
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTandem catalytic allylic C–H amination and asymmetric [2,3]-rearrangement via bimetallic relay catalysis†
Zhenwei
Wu‡
ab,
Xi
Yang‡
b,
Fangqing
Zhang
a,
Yangbin
Liu
*ab and
Xiaoming
Feng
 *bc
*bc
aState Key Laboratory of Chemical Oncogenomics, Guangdong Provincial Key Laboratory of Chemical Genomics, Peking University Shenzhen Graduate School, Shenzhen, Guangdong 518055, P. R. China. E-mail: liuyb@szbl.ac.cn
bInstitute of Chemical Biology, Shenzhen Bay Laboratory, Shenzhen, 518055, P. R. China
cKey Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, P. R. China. E-mail: xmfeng@scu.edu.cn
First published on 24th July 2024
Abstract
A bimetallic relay catalysis protocol for tandem allylic C–H amination and asymmetric [2,3]-sigmatropic rearrangement has been developed with the use of an achiral Pd0 catalyst and a chiral N,N′-dioxide–MgII complex in a one-pot operation. A series of anti-α-amino derivatives containing two stereogenic centers were prepared from readily available allylbenzenes and glycine pyrazolamide with good yields and high stereoselectivities. Moreover, the synthetic potential of this protocol was further demonstrated by the product transformations, and a catalytic cycle was proposed to illustrate the reaction process.
Introduction
Palladium-catalysed enantioselective functionalization of allylic C–H bonds represents a versatile and step-economical synthetic technology in comparison with classic asymmetric allylic substitution reactions (Tsuji-Trost), providing a straightforward approach to enable readily available α-alkenes to be used as allylating reagents without a tedious pre-functionalized process (Scheme 1a).1,2 Generally, chiral palladium complexes are able to facilitate the allylic C–H cleavage with the generation of π-allyl-palladium intermediates, which favour an outer-sphere nucleophilic substitution at the less-hindered terminus.3 In sharp contrast, advances in branch- and enantioselective allylic C–H oxidative functionalization of aryl- or alkyl-substituted α-alkenes are relatively limited. To achieve this less favoured regioselectivity, Gong's group has recently demonstrated that palladium-catalysed allylic C–H oxidative alkylation reactions employing potential coordination nucleophiles (such as α-benzothiazylacetamide and 2-acylimidazole) prefer to undergo an inner-sphere pathway, allowing for the branch-selective products (Scheme 1b).4 Another powerful strategy to access branch regioselectivity is a one-pot sequence of palladium-catalysed linear allylic