
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLiquid-assisted grinding enables a direct mechanochemical functionalization of polystyrene waste†
Morgan E.
Skala
 ,
Sarah M.
Zeitler
and
Matthew R.
Golder
*
,
Sarah M.
Zeitler
and
Matthew R.
Golder
*
Department of Chemistry, Molecular Engineering & Science Institute, University of Washington, 36 Bagley Hall, Seattle, WA 98195, USA. E-mail: goldermr@uw.edu
First published on 10th June 2024
Abstract
The plastic waste crisis has grave consequences for our environment, as most single-use commodity polymers remain in landfills and oceans long after their commercial lifetimes. Utilizing modern synthetic techniques to chemically modify the structure of these post-consumer plastics (e.g., upcycling) can impart new properties and added value for commercial applications. To expand beyond the abilities of current solution-state chemical processes, we demonstrate post-polymerization modification of polystyrene via solid-state mechanochemistry enabled by liquid-assisted grinding (LAG). Importantly, this emblematic trifluoromethylation study modifies discarded plastic, including dyed materials, using minimal exogenous solvent and plasticizers for improved sustainability. Ultimately, this work serves as a proof-of-concept for the direct mechanochemical post-polymerization modification of commodity polymers, and we expect future remediation of plastic waste via similar mechanochemical reactions.
Introduction
Aromatic polymers such as polystyrene (PS) and polycarbonates make up roughly 9.2 million metric tons of the plastic products consumed each year in the United States.1–8PS is particularly difficult to recycle due to high glass transition temperatures (Tg) and food contamination; therefore, recycling requires energy intensive and cost-prohibitive methods culminating in a recycling rate of less than 1%.2,5,8 To help mitigate the increasing amounts of plastic waste and avoid conventional mechanical recycling that may not recover bulk properties, chemical upcycling can be used to transform plastic waste into value added products.1,3,9 One attractive pathway for upcycling is post-polymerization modification (PPM) which can directly edit a polymer backbone to alter physical and thermal properties. Desirable bulk properties such as surface wettability,10,11 adhesion,12,13 and enhanced thermal properties14 can be accessed even at low levels of chemical functionalization. For example, Hartwig recently demonstrated that the addition of <1 mol% polar functional groups to polyolefin waste increases elongation and toughness.10,12 PPM of plastic wastes is also theorized to only require the same low functionalization densities, <1 wt% of an acidic or basic pendant, to maintain proper rheology for ionic compatibilization.15 For aromatic polymer modification, promising methods have emerged via C–H activation9,16–18 and (photo)redox catalysis19,20 that allow for the direct functionalization of commodity plastics (Fig. 1A). While successful in modulating bulk properties, these PPM methods often require complete polymer dissolution and are thereby limited by the solubility of high molar mass plastics and/or requisite solvent volumes.9,21 In addition, dyed plastics are particularly challenging to upcycle due to photon scattering.22–24 To overcome these limitations and improve sustainability of PPM processes, mechanochemistry via solid-state ball mill grinding (BMG) presents itself as an attractive method for PPM of plastic waste. | ||
| Fig. 1 (A) Evolution of (photo)redox-catalyzed (left) and force-induced (right) aromatic polymer functionalization and (B) silver-mediated small molecule arene trifluoromethylation that inspired (C) this work's mechanochemical trifluoromethylation of bibenzyl and polystyrene (PS). | ||
In the context of polymer science, mechanochemistry is intuitively perceived to promote destructive processes.24–28 Following the early work of Staudinger,29–31 mechanochemical chain scission has been redirected to mechanoresponsive functions (e.g., color change, catalyst activation, cargo release) via activation of engineered mechanophores using ultrasonic irradiation.27,32–38 On the other hand, BMG in polymer mechanochemistry has largely focused on constructive chemistry for polymer synthesis.36,37,39,40 Less studied in polymer mechanochemistry is PPM. Reactive extrusion is an example of an industrially relevant, mechanochemical method for PPM that relies on shear forces for mixing, but typically requires elevated temperatures for melt phase thermally mediated processes.13,41–43 Force-induced polymer modifications via BMG, however, have rarely been applied to industrially relevant polymers. Instead, accounts are limited to low molar mass or low Tg materials that will not degrade under BMG conditions (i.e., oligomeric polystyrene44 and poly(ethylene glycol)45) and polymers with pre-installed functional handles that facilitate efficient modification in the solid-state (i.e., polystyrene-based copolymers containing aldehyde46 or benzyl chloride47 moieties, functionalized polyethers,48 and poly(vinyl alcohol) and poly(vinyl chloride)49) (Fig. 1A). Most commercial polymers, however, are high molar mass species and do not have such pre-installed moieties and therefore require longer reaction times for direct functionalization. The kinetics of prolonged functionalization reactions under BMG conditions are likely to be competitive with those of chain scission, resulting in excessive backbone degradation and loss of desired bulk properties.50 This degradation via BMG can be leveraged for polymer depolymerization,26,51,52 chain-end functionalization,53,54 or radical functionalization.55 For PS, while chain scission under BMG is well documented above a limiting molar mass, Mlim (ca. 7.0–13.0 kDa),50 a recent report demonstrates a more specific relationship between Tg and the rate of degradation under BMG conditions.50 In this work, Peterson, Hwang, and Choi also demonstrate that the addition of exogenous plasticizer could decrease the rate of degradation. Inspired by this finding, we hypothesized that liquid-assisted grinding (LAG) could plasticize post-consumer plastic under BMG conditions such that the rate of PPM significantly outcompeted the rate of mechanochemical chain scission. LAG, commonly used in synthetic organic mechanochemistry, is customarily defined as the ratio of a liquid additive to the mass of the reaction components within the range of 0.1–1 μL mg−1.56,57 With plasticization of the polymer substrate, the Tg and the magnitude of shear forces experienced by polymer chains would decrease and result in slower degradation rates.50,58
To demonstrate the power of PPM using LAG, we identified trifluoromethylation as an emblematic arene functionalization reaction. Electrophilic CF3 sources should be reactive enough to add directly to aromatic monomer units in PS; importantly, for this study subsequent fluorinated products could be quantified easily via19F NMR spectroscopy.19,20 Previous reports have shown TFM-PS possesses increased hydrophobicity and improved interfacial and bulk polarization compared to unfunctionalized PS, making TFM-PS useful for applications in coatings and as a potential replacement for PS as a gate dielectric in organic electronics.19,59 Recent work by Kubota and Ito on the mechanoredox fluoroalkylation of activated arenes60 inspired us to extend trifluoromethylation to PPM via BMG. Although these conditions proved to be ineffective (Table S1†), we later identified a direct arene trifluoromethylation reported by Sanford61 (Fig. 1B) as a method adaptable to mechanochemical functionalization of unactivated aromatics, such as PS model substrate bibenzyl (BB) (Fig. 1C). Inspired by these previous reports, we now demonstrate the first example of direct mechanochemical PS functionalization using BMG and investigate the impact of varying LAG conditions on competitive polymer functionalization versus degradation (Fig. 1C). Overall, we obtain PS trifluoromethyl incorporation up to 1.4 mol%, including 0.60 mol% for post-consumer dyed PS. This work serves as a proof-of-concept for sustainable aromatic polymer upcycling via LAG enabled mechanochemistry.
