
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric bifunctionalization of allenes with aryl iodides and amino acids enabled by chiral aldehyde/palladium combined catalysis†
Hao
Zhang
,
Wei
Wen
 *,
Yu-Yang
Wang
,
Ze-Xi
Lu
,
Jin-Long
Liu
,
Zhu-Lian
Wu
,
Tian
Cai
and
Qi-Xiang
Guo
*
*,
Yu-Yang
Wang
,
Ze-Xi
Lu
,
Jin-Long
Liu
,
Zhu-Lian
Wu
,
Tian
Cai
and
Qi-Xiang
Guo
*
Key Laboratory of Applied Chemistry of Chongqing Municipality, and Chongqing Key Laboratory of Soft-Matter Material Chemistry and Function Manufacturing, School of Chemistry and Chemical Engineering, Southwest University, Chongqing, 400715, China. E-mail: wenwei1989@swu.edu.cn; qxguo@swu.edu.cn
First published on 16th July 2024
Abstract
Even though catalytic asymmetric bifunctionalization of allenes has been extensively studied, almost all of the reported examples have been achieved in a two-component manner. In this study, we report a highly efficient asymmetric bifunctionalization of allenes with iodohydrocarbons and NH2-unprotected amino acid esters. The adopted chiral aldehyde/palladium combined catalytic system precisely governs the chemoselectivity, regioselectivity, and stereoselectivity of this three-component reaction. A wide range of substituted aryl iodides, allenes and amino acid esters can well participate in this reaction and deliver structurally diverse α,α-disubstituted α-amino acid esters with excellent experimental outcomes. One of the resulting products is utilized for the total synthesis of the molecule (S,R)-VPC01091.
Introduction
Following Burton and Pechmann's significant synthesis of allene compounds in 1887,1 allene swiftly emerged as a pivotal building block for the construction of allylic molecules due to its remarkable reactivity and versatile nature.2–9 Subsequently, extensive efforts have been dedicated to the development of allene-involved hydrofunctionalization10–18 and bifunctionalization19–25 reactions, profoundly enriching the field of allene chemistry. However, the corresponding asymmetric functionalization of allenes was conspicuously absent at the close of the last century.26 In particular, investigations into the catalytic asymmetric hydrofunctionalization of allenes commenced in 2003 with Trost and colleagues' disclosure of a palladium-catalyzed hydroalkylation reaction of alkoxylallenes with Meldrum's acids and azlactones.27 Through this reaction modality, numerous asymmetric reactions for the synthesis of chiral allylic compounds, encompassing alcohols, amines, aldehydes, ketones, amino acids, and beyond, have been developed.28–39The catalytic asymmetric bifunctionalization of allenes fully exploits the multiple reactivity offered by the adjacent C–C double bonds of allenes. Following the groundbreaking work on a palladium-catalyzed enantioselective carboannulation of allenes with ortho-substituted aryl halides as reported by Larock et al.,40 this transformation has been extensively explored for the fabrication of intricately structured chiral molecules. However, the majority of these reactions have taken place in a two-component fashion through combining two of the required three reaction sites—namely, the allene, the halide, and the nucleophile—within a single substrate (Fig. 1a).41–49 Undoubtedly, dispersing the three reaction sites among individual substrates, or in other words, developing a multicomponent reaction, could significantly expand the range of substrates amenable to this bifunctionalization reaction. In fact, as early as 1998, Hiroi et al. had realized this concept with a palladium-catalyzed asymmetric reaction involving racemic allene, iodobenzene, and malonate carbanion (Fig. 1b).50,51 However, this work represented the sole successful example that can achieve the catalytic enantioselective bifunctionalization of allenes in a three-component manner. The complexity of regulating the reaction sequences, regioselectivity, and stereoselectivity has been a key factor contributing to this situation. Consequently, the creation of highly efficient multicomponent reactions involving an allene, an electrophile, and a nucleophile through asymmetric catalysis presents a captivating and challenging endeavor for synthetic chemists.