C–H functionalization, followed by branched allylic substitution (Scheme 1c). As such, Sharma and Hartwig describe an elegant branch- and enantioselective functionalization of allylic C–H bonds of α-alkenes by a sequential combination of palladium-catalysed C–H oxidation and iridium-catalysed asymmetric substitution (Scheme 1d).5 Despite this impressive progress, a sequential or relay process to achieve branch- and enantioselective allylic C–H oxidative functionalization is still in its infancy thus far. Further efforts to expand the combination of two different reaction types in one-pot show a highly valuable but challenging avenue of research.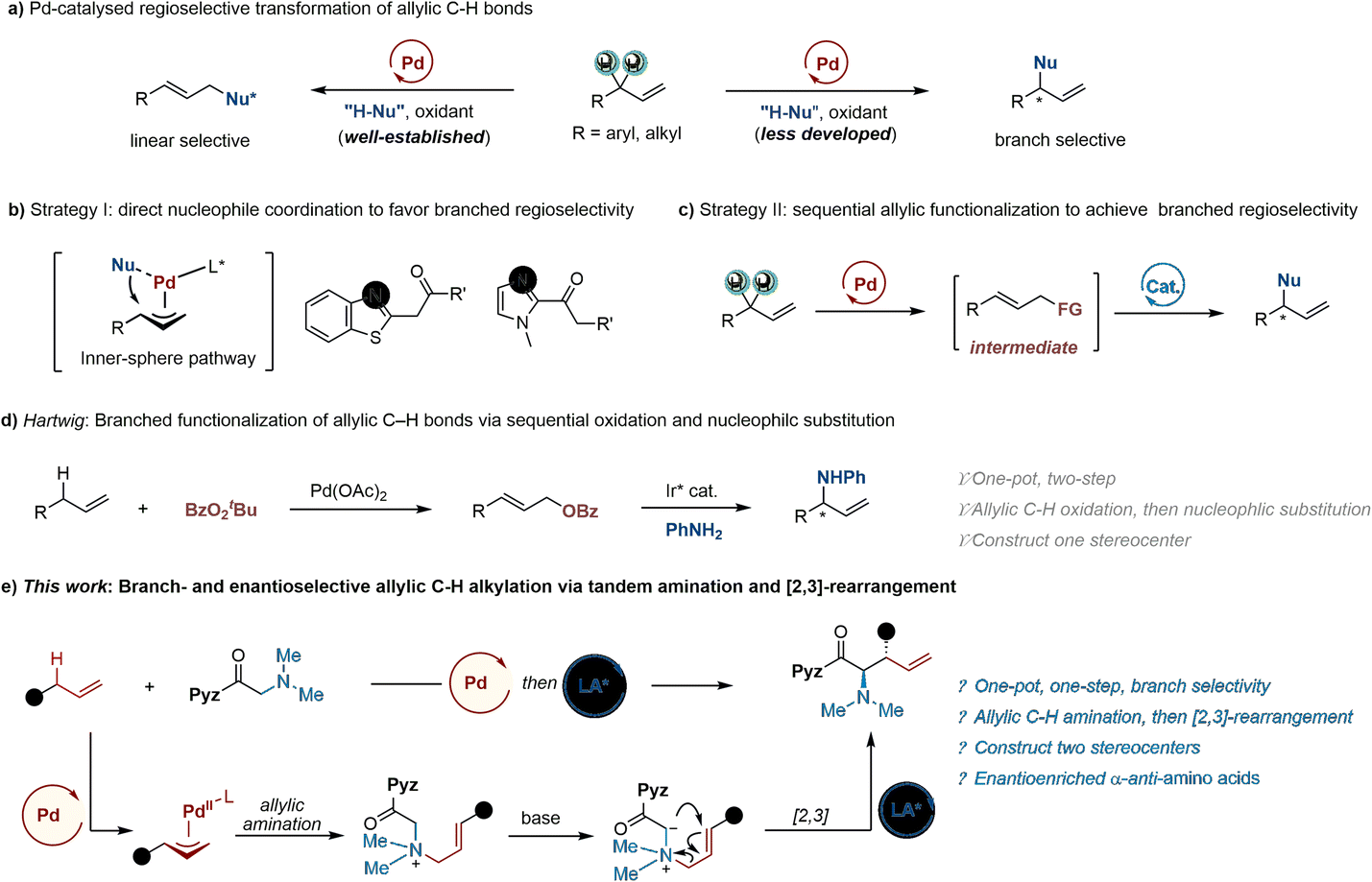 | ||
| Scheme 1 Palladium-catalysed asymmetric allylic C–H functionalization of α-alkenes. FG = functional group, Nu = nucleophile, Pyz = pyrazole, LA* = chiral Lewis acid catalyst. | ||
The chiral α-amino acids are a class with versatile structure fragments that are prevalent in numerous bioactive natural products and pharmaceuticals.6 Of the established synthetic methods for enantioenriched α-amino acid derivatives, the asymmetric [2,3]-Stevens rearrangement of glycine-derived allylic ammonium ylides is a powerful transformation, and elegant examples have been reported by the groups of Tambar and Smith, and in our previous work.7 In all these studies, preactivated allylating reagents, such as allylic bromides, carbonates and phosphates, are often required to realize the allylic quaternary ammonium salts. To the best of our knowledge, there are no examples of asymmetric [2,3]-Stevens rearrangements with simple α-alkenes as the allylic precursors. Herein, we report the use of bimetallic relay catalysis for branch- and enantioselective allylic C–H amination/[2,3]-rearrangement processes (Scheme 1e). Firstly, the allylic C–H activation of α-alkenes was catalysed by a suitable achiral palladium/oxidant system, which leads to the generation of highly electrophilic π-allyl-palladium intermediates, which can undergo allylic amination and deprotonation to afford the crucial rearrangement precursors-ammonium ylides. Then, the asymmetric [2,3]-rearrangement of the allylic ammonium ylides may be achieved by a chiral Lewis acid complex. This protocol can provide an efficient one-step conversion of the α-alkenes and glycine derivatives into enantiomerically enriched unnatural α-anti-amino acids bearing two contiguous stereocenters. Notably, preactivated allylating reagents are not required in the current method, thus enabling step-economy and convenient synthesis.
