method also tolerates ortho-substitution as illustrated by the use of o-chlorotoluene and o-xylene (3i and 3j) for C–H alkenylation. Secondary benzylic C–H sites in ethylbenzene also undergo C–H alkenylation (3l), yet exhibit lower yields due to the formation of styrene in 20% yield via β-H-atom abstraction from the ethylbenzene radical (PhCH(˙)Me) (ESI, Section 4 and Scheme S5†). Additionally, N,N-dimethyl aniline (3o) proved amenable to α-N C–H functionalization, albeit in lower yield (22%). We also examined indoles with heteroaryl-Me groups, but unfortunately no C–H alkenylation occurs, even with N-Boc protected indoles.
| a Detailed reaction conditions in ESI. Yields determined by GCMS. Isolated yield in parenthesis. †80 °C. ≠ 100 °C. *10 equiv. R–H substrate used. |
|---|
|
To highlight potential advantages of prenylation via benzylic C–H alkenylation, we performed C–H alkenylation on three pharmaceutically relevant compounds (3p–3r). Notably, our method exhibited high selectivity for benzylic C–H bonds. For example, in the case of nabumetone (3p) which possesses multiple sp3 C–H bonds, exclusive benzylic C–H functionalization occurs. Entry 3r yielded a product closely related to its o-OMe derivative with HIV inhibitory properties, highlighting the potential of this direct C–H alkenylation method to condense lengthy syntheses.15 Yet we recognize the rather modest yields for the C–H functionalization step with these compounds (Table 2, entries 3p–3r); some substrates lead to relatively strong binding with the [CuI] catalyst that impedes C–H functionalization (Scheme S2 and Fig. S4†).
Mechanistic investigations
To gain more insight into the interaction between the alkenylboronic ester and the [CuI] catalyst as well as the formation and reactivity of the proposed [CuII]-CHCMe2 intermediate, we examined facets of the reaction mechanism by integrating both experimental and computational analyses.
Alkenylboronic ester binding to copper(I) catalyst
Given the need for mild heating to encourage C–H alkenylation, we considered the possibility of equilibrium binding of the dimethylethenylboronic ester 2a with the [CuI] catalyst (Fig. 3a). The crystal structure of the [CuI](η2-2a) adduct reveals an interaction between the π electrons of the alkene and the [CuI] catalyst (Fig. 3b). Higher temperatures promote the dissociation of this adduct, leading to increased concentrations of the dissociated species. A van't Hoff plot reveals thermodynamic parameters corresponding to this equilibrium: ΔGexp (298) = 6.7 ± 1.0, ΔHexp = 11.1 ± 0.5 kcal mol−1, and ΔSexp = 14.4 ± 1.6 e.u. (Fig. 3c and S2†). | ||
| Fig. 3 (a) Reversible binding of 2a to [CuI] catalyst in benzene, (b) X-ray structure of alkenylboronic ester adduct, and (c) van't Hoff plot. | ||
Formation and reactivity of alkenyl intermediate [CuII]–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CMe2
CMe2
Mixing [CuII]-OtBu with 2a in fluorobenzene produces a substantial amount of 2,5-dimethylhexa-2,4-diene (alkenyl dimer) (84% yield, Fig. 4a). This could proceed via a [CuII]-CHCMe2 intermediate that undergoes bimolecular C–C coupling to give the observed alkenyl dimer, much as [CuII]–CCAr and [CuII]-Ar intermediates readily form ArCC-CCAr37 and Ar–Ar38 species. In situ UV-vis analysis was employed to monitor the reaction intermediate of [CuII]-OtBu and 2a (Fig. S6†). Upon introducing 2a into the [CuII]-OtBu solution at −38 °C, a rapid reaction occurred, leading to a decrease in the absorption band of [CuII]-OtBu without the detection of any newly generated [CuII] species. Based on previous bimolecular C–C bond formation via [CuII]–CCAr37 and [CuII]-Ar38 species, we propose that the [CuII]-CHCMe2 intermediate similarly undergoes rapid bimolecular C–C coupling to form Me2CCH–CHCMe2. Indeed, it is possible for a conjugated diene to bridge between two β-diketiminato [CuI] to form a [CuI]2(μ-diene) species (Fig. S8†).