Results & discussion
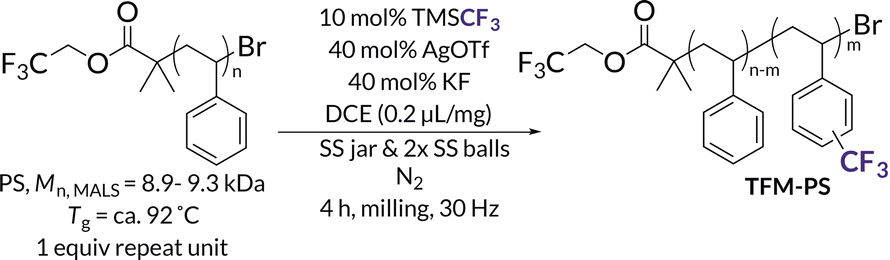
With up to 5.5 mol% functionalization obtained using the BB model (see Tables S2–S4† for mechanochemical reaction optimization and corresponding control reactions), we translated the methodology from small molecules to well-defined polymer substrates. We synthesized low molar mass PS (Mn,MALS = ca. 9.0 kDa) via atom transfer radical polymerization (ATRP)62 utilizing a trifluoroethyl bromoisobutyrate initiator for facile chain-end analysis and PPM quantification via19F NMR spectroscopy.19 The 19F NMR spectra for TFM-PS revealed a single broad peak (ca. −62.1 ppm) which is consistent with analytical data from prior reports of radical PS trifluoromethylations (Fig. S58–S65†).19–21,63 To determine the optimal mechanochemical parameters needed for lower molar mass PS functionalization, we varied the size of the milling jar, size and number of milling balls, and reaction scale for an optimized charge ratio (Table 1). The highest density of trifluoromethylation resulted from the mechanochemical parameters optimized on BB (Table 1, entires 1 and 2). Neither increasing the number of milling balls nor increasing jar size resulted in improved trifluoromethyl radical addition (Table 1, entries 3 and 5, respectively). Increasing the diameter of the milling balls from 8 mm to 10 mm did result in a slight increase in trifluoromethylation density, but also caused more chain scission (i.e., lower Mn), presumably due to increased shear forces (Table 1, Entry 4). Additionally, the reaction was successfully scaled up, albeit with a higher experimental Mn (Table 1, entry 6). Notably, the functionalization density does not entirely account for the general increase in TFM-PSMn. The second compounding factor is a high molar mass shoulder observed at all functionalization densities that is not seen when PS is milled without additives (Fig. 2). Isolation and characterization of both the parent and shoulder peaks by preparative gel permeation chromatography revealed the shoulder peak maintained a molar mass roughly twice that of the parent PS (Fig. S4,† parent Mn = 8.9 kDa and shoulder Mn = 18.6 kDa), suggestive of dimerization through chain coupling events (Fig. S5†). Recently, interchain coupling was reported during the solution-state degradation of PS in the presence of small amounts of triflic acid,64 a byproduct of the silver-mediated trifluoromethylation methodology utilized in this work, that may be a cause of dimerization.61 To ensure these deleterious chain coupling/branching reactions do not impact our chain-end analysis, we first calculated functionalization densities using 19F NMR spectroscopy based on chain-end analysis (eqn (S3)†). When compared to those obtained using 4,4′-difluorobenzophenone as an internal standard (eqn (S4)†), both methods were generally in agreement with one another (Table S5†).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Jar size (mL) | Ball size (mm) | Initial Mnf (kDa) | m (mol%) | TFM-PS M n (kDa) | Final Đf |
| a Reaction conditions: PS repeat unit = 0.81 mmol, AgOTf = 0.32 mmol, KF = 0.32 mmol, TMSCF3 = 0.08 mmol, DCE (for LAG) = 37.4 μL (0.2 μL mg−1), jar temperature = 20 °C–36 °C. b 3× SS balls were used. c 4 SS balls were used. d Reaction conditions: PS repeat unit = 1.62 mmol, AgOTf = 0.65 mmol, KF = 0.65 mmol, TMSCF3 = 0.16 mmol, DCE (for LAG) = 79.3 μL (0.2 μL mg−1). e Functionalization density (m) was determined by 19F NMR spectroscopy using 4,4′-difluorobenzophenone as an internal standard. Based on TMSCF3 as the limiting reagent, a maximum m is 10 mol%. f M n and Đ were determined by GPC-MALS-RI. | ||||||
| 1 | 5 | 8 | 8.9 | 1.1 | 9.0 | 1.05 |
| 2 | 5 | 8 | 9.3 | 1.2 | 9.6 | 1.17 |
| 3 | 5 | 5b | 9.3 | 0.76 | 9.5 | 1.07 |
| 4 | 5 | 10 | 9.3 | 1.6 | 9.1 | 1.07 |
| 5 | 25 | 8c | 9.3 | 0.97 | 10.2 | 1.09 |
| 6d | 25 | 8c | 8.9 | 0.86 | 10.2 | 1.10 |
 | ||
| Fig. 2 GPC-RI traces comparing unmilled parent PS, 1.3 mol% TFM-PS containing a high molar mass shoulder, and PS milled without additives for 4 h. | ||