Results and discussion
In the last two decades, Feng chiral N,N′-dioxide ligands have been applied as privileged chiral Lewis acid catalysts in diverse asymmetric transformations,8 which show good compatibility with late transition metals and oxidative reaction conditions.9 As such, we have recently developed bimetallic synergistic catalysis with an achiral Pd(0) catalyst and a chiral N,N′-dioxide–Co(II) complex with good compatibility to achieve asymmetric allylic C–H alkylation of α-alkenes.9i This precedent encouraged us to investigate the feasibility of merging a palladium/oxidant system and a chiral N,N′-dioxide-Lewis acid system using relay catalysis for asymmetric allylic C–H functionalization. Pyrazoleamides are a class of versatile compounds, which can be readily converted into carboxylic acid derivatives and can coordinate tightly with chiral Lewis acid catalysts via a bidentate coordination.10 Thus, glycine pyrazoleamide 2 bearing a tertiary amine moiety was selected as the model substrate. Initially, various palladium precatalysts combining 2,5-di-tert-butylquinone (2,5-DTBQ) as an oxidant were evaluated for the tandem allylic C–H amination/[2,3]-rearrangement in the presence of Mg(OTf)2 and chiral N,N′-dioxide (L3-PrAd) and Na2CO3 as a base (Table 1). It was found that Pd2(dba)3 was superior to other commonly used Pd(PPh3)4, and Pd(TFA)2 and gave the desired branch-selective allylic C–H alkylation product 3a with a higher yield, diastereo- and enantio-selectivity (entry 3, 55% yield, 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, 78:22 er). This result demonstrated that achiral palladium/chiral Lewis acid relay catalysis could be a potential stereochemical control strategy for allylic C–H functionalization, by enabling the asymmetric version of such a one-pot transformation. Other Lewis acids, such as Sc(OTf)3 and Ni(OTf)2, were also examined and no enhanced results were obtained (entries 4 and 5, for more details see the ESI†). Next, an extensive investigation of chiral ligands was carried out, including the change of the amide moieties bearing a distinct steric hindrance and diverse chiral α-amino acid skeletons. The L-ramipril derived L3-RaAd was confirmed to be the best option for the control of stereoselectivity (entry 10, 47% yield, 3:1 dr, 82:18 er). Increasing the amount of 2,5-DTBQ to 1.5 equivalent and changing the palladium source to the Pd2(dba)3·chloroform adduct could slightly improve the reactivity and diastereo-selectivity (entries 11 and 12). Moreover, the enantioselectivity was further increased when the reaction was conducted at 20 °C, and the yield could be maintained when the reaction time was prolonged (entry 13). Considering the [2,3]-rearrangement of the allylic ammonium salts as a base-promoted reaction, we investigated a series of bases such as K2CO3, LiOAc and iPrNEt (or more details see the ESI†). Interestingly, in the absence of any external base, such a tandem reaction still proceeded well and gave product 3a with a good yield and a higher level of enantioselectivity (entry 14, 56% yield, 5:1 dr, 93:7 er). This suggested that the phenoxide reduced by the benzoquinone could be used as a base formed in situ to convert the ammonium salt into ammonium ylide. Furthermore, the addition of NaBAr4F as an additive allowed the delivery of the optimized outcomes (entry 15, 67% yield, 8:1 dr, 95:5 er). We speculate that the complex of Mg(OTf)2 with the chiral N,N′-dioxide ligand follows an anion exchange with NaBArF4, leading to an enhancement of its Lewis acidity. This stronger Lewis acid complex can promote the deprotonation event of the pyrazolamide ammonium salt to form an active ammonium ylide and thereby increases the [2,3]-rearrangement yield. In addition, the large steric hindrance of the BArF4 anion may also contribute to the improved diastereo- and enantio-selectivity in the catalytic process.
:1 dr, 78:22 er). This result demonstrated that achiral palladium/chiral Lewis acid relay catalysis could be a potential stereochemical control strategy for allylic C–H functionalization, by enabling the asymmetric version of such a one-pot transformation. Other Lewis acids, such as Sc(OTf)3 and Ni(OTf)2, were also examined and no enhanced results were obtained (entries 4 and 5, for more details see the ESI†). Next, an extensive investigation of chiral ligands was carried out, including the change of the amide moieties bearing a distinct steric hindrance and diverse chiral α-amino acid skeletons. The L-ramipril derived L3-RaAd was confirmed to be the best option for the control of stereoselectivity (entry 10, 47% yield, 3:1 dr, 82:18 er). Increasing the amount of 2,5-DTBQ to 1.5 equivalent and changing the palladium source to the Pd2(dba)3·chloroform adduct could slightly improve the reactivity and diastereo-selectivity (entries 11 and 12). Moreover, the enantioselectivity was further increased when the reaction was conducted at 20 °C, and the yield could be maintained when the reaction time was prolonged (entry 13). Considering the [2,3]-rearrangement of the allylic ammonium salts as a base-promoted reaction, we investigated a series of bases such as K2CO3, LiOAc and iPrNEt (or more details see the ESI†). Interestingly, in the absence of any external base, such a tandem reaction still proceeded well and gave product 3a with a good yield and a higher level of enantioselectivity (entry 14, 56% yield, 5:1 dr, 93:7 er). This suggested that the phenoxide reduced by the benzoquinone could be used as a base formed in situ to convert the ammonium salt into ammonium ylide. Furthermore, the addition of NaBAr4F as an additive allowed the delivery of the optimized outcomes (entry 15, 67% yield, 8:1 dr, 95:5 er). We speculate that the complex of Mg(OTf)2 with the chiral N,N′-dioxide ligand follows an anion exchange with NaBArF4, leading to an enhancement of its Lewis acidity. This stronger Lewis acid complex can promote the deprotonation event of the pyrazolamide ammonium salt to form an active ammonium ylide and thereby increases the [2,3]-rearrangement yield. In addition, the large steric hindrance of the BArF4 anion may also contribute to the improved diastereo- and enantio-selectivity in the catalytic process.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | [Pd] | Lewis acid | Ligand | Base | Yield (%) | dr | er |
|
a Reaction conditions: 1a (0.2 mmol, 2.0 equiv.), 2 (0.1 mmol, 1.0 equiv.), [Pd]/PPh3 (1:2, 4 mol%), 2,5-DTBQ (0.11 mmol, 1.1 equiv.), Lewis acid (10 mol%), Ligand (10 mol%), and Na2CO3 (0.12 mmol, 1.2 equiv.) in MeCN (2.0 mL) at 50 °C for 24 h. The yield and diastereomeric ratio (dr) were determined using 1H-NMR analysis of the crude reaction mixtures using 4-methylanisole as the internal standard. The enantiomeric ratio (er) was determined using chiral HPLC analysis. Compound 3a was quantitatively converted into its methyl ester derivative with MeOH at 60 °C to determine its er value.
