
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformational modulation and polymerization-induced folding of proteomimetic peptide brush polymers†
Julia
Oktawiec
 a,
Omar M.
Ebrahim
a,
Yu
Chen
b,
Kaylen
Su
c,
Christopher
Sharpe
b,
Nathan D.
Rosenmann
b,
Clara
Barbut
a,
Steven J.
Weigand
d,
Matthew P.
Thompson
a,
James
Byrnes
e,
Baofu
Qiao
c and
Nathan C.
Gianneschi
*abf
a,
Omar M.
Ebrahim
a,
Yu
Chen
b,
Kaylen
Su
c,
Christopher
Sharpe
b,
Nathan D.
Rosenmann
b,
Clara
Barbut
a,
Steven J.
Weigand
d,
Matthew P.
Thompson
a,
James
Byrnes
e,
Baofu
Qiao
c and
Nathan C.
Gianneschi
*abf
aDepartment of Chemistry, Northwestern University, Evanston, IL 60208, USA. E-mail: nathan.gianneschi@northwestern.edu
bDepartment of Materials Science and Engineering, Northwestern University, Evanston, IL 60208, USA
cDepartment of Natural Sciences, Baruch College, City University of New York, New York, NY 10010, USA
dDuPont−Northwestern−Dow Collaborative Access Team (DND-CAT) Synchrotron Research Center, Northwestern University, Argonne, IL 60208, USA
eBeamline 16ID, NSLS-II, Brookhaven National Laboratory, Upton, NY 11973, USA
fInternational Institute for Nanotechnology, Chemistry of Life Processes Institute, Simpson Querrey Institute, Lurie Cancer Center, Department of Biomedical Engineering, and Department of Pharmacology, Northwestern University, Evanston, IL 60208, USA
First published on 29th July 2024
Abstract
Peptide-brush polymers generated by graft-through living polymerization of peptide-modified monomers exhibit high proteolytic stability, therapeutic efficacy, and potential as functional tandem repeat protein mimetics. Prior work has focused on polymers generated from structurally disordered peptides that lack defined conformations. To obtain insight into how the structure of these polymers is influenced by the folding of their peptide sidechains, a set of polymers with varying degrees of polymerization was prepared from peptide monomers that adopt α-helical secondary structure for comparison to those having random coil structures. Circular dichroism and nuclear magnetic resonance spectroscopy confirm the maintenance of the secondary structure of the constituent peptide when polymerized. Small-angle X-ray scattering (SAXS) studies reveal the solution-phase conformation of PLPs in different solvent environments. In particular, X-ray scattering shows that modulation of solvent hydrophobicity, as well as hydrogen bonding patterns of the peptide sidechain, plays an important role in the degree of globularity and conformation of the overall polymer, with polymers of helical peptide brushes showing less spherical compaction in conditions where greater helicity is observed. These structural insights into peptide brush folding and polymer conformation inform the design of these proteomimetic materials with promise for controlling and predicting their artificial fold and morphology.
Introduction
The ability to discover and develop peptides to specifically target and bind proteins has resulted in an increasing interest in peptide-based or peptidomimetic therapeutics.1–4 However, cellular penetration, resistance to proteolysis,5 and multivalent, high avidity binding to intracellular targets remain challenges facing the translational potential of peptides generally. To this end, cyclization,6–8 lipidation,9–11 and peptide-stapling approaches12–14 have been and remain key strategies for enabling the development of peptides and peptidomimetics.15,16 An emerging strategy involves coupling a polymerizable moiety to a peptide of interest to yield a peptidyl-macromonomer that is then subjected to graft-through polymerization using ring-opening metathesis polymerization (ROMP)17,18 or controlled radical polymerization chemistry, including photoinduced reversible addition−fragmentation transfer (RAFT) radical polymerization.19,20 The resulting peptide-brush polymers show enhanced resistance to proteolysis,18 as well as the ability to internalize into cells and to access intracellular, cytosolic targets with high affinities.21–24 The observed elevated resistance to proteolysis is ascribed to the collapsed globular structures of the polymers observed in water that arise, in particular in the case of brush polymers generated from norbornenyl-peptides (polynorbornenes), due to backbone hydrophobicity driving an entropically-favorable collapse. This collapse leads to sterically crowded peptide brushes, rendering them less accessible to proteases, while preserving their ability to interact with therapeutic targets and cellular membranes due to polymer dynamics.18,20 As a result of these collective, emergent properties, we refer to these materials as Protein-Like Polymers (PLPs).22,25Despite initial success in implementing this strategy to achieve desired biological properties and outcomes, an in-depth understanding of how the polymer backbone influences the folding and conformation of such peptide brushes, and vice versa, is needed. One related example involves brush polymers of poly(γ-benzyl-L-glutamate) generated using norbornene monomers with variable lengths of poly(γ-benzyl-L-glutamate) segments.26 The helicity of these polymers was assessed using 1H NMR spectroscopy in 2% TFA-d/CDCl3, and it was found that brush polymers had slightly lower degrees of helicity than the poly(γ-benzyl-L-glutamate) segments by themselves. The interruption of folding was reduced when grafting density was lessened. Similar polymers generated by post-polymerization modifications have been shown to form rod-like structures by dry-state atomic force microscopy.27 In general, experimental and computational studies find that while hydrophobic environments can promote helicity of peptide amphiphiles,28,29 steric crowding can also interrupt the uniform folding of peptides into helices.30
Our motivation to understand these effects lies in the ability to control folding and structure in a cooperative way, which will influence the utility of these materials in a range of settings. Greater structural insight into more complex peptide-brush polymers would allow for the determination of structure–function relationships for the targeted design of peptide-brush polymer tools for engaging specific protein targets. In particular, it may be important to preserve the organization of functional groups on the face of helical peptide side chains, so that they can readily inhibit particular protein–protein interactions involved in disease progression.31,32 Herein, a particular focus was to establish whether helicity could be preserved or enhanced in aqueous environments when an α-helical peptide is incorporated onto a polymer backbone. Approaches described here were inspired, in part, by efforts to characterize the folding of single-chain nanoparticle polymers using techniques including small-angle X-ray scattering (SAXS).33–39
Towards this end, polymers were designed to contain brushes of peptides with a classical secondary structure, the α-helix, as well as a peptide with an unstructured, random coil configuration (Fig. 1). Using these peptides as macromonomers allows the use of techniques probing the perturbation of peptide folding, such as circular dichroism (CD) spectroscopy and nuclear magnetic resonance (NMR) spectroscopy. A set of polymers incorporating these peptide monomers was generated and studied using spectroscopy and X-ray scattering, coupled with atomistic metadynamics simulations, to gain a granular understanding of peptide folding and polymer conformation.