 | ||
| Fig. 4 Reaction of [Cl2NN]Cu-OtBu and 2a (a) in C6H5F, (b) in the presence of Gomberg's dimer in C6H5F, and (c) in toluene. +isolated yield. | ||
To further confirm the reaction intermediate obtained upon addition of alkenylboronic ester 2a with [CuII]-OtBu as [CuII]-CHCMe2, reaction in the presence of Gomberg's dimer (that dissociates to produce the trityl radical Ph3C˙) provides Ph3C–CHMe2 in 35% yield (Fig. 4b). Additionally, a minimal amount of 3a also forms when the reaction occurs in toluene (Fig. 4c). Product 3a can arise from the sequential steps of toluene radical formation through HAT, followed by subsequent radical capture (Fig. 5c).
 | ||
| Fig. 5 DFT calculation of (a) [CuI] binds to alkenes, 2a and 3a. (b) spin density plot of [CuII]-CHCMe2. (c) Possible reaction pathways for [CuII]-CHCMe2. Free energies in kcal mol−1 at 298.15 K. For more details, see Schemes S3 and S4. | ||
Computational analysis and insights
We employed density functional theory (DFT) to better understand and interpret the above experimental findings. Remarkably, the experimental thermodynamic parameters derived from the van't Hoff plot for the binding of the alkenylboronic ester to [CuI], closely correspond to the predictions from density functional theory (DFT), with ΔGDFT = 7.5 kcal mol−1, ΔHDFT = 11.5 kcal mol−1, and ΔSDFT = 12.1 e.u. (Fig. 5a). The strong agreement between experimental data (ΔGexp = 6.7 ± 1.0 kcal mol−1) and DFT results supports the reliability of the thermodynamic data from calculations. Furthermore, the alkenylated product 3a exhibits a lower affinity for binding to [CuI] compared to 2a (ΔGDFT = 5.9 kcal mol−1) (Scheme S4†).This result is consistent with the need for mild heating (60 °C) to disrupt the interaction between alkenyl precursors or products 2a or 3a and the [CuI] catalyst, thereby initiating the catalytic cycle. Upon dissociation of the alkene from the [CuI] catalyst, the [CuI] complex undergoes oxidation by tBuOOtBu which requires on an accessible coordination site. Furthermore, the formation of [CuII]-CHCMe2via transmetalation of [CuII]-OtBu with 2a is exergonic (ΔG = −3.5 kcal mol−1) with a modest reaction barrier (ΔG‡ = 13.6 kcal mol−1) (Scheme S3†).
A spin density plot of the [CuII]-CHCMe2 intermediate indicates 31% localization on Cα (Fig. 5b). The radical nature of the alkenyl group bound to the copper(II) center in [CuII]-CHCMe2 also accounts for its propensity to dimerize to form Me2CCH–CHCMe2, a highly favorable reaction (ΔGrxn = −69.1 kcal mol−1). A relaxed energy scan for dimerization further reveals essentially no barrier for this process (Fig. S22†). Similarly, the radical capture by trityl radical pathway is also a highly favourable reaction (ΔG = −39.0 kcal mol−1) (Fig. 5c(ii)). We note that loss of the alkenyl radical ˙CHCMe2 from [CuII]-CHMe2 is significantly uphill in free energy (ΔG = 36.0 kcal mol−1; Fig. S21†); accordingly we do not anticipate the direct involvement of the ˙CHCMe2 radical in these copper-catalyzed reactions.
Due to the radical character on Cα, we investigated whether [CuII]-CHCMe2 could abstract a H-atom from a benzylic C–H bond. This HAT process is favourable with a relatively low reaction barrier (ΔG = −8.2 kcal mol; ΔG‡ = 12.9 kcal mol; Fig. 5v(iv)). This calculation result is consistent with the experiment, where a trace amount of alkenylation product, 3a, was observed in the absence of tBuO˙ (Fig. 4c). After [CuII]-CHCMe2 abstracts an H atom from toluene, the resulting toluene radical undergoes capture by [CuII]-CHCMe2 to form 3a (Fig. 5c(iii)). Calculations also rationalize the formation of styrene as a byproduct in the C–H alkenylation of ethylbenzene. Both radical capture and H-atom abstraction of a β-H of the 2° ethylbenzene radical PhCH(˙)Me are extremely favorable (ΔG = −55.0 and −48.3 kcal mol−1, respectively; Scheme S5†).
Conclusions
This report illustrates the use of alkenylboronic esters in catalytic benzylic C–H functionalization for sp3–sp2 C–C bond construction. The Me2CCH–B(OR)2 reagent 2a exhibits a broad C–H substrate scope across typical 1° benzylic C–H bonds. Importantly, it significantly expands the scope for benzylic C–H alkenylation as it does not require highly acidified ArCF2-H bonds.25 Importantly, this study illustrates how sp3 C–H alkenylation can lead to formation of the prenyl group known to engender biological activity in small molecules.