With mechanochemical parameters optimized, we next focused on LAG conditions for well-defined higher molar mass PS. PS (Mn,MALS = 26.0 kDa), also synthesized via ATRP, is well above the Mlim of PS and therefore allows better visualization of how Mn and Đ are impacted by DCE LAG and chain scission kinetics (see Tables S6 and S7† for evaluation of different LAG solvents). LAG volumes were then varied from 0.2 μL mg−1 to 1.2 μL mg−1 (Fig. 3A). At 0.2 μL mg−1 LAG, while Mn decreases and dispersity increases as evidenced by low molar mass tailing in the GPC trace, the highest CF3 density (1.3 mol%) is also observed. As LAG volume increases above 0.2 μL mg−1, only a slight shift to lower molar masses and minimal changes in dispersity compared to the initial polymer are observed. We hypothesize this lack of chain scission is due to increased plasticization above 0.2 μL mg−1 of LAG solvent. Additionally, increasing LAG volume results in decreased interchain coupling as evidenced by the decreasing Mn and shoulder peak area; these results are likely due to increasingly more dilute concentrations of triflic acid. The highest functionalization density with minimal degradation of TFM-PS was achieved at 0.4 μL mg−1 LAG volume. At this LAG loading, plasticization appears to hinder the degradation kinetics while maintaining productive impact forces needed for efficient functionalization. At LAG loadings above 0.4 μL mg−1, we attribute decreases in trifluoromethylation to dissipation of force in the reaction mixture. To assess the impact of plasticization and stress dissipation to the solid reagents on mechanochemical chain scission, we milled 26.0 kDa PS with either just the solid reagents (AgOTf and KF) or just 0.4 μL mg−1 LAG solvent (DCE) and liquid reagent (TMSCF3) for 4 h, the length of the standard reaction time (Fig. 3B). When milled with only solid reactants, PS saw slightly less chain scission occur (Mn,final = 8.3 kDa) than the initial PS milled alone (Mn = 7.0 kDa), likely due to powder cushioning effects.65 When PS was milled with only the LAG solvent and liquid additive, virtually no degradation or deleterious side reactions occur as assessed by GPC. To further confirm this result was due to plasticization, we used differential scanning calorimetry (DSC) to determine the Tg of the 26.0 kDa PS after milling with only 0.4 μL mg−1 of the LAG solvent and the liquid additive (Fig. 3C). The DSC curves of this liquid-milled PS compared to the initial 26.0 kDa PS reveal a decrease in Tg by ca. 30 °C, indicative of plasticization. Based on these results, we determined that plasticization suppresses Tg and diminishes the force experienced by polymer chains, thus decreasing the rate of chain scission.
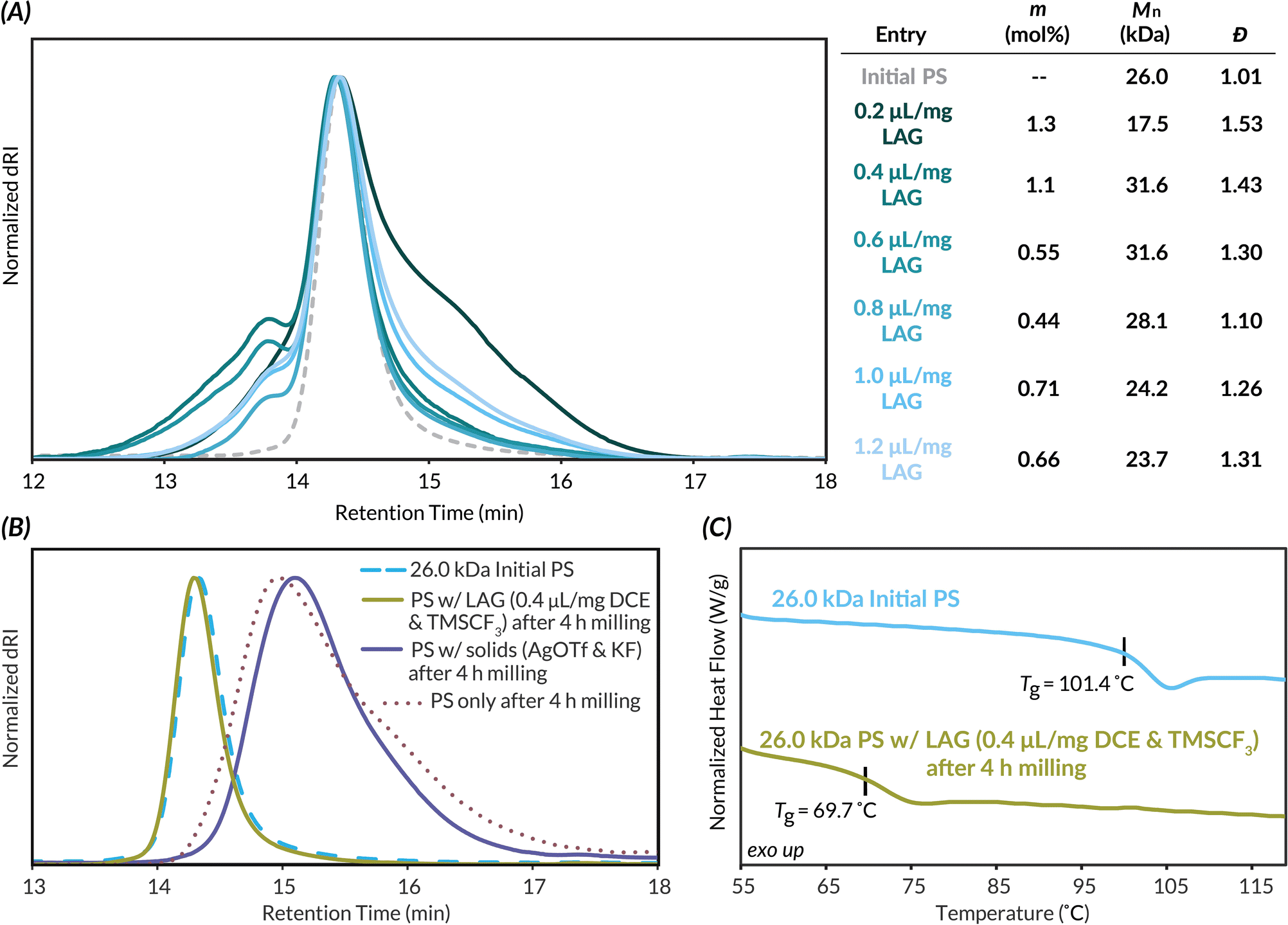 | ||
| Fig. 3 (A) GPC-RI traces and corresponding analytical data highlighting the effect of increasing LAG volume on functionalization density (m), Mn, and dispersity of 26.0 kDa PS trifluoromethylation. Based on TMSCF3 as the limiting reagent, a maximum m is 10 mol%. (B) GPC-RI traces of a control study demonstrating the impact of LAG on PS chain scission kinetics. (C) DSC curves showing a decrease in Tg for 26.0 kDa PS after pre-milling with DCE (0.4 μL mg−1) and TMSCF3 (12.4 μL) for 4 h. | ||
With BMG functionalization now demonstrated on well-defined PS, we turned our attention to modifying commercial and post-consumer plastic waste, particularly expanded polystyrene (EPS) foam waste (EPS Foam) and dyed PS coffee cup lids (PS Lid). To optimize grafting density while minimizing degradation, LAG amounts were first evaluated on Precipitated EPS Foam waste, which was precipitated as an attempt to control for the impact of inherent plasticizers and additives present in the “native” post-consumer plastics (Table 2). LAG amounts between 0.2 and 0.6 μL mg−1 resulted in 0.68–0.89 mol% CF3 incorporation, retaining a similar efficiency to the PPM of well-defined PS, although significant degradation was now observed, likely due to higher molar mass starting material (Table 2, entries 1–3). Milling with 0.8 μL mg−1 of DCE resulted in the highest amount of functionalization, 1.1 mol%, with significantly less chain scission than with lower LAG volumes (Table 2, entry 4). At 1.0 and 1.2 μL mg−1 LAG, trifluoromethylation incorporation begins to decrease alongside minimal changes in Mn, likely due to less effective applied force (Table 2, entries 5 and 6).
|
|
||||
|---|---|---|---|---|
| Entry | LAG (μL mg−1) | m (mol%) | M n (kDa) | Đ |
| a Reaction conditions: PS repeat unit = 0.81 mmol, AgOTf = 0.32 mmol, KF = 0.32 mmol, TMSCF3 = 0.08 mmol, DCE (for LAG) = 0.2–1.2 μL mg−1, internal jar temperature = 20 °C−36 °C. b Functionalization density (m) was determined by 19F NMR spectroscopy using 4,4′-difluorobenzophenone as an internal standard. Based on TMSCF3 as the limiting reagent, a maximum m is 10 mol%. c M n and Đ were determined by GPC-MALS-RI. | ||||