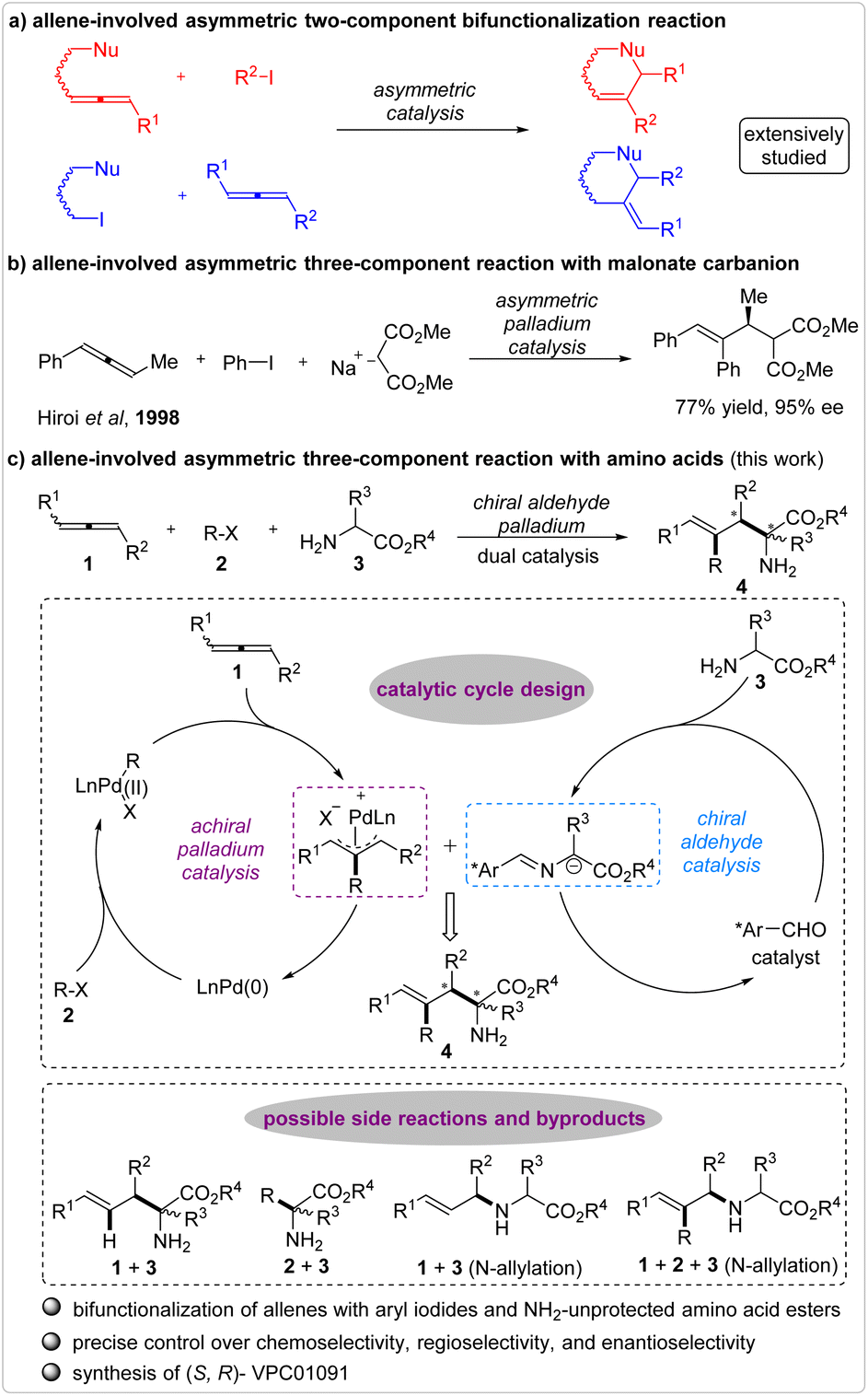 | ||
| Fig. 1 The catalytic asymmetric bifunctionalization of allene. | ||
Drawing from the pioneering work unveiled by Hiroi,50 and building on our investigation of the catalytic asymmetric allylation of NH2-unprotected amino acids with various allylation reagents,52,53 we envisioned a three-component reaction involving allenes, halohydrocarbons, and amino acid esters, facilitated by the synergy of chiral aldehyde/palladium combined catalysis (Fig. 1c).54–56 This process would yield enantioenriched α,α-disubstituted nonproteinogenic amino acid derivatives with distinctive structures.57–63 To our knowledge, the catalytic asymmetric bifunctionalization of allenes has never been employed for the construction of chiral amino acid derivatives. In our proposed reaction, the palladium catalytic cycle commences with the oxidation addition of Pd(0) to a halohydrocarbon, forming a weakly nucleophilic carbon–Pd(II) intermediate. Subsequent addition of this intermediate to an allene generates an active electrophilic π-allyl palladium species for the ensuing coupling process. The chiral aldehyde catalyst plays a pivotal role in the generation of nucleophilic carbanion intermediates from amino acid esters. However, significant challenges are evident in this reaction. Apart from the aforementioned complexities associated with controlling the reaction sequences, regioselectivity, and stereoselectivity, the chemoselectivity of N-allylation and C-allylation further complicates this reaction. Here, we documented our efforts to investigate this catalytic asymmetric three-component reaction, yielding a diverse range of optically active nonproteinogenic α-amino acid esters while precisely controlling the corresponding chemoselectivity, regioselectivity, and enantioselectivity. Furthermore, leveraging this methodology, one of the stereoisomer of VPC01091 was succinctly synthesized.