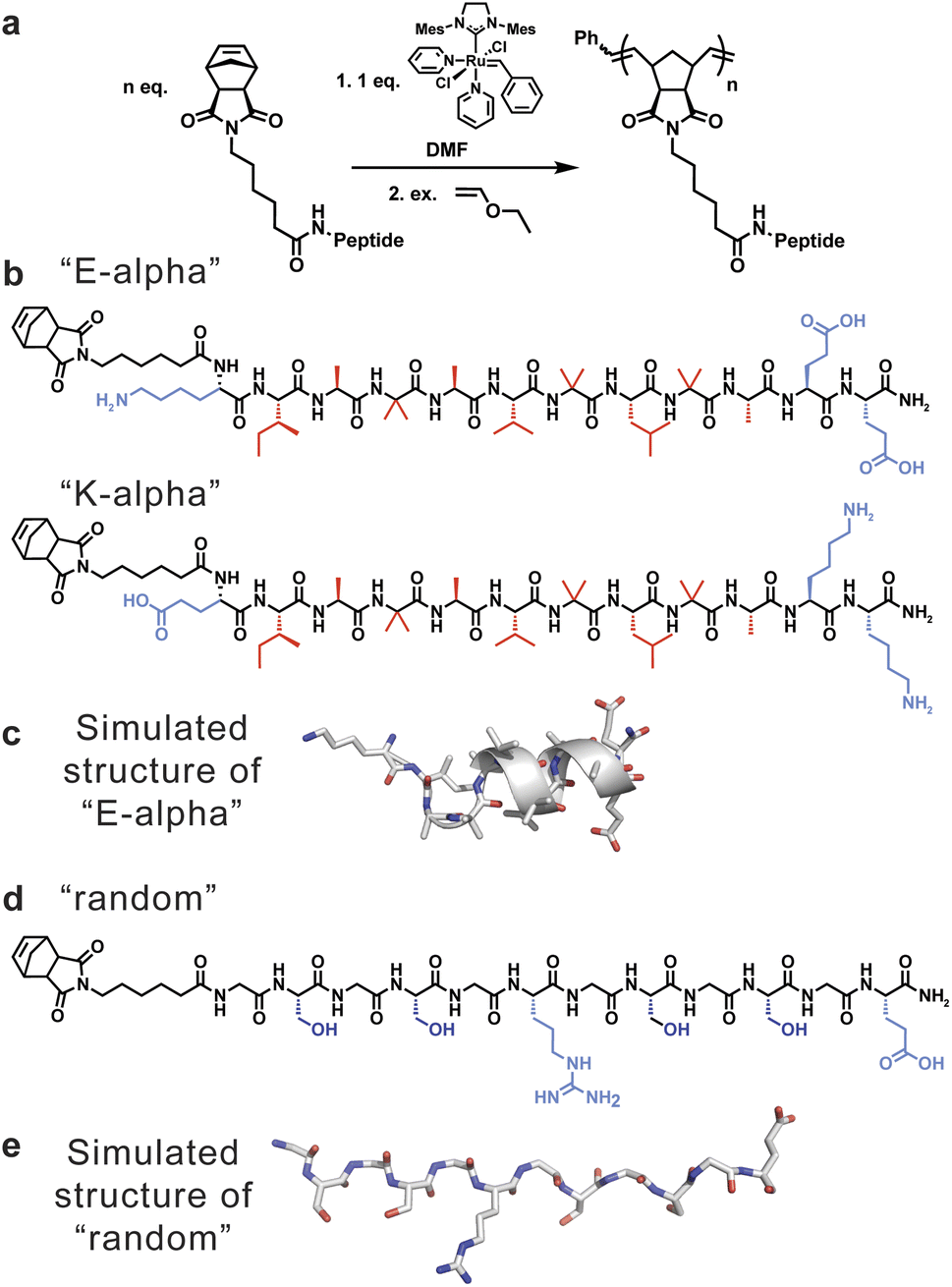 | ||
| Fig. 1 (a) Scheme showing the synthesis of peptide-brush polymers using graft-through ROMP. (b) Peptide sequences of α-helical monomers utilized to generate peptide-brush polymers, including E-alpha (sequence = NorAhaKIABAVBLBAEE) and K-alpha (sequence = NorAhaEIABAVBLBAKK), with hydrophobic residues indicated in red and hydrophilic residues indicated in blue. (c) Model of E-alpha peptide obtained from molecular dynamics simulations. (d) Peptide sequence of random coil peptide monomer utilized to generate peptide-brush polymers (sequence = NorAhaGSGSGRGSGSGE), with hydrophilic residues indicated in blue. (e) Model of random peptide obtained using AlphaFold2.40 | ||
Results and discussion
Polymer design and synthesis
To understand the influence of secondary structure on polymer dynamics, and vice versa, two sets of peptides were chosen (Fig. 1). Firstly, we studied polymers derived from a helical peptide. We employed as a guide a crystal structure of a peptide (Boc-WIABIVBLBP-OMe),41 which is α-helical due to the high content of 2-aminoisobutyric acid (Aib, B) residues. This unnatural amino acid, with its disubstituted alpha carbon, is known to have a propensity to favor helical conformations in peptide sequences that contain it.42 Specifically, the disubstitution greatly reduces the rotational space accessible by the amino acid to orientations that favor helicity. We then mutated the sequence to further promote helicity by specifically replacing the tryptophan and proline with lysine and alanine, and added additional glutamic acid residues for solubility, to generate the sequence KIABAVBLBAEE (“E-alpha”). The amino acids lysine, alanine, and glutamic acid were chosen as they are known to be found in and promote helical arrangements in proteins, hypothesized to be in part due to their side chain sterics and hydrophobicity.43–46 We also synthesized a sequence flipping the charged peptides to match the macrodipole of a helix (EIABAVBLBAKK, “K-alpha”), as it is established that positively charged residues at the negatively-charged C-terminal end promote greater helix stability.47,48 A third peptide was synthesized by replacing the helix-directing amino acid Aib with alanine (EIAAAVALAAKK, “A-alpha”). Secondly, we designed a random coil peptide sequence with a high proportion of glycine and serine, with one arginine and one glutamic acid to promote water solubility (GSGSGRGSGSGE, “random”). This particular peptide is thus water soluble without being likely to fold into any defined secondary structure, and its lack of defined structure was further supported using AlphaFold2 (Fig. 1e), as well as CD spectroscopy, discussed below.40 The differences in physical properties and folding propensities between the two sets of peptides were thus chosen to provide a strong contrast to probe the influence of secondary structure on polymer conformation.The chosen peptides were synthesized with exo-norbornene-2,3-dicarboxyimide attached via an aminohexanoic acid linker (denoted as “NorAha”) at the N-terminus using established solid-phase peptide synthesis (SPPS) chemistry (ESI Section 2 and ESI Fig. S1–S8†).49 After purification, the resulting peptide macromonomers were polymerized via ROMP to degrees of polymerization (DPs) of 15, 30, and 45 (ESI Section 3†). Additional DPs of 7 and 90 were prepared for the helical sequence, E-alpha. All polymerizations were monitored by 1H NMR spectroscopy (ESI Fig. S9–S11†). The molecular weights and polymer dispersities were determined by size exclusion chromatography coupled with multi-angle light scattering (SEC-MALS) and via sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) (Fig. 2, Table 1, and ESI Fig. S12–S14†). The resulting polymers were analyzed by dynamic light scattering revealing the polymers were not aggregated, with radii of hydration in aqueous solutions ranging from 2 to 4 nanometers (ESI Table S1†). We note that the A-alpha sequence could not be polymerized due to intractable aggregation of the monomer during polymerization. However, this peptide served as a reference for CD spectroscopy (see ESI Fig. S15†).