| SM | — | — | 101 | 1.87 |
| 1 | 0.2 | 0.89 | 16.0 | 2.25 |
| 2 | 0.4 | 0.86 | 55.6 | 2.64 |
| 3 | 0.6 | 0.68 | 32.3 | 4.96 |
| 4 | 0.8 | 1.1 | 71.0 | 1.45 |
| 5 | 1.0 | 0.89 | 79.9 | 1.66 |
| 6 | 1.2 | 0.61 | 66.6 | 1.69 |

With optimized LAG conditions in hand (0.8–1.0 μL mg−1 DCE), we subjected high molar mass (Mn = 132 kDa) Commercial PS and post-consumer PS waste to mechanochemical trifluoromethylation conditions (Table 3). We refer to the crude post-consumer waste (i.e., containing all additives and plasticizers) as Native EPS Foam and Native PS Lid and the purified PS as Precipitated EPS Foam and Precipitated PS lLid. Utilizing LAG, we achieved trifluoromethylation loadings of 0.65 mol%, 0.29 mol%, and 0.60 mol% of Commercial PS, Native EPS Foam, and Native PS Lid, respectively (Table 3, entries 1–3). As assessed by GPC-MALS, less degradation was seen during mechanochemical trifluoromethylation for Commercial PS and both post-consumer “native” PS substrates relative to “precipitated” PS substrates (Table 3). These results are supported by differential scanning calorimetry (DSC) experiments that reveal glass transitions of 93 °C–94 °C for the Native EPS Foam and Native PS Lid (Fig. S72 and S74†); these values are lower than the Tg's of their respective precipitated forms66 (Fig. S73 and S75†) and indicate the presence of plasticizers and/or additives in the crude polymers. When combined with LAG conditions, the result is limited backbone degradation compared to precipitated high molar mass PS. To further explore the advantages of mechanochemistry, we subjected Commercial PS, Native EPS Foam, and Native PS Lid to force-free trifluoromethylation conditions in solution (Table S8,† entries 5–9). In all cases, we noted, at best, trace functionalization while also obtaining insoluble cross-linked materials (Fig. S8†) from Native EPS Foam and Commercial PS. Overall, these efforts highlight the efficacy of our mechanochemical methodology over solution-state methods (see Table S8† for solution-state reactions of additional substrates).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | LAG (μL mg−1) | Initial Mnc (kDa) | Initial Đc | m (mol%) | TFM-PS M n (kDa) | Final Đc |
| a Reaction conditions: PS repeat unit = 0.81 mmol, AgOTf = 0.32 mmol, KF = 0.32 mmol, TMSCF3 = 0.08 mmol, DCE (for LAG) = 0.8–1.0 μL mg−1, jar temperature = 20 °C–36 °C. b Functionalization density (m) was determined by 19F NMR spectroscopy using 4,4′-difluorobenzophenone as an internal standard. Based on TMSCF3 as the limiting reagent, a maximum m is 10 mol%. c M n and Đ were determined by GPC-MALS-RI. | |||||||
| 1 | Native EPS Foam | 0.8 | 119 | 1.82 | 0.29 | 83.6 | 2.57 |
| 2 | Native PS Lid | 1.0 | 113 | 1.78 | 0.60 | 114 | 2.62 |
| 3 | Commercial PS | 1.0 | 132 | 2.19 | 0.65 | 93.2 | 1.65 |
| 4 | Precipitated EPS Foam | 0.8 | 101 | 1.87 | 1.1 | 71.0 | 1.45 |
| 5 | Precipitated PS Lid | 1.0 | 131 | 1.72 | 0.46 | 13.3 | 3.78 |

Despite the benefits of LAG for reducing mechanochemical chain scission during PS trifluoromethylation, for commercially relevant high molar mass samples, we generally still observe a decrease in Mn during trifluoromethylation (Table 3); such changes can drastically impact downstream bulk properties. At the outset, we hypothesized that the intrinsic plasticizers67 in “native” PS waste could reduce the rate of mechanochemical chain scission through Tg depression, but such phenomena may vary across different post-consumer substrates. Indeed, in the case of Native PS Lid (Table 3, entry 2) no change in Mn was observed, while Native EPS Foam (Table 3, entry 1) showed a decrease in Mn by ca. 35 kDa. However, any potential benefits of these plasticizers still require synergistic LAG solvent; mechanochemical trifluoromethylation experiments with Native EPS Foam and Native PS Lid run in the absence of LAG solvent (Fig. S9 and S10†) leads to low molar mass PS approaching Mlim (ca. 9.0 kDa) for both substrates. We also performed identical experiments on Precipitated EPS Foam without LAG, but with the addition of 2 or 4 wt% of exogenous plasticizers dioctyl terephthalate (DOTP) and tributyl citrate (TBC) (Fig. S11 and S12†). Interestingly, we still achieved ca. 0.5–1 mol% functionalization across these representative examples, suggesting that “plasticizers” support our mechanochemical trifluoromethylation chemistry.