A combination of experimental and computational studies support that this sp3 C–H alkenylation protocol proceeds via a copper(II) alkenyl intermediate [CuII]-CHCMe2 (Scheme 2). This [CuII]-CHCMe2 intermediate promotes C–C bond formation to form R–CHCMe2 products in the capture of an alkyl radical R˙ derived from H-atom abstraction from R–H via the tBuO˙ radical generated upon reaction of tBuOOtBu with [CuI]. Facile bimolecular C–C coupling between [CuII]-CHCMe2 species results in a competing pathway to form the diene Me2CCH–CHCMe2. Yet, the copper(II) alkenyl intermediate can also directly transform substrates R–H into R–CHCMe2, albeit in low yield, due to the ability of the [CuII]-CHCMe2 intermediate to abstract a H-atom from substrates R–H to form R˙. This unusual chemical pathway available uncovered for copper(II) alkenyls offers additional opportunities to construct catalytic C–H alkenylation protocols via selective H-atom abstraction of substrates R–H via metal-centred intermediates en route to functionalized species R–CHCRR′.
 | ||
| Scheme 2 Catalytic cycle for sp3 C–H alkenylation with competing pathways via the [CuII]-CHCMe2 intermediate. | ||
Data availability
All synthetic procedures, characterization data, spectroscopic data, computational data, supplementary figures and tables, and detailed crystallographic information can be found in the ESI. Crystallographic data are available via the Cambridge Crystallographic Data Centre (CCDC): 2268882 and 2329536.Author contributions
T.-A. C. and T. H. W. conceived project. T.-A. C. carried out experimental and computational works. T.-A. C. and R. J. S. collected, solved, and refined crystallographic data. T. H. W. supervised the experimental and computational work. T.-A. C. and T. H. W. wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to NSF (CHE-1955942 and CHE-2303206) for supporting this work. The Rigaku Synergy S Diffractometer was purchased with support of the NSF MRI program (CHE-1919565). Dr Daniel Holmes for help in variable temperature NMR experiments, and Dr Anthony Schilmiller for insightful discussions regarding HRMS analyses as well as Nathan Slater and Dr Xingling Pan for synthetic assistance.Notes and references
- M. E. G. Mosquera, G. Jiménez, V. Tabernero, J. Vinueza-Vaca, C. García-Estrada, K. Kosalková, A. Sola-Landa, B. Monje, C. Acosta, R. Alonso and M. Á. Valera, Sustainable Chem., 2021, 2, 467–492 CrossRef CAS.
- J. D. Ochocki and M. D. Distefano, Med. Chem. Commun., 2013, 4, 476–492 RSC.
- K. Šmejkal, Phytochem. Rev., 2014, 13, 245–275 CrossRef.
- M. E. Tanner, Nat. Prod. Rep., 2014, 32, 88–101 RSC.
- X. Yang, Y. Jiang, J. Yang, J. He, J. Sun, F. Chen, M. Zhang and B. Yang, Trends Food Sci. Technol., 2015, 44, 93–104 CrossRef CAS.
- R. Ciochina and R. B. Grossman, Chem. Rev., 2006, 106, 3963–3986 CrossRef CAS.
- P.-H. Liang, T.-P. Ko and A. H.-J. Wang, Eur. J. Biochem., 2002, 269, 3339–3354 CrossRef CAS PubMed.
- D. P. Labbé, S. Hardy and M. L. Tremblay, Prog. Mol. Biol. Transl. Sci., 2012, 106, 253–306 Search PubMed.
- C. C. Palsuledesai and M. D. Distefano, ACS Chem. Biol., 2015, 10, 51–62 CrossRef CAS.
- E. S. Marakasova, N. K. Akhmatova, M. Amaya, B. Eisenhaber, F. Eisenhaber, M. L. van Hoek and A. V. Baranova, Mol. Biol., 2013, 47, 622–633 CrossRef CAS.
- B. Botta, A. Vitali, P. Menendez, D. Misiti and G. D. Monache, Curr. Med. Chem., 2005, 12, 713–739 CrossRef CAS.
- M. M. M. Pinto, M. E. Sousa and M. S. J. Nascimento, Curr. Med. Chem., 2005, 12, 2517–2538 CrossRef CAS PubMed.
- K. Yazaki, K. Sasaki and Y. Tsurumaru, Phytochemicals, 2009, 70, 1739–1745 CrossRef CAS.
- X. Chen, E. Mukwaya, M.-S. Wong and Y. Zhang, Pharm. Biol., 2014, 52, 655–660 CrossRef.