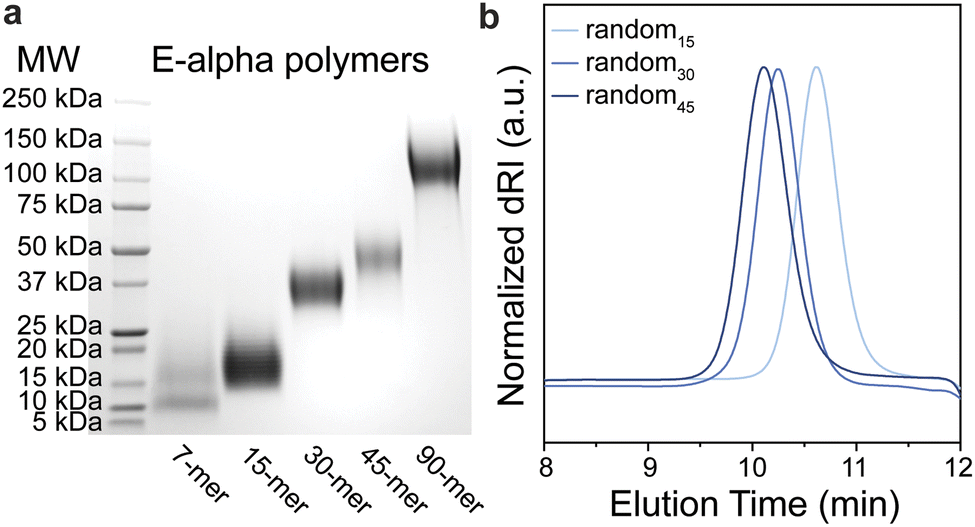 | ||
| Fig. 2 (a) SDS-PAGE gel of E-alpha polymers and the molecular weight ladder (MW), showing the approximate molecular weight distributions of each polymer. (b) Aqueous SEC-MALS of random peptide-based polymers (blue traces). | ||
| Polymer sample | Theoretical Mn (kDa) | Experimental Mw/Mna (kDa) | Đ (Mw/Mn)a | M W (kDa) |
|---|---|---|---|---|
| a M w and Mn values for E-alpha PLPs could not be determined as SDS–PAGE, the only method by which the molecular weight of the polymer sample could be measured, does not allow for the determination of these values. We note that dispersities determined using SEC-MALS are an underestimation due to limitations of the technique.50 Further characterization details are provided in ESI Section 2, and additional polymer characterization is listed in ESI Table S1. b MW values determined by estimating the middle of the sample band via visual inspection of SDS-PAGE data (Fig. 2 and S11–S14). | ||||
| E-alpha7 | 10 | — | — | 11 |
| E-alpha15 | 22 | — | — | 18 |
| E-alpha30 | 44 | — | — | 37 |
| E-alpha45 | 66 | — | — | 50 |
| E-alpha90 | 131 | — | — | 120 |
| random15 | 19 | 18.8/18.6 | 1.01 | 20 |
| random30 | 38 | 40.7/40.5 | 1.01 | 30 |
| random45 | 56 | 53.0/51.5 | 1.03 | 50 |
| random90 | 113 | — | — | 95 |
Characterization of peptide brush folding by CD spectroscopy
The secondary structures of free peptide monomers and polymers were determined by CD spectroscopy (Fig. 3 and ESI Section 4, see ESI Fig. S15–S22†). Generally, the polymers displayed consistent structures to those of the monomeric peptides, with the glycine–serine repeat peptide and polymers showing characteristic features of random coil structure (Fig. 3). Intriguingly, in aqueous buffer, the polymers of the designed helical peptides E-alpha and K-alpha maintain a strong helical signal, with a strong maximum at 195 nm, and two minima at approximately 205 and 220 nm, while the peptide monomers are almost completely unfolded (10–16% helicity by analysis of CD data detailed in ESI Section 4, ESI Tables S2 and S3†). The intensity of helicity of the sample is clearly related to the degree of polymerization, with shorter polymers having less helical character (5% and 24% for E-alpha7 and E-alpha15 respectively), and longer polymers having greater degrees of helicity (43% and 46% for E-alpha45 and E-alpha90 respectively). The peptide monomers unfold in water because the solvent competes with the intramolecular hydrogen bonding necessary for helical folding. In contrast, the increase of helicity with increasing polymer lengths suggests that the organization of peptide units along the polymer backbone generates a local hydrophobic environment, ascribed to the norbornene linker as well as the large proportion of hydrophobic side chains on E-alpha. This hydrophobic environment promotes the intramolecular hydrogen bonding of the peptide over solvation of the backbone amides by water.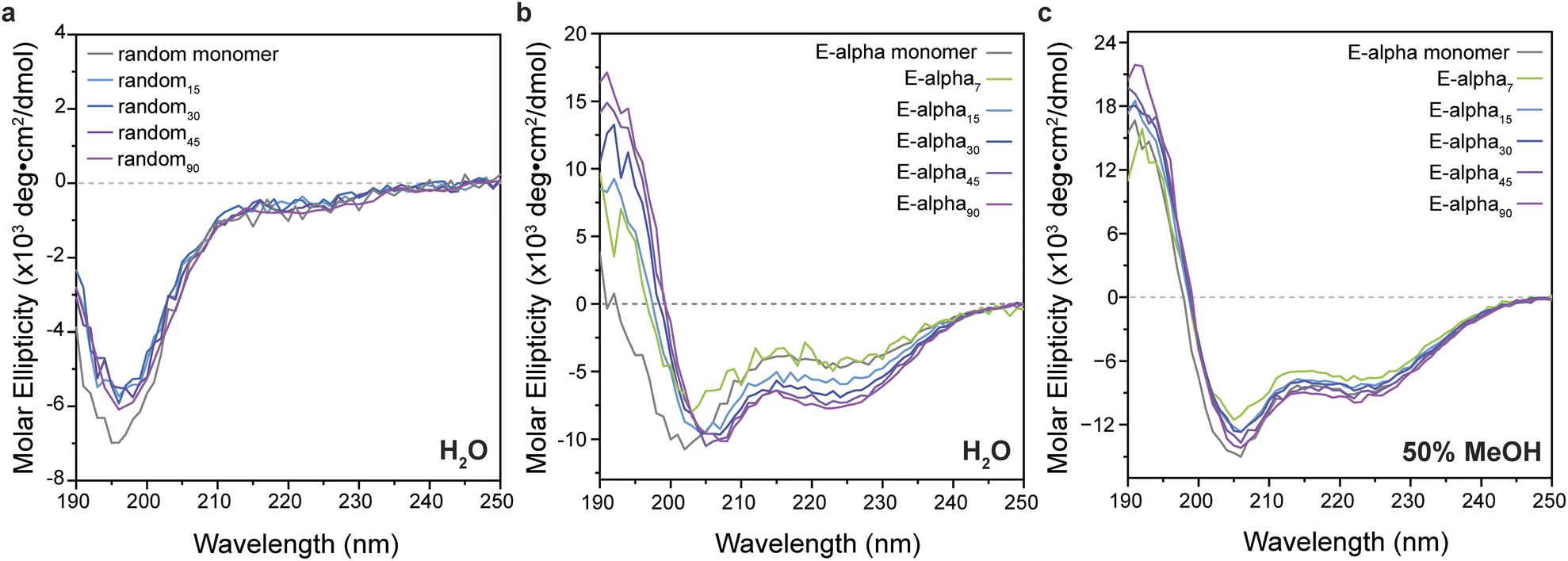 | ||
| Fig. 3 Circular dichroism (CD) spectra of (a) random coil peptide-based monomer and polymers in water and (b) α-helical peptide (E-alpha)-based monomer and polymers in 50 mM sodium carbonate buffer, and (c) helical peptide-based monomer and polymers in 50% methanol with 50 mM sodium carbonate buffer. | ||
To confirm the influence of hydrophobic solvation on peptide brush conformation, CD spectra were obtained of the polymers of designed helical peptides in 50% methanol and 100% methanol. All samples displayed a strong helical signal (Fig. 3 and S18† respectively), similar in intensity to the signal of the monomer when all are plotted relative to the concentration of the monomer unit.51 Additionally, the α-helical signal in 100% methanol (at 193 nm, 29 × 103 deg cm2 dmol−1) is stronger than in 50% methanol (at 193 nm, 13–16 × 103 deg cm2 dmol−1), indicating that the more hydrophobic solvent does promote the intramolecular hydrogen bonding of the peptide backbone amides necessary for the α-helical fold, rather than solvating the backbone amides and causing them to unfold. However, the intensity of the samples' CD signal in water is still lower than those observed in methanol, likely due to the dynamic nature of the polymer despite its globular, crowded conformations. This result is consistent with previous simulations and enzymatic assays,18,20,21 where the polymer evades proteolysis of otherwise susceptible peptides.