Results and discussion
Optimization of reaction conditions
Our investigation commenced with the assessment of the reaction involving allene 1a, iodobenzene 2a, and amino acid ester 3a, under the influence of a combined catalytic system employing chiral aldehyde CA-1 and palladium Pd(PPh3)4. The introduction of the Lewis acid ZnCl2 and base TMG (1,1,3,3-tetramethylguanidine) expedited the processes of Schiff base formation and deprotonation. As anticipated, the desired product 4a was yielded at an 83% yield and 81% ee. Encouraged by these outcomes, a comprehensive catalyst screening and optimization of reaction conditions were conducted. Chiral aldehyde catalyst screening revealed that CA-1 and CA-5 delivered comparable yields and enantioselectivities (Table 1, entries 1 and 5). With CA-1 as the organocatalyst, a variety of achiral and chiral ligands for palladium catalysis was evaluated, establishing that the achiral ligand dppp produced optimal results (Table 1, entry 1). Subsequent assessments of Lewis acids and reaction solvents failed to yield improved experimental outcomes (see ESI†). Upon substituting the base TMG with TDMAIP, product 4a was obtained with a 90% ee (Table 1, entry 19). To enhance the corresponding yields, chiral aldehyde catalysts were reassessed with the introduction of the base TDMAIP, revealing that the utilization of CA-5 yielded product 4a at an 80% yield and 90% ee (Table 1, entry 20). Additionally, palladium sources were scrutinized, with [Pd(C3H5)Cl]2 yielding superior results compared to Pd(PPh3)4 (Table 1, entry 22). Lowering the reaction temperature and diluting the reactant concentration led to the production of product 4a at an 89% yield and 90% ee (Table 1, entry 24). Based on these findings, the optimal reaction conditions for this three-component asymmetric reaction were determined.|
|
|||||
|---|---|---|---|---|---|
| Entry | CA | [Pd] | L | y (%) | eeb (%) |
| a Yields of isolated products. b Determined by chiral HPLC. c Using TDMAIP as the base. d With 5 mol% palladium. e At 55 °C. f With 1.5 mL toluene. TDMAIP = tris(dimethylamino)iminophosphorane. | |||||
| 1 | CA-1 | Pd(PPh3)4 | L1 | 83 | 81 |
| 2 | CA-2 | Pd(PPh3)4 | L1 | 39 | 28 |
| 3 | CA-3 | Pd(PPh3)4 | L1 | 34 | 12 |
| 4 | CA-4 | Pd(PPh3)4 | L1 | 71 | 20 |
| 5 | CA-5 | Pd(PPh3)4 | L1 | 81 | 80 |
| 6 | CA-6 | Pd(PPh3)4 | L1 | 74 | 68 |
| 7 | CA-7 | Pd(PPh3)4 | L1 | 34 | 70 |
| 8 | CA-8 | Pd(PPh3)4 | L1 | 19 | 95 |
| 9 | CA-9 | Pd(PPh3)4 | L1 | 32 | 96 |
| 10 | CA-10 | Pd(PPh3)4 | L1 | 28 | 89 |
| 11 | CA-11 | Pd(PPh3)4 | L1 | 16 | 84 |
| 12 | CA-1 | Pd(PPh3)4 | L2 | 24 | 62 |
| 13 | CA-1 | Pd(PPh3)4 | L3 | 19 | 50 |
| 14 | CA-1 | Pd(PPh3)4 | L4 | 10 | 32 |
| 15 | CA-1 | Pd(PPh3)4 | L5 | 19 | 56 |
| 16 | CA-1 | Pd(PPh3)4 | L6 | 34 | 34 |
| 17 | CA-1 | Pd(PPh3)4 | L7 | 67 | 86 |
| 18 | ent- CA-1 | Pd(PPh3)4 | L7 | 21 | −29 |
| 19c | CA-1 | Pd(PPh3)4 | L1 | 70 | 90 |
| 20c | CA-5 | Pd(PPh3)4 | L1 | 80 | 90 |
| 21c | CA-5 | Pd(dppe)2 | L1 | 52 | 84 |
| 22c,d | CA-5 | [Pd(C3H5)Cl]2 | L1 | 93 | 89 |
| 23c,d,e | CA-5 | [Pd(C3H5)Cl]2 | L1 | 83 | 90 |
| 24c,d,e,f | CA-5 | [Pd(C3H5)Cl]2 | L1 | 89 | 90 |

The substrate scopes
After establishing the optimal reaction conditions, we proceeded to examine the substrate scope of this reaction. Initially, various amino acid esters were utilized as reaction partners with allene 1a and iodobenzene 2a (Fig. 2). The results indicated that amino acid esters containing saturated alkyls performed well in this reaction, yielding the corresponding products 4a–4e with excellent yields and enantioselectivities. Ethyl esters generated from methionine and α-allyl α-amino acid also exhibited good reactivity in this reaction, producing products 4f and 4g with favorable experimental outcomes. Subsequently, phenylalanines, homophenylalanine, glutamic acid and lysine-derived amino acid esters were employed as reactants, all of which led to the formation of the respective products with good-to-excellent yields and excellent enantioselectivities (Fig. 2, 4h–4t). Glycine ethyl ester could participate in this reaction, but no enantioselective bias was observed in the product (Fig. 2, 4u). | ||