Given that LAG is clearly necessary for maintaining high molar mass polymers during mechanochemical PPM, we then subjected Precipitated EPS Foam to trifluoromethylation conditions with exogenous plasticizer (2 or 4 wt% of DOTP or TBC) and 0.8 μL mg−1 DCE LAG (Fig. 4). We investigated two different plasticizers because LAG additive polarity can alter reaction kinetics and/or reactivity.68,69 At just 2 wt% DOTP we maintain functionalization density (1.4 mol%) but now observe less chain scission, with a loss of ca. 15 kDa in Mn (Fig. 4A). Use of 2 wt% TBC increases Mnca. 50 kDa with similar functionalization (1.2 mol%). At 4 wt% DOTP and TBC, functionalization density decreases slightly with similar plasticizer dependent outcomes in the final product molar mass (Fig. 4B).
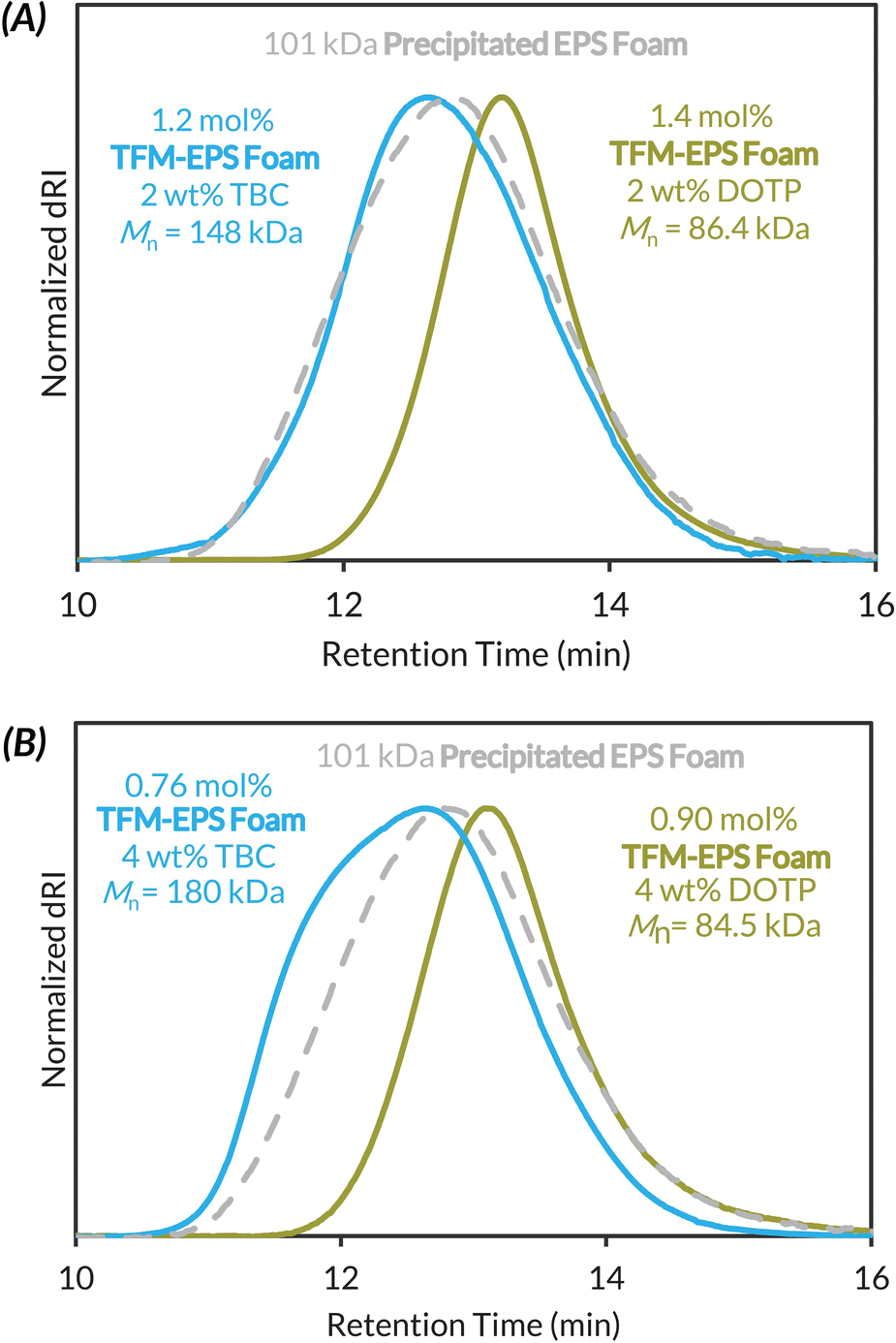 | ||
| Fig. 4 GPC-RI traces and corresponding analytical data highlighting the addition of plasticizers with 0.8 μL mg−1 LAG and their effect on functionalization density, Mn, and dispersity of Precipitated EPS Foam trifluoromethylation: (A) 2 wt% DOTP or 2 wt% TBC; (B) 4 wt% DOTP or 4 wt% TBC. | ||
At 8 wt% TBC loading we recovered mostly insoluble cross-linked material, while at 8 wt% DOTP we maintain similar degradation kinetics as at 2 and 4 wt% DOTP (Table S9†). Because added TBC often led to insoluble material, we opted to study the impact of 2 wt% DOTP + LAG on the mechanochemical functionalization of native post-consumer waste and Commercial PS.