The helical CD signal of the polymer is also relatively temperature stable in water. When the 30-mer is heated to 90 °C, while some intensity at 220 nm is lost, the polymer's degree of secondary structure is completely regained when the sample is cooled back to room temperature (ESI Fig. S21†). These variable temperature curves do not show a clear sigmoidal melting transition (ESI Fig. S22†), similar to other variable temperature CD studies performed on helical peptides in water,52,53 indicating that the folding of the polymer does not behave in a cooperative, two-state fashion. Additionally, despite the loss of some helicity at elevated temperatures, the polymer backbone allows the peptide to reversibly recover its folding patterns when cooled back to room temperature.
To quantitively examine the probability of the α-helix, we conducted metadynamics simulations. In metadynamics simulations, a history-dependent Gaussian bias potential is introduced to the Hamiltonian of the system as a function of collective variables (CVs).54 The added Gaussian bias potential fills the underlying free energy basins and reflects the free energy surface as a function of the CVs. In well-tempered metadynamics, the height of the Gaussian bias potential continuously decreases over the simulation time to ensure the convergence of the final bias potential to the actual FES.55 In this work, the CV of alphaRMSD56 was used, which calculated the number, and consequently the probability, of α-helix structure for the peptide. Well-tempered metadynamics simulations supported the probability of α-helix of around 10% for the E-alpha peptide-based macromonomer (Fig. 4 and S23–S25†). Further analyses showed that the central portion of the peptide strongly favors the formation of α-helix configuration (inset of Fig. 4 and S24†). These results correlate well with the experimental data, which supports 10% helical content in the peptide monomer by CD spectroscopy (Tables S2 and S3†). Although metadynamics simulations of the secondary structure of the E-alpha15 polymer did not converge despite considerable computational time (ESI Section 5†), hindering computational confirmation of the helicity observed, the consistency observed with monomer folding indicates that the polymer environment likely significantly alters the folding of the peptide sidechains.
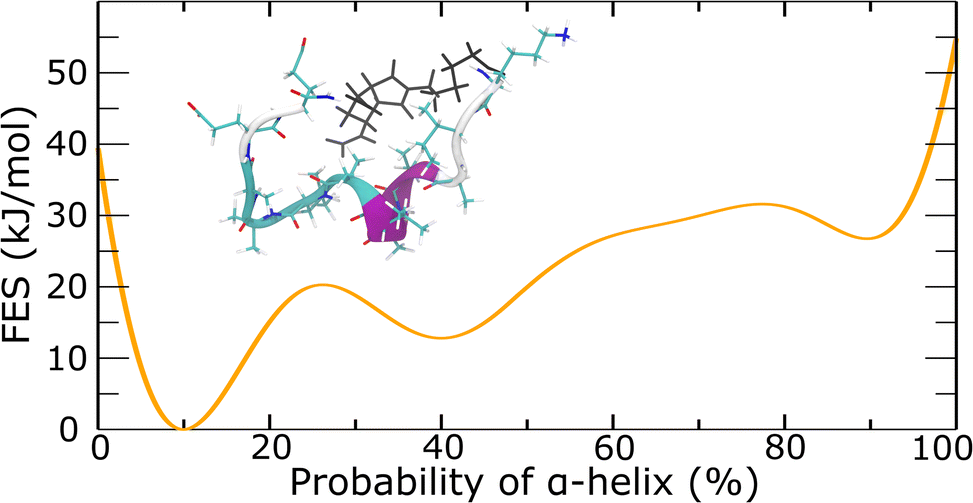 | ||
| Fig. 4 Free energy surface as a function of the probability of α-helix for E-alpha obtained from well-tempered metadynamics simulations. A probability of 10% is the most energetically stable with a characteristic structure provided in the inset. The α-helix/turn/random coil secondary structures of the peptide backbone are colored in magenta/cyan/white, respectively; NorAha atoms are colored in black. | ||
NMR spectroscopy reveals features of α-helical folding by peptide brushes
In conjunction with CD spectroscopy, NMR spectroscopic studies of peptides and proteins provide critical insight into their structure. For example, the rate of exchange of amide protons with solvent protons is slowed down by intramolecular hydrogen bonding, such as that which results in folded peptide or protein structures.57 As a result, a qualitative understanding of solvent-exposed regions of samples can be obtained by experiments where samples are dissolved in deuterium oxide, and the disappearance of peaks corresponding to exchangeable protons, like amide NH peaks, is monitored.58,59 Specifically, the hydrogen bonding between peptide amide groups that is necessary for α-helical folding slows down the rate at which such groups will exchange with deuterium oxide, resulting in the slower disappearance of such peaks, and thus identification of unfolded and folded regions within samples. Such experiments were conducted using D2O and CD3OD on the helical peptide-based monomer K-alpha and its resulting 15-mer polymer (Fig. 5 and S26–S43†). We focused on these and subsequent NMR studies of the K-alpha monomer and K-alpha15 polymer due to the basic conditions needed to dissolve the E-alpha peptide-based samples, which hinder analysis of the amide NH peaks due to their rapid exchange in basic solution.60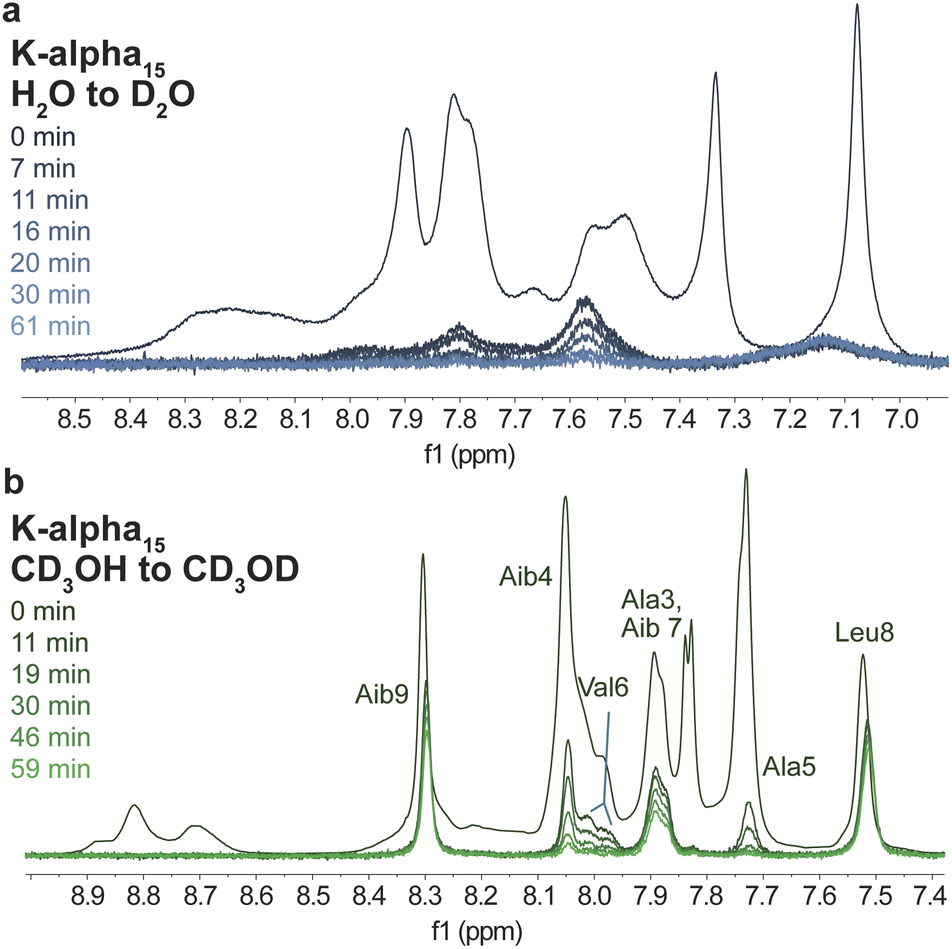 | ||
| Fig. 5 1H NMR spectra of the K-alpha15 polymer. (a) Dissolved in 100% D2O (blue traces). (b) Dissolved in 100% CD3OD (green traces). Analysis shows the disappearance of NH amide peaks of the peptide brush sequence upon exchange with the deuterated solvent. The spectra at 0 min were obtained from separate experiments where samples were dissolved in 90% H2O with 10% D2O or 100% CD3OH. The sample dissolved in CD3OD shows a slower loss of peaks, likely due to the greater folding observed in this solvent. | ||
Samples were dissolved and then monitored by 1H NMR spectroscopy over time, showing that in D2O, samples generally show rapid disappearance of all NH peaks within one hour (Fig. 5 and S43†). In contrast, in CD3OD both peptide monomer and polymer retain many amide signals, with the peptide monomer displaying less rapid amide peak disappearance in comparison to the polymer (Fig. 5 and S43†). Specifically, the polymer retains only three amide peaks after one hour, whereas the monomer retains eleven peaks. The retained polymer peaks correspond to the NH moieties of Aib7, Leu8, and Aib9, while in the monomer case, only the Glu1 NH peak disappears. The large difference between the monomer and polymer in methanol indicates that although peptides appear to be folded to the same degree by CD spectroscopy, the polymeric system appears to be more dynamic than the monomer in methanol, leading to an increased number of interactions with the solvent that result in a more rapid loss of signal. We note that more internal amino acids in the polymer case are retained (Aib7 to Aib9), with a gradual loss of intensity of residues next to this segment (Aib4, Ala5, and Val6). The amino acids that retain intensity correspond well to residues identified in metadynamics simulations of the E-alpha peptide monomer that are more likely to be helical, specifically, B4 to L8 (Fig. 4 and 5). In contrast, the NMR spectra of the peptide monomer only shows loss of the Glu1 NH peak, which is right next to the norbornene unit, and is consistent with observations that helices unfold by fraying at the peptide ends.61,62 The amino acids at the ends of the helix are thus more likely to have interactions with solvent and thus show loss of intensity in the CD3OD experiment. The dynamicity of the K-alpha15 polymer is replicated in exchange studies performed on the random peptide and random15 polymer (ESI Fig. S42†), where the polymer shows more rapid exchange than the peptide.