| Fig. 2 Substrate scope of amino acid esters. | ||
Subsequently, a variety of aryl and alkenyl iodides were examined (Fig. 3a). Phenyl iodides comprising of ortho-, meta-, or para-substituted phenyl groups demonstrated good participation in this reaction, resulting in the corresponding products with good-to-excellent yields and high-to-excellent enantioselectivities (Fig. 3, 5a–5n). Similar experimental outcomes were observed when phenyl iodides containing two substituents were utilized as reactants (Fig. 3, 5o–5t). Other aryl iodides, such as 2-iodothiophene, 2-iodonaphthalene, and 2-iodo-9H-fluorene, acted as good reaction partners with allene 1a and phenylalanine ethyl ester 3b, producing the corresponding products 5u–5w with good yields and excellent enantioselectivities. The reactivity of alkenyl iodide substrates was also examined, however, low yield was observed. For instance, (E)-(2-iodovinyl)benzene could partake in this reaction but the corresponding product 5z was only yielded in 40% with 78% ee. Aryl iodides bearing polar groups such as ester and thioether also exhibited good reactivity in this reaction, leading to products 5aa and 5ab in good yields and excellent enantioselectivities.
 | ||
| Fig. 3 Substrate scope of aryl iodides and allenes. | ||
We then investigated the substrate scope of substituted allenes in reaction with iodobenzene 2a and phenylalanine ethyl ester 3b (Fig. 3b). Our findings indicated that allenes bearing substituted phenyl groups performed well in this reaction. For instance, all phenyl allenes containing ortho-, meta-, or para-substituents displayed good reactivity, producing the corresponding products 6a–6n with good-to-excellent yields and excellent enantioselectivities. Positive experimental outcomes were also observed for 2-naphthyl- and 2-thienyl-substituted allenes (Fig. 3, 6o–6p). In comparison, the reactivity of alkyl-substituted allenes notably decreased, with buta-2,3-dien-1-ylbenzene and penta-3,4-dien-1-ylbenzene generating the corresponding products in yields of 45% and 41%, respectively (Fig. 3, 6q and 6r). Additionally, the enantioselectivity produced by penta-3,4-dien-1-ylbenzene was lower than that of other allenes. Aryl-substituted allenes bearing polar groups including ester and thioether could participate in this reaction, giving products 6s and 6t in moderate yields and excellent enantioselectivities.
Mechanism studies
In order to determine the E/Z conformation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and the absolute configuration of the chiral carbon center, the product 4a was transformed into the chiral oxazolinone 8 for the preparation of a single crystal (see the ESI†). The X-ray crystal analysis of 8 revealed that the alkene unit adopts a Z-conformation and the absolute configuration of the chiral carbon center is S. Consequently, the stereocenters of 4a were deduced to be (Z,S). Considering the (Z,S) stereocenters induced by the combined chiral aldehyde/palladium catalytic system and our previous studies on the mechanism of the same catalytic system that enabled asymmetric allylation and benzylation reactions,64,65 potential catalytic cycles and transition states were proposed as follows. Under the chiral aldehyde catalysis, the chiral aldehyde condensed with amino acid ester 3a and then coordinated with Lewis acid ZnCl2 to form the Zn-Schiff base complex I. Upon deprotonation by the base, this Schiff base converted into the active nucleophilic intermediate enolate II. In the palladium catalysis, iodobenzene 2a underwent oxidative addition to generate the corresponding nucleophilic carbon-Pd(II) intermediate. Upon addition of this intermediate to allene 1a, the active electrophilic π-allyl palladium species III was produced. Through a ligand exchange process, the enolate II and the π-allyl palladium III fused together to form the potential transition state IV, wherein the enolate provided its si-face to attack the π-allyl palladium species, leading to the formation of the Schiff base V and releasing Pd(0). The chiral aldehyde catalytic cycle concluded with the hydrolysis or amine exchange of the Schiff base V with 3a (Fig. 4).