b 2,5-DTBQ (1.5 equiv.).
c At 20 °C for 72 h.
d NaBArF4 {NaB[3,5-(F3C)2C6H3]4} (15 mol%) was added. N.D. = no detection. 2,5-DTBQ = 2,5-di-tert-butyl quinone.
|
|||||||
| 1 | Pd(PPh3)4 | Mg(OTf)2 | L3-PrAd | Na2CO3 | Trace | N.D. | N.D. |
| 2 | Pd(TFA)2 | Mg(OTf)2 | L3-PrAd | Na2CO3 | 48 | 3.5:1 |
76:24/54:46 |
| 3 | Pd2dba3 | Mg(OTf)2 | L3-PrAd | Na2CO3 | 55 | 5:1 |
78:22/62:38 |
| 4 | Pd2dba3 | Sc(OTf)3 | L3-PrAd | Na2CO3 | Trace | N.D. | N.D. |
| 5 | Pd2dba3 | Ni(OTf)2 | L3-PrAd | Na2CO3 | Trace | N.D. | N.D. |
| 6 | Pd2dba3 | Mg(OTf)2 | L3-PrcH | Na2CO3 | 48 | 3:1 |
60:40/57:43 |
| 7 | Pd2dba3 | Mg(OTf)2 | L3-PrCHPh2 | Na2CO3 | 30 | 3:1 |
56:44/57:43 |
| 8 | Pd2dba3 | Mg(OTf)2 | L3-PrMe2 | Na2CO3 | 35 | 2:1 |
56:44/53:47 |
| 9 | Pd2dba3 | Mg(OTf)2 | L3-PiAd | Na2CO3 | 44 | 2:1 |
54:46/51:49 |
| 10 | Pd2dba3 | Mg(OTf)2 | L3-RaAd | Na2CO3 | 47 | 3:1 |
82:18/62:38 |
| 11b | Pd2dba3 | Mg(OTf)2 | L3-RaAd | Na2CO3 | 60 | 4.5:1 |
83:17/60:40 |
| 12b | Pd2(dba)3·CHCl3 | Mg(OTf)2 | L3-RaAd | Na2CO3 | 65 | 4.5:1 |
84:16/60:40 |
| 13bc | Pd2(dba)3·CHCl3 | Mg(OTf)2 | L3-RaAd | Na2CO3 | 62 | 5:1 |
87:13/63:37 |
| 14bc | Pd2(dba)3·CHCl3 | Mg(OTf)2 | L3-RaAd | — | 56 | 5:1 |
93:7/60:40 |
| 15bcd | Pd2(dba)3·CHCl3 | Mg(OTf)2 | L3-RaAd | — | 67 | 8:1 |
95:5/— |

With the optimal conditions identified, the scope of the α-alkenes was explored for the asymmetric tandem allylic C–H amination/[2,3]-rearrangement (Table 2). We found that allylbenzenes containing alkyl and phenyl substitutions on the aromatic ring were well tolerated and gave the desired branch products in moderate to good yields with high diastereo- and enantio-selectivities (3a–3e). The presence of alkoxy and phenoxy substituents was able to give the corresponding products with good yields with good enantioselectivities (3f–3h). Various halogen-substituents at different positions of the aryl ring were also compatible to generate 3i–3n with satisfactory results, even for the dichloro-substituted substrate. Other α-alkenes bearing electron-withdrawing functional groups (–CO2Me, and –CO2Bn) were also amenable to this process, demonstrating good reaction efficiency and moderate diastereomeric and enantiomeric ratios (3o and 3p). Generally, the allylbenzenes with an electron-rich group provided the final allylic C–H alkylation product with a slightly higher level of enantioselectivity than that with an electron-deficient group. Furthermore, the introduction of 1,3-dioxopentane to the aryl substituents led to acceptable outcomes (3q). When α-alkenes were replaced by fused- and heteroaryl groups, such as naphthyl (3r), benzofuryl (3s), dibenzothiophenyl (3t), dibenzofuryl (3u), and benzothienyl (3v), this bimetallic relay catalysis could still proceed well and smoothly yielded the desired α-amino acids bearing these important heterocyclic motifs. For a conjugated styryl substituted α-alkene, the allylic alkylation product 3w was obtained with exclusive branched regioselectivity with a moderate yield and excellent diastereo- and enantio-selectivity. The allylbenzene bearing a para-cyclohexyl group was also well tolerated, providing the rearrangement product 3x with a 54% yield with 7:1 dr. Moreover, glycine pyrazoleamide derivatives with varying substituents on the nitrogen also gave the desired [2,3]-rearrangement products in 37–52% yields with moderate to good diastereo- and enantio-selectivities (3aa and 3ab). We also explored the non-aryl substituted α-alkenes with glycine pyrazoleamide under standard relay conditions. Unfortunately, no desired target product was observed from the crude 1H-NMR (3ba). Given the biomedical potential of α-amino acids, we also applied this protocol to coupling with pharmaceutical molecules, such as probenecid and naproxen, delivering the desired products with good results (3ca and 3cb). It is worth noting that this formal allylic C–H alkylation of α-alkenes allowed for the perfect branch regioselectivity (>20:1 b/l) in all cases, when compared with the most linear products obtained by palladium-catalysed allylic substitution reactions.