In addition, 2D TOCSY and NOESY NMR spectra were obtained for the K-alpha peptide monomer and K-alpha15 polymer in H2O/D2O and CD3OH to determine the degree of folding of the two samples as a function of the solvent environment, as such experiments allow for the identification of secondary structure (ESI Fig. S32–S41†). Specifically, 2D total correlation spectroscopy (TOCSY) provides correlations of atoms within the spin system, allowing for identification of which protons correspond to which amino acids, while 2D nuclear Overhauser effect spectroscopy (NOESY) provides through-space correlations between protons that are in close proximity (<5 Å). The combination of these two techniques allows for the assignment of all resonances, and then determination of close contacts, particularly those involved in intramolecular hydrogen bonding within folded regions of the peptide sequences.63,64 Qualitatively, we observe a much greater number of nuclear Overhauser effect (NOE) features in the samples dissolved in CD3OH (e.g. peptide 2D NOESY spectra in ESI Fig. S33, S34, S36 and S37;† polymer 2D NOESY spectra in ESI Fig. S39–S41†), implicating more intramolecular hydrogen bonding, and thus more peptide folding. Moreover, the polymer spectra display a significant broadening of peaks in both H2O/D2O and CD3OH relative to the peptide monomers (ESI Fig. S26–S31†). This broadening is due to both the polymer dispersity and differences in the conformation of peptide brushes along the polymer, where the repeating peptide brush experiences slightly different chemical environments, thus resulting in averaged and broadened features. Additionally, the more efficient relaxation of the larger polymer molecule relative to the peptide (18 kDa vs. 1.5 kDa) contributes to broadening.65
The K-alpha peptide monomer displays some slight evidence of helicity in H2O/D2O, and even more clear evidence of helicity in CD3OH, with strong NOE signals of NN(i, i + 1) NOEs in both being visible, as well as (i, i + 4) NOEs in CD3OH (ESI Fig. S36 and S37†).66 We also note that the J-coupling values and Hα positions of most amino acids, though likely perturbed due to being in CD3OH, are more consistent with α-helicity than random coil values.67–69
While the broadness of the polymer data in H2O/D2O hinders further analysis, the 2D NMR data of the sample dissolved in CD3OH allows for the identification of the positions of NH and Hα peaks corresponding to each amino acid of the monomer unit (ESI Fig. S31†). Of interest is that the positions of Glu1, Ile2, and Ala3 shift significantly downfield relative to their position in the peptide monomer spectra, suggesting that these NH groups experience a substantial change in their chemical environment that is consistent with their position proximal to the norbornene-based linker and polymer backbone. Interestingly, Glu1, Ile2, and Val6 broaden significantly and display two distinct peaks per amino acid, perhaps indicative of chemical exchange between an unfolded and folded peptide brush conformation. While significant overlap of the peaks of some residues (i.e. Hα of Ala3 and Ala5) hinders further analysis, NOEs between the Hα of Val6 and NH peaks of i + 1 (Aib7), i + 2 (Leu8), i + 3 (Aib9), and i + 4 (Ala10) residues can be identified, establishing the helical character of this segment in the polymer (ESI Fig. S41†).
Polymer conformation: insight from SAXS and cryo-TEM data
In addition to the information obtained by CD spectroscopy, we sought to obtain greater structural insight using small-angle X-ray scattering (SAXS). Solutions of polymers dissolved in a range of solvents were prepared and analyzed (Fig. 6; ESI Section 7, Tables S4–S11 and Fig. S44–S57†). In all cases, the peptide-brush polymers displayed approximately globular conformations, with a clear maximum near the ideal values for a compact sphere in the normalized Kratky plot, which is a peak in (qRg)2I(q)/I(0) of 1.104 at qRg ∼1.73 (Fig. 6). Consistent with studies of other types of brush polymers that find that longer polymers are more rod-like, as opposed to star-like at shorter lengths, the 90-mer polymers of the helical peptide and random coil peptide monomers have a normalized Kratky plot with a maximum at greater values than that of the ideal sphere (magenta traces in Fig. 6a, c and d).70 | ||
| Fig. 6 Normalized Kratky plots of SAXS data of random (a and b) and α-helical brush-polymers derived from the E-alpha peptide sequence (c and d) in water and 50% methanol. The dashed lines indicate the values for an ideal compact sphere (qRg = 1.73 and (qRg)2I(q)/I(0) = 1.1). In water and with the random coil sequence in 50% methanol, the Kratky plots display a peak close to the values for an ideal compact sphere, and little intensity at higher values of qRg (indicated by black arrows in panel c). However, in 50% methanol, the E-alpha-based polymers show greater intensity at higher values of qRg (black arrows in panel d), indicating more polymer mass at longer relative distances and thus greater flexibility and extension. | ||
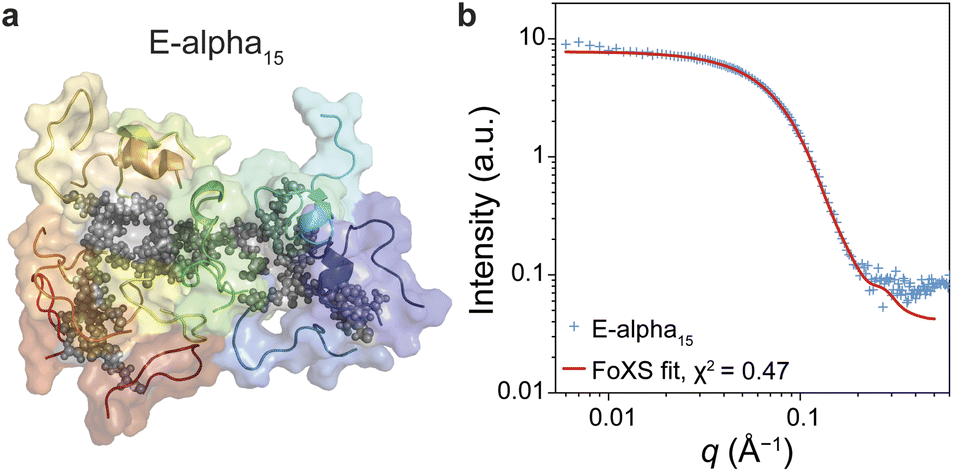 | ||
| Fig. 7 (a) Atomistic simulation structure of E-alpha15. (b) Fit of E-alpha15 structure from simulations to the experimental SAXS pattern of E-alpha15. | ||
In contrast to the results of PLPs dissolved in aqueous solutions, the E-alpha peptide brush polymers in 50% and 100% methanol solutions show much greater extension and flexibility (Fig. 6d and S47–S49†). Guinier analysis generally yields Rg values much larger than those for the same samples measured in aqueous buffers, and Guinier–Porod analysis yields larger values of s, indicative of greater rod-like character (ESI Tables S4 and S7†).71 Additionally, the normalized Kratky plots of these samples in methanol deviate significantly from an ideal bell curve representative of a compact sphere or globule, with greater intensity observed at larger values of qRg. These more extended polymer conformations observed via SAXS are consistent with the greater helicity observed in these polymers by CD and NMR spectroscopy, suggesting that the more rigid helical side chains resist collapse of the polymers into globular structures.