C bond and the absolute configuration of the chiral carbon center, the product 4a was transformed into the chiral oxazolinone 8 for the preparation of a single crystal (see the ESI†). The X-ray crystal analysis of 8 revealed that the alkene unit adopts a Z-conformation and the absolute configuration of the chiral carbon center is S. Consequently, the stereocenters of 4a were deduced to be (Z,S). Considering the (Z,S) stereocenters induced by the combined chiral aldehyde/palladium catalytic system and our previous studies on the mechanism of the same catalytic system that enabled asymmetric allylation and benzylation reactions,64,65 potential catalytic cycles and transition states were proposed as follows. Under the chiral aldehyde catalysis, the chiral aldehyde condensed with amino acid ester 3a and then coordinated with Lewis acid ZnCl2 to form the Zn-Schiff base complex I. Upon deprotonation by the base, this Schiff base converted into the active nucleophilic intermediate enolate II. In the palladium catalysis, iodobenzene 2a underwent oxidative addition to generate the corresponding nucleophilic carbon-Pd(II) intermediate. Upon addition of this intermediate to allene 1a, the active electrophilic π-allyl palladium species III was produced. Through a ligand exchange process, the enolate II and the π-allyl palladium III fused together to form the potential transition state IV, wherein the enolate provided its si-face to attack the π-allyl palladium species, leading to the formation of the Schiff base V and releasing Pd(0). The chiral aldehyde catalytic cycle concluded with the hydrolysis or amine exchange of the Schiff base V with 3a (Fig. 4).
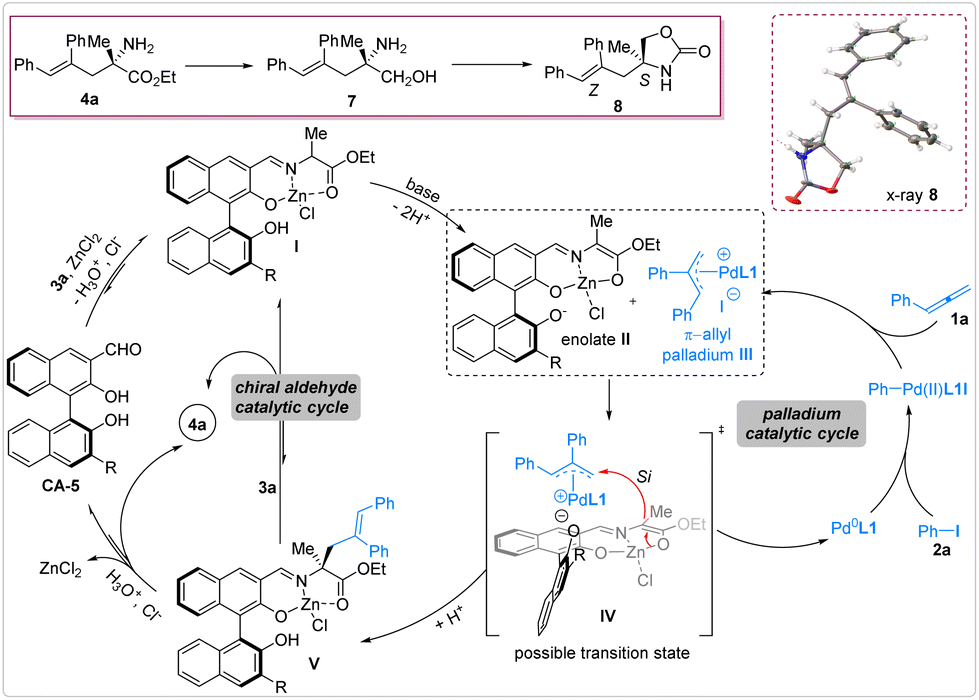 | ||
| Fig. 4 Proposed catalytic cycles and transition state. | ||
Synthetic applications
VPC01091 contains four stereoisomers and some of the them could be converted into the potent S1P3 antagonists and S1P1 receptor agonists.66 The molecular structure of VPC01091 is characterized by a 1,3-disubstituted cyclopentanamine with nonadjacent quaternary–tertiary carbon centers. Several asymmetric synthetic routes have been developed for the synthesis of this compound.67–70 Typically, compound 11 has been identified as one of the most commonly employed building blocks for the production of VPC01091, and the core five-membered carbon ring of 11 was typically constructed via olefin metathesis. Building upon this established synthetic strategy, as well as the three-component asymmetric allylation reaction we disclosed here, we hypothesized that the catalytic product 5o could serve as a promising chiral building block for the synthesis of optically active (S,R)-VPC01091. Notably, the simultaneous introduction of an allylic unit and the establishment of a chiral quaternary carbon center allowed for the streamlining of the entire synthetic pathway.With this idea in mind, we initiated an exploration of this novel synthetic pathway (Scheme 1). After protecting the amino group of 5o, an olefin metathesis reaction was performed utilizing the second-generation Grubb's catalyst, resulting in the formation of iodine-substituted amino acid ester 10 with a total yield of 66% (two steps). Subsequently, the coupling reaction of compound 10 with octylboronic acid furnished product 11 with a high yield (80%). Then, the ester group and the CC bonds of compound 11 were successively reduced, leading to the generation of the pivotal chiral building block 13 with favorable yields. Adhering to the established deprotecting strategy in literature,70 the (S,R)-VPC01091 was successfully prepared. Notably, commencing from easily accessible starting materials, the total synthesis of (S,R)-VPC01091 was achieved within 7 steps, which represented the most concise synthesis route at present. Furthermore, by altering the absolute configuration of the chiral catalyst, the synthesis of other stereoisomers of VPC01091 is anticipated to be accomplished. The utilization of mild reaction conditions in this synthetic pathway rendered it favorable for a potential large-scale synthesis.