With the ability to effectively maintain high Mn polymers through addition of LAG solvent and plasticizer, we set out to optimize the mechanochemical trifluoromethylation of commercial and post-consumer “native” substrates (Fig. 5). Although Native EPS Foam contains plasticizer, we still observed a decrease in Mn by ca. 35 kDa (Table 3, entry 1). However, the addition of just 2 wt% DOTP with LAG (1.0 μL mg−1 DCE) provided efficient functionalization (1.0 mol%) with a slight increase in Mn and dispersity relative to the initial substrate. Without exogenous plasticizer but with 1.0 μL mg−1 LAG, Native PS Lid still maintains similar trifluoromethylation densities (0.60 mol%) with a slight increase in dispersity. Because Native PS Lid performs so well without exogenous plasticizer, we investigated the supernatant content after precipitation. While NMR spectroscopy and GC reveal a complex mixture of additives, we estimate they only account for ca. 1–2 wt% (Fig. S78 and S79†). Commercial PS results in small amounts of chain scission (loss of ca. 30 kDa) with 2 wt% DOTP and 0.8 μL mg−1 LAG. We attribute this increased chain scission to the higher dispersity of Commercial PS (Đ = 2.19).50 To increase Mnvia chain coupling/branching (132 kDa to 373 kDa), 2 wt% TBC can be used instead (Fig. S13†). These results are promising for future mechanochemical functionalizations of post-consumer plastic waste that already contains additives and/or high degrees of plasticizers. Additives that might otherwise hinder solution-state PPM reactions can potentially tune reactivity under mechanochemical conditions.
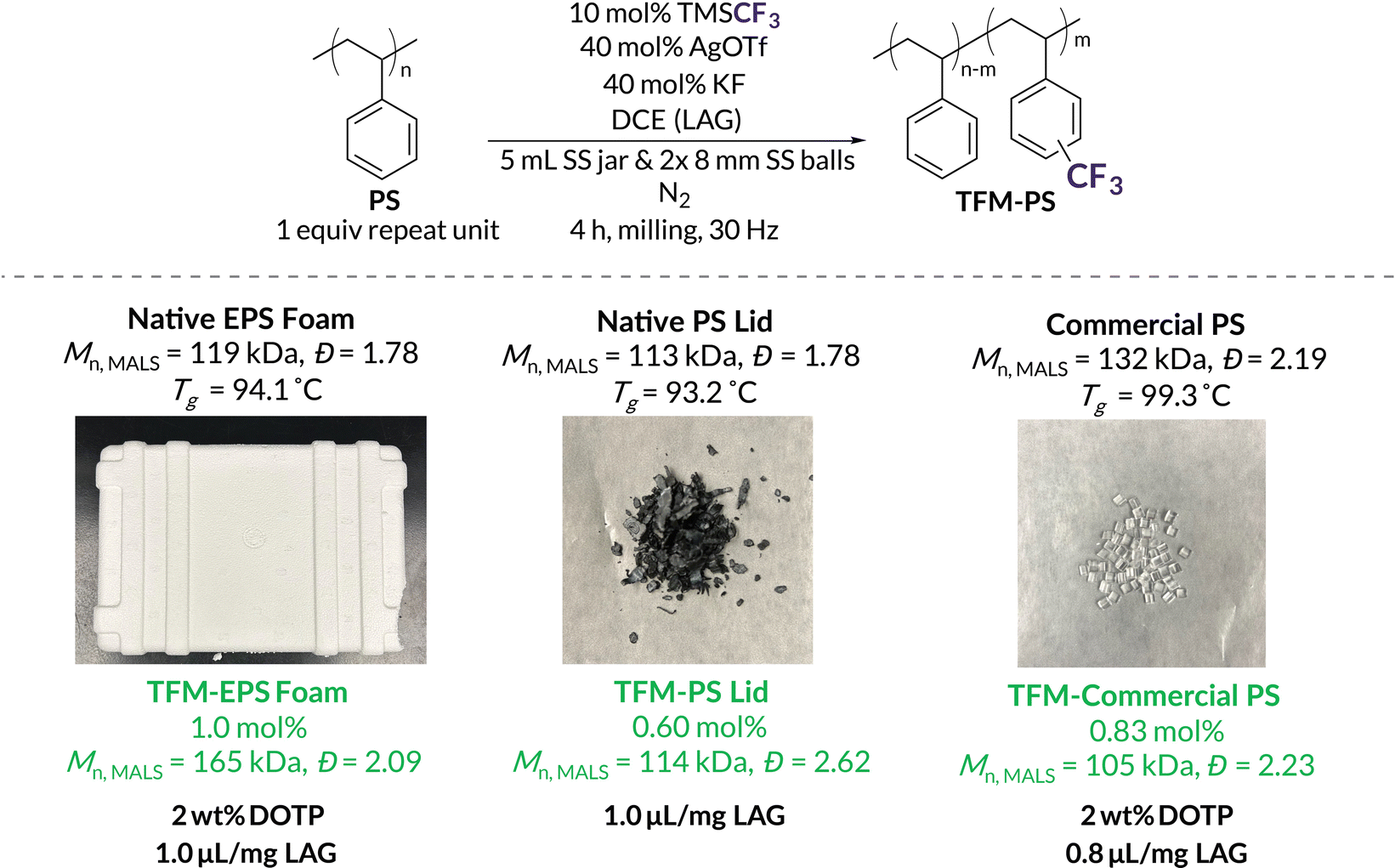 | ||
| Fig. 5 Optimized mechanochemical trifluoromethylations and corresponding analytical data of Native EPS Foam, Native PS Lid, and Commercial PS. Based on TMSCF3 as the limiting reagent, a maximum m is 10 mol%. | ||
Conclusions
In summary, we developed methodology for the direct mechanochemical trifluoromethylation of PS under BMG conditions utilizing LAG to limit mechanochemical chain scission. Initially, we evaluated and identified a silver-mediated trifluoromethylation that maintained similar efficiency of 5.5 mol% grafting density compared to the solution-state method under milder conditions (e.g., shorter reaction times, ambient temperature). After translation to PS substrates, we obtained trifluoromethylation incorporation up to 1.4 mol% without significant changes in Mn or Đ and studied the effects of LAG on these processes. Additionally, we confirmed that LAG conditions decreased the Tg of PS, demonstrating that LAG and exogenous liquid additives act as a plasticizer under BMG conditions. Through this plasticization effect, we can simultaneously decrease the rate of mechanochemical chain scission without dramatically compromising the rate of functionalization. To prevent mechanochemical chain scission on high molar mass PS, we incorporated 2 wt% of DOTP to optimized trifluoromethylation conditions to maintain high Mn and ca. 1 mol% grafting of post-consumer and commercial substrates. Notably, these examples include the functionalization of Native EPS Foam and dyed, Native PS Lid. Together, these findings serve as a proof-of-concept for the outlook of direct mechanochemical PPM; our work reveals the importance of future mechanochemical upcycling methods utilizing synergistic LAG and exogenous plasticizers with a focus on dyed materials and other challenging post-consumer plastic waste without employing copious volumes of exogenous organic solvent.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