It could be hypothesized that more hydrophobic solvent mixtures contribute to extension of the polymer by solubilizing the hydrophobic polymer backbone. However, the SAXS data and normalized Kratky plots of scattering from the random coil PLPs retain characteristic globular features and show little change relative to the Kratky plots of the polymers in water (Fig. 6a and b), in stark contrast to the results obtained with the helical peptide brush polymers in methanol. This result suggests that the solvation of the hydrophobic polymer backbone is not the key factor influencing conformation, but rather, the properties of the peptide brush play a significant role. Importantly, this result suggests that peptide structure can be designed to influence brush polymer conformations and their prevalence in particular environments.
Atomistic metadynamics simulations, while unable to equilibrate sufficiently, did provide structural models of the E-alpha15 polymer that were consistent with CD spectroscopy data in that a portion of the peptide sidechains was folding as α-helices (Fig. 4). We wished to corroborate whether the models were consistent with the SAXS data provided. Accordingly, we fit the experimental SAXS pattern of E-alpha15 in aqueous buffer to one such model using the FoXS server,72,73 resulting in a goodness-of-fit parameter of χ2 = 0.47 (Fig. 8). This correspondence between the in silico model and the experimental data indicates that the model may be a reasonable representation of the polymer's conformation at longer length-scales (>1 nm), and lends further insight into the structure of these polymers complementary to the structural data obtained from spectroscopic studies.
 | ||
| Fig. 8 (a) Cryo-TEM micrograph of unstained E-alpha90 frozen in aqueous sodium carbonate buffer. (b) Size distribution analysis of particles found in the micrograph. Average particle size is 16 ± 1 nm. The image was acquired with a total accumulated dose of 16.49 e− Å−2. | ||
To provide additional insight into polymer conformation in solution, cryogenic transmission electron microscopy (cryo-TEM) was employed (ESI Section 8†). This technique has been successfully utilized to visualize conjugated polymers,74 self-assembling amphiphilic polymers,75,76 and dendritic polymers.77 The size of lower molecular weight polymers in this work, of up to approximately 45 kDa, suggested that these samples might be challenging to observe via cryo-TEM.78,79 However, we anticipated that alpha90, which has an approximate molecular weight of 120 kDa could provide enough contrast. Indeed, the resulting micrographs of aqueous, unstained E-alpha90 revealed distinct, low-dispersity particles aligning closely with the size range predicted by the SAXS data, averaging around 16 ± 1 nm in diameter (Fig. 8). Particle shapes appear roughly spherical, which is likewise consistent with the observed sizes, which are more collapsed than would be anticipated based on the polymer length, as well as the SAXS data that suggests a collapsed polymer conformation. We note that these micrographs are similar to cryo-TEM results obtained for single-chain nanoparticles37 and PEGylated bottlebrush polymers.80
Conclusions
In conclusion, we describe a structural interrogation of peptide-brush polymers incorporating α-helical peptide-based monomers, as well as a random coil-based peptide monomer. These peptides and polymers were investigated using CD spectroscopy, NMR spectroscopy, small-angle X-ray scattering, and atomistic metadynamics simulations, confirming key differences in peptide folding on the polymer backbone, as well as the overall polymer conformation. Specifically, the polymer environment appears to enforce a greater degree of helicity than is present in peptide monomers, which increases in magnitude as the degree of polymerization increases. NMR studies confirm the helicity for the polymer structures and that helical folding is concentrated in central residues of the peptide sequence in both monomer and polymer brushes. SAXS analysis confirms that while both sets of polymers are compact and globular in aqueous environments, the α-helical peptide-brush polymers are more extended and flexible in more hydrophobic solvents, consistent with the greater helicity observed in these environments. This result suggests that peptide brushes that are completely folded may resist forming compact polymeric structures, which would make the peptides more accessible to protein targets of interest as well as proteolytic enzymes. Overall, this demonstration of the tunability of polymer conformation and peptide brush folding is a key insight guiding future designs as we seek to mimic both functional and structural aspects of proteins and to engage natural proteins in specific ways for the development of peptide brush polymers as proteomimetic therapeutics and functional materials.Data availability
The data supporting this article has been included in the ESI.†Author contributions
J. O. and N. C. G. conceived of the project and wrote the manuscript. J. O. made and purified all compounds, with help from C. B. and M. T. J. O. measured and analyzed all CD and NMR data. J. O. measured all SAXS samples with help from C. S., N. D. R., S. J. W., and J. B. SAXS data was analyzed by J. O., with input from O. M. E., C. S., N. D. R., S. J. W., and J. B. K. S. and B. Q. completed computational work. Y. C. performed all cryo-TEM measurements and analyses, with input from N. D. R. All authors contributed to editing of the manuscript and ESI.†Conflicts of interest
Intellectual property related to the materials described in this manuscript is currently under license to Grove Biopharma, a company co-founded by N. C. G.Acknowledgements
This work was supported by the National Science Foundation (NSF) under award numbers DMR-2004899 and DMR 2403955. J. O. acknowledges support through an NIH NRSA F32 fellowship from the National Institute of General Medical Sciences (NIGMS; F32GM143925). O. E. acknowledges support from the NSF Graduate Research Fellowship (Grant No. 2234667). N. R. acknowledges support from the Ryan Fellowship and the International Institute for Nanotechnology at Northwestern University (NU). This work made use of the IMSERC MS and NMR facility at NU, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633), and NU; the NMR facility has also support from NIH 1S10OD012016-01/1S10RR019071-01A1. Work was performed at the BioCryo and Keck-II facilities of NU's NUANCE Center, which have received support from the SHyNE Resource (NSF ECCS-2025633), the IIN, and Northwestern's MRSEC program (NSF DMR-2308691). Additionally, this work used instruments in the Keck Biophysics Facility, a shared resource of the Robert H. Lurie Comprehensive Cancer Center of NU supported in part by the NCI Cancer Center Support Grant P30 CA060553. SAXS data was measured at Beamline 16ID (LiX) of the National Synchrotron Light Source II, part of the Center for BioMolecular Structure (CBMS), which is supported by the NIH NIGMS (P30GM133893) and by the Department of Energy (DOE) Office of Biological and Environmental Research (KP1605010). LiX also received additional support from NIH Grant S10 OD012331. Work performed at the CBMS is supported in part by the U.S. DOE Office of Science, Office of Basic Energy Sciences Program under contract number DE-SC0012704. Additional SAXS data was collected at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of the Advanced Photon Source (APS). DND-CAT is supported by NU, The Dow Chemical Company, and DuPont de Nemours, Inc. The APS is a U.S. DOE Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Data at DND-CAT was collected using an instrument funded by the NSF (Award Number 0960140). K. S. and B. Q. acknowledge the Texas Advanced Computing Center (TACC) at the University of Texas at Austin for the computational resources used in this work. This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union's Horizon 2020 research and innovation program under the SINE2020 project, grant agreement No. 654000. Images of peptides and polymers in Fig. 1 and 7 were prepared using PyMol v. 2.5.0. We thank Dr Lin Yang, Dr Or Berger, Dr Nicholas Hampu, Prof. Hao Sun, Prof. Andy Nguyen, Navjit Paul, Madeline Hopps, Mara Fattah, and Dr Yongbo Zhang for helpful discussions and experimental assistance in the course of this study.Notes and references
- B. L. Bray, Nat. Rev. Drug Discovery, 2003, 2, 587–593 CrossRef CAS PubMed.