 | ||
| Scheme 1 The total synthesis of (S,R)-VPC01091. | ||
Conclusions
In conclusion, we have described a highly efficient catalytic asymmetric three-component reaction involving allenes, aryl iodides and NH2-unprotected amino acid esters. The employed chiral aldehyde/palladium combining catalytic system precisely governed the chemoselectivity, regioselectivity and enantioselectivity. A wide range of allenes, aryl iodides and NH2-unprotected amino acid esters could participate in this reaction well and afford corresponding optically active nonproteinogenic α-amino acid esters with exceptional experimental outcomes. A reasonable transition state was proposed to illustrate the observed stereoselective control results. This method could be used for the synthesis of the key chiral intermediate leading to a potential S1P3 antagonist and S1P1 receptor agonist.Data availability
All data supporting the findings of this study are available within the article and its ESI† file.Author contributions
W. W. and G. Q. X. conceived this project. Z. H.; W. Y. Y.; L. Z. X. and L. J. L. carried out the experiments. W. Z. L. and C. T. performed the HRMS analysis. G. Q. X. wrote the manuscript. All authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from NSFC (22371233, 22071199), and the Innovation Research 2035 Pilot Plan of Southwest University (SWU-XDZD22011).Notes and references
- B. S. Burton and H. v. Pechmann, Ber. Dtsch. Chem. Ges., 1887, 20, 145–149 CrossRef.
- S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074–3112 CrossRef CAS.
- R. Blieck, M. Taillefer and F. Monnier, Chem. Rev., 2020, 120, 13545–13598 CrossRef CAS.
- P. Koschker and B. Breit, Acc. Chem. Res., 2016, 49, 1524–1536 CrossRef CAS PubMed.
- Y. Yamamoto, M. Al-Masum and N. Asao, J. Am. Chem. Soc., 1994, 116, 6019–6020 CrossRef CAS.
- B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS PubMed.
- J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461–1475 CrossRef CAS PubMed.
- I. Shimizu and J. Tsuji, Chem. Lett., 1984, 13, 233–236 CrossRef.
- B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2944 CrossRef CAS PubMed.
- B. M. Trost, J. Xie and J. D. Sieber, J. Am. Chem. Soc., 2011, 133, 20611–20622 CrossRef CAS PubMed.
- H. Zhou, Z. Wei, J. Zhang, H. Yang, C. Xia and G. Jiang, Angew. Chem., Int. Ed., 2017, 56, 1077–1081 CrossRef CAS PubMed.
- H. Zhou, Y. Wang, L. Zhang, M. Cai and S. Luo, J. Am. Chem. Soc., 2017, 139, 3631–3634 CrossRef CAS PubMed.
- Z. Yang and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 27288–27292 CrossRef CAS.
- M. Zhu, Q. Zhang and W. Zi, Angew. Chem., Int. Ed., 2021, 60, 6545–6552 CrossRef CAS.
- W. Chai, B. Guo, Q. Zhang and W. Zi, Chem. Catal., 2022, 2, 1428–1439 CrossRef CAS.
- T. M. Beck and B. Breit, Angew. Chem., Int. Ed., 2017, 56, 1903–1907 CrossRef CAS PubMed.
- J. D. Osborne, H. E. Randell-Sly, G. S. Currie, A. R. Cowley and M. C. Willis, J. Am. Chem. Soc., 2008, 130, 17232–17233 CrossRef CAS PubMed.