- S. Tamura, T. Fujitani, M. Kaneko and N. Murakami, Bioorg. Med. Chem. Lett., 2010, 20, 3717–3720 CrossRef PubMed.
- G. Comte, J.-B. Daskiewicz, C. Bayet, G. Conseil, A. Viornery-Vanier, C. Dumontet, A. Di Pietro and D. Barron, J. Med. Chem., 2001, 44, 763–768 CrossRef.
- R. Badhan and J. Penny, Eur. J. Med. Chem., 2006, 41, 285–295 CrossRef PubMed.
- Y. Yang and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 10642–10645 CrossRef PubMed.
- Y. Yang, T. J. L. Mustard, P. H.-Y. Cheong and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 14098–14102 CrossRef.
- J. L. Farmer, H. N. Hunter and M. G. Organ, J. Am. Chem. Soc., 2012, 134, 17470–17473 CrossRef.
- M. A. Tarselli, A. Liu and M. R. Gagné, Tetrahedron, 2009, 65, 1785–1789 CrossRef PubMed.
- Y. J. Zhang, E. Skucas and M. J. Krische, Org. Lett., 2009, 11, 4248–4250 CrossRef PubMed.
- S.-Y. Chen, Q. Li and H. Wang, J. Org. Chem., 2017, 82, 11173–11181 CrossRef.
- R. Zeng, C. Fu and S. Ma, J. Am. Chem. Soc., 2012, 134, 9597–9600 CrossRef PubMed.
- K. Chakrabarti, M. M. W. Wolfe, S. Guo, J. W. Tucker, J. Lee and N. K. Szymczak, Chem. Sci., 2024, 15, 1752–1757 RSC.
- S. Guo, Y. Yuan and J. Xiang, New J. Chem., 2015, 39, 3093–3097 RSC.
- Z. Fang, C. Wei, J. Lin, Z. Liu, W. Wang, C. Xu, X. Wang and Y. Wang, Org. Biomol. Chem., 2017, 15, 9974–9978 RSC.
- Z. Cui, X. Shang, X.-F. Shao and Z.-Q. Liu, Chem. Sci., 2012, 3, 2853–2858 RSC.
- H. Yang, P. Sun, Y. Zhu, H. Yan, L. Lu, X. Qu, T. Li and J. Mao, Chem. Commun., 2012, 48, 7847–7849 RSC.
- H. Yang, H. Yan, P. Sun, Y. Zhu, L. Lu, D. Liu, G. Rong and J. Mao, Green Chem., 2013, 15, 976–981 RSC.
- D. L. Golden, S.-E. Suh and S. S. Stahl, Nat. Rev. Chem, 2022, 6, 405–427 CrossRef PubMed.
- Z. Zhang, P. Chen and G. Liu, Chem. Soc. Rev., 2022, 51, 1640–1658 RSC.
- E. S. Jang, C. L. McMullin, M. Käβ, K. Meyer, T. R. Cundari and T. H. Warren, J. Am. Chem. Soc., 2014, 136, 10930–10940 CrossRef.
- M. S. Kharasch and G. Sosnovsky, J. Am. Chem. Soc., 1958, 80, 756 CrossRef.
- M. S. Kharasch, G. Sosnovsky and N. C. Yang, J. Am. Chem. Soc., 1959, 81, 5819–5824 CrossRef.
- A. Bakhoda, O. E. Okoromoba, C. Greene, M. R. Boroujeni, J. A. Bertke and T. H. Warren, J. Am. Chem. Soc., 2020, 142, 18483–18490 CrossRef.
- S. Kundu, C. Greene, K. D. Williams, T. K. Salvador, J. A. Bertke, T. R. Cundari and T. H. Warren, J. Am. Chem. Soc., 2017, 139, 9112–9115 CrossRef.
- W. Xie, J. Heo, D. Kim and S. Chang, J. Am. Chem. Soc., 2020, 142, 7487–7496 CrossRef PubMed.
- A. Vasilopoulos, S. L. Zultanski and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 7705–7708 CrossRef PubMed.
- B. C. Figula, T.-A. Chen, J. A. Bertke and T. H. Warren, ACS Catal., 2022, 12, 11854–11859 CrossRef CAS.
- R. T. I. Gephart, C. L. McMullin, N. G. Sapiezynski, E. S. Jang, M. J. B. Aguila, T. R. Cundari and T. H. Warren, J. Am. Chem. Soc., 2012, 134, 17350–17353 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. Detailed experimental procedures are provided. CCDC 22688822329536. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03430a |
| This journal is © The Royal Society of Chemistry 2024 |