| a Reaction conditions: 1 (0.4 mmol, 2.0 equiv.), 2 (0.2 mmol, 1.0 equiv.), Pd2(dba)3·CHCl3 (4 mol%), PPh3 (8 mol%), 2,5-DTBQ (1.5 equiv.), Mg(OTf)2 (10 mol%), L3-RaAd (10 mol%), NaBArF4 (15 mol%) in MeCN (4.0 mL) at 20 °C for 48–72 h. Isolated yield. Values of dr and b/l were determined using 1H-NMR analysis. The enantiomeric ratio (er) was determined by chiral HPLC analysis. b er is the value of the product's methyl ester derivative. c Reaction performed at 10 °C, and isolated yield of the major diastereomer. d Reaction performed at 50 °C. |
|---|

|
To further demonstrate the application of this methodology, simple transformations were performed to convert them to other enantioenriched building blocks. As shown in Scheme 2, the pyrazoleamide moiety as a readily modifiable functional group could easily be converted under mild conditions. Reduction of product 3e with NaBH4 gave the corresponding alcohol 4 with an 85% yield with no erosion of stereoselectivity (>20:1 dr, 95:5 er). Dipeptide 5 was efficiently accessed from the direct condensation of product 3e with an optically pure α-amino acid, demonstrating its potential for the synthesis of valuable peptides. The corresponding α-amino acid 6 was obtained as its hydrochloride upon treatment of 3e with LiOH·H2O in THF/H2O. An almost quantitative transesterification with methanol provided α-amino ester 7 with a 90% isolated yield with 96:4 er. Subsequently, hydrogenation of the terminal alkene moiety at room temperature (H2, Pd/C) gave 8 with a 91% yield. Primary alcohol 9 was generated using a sequence hydroboration-oxidation (9-BBN and then H2O2) and good results were obtained.
 | ||
| Scheme 2 Synthetic transformation of the allylation products. | ||
In order to gain insight into the possible catalytic mechanism, a series of control experiments were conducted. Firstly, we determined the reaction constants of two parallel reactions with allylbenzene 3e and deuterated 3e-d2. A significant primary kinetic isotope effect (KIE, kH/kD = 2.9) was observed, suggesting that the allylic C–H cleavage might be the rate-determining step (Scheme 3a). To confirm the [2,3]-rearrangement process, allylic ammonium salt 10 was prepared using glycine and cinnamyl bromide, and then this was investigated using control experiments (Scheme 3b). Firstly, it was subjected to the standard conditions and no desired product 3a was detected, and this was probably due to the absence of external bases to facilitate the formation of ammonium ylide. Then, the [2,3]-rearrangement of allylic ammonium salt 10 was promoted by the addition of Na2CO3 or NaOAr alone as the base, both of which were able to give the desired rearrangement product 3a with a 19% or 64% yield with 1:1 dr. These results indicate that a suitable base is necessary to generate the active ammonium ylide. Furthermore, the chiral Mg(II) complex was employed to induce an asymmetric [2,3]-rearrangement of allylic ammonium salt in the presence of the equivalent base, only resulting in low diastereo- and enantio-selectivities. We speculate that a stoichiometric amount of base (such as Na2CO3 and NaOAr) may lead to a racemic background reaction in the [2,3]-rearrangement of the allylic ammonium salt (10), thus giving the product 3a with decreased diastereo- or enantio-selectivities. In the relay catalytic process, the phenoxide (ArO−) as the base, is generated in situ in a catalytic amount to enable slow release of the ammonium ylide, which is potentially captured by the chiral N,N′-dioxide–Mg(II) complex and undergoes a high level of enantioselective [2,3]-Stevens rearrangement. We also attempted to observe the ammonium salts (10) formed by allylic C–H amination of allylbenzenes with glycine pyrazoleamides using in situ1H-HMR analysis. However, it was difficult to detect such ammonium intermediates, probably due to the unfavourable equilibrium for palladium-catalysed ammonium salt formation and the fast [2,3]-rearrangement of the ammonium salts.7a,b,e
 | ||