- J.-L. Xu, Z.-Y. Xu, Z.-L. Wang, W.-W. Ma, X.-Y. Sun, Y. Fu and Y.-H. Xu, J. Am. Chem. Soc., 2022, 144, 5535–5542 CrossRef CAS.
- S. Ma, Z. Shi and S. Wu, Tetrahedron: Asymmetry, 2001, 12, 193–195 CrossRef CAS.
- X. Xie and S. Ma, Chem. Commun., 2013, 49, 5693–5695 RSC.
- M. Li, A. Hawkins, D. M. Barber, P. Bultinck, W. Herrebout and D. J. Dixon, Chem. Commun., 2013, 49, 5265–5267 RSC.
- H. Luo, Z. Yang, W. Lin, Y. Zheng and S. Ma, Chem. Sci., 2018, 9, 1964–1969 RSC.
- H. Li, I. Khan, Q. Li and Y. J. Zhang, Org. Lett., 2022, 24, 2081–2086 CrossRef CAS.
- Q.-W. Shen, W. Wen and Q.-X. Guo, Org. Lett., 2023, 25, 3163–3167 CrossRef CAS.
- S. Inuki, A. Iwata, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2011, 76, 2072–2083 CrossRef CAS PubMed.
- B. M. Trost, A. B. C. Simas, B. Plietker, C. Jäkel and J. Xie, Chem.–Eur. J., 2005, 11, 7075–7082 CrossRef CAS PubMed.
- B. M. Trost, C. Jäkel and B. Plietker, J. Am. Chem. Soc., 2003, 125, 4438–4439 CrossRef CAS.
- W. Lim, J. Kim and Y. H. Rhee, J. Am. Chem. Soc., 2014, 136, 13618–13621 CrossRef CAS PubMed.
- S. Ganss and B. Breit, Angew. Chem., Int. Ed., 2016, 55, 9738–9742 CrossRef CAS PubMed.
- P. Steib and B. Breit, Angew. Chem., Int. Ed., 2018, 57, 6572–6576 CrossRef CAS.
- H. Kim and Y. H. Rhee, J. Am. Chem. Soc., 2012, 134, 4011–4014 CrossRef CAS.
- H. Kim, W. Lim, D. Im, D.-g. Kim and Y. H. Rhee, Angew. Chem., Int. Ed., 2012, 51, 12055–12058 CrossRef PubMed.
- S. H. Jang, H. W. Kim, W. Jeong, D. Moon and Y. H. Rhee, Org. Lett., 2018, 20, 1248–1251 CrossRef CAS PubMed.
- R. Zimmer, C. U. Dinesh, E. Nandanan and F. A. Khan, Chem. Rev., 2000, 100, 3067–3126 CrossRef CAS.
- G. Helmchen and A. Pfaltz, Acc. Chem. Res., 2000, 33, 336–345 CrossRef CAS.
- J. D. Weaver, A. Recio III, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846–1913 CrossRef CAS PubMed.
- S. Biswas, M. M. Parsutkar, S. M. Jing, V. V. Pagar, J. H. Herbort and T. V. RajanBabu, Acc. Chem. Res., 2021, 54, 4545–4564 CrossRef CAS.
- G. Li, X. Huo, X. Jiang and W. Zhang, Chem. Soc. Rev., 2020, 49, 2060–2118 RSC.
- J. Le Bras and J. Muzart, Chem. Soc. Rev., 2014, 43, 3003–3040 RSC.
- R. C. Larock, N. G. Berrios-Pena and C. A. Fried, J. Org. Chem., 1991, 56, 2615–2617 CrossRef CAS.
- J. M. Zenner and R. C. Larock, J. Org. Chem., 1999, 64, 7312–7322 CrossRef CAS.
- Q. Yang, X. Jiang and S. Ma, Chem.–Eur. J., 2007, 13, 9310–9316 CrossRef CAS PubMed.
- W. Shu, Q. Yang, G. Jia and S. Ma, Tetrahedron, 2008, 64, 11159–11166 CrossRef CAS.
- W. Shu, Q. Yu and S. Ma, Adv. Synth. Catal., 2009, 351, 2807–2810 CrossRef CAS.
- P. Renzi, E. Azzi, E. Bessone, G. Ghigo, S. Parisotto, F. Pellegrino and A. Deagostino, Org. Chem. Front., 2022, 9, 906–916 RSC.
- F. Yan, H. Liang, J. Song, J. Cui, Q. Liu, S. Liu, P. Wang, Y. Dong and H. Liu, Org. Lett., 2017, 19, 86–89 CrossRef CAS PubMed.
- R. Grigg, S. McCaffrey, V. Sridharan, C. W. G. Fishwick, C. Kilner, S. Korn, K. Bailey and J. Blacker, Tetrahedron, 2006, 62, 12159–12171 CrossRef CAS.