- J. Vagner, H. Qu and V. J. Hruby, Curr. Opin. Chem. Biol., 2008, 12, 292–296 CrossRef CAS PubMed.
- K. Fosgerau and T. Hoffmann, Drug Discov. Today, 2015, 20, 122–128 CrossRef CAS PubMed.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- O. Al Musaimi, L. Lombardi, D. R. Williams and F. Albericio, Pharmaceuticals, 2022, 15, 1283 CrossRef CAS PubMed.
- L. R. Malins, J. N. deGruyter, K. J. Robbins, P. M. Scola, M. D. Eastgate, M. R. Ghadiri and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 5233–5241 CrossRef CAS PubMed.
- H. Kessler, Angew. Chem., Int. Ed., 1982, 21, 512–523 CrossRef.
- R. M. Kohli, C. T. Walsh and M. D. Burkart, Nature, 2002, 418, 658–661 CrossRef CAS PubMed.
- J. Lau, P. Bloch, L. Schäffer, I. Pettersson, J. Spetzler, J. Kofoed, K. Madsen, L. B. Knudsen, J. McGuire, D. B. Steensgaard, H. M. Strauss, D. X. Gram, S. M. Knudsen, F. S. Nielsen, P. Thygesen, S. Reedtz-Runge and T. Kruse, J. Med. Chem., 2015, 58, 7370–7380 CrossRef CAS PubMed.
- P. Berndt, G. B. Fields and M. Tirrell, J. Am. Chem. Soc., 1995, 117, 9515–9522 CrossRef CAS.
- B. Chen, Y. Sun, J. Niu, G. K. Jarugumilli and X. Wu, Cell Chem. Biol., 2018, 25, 817–831 CrossRef CAS PubMed.
- Y.-W. Kim, T. N. Grossmann and G. L. Verdine, Nat. Protoc., 2011, 6, 761–771 CrossRef CAS PubMed.
- Y. Wang and D. H.-C. Chou, Angew. Chem., Int. Ed., 2015, 54, 10931–10934 CrossRef CAS PubMed.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- W. S. Horne and T. N. Grossmann, Nat. Chem., 2020, 12, 331–337 CrossRef CAS PubMed.
- W. S. Horne, Expet Opin. Drug Discov., 2011, 6, 1247–1262 CrossRef CAS PubMed.
- J. K. Kammeyer, A. P. Blum, L. Adamiak, M. E. Hahn and N. C. Gianneschi, Polym. Chem., 2013, 4, 3929–3933 RSC.
- A. P. Blum, J. K. Kammeyer, J. Yin, D. T. Crystal, A. M. Rush, M. K. Gilson and N. C. Gianneschi, J. Am. Chem. Soc., 2014, 136, 15422–15437 CrossRef CAS PubMed.
- H. Sun, W. Choi, N. Zang, C. Battistella, M. P. Thompson, W. Cao, X. Zhou, C. Forman and N. C. Gianneschi, Angew. Chem., Int. Ed., 2019, 58, 17359–17364 CrossRef CAS PubMed.
- H. Sun, B. Qiao, W. Choi, N. Hampu, N. C. McCallum, M. P. Thompson, J. Oktawiec, S. Weigand, O. M. Ebrahim, M. O. de la Cruz and N. C. Gianneschi, ACS Cent. Sci., 2021, 7, 2063–2072 CrossRef CAS PubMed.
- A. P. Blum, J. K. Kammeyer and N. C. Gianneschi, Chem. Sci., 2016, 7, 989–994 RSC.
- A. P. Blum, D. A. Nelles, F. J. Hidalgo, M. A. Touve, D. S. Sim, A. A. Madrigal, G. W. Yeo and N. C. Gianneschi, Angew. Chem., Int. Ed., 2019, 58, 15646–15649 CrossRef CAS PubMed.
- W. Choi, A. K. Nensel, S. Droho, M. A. Fattah, S. Mokashi-Punekar, D. I. Swygart, S. T. Burton, G. W. Schwartz, J. A. Lavine and N. C. Gianneschi, Sci. Adv., 2023, 9, eadi8534 CrossRef CAS PubMed.
- K. P. Carrow, H. L. Hamilton, M. P. Hopps, Y. Li, B. Qiao, N. C. Payne, M. P. Thompson, X. Zhang, A. Magassa, M. Fattah, S. Agarwal, M. P. Vincent, M. Buyanova, P. A. Bertin, R. Mazitschek, M. Olvera de la Cruz, D. A. Johnson, J. A. Johnson and N. C. Gianneschi, Adv. Mater., 2024, 36, 2311467 CrossRef CAS PubMed.
- C. E. Callmann, M. P. Thompson and N. C. Gianneschi, Acc. Chem. Res., 2020, 53, 400–413 CrossRef CAS PubMed.
- J. Wang, H. Lu, Y. Ren, Y. Zhang, M. Morton, J. Cheng and Y. Lin, Macromolecules, 2011, 44, 8699–8708 CrossRef CAS.
- R. Baumgartner, H. Fu, Z. Song, Y. Lin and J. Cheng, Nat. Chem., 2017, 9, 614–622 CrossRef CAS PubMed.
- R. Marullo, M. Kastantin, L. B. Drews and M. Tirrell, Biopolymers, 2013, 99, 573–581 CrossRef CAS PubMed.
- Y.-C. Yu, M. Tirrell and G. B. Fields, J. Am. Chem. Soc., 1998, 120, 9979–9987 CrossRef CAS.
- M. Kastantin and M. Tirrell, Macromolecules, 2011, 44, 4977–4987 CrossRef CAS PubMed.
- M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed., 2015, 54, 8896–8927 CrossRef CAS PubMed.
- R. R. Araghi and A. E. Keating, Curr. Opin. Struct. Biol., 2016, 39, 27–38 CrossRef PubMed.
- R. Upadhya, N. S. Murthy, C. L. Hoop, S. Kosuri, V. Nanda, J. Kohn, J. Baum and A. J. Gormley, Macromolecules, 2019, 52, 8295–8304 CrossRef CAS PubMed.
- E. B. Berda, E. J. Foster and E. W. Meijer, Macromolecules, 2010, 43, 1430–1437 CrossRef CAS.
- H. K. Murnen, A. R. Khokhlov, P. G. Khalatur, R. A. Segalman and R. N. Zuckermann, Macromolecules, 2012, 45, 5229–5236 CrossRef CAS.
- G. M. ter Huurne, M. A. J. Gillissen, A. R. A. Palmans, I. K. Voets and E. W. Meijer, Macromolecules, 2015, 48, 3949–3956 CrossRef CAS.
- T. Terashima, T. Mes, T. F. A. De Greef, M. A. J. Gillissen, P. Besenius, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742–4745 CrossRef CAS PubMed.