| Scheme 3 Mechanistic investigation. (a) Determination of the kinetic isotope effect (KIE) with allylbenzene 3e and 3e-d2. (b) Control experiments to investigate the [2,3]-rearrangement of the allylic ammonium salt 10. (c) Proposed mechanism. | ||
Based on the mechanistic experimental results and our previous work,7e,9i a plausible relay catalytic cycle for the tandem asymmetric allylic C–H amination/[2,3]-rearrangement is proposed (Scheme 3c). The active Pd0/PPh3 catalyst coordinates with allylbenzene 1a and 2,5-DTBQ to form an intermediate I, followed by a concerted proton and two-electron transfer process to generate a π-allyl-palladium complex II. Subsequently, nucleophilic attack of glycine pyrazoleamide can provide the key ammonium salt III and regenerates the palladium complex. The chiral N,N′-dioxide–Mg(II) complex activates the corresponding ammonium salt III in a bidentate coordination model, and then promotes its deprotonation into ammonium ylide IV using the in situ generated phenoxide (ArO−) as the base. According to the anti-selective product, we suppose that the enantioselective [2,3]-rearrangement proceeds in an exo-transition state leading to good regio-, diastereo- and enantio-controls.
Conclusions
In conclusion, we have developed an asymmetric bimetallic relay catalysis strategy for Pd-catalysed allylic C–H direct functionalization. This methodology successfully combines an achiral Pd0 complex-catalysed allylic C–H amination with a chiral N,N′-dioxide–Mg(OTf)2 complex-controlled enantioselective [2,3]-rearrangement in a one-pot operation. A diversity of valuable enantioenriched anti-α-amino acid derivatives were prepared from readily available α-alkenes and glycine pyrazoleamide with excellent linear-selectivity, and moderate to high diastereo- and enantio-selectivities. The synthetic utility is also demonstrated in the further transformations of the α-amino acid products. We believe that this relay catalysis strategy will have wider application prospects in the field of enantioselective allylic C–H functionalization of simple alkenes.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Z. W. W. and X. Y. performed the experiments and prepared the paper and the ESI.† F. Q. Z. performed some experiments. Y. B. L. and X. M. F. conceived the concept, directed the project and helped with modifying the paper and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the generous financial support received from the National Natural Science Foundation of China (22001177 and 22188101), the Shenzhen Bay Laboratory Startup Fund (S201100003), the Major Program of Shenzhen Bay Laboratory (S211101001-1), the Shenzhen Bay Qihang Fellow Program (QH23001), and the Guangdong Pearl River Talent Program (2021QN020268).Notes and references
- For selected reviews about palladium-catalyzed allylic substitution reactions, see: (a) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2944 CrossRef CAS PubMed; (b) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258–297 CrossRef CAS PubMed; (c) Y. Liu, S.-J. Han, W. B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS PubMed; (d) O. Pàmies, J. Margalef, S. Cañellas, J. James, E. Judge, P. J. Guiry, C. Moberg, J. E. Bäckvall, A. Pfaltz, M. A. Pericàs and M. Diéguez, Chem. Rev., 2021, 121, 4373–4505 CrossRef PubMed; (e) X. Wang, Y. Peng, L. Zhao, X. Huo and W. Zhang, Sci. China: Chem., 2023, 66, 2238–2255 CrossRef CAS.
- For selected reviews about transition metal-catalyzed allylation reactions, see: (a) J. T. Mohr and B. M. Stoltz, Chem.–Asian J., 2007, 2, 1476–1491 CrossRef CAS PubMed; (b) J. D. Weaver, A. Recio III, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846–1913 CrossRef CAS PubMed; (c) N. A. Butt and W. Zhang, Chem. Soc. Rev., 2015, 44, 7929–7967 RSC; (d) J. James, M. Jackson and P. J. Guiry, Adv. Synth. Catal., 2019, 361, 3016–3049 CrossRef CAS; (e) T. B. Wright and P. A. Evans, Chem. Rev., 2021, 121, 9196–9242 CrossRef CAS PubMed; (f) Z. W. Wu, X. M. Feng and Y. B. Liu, Synthesis, 2024 DOI:10.1055/s-0042-1751568.