M. E. S. and M. R. G. conceived of the idea. M. E. S. and S. M. Z. conducted synthetic experiments and analyzed physical properties. M. E. S. and M. R. G. wrote the manuscript; all authors discussed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by generous start-up funds from the University of Washington. M. E. S. acknowledges the University of Washington Clean Energy Institute for a graduate research fellowship. Differential scanning calorimetry instrumentation is funded by the Student Technology Fund (STF) at the University of Washington. The authors thank Prof. Dianne Xiao for use of thermogravimetric analysis instrumentation. This material is based in part upon work supported by the state of Washington through the University of Washington Clean Energy Institute. NMR spectroscopy resources are supported under NIH S10 OD030224-01A1.Notes and references
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. De Los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084–1129 RSC.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y.-X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- Z. R. Hinton, M. R. Talley, P. A. Kots, A. V. Le, T. Zhang, M. E. Mackay, A. M. Kunjapur, P. Bai, D. G. Vlachos, M. P. Watson, M. C. Berg, T. H. Epps and L. T. J. Korley, Annu. Rev. Mater. Res., 2022, 52, 249–280 CrossRef CAS.
- S. R. Nicholson, N. A. Rorrer, A. C. Carpenter and G. T. Beckham, Joule, 2021, 5, 673–686 CrossRef CAS.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- T. H. Epps, L. T. J. Korley, T. Yan, K. L. Beers and T. M. Burt, JACS Au, 2022, 2, 3–11 CrossRef PubMed.
- X. Zhao, B. Boruah, K. F. Chin, M. Đokić, J. M. Modak and H. S. Soo, Adv. Mater., 2022, 34, 2100843 CrossRef CAS PubMed.
- J. B. Williamson, S. E. Lewis, R. R. Johnson, I. M. Manning and F. A. Leibfarth, Angew. Chem., Int. Ed., 2019, 58, 8654–8668 CrossRef CAS PubMed.
- J. X. Shi, N. R. Ciccia, S. Pal, D. D. Kim, J. N. Brunn, C. Lizandara-Pueyo, M. Ernst, A. M. Haydl, P. B. Messersmith, B. A. Helms and J. F. Hartwig, J. Am. Chem. Soc., 2023, 145, 21527–21537 CrossRef CAS PubMed.
- M. N. Hodges, M. J. Elardo, J. Seo, A. F. Dohoda, F. E. Michael and M. R. Golder, Angew. Chem., Int. Ed., 2023, 62, e202303115 CrossRef CAS PubMed.
- N. R. Ciccia, J. X. Shi, S. Pal, M. Hua, K. G. Malollari, C. Lizandara-Pueyo, E. Risto, M. Ernst, B. A. Helms, P. B. Messersmith and J. F. Hartwig, Science, 2023, 381, 1433–1440 CrossRef CAS PubMed.
- J. B. Williamson, C. G. Na, R. R. Johnson, W. F. M. Daniel, E. J. Alexanian and F. A. Leibfarth, J. Am. Chem. Soc., 2019, 141, 12815–12823 CrossRef CAS PubMed.
- J. Li and H.-M. Li, Eur. Polym. J., 2005, 41, 823–829 CrossRef CAS.
- G. H. Fredrickson, S. Xie, J. Edmund, M. L. Le, D. Sun, D. J. Grzetic, D. L. Vigil, K. T. Delaney, M. L. Chabinyc and R. A. Segalman, ACS Polym. Au, 2022, 2, 299–312 CrossRef CAS PubMed.
- E. Blasco, M. B. Sims, A. S. Goldmann, B. S. Sumerlin and C. Barner-Kowollik, Macromolecules, 2017, 50, 5215–5252 CrossRef CAS.
- B. Xue, P.-P. Huang, M.-Z. Zhu, S.-Q. Fu, J.-H. Ge, X. Li and P.-N. Liu, ACS Macro Lett., 2022, 11, 1252–1257 CrossRef CAS PubMed.
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- S. E. Lewis, B. E. Wilhelmy and F. A. Leibfarth, Chem. Sci., 2019, 10, 6270–6277 RSC.
- S. E. Lewis, B. E. Wilhelmy and F. A. Leibfarth, Polym. Chem., 2020, 11, 4914–4919 RSC.
- Y. Hayakawa, N. Terasawa and H. Sawada, Polymer, 2001, 42, 4081–4086 CrossRef CAS.
- S. Oh and E. E. Stache, J. Am. Chem. Soc., 2022, 144, 5745–5749 CrossRef CAS PubMed.
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed.
- A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondruschka, Chem. Soc. Rev., 2011, 40, 2317 RSC.
- E. Jung, D. Yim, H. Kim, G. I. Peterson and T. Choi, J. Polym. Sci., 2023, 61, 553–560 CrossRef CAS.