- R. Grigg and V. Sridharan, J. Organomet. Chem., 1999, 576, 65–87 CrossRef CAS.
- R. C. Larock and J. M. Zenner, J. Org. Chem., 1995, 60, 482–483 CrossRef CAS.
- K. Hiroi, F. Kato and A. Yamagata, Chem. Lett., 1998, 27, 397–398 CrossRef.
- F. Kato and K. Hiroi, Chem. Pharm. Bull., 2004, 52, 95–103 CrossRef CAS.
- J.-H. Liu, Q. Zhou, Y. Lin, Z.-L. Wu, T. Cai, W. Wen, Y.-M. Huang and Q.-X. Guo, ACS Catal., 2023, 13, 6013–6022 CrossRef CAS.
- H. Zhang, W. Wen, Z.-X. Lu, Z.-L. Wu, T. Cai and Q.-X. Guo, Org. Lett., 2024, 26, 153–159 CrossRef CAS PubMed.
- W. Wen and Q.-X. Guo, Synthesis, 2023, 55, 719–732 CrossRef CAS.
- W. Wen and Q.-X. Guo, Acc. Chem. Res., 2024, 57, 776–794 CrossRef CAS.
- Z. Su, B. Tan, H. He, K. Chen, S. Chen, H. Lei, T.-G. Chen, S.-F. Ni and Z. Li, Angew. Chem., Int. Ed., 2024, e202402038 CAS.
- D. Ma, K. Ding, H. Tian, B. Wang and D. Cheng, Tetrahedron: Asymmetry, 2002, 13, 961–969 CrossRef CAS.
- H. Bräuner-Osborne, J. Egebjerg, E. Ø. Nielsen, U. Madsen and P. Krogsgaard-Larsen, J. Med. Chem., 2000, 43, 2609–2645 CrossRef PubMed.
- X. Huo, J. Zhang, J. Fu, R. He and W. Zhang, J. Am. Chem. Soc., 2018, 140, 2080–2084 CrossRef CAS PubMed.
- L. Wei, S.-M. Xu, Q. Zhu, C. Che and C.-J. Wang, Angew. Chem., Int. Ed., 2017, 56, 12312–12316 CrossRef CAS.
- T. Kanayama, K. Yoshida, H. Miyabe and Y. Takemoto, Angew. Chem., Int. Ed., 2003, 42, 2054–2056 CrossRef CAS PubMed.
- T. Kanayama, K. Yoshida, H. Miyabe, T. Kimachi and Y. Takemoto, J. Org. Chem., 2003, 68, 6197–6201 CrossRef CAS PubMed.
- R. He, X. Huo, L. Zhao, F. Wang, L. Jiang, J. Liao and W. Zhang, J. Am. Chem. Soc., 2020, 142, 8097–8103 CrossRef CAS.
- J.-H. Liu, W. Wen, J. Liao, Q.-W. Shen, Y. Lin, Z.-L. Wu, T. Cai and Q.-X. Guo, Nat. Commun., 2022, 13, 2509 CrossRef CAS PubMed.
- L. Chen, M.-J. Luo, F. Zhu, W. Wen and Q.-X. Guo, J. Am. Chem. Soc., 2019, 141, 5159–5163 CrossRef CAS.
- R. Zhu, A. H. Snyder, Y. Kharel, L. Schaffter, Q. Sun, P. C. Kennedy, K. R. Lynch and T. L. Macdonald, J. Med. Chem., 2007, 50, 6428–6435 CrossRef CAS PubMed.
- V. C. de Oliveira, J. M. de Oliveira, V. H. Menezes da Silva, I. U. Khan and C. R. D. Correia, Adv. Synth. Catal., 2020, 362, 3395–3406 CrossRef CAS.
- G. A. Wallace, T. D. Gordon, M. E. Hayes, D. B. Konopacki, S. R. Fix-Stenzel, X. Zhang, P. Grongsaard, K. P. Cusack, L. M. Schaffter, R. F. Henry and R. H. Stoffel, J. Org. Chem., 2009, 74, 4886–4889 CrossRef CAS PubMed.
- S. R. Fix-Stenzel, M. E. Hayes, X. Zhang, G. A. Wallace, P. Grongsaard, L. M. Schaffter, S. M. Hannick, T. S. Franczyk, R. H. Stoffel and K. P. Cusack, Tetrahedron Lett., 2009, 50, 4081–4083 CrossRef CAS.
- I. U. Khan, S. Kattela, A. Hassan and C. R. D. Correia, Org. Biomol. Chem., 2016, 14, 9476–9480 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2326391. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03398a |
| This journal is © The Royal Society of Chemistry 2024 |