- For selected reviews about direct asymmetric allylic C–H bond functionalization, see: (a) R. Wang, Y. Luan and M. Ye, Chin. J. Chem., 2019, 37, 720–743 CrossRef CAS; (b) R. A. Fernandes and J. L. Nallasivam, Org. Biomol. Chem., 2019, 17, 8647–8672 RSC; (c) P.-S. Wang and L.-Z. Gong, Acc. Chem. Res., 2020, 53, 2841–2854 CrossRef CAS PubMed; (d) K. Yang, M. Song, H. Liu and H. Ge, Chem. Sci., 2020, 11, 12616–12632 RSC; (e) P.-S. Wang and L.-Z. Gong, Chin. J. Chem., 2023, 41, 1841–1848 CrossRef CAS; (f) Y. C. Wang, J. B. Liu and Z.-T. He, Chin. J. Org. Chem., 2023, 43, 2614–2627 CrossRef CAS; (g) H.-Y. Lu and Z.-T. He, Chin. Chem. Lett., 2023, 34, 108105 CrossRef CAS ; For selected examples, see:; (h) B. M. Trost, E. J. Donckele, D. A. Thaisrivongs, M. Osipov and J. T. Masters, J. Am. Chem. Soc., 2015, 137, 2776–2784 CrossRef CAS PubMed; (i) R.-B. Hu, C.-H. Wang, W. Ren, Z. Liu and S.-D. Yang, ACS Catal., 2017, 7, 7400–7404 CrossRef CAS; (j) R. Ma and M. C. White, J. Am. Chem. Soc., 2018, 140, 3202–3205 CrossRef CAS PubMed; (k) W. Liu, S. Z. Ali, S. E. Ammann and M. C. White, J. Am. Chem. Soc., 2018, 140, 10658–10662 CrossRef CAS PubMed.
- For selected examples about palladium-catalyzed allylic C–H alkylation reactions for the formation of branched product, see: (a) L.-F. Fan, S.-W. Luo, S.-S. Chen, T.-C. Wang, P.-S. Wang and L.-Z. Gong, Angew. Chem., Int. Ed., 2019, 58, 16806–16810 CrossRef CAS PubMed; (b) H.-C. Lin, P.-P. Xie, Z.-Y. Dai, S.-Q. Zhang, P.-S. Wang, Y.-G. Chen, T.-C. Wang, X. Hong and L.-Z. Gong, J. Am. Chem. Soc., 2019, 141, 5824–5834 CrossRef CAS PubMed; (c) T.-C. Wang, L.-F. Fan, Y. Shen, P.-S. Wang and L.-Z. Gong, J. Am. Chem. Soc., 2019, 141, 10616–10620 CrossRef CAS PubMed; (d) T.-C. Wang, P.-S. Wang and L.-Z. Gong, Sci. China: Chem., 2020, 63, 454–459 CrossRef CAS; (e) Z.-S. Nong, L. Zhu, T.-C. Wang, L.-F. Fan, P.-S. Wang and L.-Z. Gong, Nat. Synth., 2022, 1, 487–496 CrossRef; (f) Z.-S. Nong, X.-R. Chen, P.-S. Wang, X. Hong and L.-Z. Gong, Angew. Chem., Int. Ed., 2023, 62, e202312547 CrossRef CAS PubMed.
- A. Sharma and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 17983–17989 CrossRef CAS PubMed.
- (a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (b) J. Mayol-Llinàs, A. Nelson, W. Farnaby and A. Ayscough, Drug Discovery Today, 2017, 22, 965–969 CrossRef PubMed.
- (a) A. Soheili and U. K. Tambar, J. Am. Chem. Soc., 2011, 133, 12956–12959 CrossRef CAS PubMed; (b) A. Nash, A. Soheili and U. K. Tambar, Org. Lett., 2013, 15, 4770–4773 CrossRef CAS PubMed; (c) T. H. West, D. S. B. Daniels, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2014, 136, 4476–4479 CrossRef CAS PubMed; (d) T. H. West, S. S. M. Spoehrle, K. Kasten, J. E. Taylor and A. D. Smith, ACS Catal., 2015, 5, 7446–7479 CrossRef CAS; (e) S. S. M. Spoehrle, T. H. West, J. E. Taylor, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2017, 139, 11895–11902 CrossRef CAS PubMed; (f) Q. C. Lin, B. W. Hu, X. Xu, S. X. Dong, X. H. Liu and X. M. Feng, Chem. Sci., 2020, 11, 3068–3073 RSC.