- B. A. Versaw, T. Zeng, X. Hu and M. J. Robb, J. Am. Chem. Soc., 2021, 143, 21461–21473 CrossRef CAS PubMed.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed.
- H. Staudinger and H. F. Bondy, Ber. Dtsch. Chem. Ges. A, 1930, 63, 734–736 CrossRef.
- H. Staudinger and W. Heuer, Ber. Dtsch. Chem. Ges. A, 1934, 67, 1159–1164 CrossRef.
- H. Staudinger and E. O. Leupold, Ber. Dtsch. Chem. Ges. A, 1930, 63, 730–733 CrossRef.
- M. E. McFadden and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 11388–11392 CrossRef CAS PubMed.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- Y. Lin, T. B. Kouznetsova and S. L. Craig, J. Am. Chem. Soc., 2020, 142, 2105–2109 CrossRef CAS PubMed.
- J. A. Leitch and D. L. Browne, Chem.–Eur. J., 2021, 27, 9721–9726 CrossRef CAS PubMed.
- P. Chakma, S. M. Zeitler, F. Baum, J. Yu, W. Shindy, L. D. Pozzo and M. R. Golder, Angew. Chem., Int. Ed., 2023, 62, e202215733 CrossRef CAS PubMed.
- S. M. Zeitler, P. Chakma and M. R. Golder, Chem. Sci., 2022, 13, 4131–4138 RSC.
- C. L. Brown and S. L. Craig, Chem. Sci., 2015, 6, 2158–2165 RSC.
- G. S. Lee, B. R. Moon, H. Jeong, J. Shin and J. G. Kim, Polym. Chem., 2019, 10, 539–545 RSC.
- A. Krusenbaum, S. Grätz, G. T. Tigineh, L. Borchardt and J. G. Kim, Chem. Soc. Rev., 2022, 51, 2873–2905 RSC.
- A. Breuillac, F. Caffy, T. Vialon and R. Nicolaÿ, Polym. Chem., 2020, 11, 6479–6491 RSC.
- G. Moad, Prog. Polym. Sci., 1999, 24, 81–142 CrossRef CAS.
- Y.-J. Sun, R. J. G. Willemse, T. M. Liu and W. E. Baker, Polymer, 1998, 39, 2201–2208 CrossRef CAS.
- D. Chatterjee, A. Sajeevan, S. Jana, R. S. Birajdar, S. H. Chikkali, S. Sivaram and S. S. Gupta, ACS Catal., 2024, 14, 7173–7181 CrossRef CAS.
- M. Y. Malca, P.-O. Ferko, T. Friščić and A. Moores, Beilstein J. Org. Chem., 2017, 13, 1963–1968 CrossRef CAS PubMed.
- N. Ohn and J. G. Kim, ACS Macro Lett., 2018, 7, 561–565 CrossRef CAS PubMed.
- M. Ashlin and C. E. Hobbs, Macromol. Chem. Phys., 2019, 220, 1900350 CrossRef CAS.
- J. W. Lee, J. Park, J. Lee, S. Park, J. G. Kim and B.-S. Kim, ChemSusChem, 2021, 14, 3801–3805 CrossRef CAS PubMed.
- B. G. Fiss, L. Hatherly, R. S. Stein, T. Friščić and A. Moores, ACS Sustain. Chem. Eng., 2019, 7, 7951–7959 CrossRef CAS.
- G. I. Peterson, W. Ko, Y.-J. Hwang and T.-L. Choi, Macromolecules, 2020, 53, 7795–7802 CrossRef CAS.
- S. Aydonat, A. H. Hergesell, C. L. Seitzinger, R. Lennarz, G. Chang, C. Sievers, J. Meisner, I. Vollmer and R. Göstl, Polym. J., 2024, 56, 249 CrossRef CAS.
- J. Zhou, T. Hsu and J. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300768 CrossRef CAS PubMed.
- R. Schwarz and C. E. Diesendruck, Adv. Sci., 2023, 10, 2304571 CrossRef CAS PubMed.
- K. Kubota, N. Toyoshima, D. Miura, J. Jiang, S. Maeda, M. Jin and H. Ito, Angew. Chem., Int. Ed., 2021, 60, 16003–16008 CrossRef CAS PubMed.
- K. Kubota, J. Jiang, Y. Kamakura, R. Hisazumi, T. Endo, D. Miura, S. Kubo, S. Maeda and H. Ito, J. Am. Chem. Soc., 2024, 146, 1062–1070 CrossRef CAS PubMed.
- G. A. Bowmaker, Chem. Commun., 2013, 49, 334–348 RSC.
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- J. Csernica and A. Brown, J. Chem. Educ., 1999, 76, 1526 CrossRef CAS.
- E. Plunkett, T. S. Kale, Q. Zhang, H. E. Katz and D. H. Reich, Appl. Phys. Lett., 2019, 114, 023301 CrossRef.
- Y. Pang, J. W. Lee, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2020, 59, 22570–22576 CrossRef CAS PubMed.
- Y. Ye, S. H. Lee and M. S. Sanford, Org. Lett., 2011, 13, 5464–5467 CrossRef CAS PubMed.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- B. Briou, O. Gimello, C. Totee, T. Ono and B. Ameduri, Chem.–Eur. J., 2020, 26, 16001–16010 CrossRef CAS PubMed.
- A. Thigale, C. Trant, J. Ahn, M. Nelson, C. Y. Ryu, C. Bae and S. Lee, Macromolecules, 2023, 56, 5117–5126 CrossRef CAS.
- S. Cayirli, Physicochem. Probl. Miner. Process., 2018, 54, 751–762 CAS.
- J. Rieger, J. Therm. Anal., 1996, 46, 965–972 CrossRef CAS.
- A. A. Cuthbertson, C. Lincoln, J. Miscall, L. M. Stanley, A. K. Maurya, A. S. Asundi, C. J. Tassone, N. A. Rorrer and G. T. Beckham, Green Chem., 2024 10.1039/D4GC00659C.
- J. L. Howard, Y. Sagatov, L. Repusseau, C. Schotten and D. L. Browne, Green Chem., 2017, 19, 2798–2802 RSC.
- J. L. Howard, M. C. Brand and D. L. Browne, Angew. Chem., Int. Ed., 2018, 57, 16104–16108 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03362k |
| This journal is © The Royal Society of Chemistry 2024 |