- M. H. Barbee, Z. M. Wright, B. P. Allen, H. F. Taylor, E. F. Patteson and A. S. Knight, Macromolecules, 2021, 54, 3585–3612 CrossRef CAS.
- C. K. Lyon, A. Prasher, A. M. Hanlon, B. T. Tuten, C. A. Tooley, P. G. Frank and E. B. Berda, Polym. Chem., 2015, 6, 181–197 RSC.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed.
- I. L. Karle, M. Sukumar and P. Balaram, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 9284–9288 CrossRef CAS PubMed.
- I. L. Karle and P. Balaram, Biochemistry, 1990, 29, 6747–6756 CrossRef CAS PubMed.
- P. Y. Chou and G. D. Fasman, Biochemistry, 1974, 13, 222–245 CrossRef CAS PubMed.
- O. D. Monera, T. J. Sereda, N. E. Zhou, C. M. Kay and R. S. Hodges, J. Pept. Sci., 1995, 1, 319–329 CrossRef CAS PubMed.
- M. Blaber, X. Zhang and B. W. Matthews, Science, 1993, 260, 1637–1640 CrossRef CAS PubMed.
- A. Chakrabartty, T. Kortemme and R. L. Baldwin, Protein Sci., 1994, 3, 843–852 CrossRef CAS PubMed.
- J. M. Scholtz, H. Qian, V. H. Robbins and R. L. Baldwin, Biochemistry, 1993, 32, 9668–9676 CrossRef CAS PubMed.
- K. M. Armstrong and R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 11337–11340 CrossRef CAS PubMed.
- H. D. Maynard, S. Y. Okada and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 1275–1279 CrossRef CAS PubMed.
- J. B. Matson, A. Q. Steele, J. D. Mase and M. D. Schulz, Polym. Chem., 2023, 15, 127–142 RSC.
- N. J. Greenfield, Nat. Protoc., 2007, 1, 2876–2890 CrossRef PubMed.
- N. J. Greenfield, Nat. Protoc., 2007, 1, 2527–2535 CrossRef PubMed.
- J. M. Scholtz, S. Marqusee, R. L. Baldwin, E. J. York, J. M. Stewart, M. Santoro and D. W. Bolen, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 2854–2858 CrossRef CAS PubMed.
- A. Barducci, M. Bonomi and M. Parrinello, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2011, 1, 826–843 CrossRef CAS.
- A. Barducci, G. Bussi and M. Parrinello, Phys. Rev. Lett., 2008, 100, 020603 CrossRef PubMed.
- F. Pietrucci and A. Laio, J. Chem. Theory Comput., 2009, 5, 2197–2201 CrossRef CAS PubMed.
- C. E. Dempsey, Prog. Nucl. Magn. Reson. Spectrosc., 2001, 39, 135–170 CrossRef CAS.
- A. Hvidt and S. O. Nielsen, Adv. Protein Chem., 1966, 21, 287–386 CrossRef CAS PubMed.
- S. W. Englander, L. Mayne, Y. Bai and T. R. Sosnick, Protein Sci., 1997, 6, 1101–1109 CrossRef CAS PubMed.
- K. Wüthrich and G. Wagner, J. Mol. Biol., 1979, 130, 1–18 CrossRef PubMed.
- R. A. G. D. Silva, J. Kubelka, P. Bour, S. M. Decatur and T. A. Keiderling, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8318–8323 CrossRef CAS PubMed.
- D. Diana, B. Ziaco, G. Scarabelli, C. Pedone, G. Colombo, L. D. D'Andrea and R. Fattorusso, Chem.–Eur. J., 2010, 16, 5400–5407 CrossRef CAS PubMed.
- A. Bax, Annu. Rev. Biochem., 1989, 58, 223–256 CrossRef CAS PubMed.
- K. Wüthrich, NMR of Proteins and Nucleic Acids, Wiley-Interscience, 1986 Search PubMed.
- A. D. Bain, D. R. Eaton, A. E. Hamielec, M. Mlekuz and B. G. Sayer, Macromolecules, 1989, 22, 3561–3564 CrossRef CAS.
- We note here usage of nomenclature to refer to relationships between nearby amino acids in a peptide or protein sequence. Specifically, (i, i + n) is used to refer to a pair of amino acids in a sequence that are n amino acids apart from each other. This nomenclature is particularly useful in the evaluation of α-helicity in examining NOEs, as the i + 4 amino acid will be hydrogen-bonding to amino acid i in a helical arrangement. Additionally, “NN” is used here to refer to a pair of amide protons (NH). Hα is used to refer to the proton on the alpha carbon of the amino acid.
- S. Schwarzinger, G. J. A. Kroon, T. R. Foss, J. Chung, P. E. Wright and H. J. Dyson, J. Am. Chem. Soc., 2001, 123, 2970–2978 CrossRef CAS PubMed.
- S. P. Mielke and V. V. Krishnan, Prog. Nucl. Magn. Reson. Spectrosc., 2009, 54, 141–165 CrossRef CAS PubMed.
- D. S. Wishart, B. D. Sykes and F. M. Richards, J. Mol. Biol., 1991, 222, 311–333 CrossRef CAS PubMed.
- S. L. Pesek, X. Li, B. Hammouda, K. Hong and R. Verduzco, Macromolecules, 2013, 46, 6998–7005 CrossRef CAS.
- Y. Wei and M. J. A. Hore, J. Appl. Phys., 2021, 129, 171101 CrossRef CAS.
- D. Schneidman-Duhovny, M. Hammel and A. Sali, Nucleic Acids Res., 2010, 38, 540–544 CrossRef PubMed.
- D. Schneidman-Duhovny, M. Hammel, J. A. Tainer and A. Sali, Biophys. J., 2013, 105, 962–974 CrossRef CAS PubMed.
- Y. Y. Zhou, Y. C. Xu, Z. F. Yao, J. Y. Li, C. K. Pan, Y. Lu, C. Y. Yang, L. Ding, B. F. Xiao, X. Y. Wang, Y. Shao, W. Bin Zhang, J. Y. Wang, H. Wang and J. Pei, Nat. Commun., 2023, 14, 3340 CrossRef CAS PubMed.
- S. E. Smeltzer, C. A. Sanders, Y. Liu, S. R. George, C. Amiri, A. Gernandt, B. Reck and M. F. Cunningham, Macromolecules, 2023, 56, 1601–1614 CrossRef CAS.
- J. Y. Lee, Y. Song, M. G. Wessels, A. Jayaraman, K. L. Wooley and D. J. Pochan, Macromolecules, 2020, 53, 8581–8591 CrossRef CAS.
- Y. Bae, M. Y. Ha, K. T. Bang, S. Yang, S. Y. Kang, J. Kim, J. Sung, S. Kang, D. Kang, W. B. Lee, T. L. Choi and J. Park, Adv. Mater., 2022, 34, 2202353 CrossRef CAS PubMed.
- M. A. Herzik, M. Wu and G. C. Lander, Nat. Commun., 2019, 10, 1032 CrossRef PubMed.
- Y. Zhang, R. Tammaro, P. J. Peters and R. B. G. Ravelli, J. Chem. Inf. Model., 2020, 60, 2605–2613 CrossRef CAS PubMed.
- A. Detappe, H. V. T. Nguyen, Y. Jiang, M. P. Agius, W. Wang, C. Mathieu, N. K. Su, S. L. Kristufek, D. J. Lundberg, S. Bhagchandani, I. M. Ghobrial, P. P. Ghoroghchian and J. A. Johnson, Nat. Nanotechnol., 2023, 18, 184–192 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General considerations; synthesis and characterization of peptides and polymers; methods of spectroscopy, scattering, and simulations. See DOI: https://doi.org/10.1039/d4sc03420a |
| This journal is © The Royal Society of Chemistry 2024 |