- For selected reviews about the properties of chiral N,N′-dioxide-Lewis acid complexes, see: (a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574–587 CrossRef CAS PubMed; (b) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298–302 RSC; (c) X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621–2631 CrossRef CAS PubMed; (d) M.-Y. Wang and W. Li, Chin. J. Chem., 2021, 39, 969–984 CrossRef CAS; (e) Y. B. Liu, X. H. Liu and X. M. Feng, Chem. Sci., 2022, 13, 12290–12308 RSC; (f) S. X. Dong, X. H. Liu and X. M. Feng, Acc. Chem. Res., 2022, 55, 415–428 CrossRef CAS PubMed; (g) S. X. Dong, W. L. Cao, M. P. Pu, X. H. Liu and X. M. Feng, CCS Chem., 2023, 5, 2717–2735 CrossRef CAS; (h) Y.-M. He, Y.-Z. Cheng, Y. Duan, Y.-D. Zhang, Q.-H. Fan, S.-L. You, S. Luo, S.-F. Zhu, X.-F. Fu and Q.-L. Zhou, CCS Chem., 2023, 5, 2685–2716 CrossRef CAS; (i) D.-F. Chen and L.-Z. Gong, Org. Chem. Front., 2023, 10, 3676–3683 RSC ; For selected examples, see:; (j) T. Y. Zhan, L. K. Yang, Q. Y. Chen, R. Weng, X. H. Liu and X. M. Feng, CCS Chem., 2023, 5, 2101–2110 CrossRef CAS; (k) J. He, W. Liu, J. Zhang, Z. W. Zhong, L. L. Lin and X. M. Feng, Sci. China: Chem., 2023, 66, 2803–2809 CrossRef CAS; (l) W. Yang, Z. D. Yang, L. Chen, Y. C. Lu, C. F. Zhang, Z. S. Su, X. H. Liu and X. M. Feng, Chin. Chem. Lett., 2023, 34, 107791 CrossRef CAS; (m) Z. Zeng, L. Q. Yang, Z. J. Xiao, L. Chen, Z. W. Zhong, F. Wang, X. H. Liu, S. X. Dong and X. M. Feng, ACS Catal., 2024, 14, 2908–2916 CrossRef CAS; (n) N. Xu, M. P. Pu, H. Yu, G. F. Yang, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2024, 63, e202314256 CrossRef CAS PubMed; (o) L. Ge, H. K. Wang, Y. B. Liu and X. M. Feng, J. Am. Chem. Soc., 2024, 146, 13347–13355 CrossRef CAS PubMed; (p) Z. D. Tan, Y. B. Liu and X. M. Feng, Sci. Adv., 2024, 10, eadn9738 CrossRef CAS PubMed; (q) M. Chen, L. Q. Yang, Y. Z. Li, Y. H. Qu, G. H. Pan, X. M. Feng and X. H. Liu, Sci. China: Chem., 2024, 67, 542–550 CrossRef CAS.
- For selected examples about bimetallic catalysis, see: (a) Y. S. Chen, S. X. Dong, X. Xu, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2018, 57, 16554–16558 CrossRef CAS PubMed; (b) C. R. Xu, K. X. Wang, D. W. Li, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2019, 58, 18438–18442 CrossRef CAS PubMed; (c) S. L. Ge, W. D. Cao, T. F. Kang, B. W. Hu, H. Zhang, Z. S. Su, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2019, 58, 4017–4021 CrossRef CAS PubMed; (d) Y. S. Chen, Y. Liu, Z. J. Li, S. X. Dong, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2020, 59, 8052–8056 CrossRef CAS PubMed; (e) X. Y. Hu, X. X. Tang, X. Y. Zhang, L. L. Lin and X. M. Feng, Nat. Commun., 2021, 12, 3012 CrossRef PubMed; (f) C. R. Xu, J. L. Qiao, S. X. Dong, Y. Q. Zhou, X. H. Liu and X. M. Feng, Chem. Sci., 2021, 12, 5458–5463 RSC; (g) Y. Xu, H. K. Wang, Z. Yang, Y. Q. Zhou, Y. B. Liu and X. M. Feng, Chem, 2022, 8, 2011–2022 CrossRef CAS; (h) W. Wang, F. Q. Zhang, Y. B. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2022, 61, e202208837 CrossRef CAS PubMed; (i) H. K. Wang, Y. Xu, F. Q. Zhang, Y. B. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2022, 61, e202115715 CrossRef CAS PubMed.
- (a) X. B. Lin, Y. Tang, W. Yang, F. Tan, L. L. Lin, X. H. Liu and X. M. Feng, J. Am. Chem. Soc., 2018, 140, 3299–3305 CrossRef CAS PubMed; (b) X. B. Lin, W. Yang, W. K. Yang, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2019, 58, 13492–13498 CrossRef CAS PubMed; (c) W. Yang, M. P. Pu, X. B. Lin, M. Chen, Y. J. Song, X. H. Liu, Y. D. Wu and X. M. Feng, J. Am. Chem. Soc., 2021, 143, 9648–9656 CrossRef CAS PubMed; (d) S. Meninno, F. Franco, M. Benaglia and A. Lattanzi, Adv. Synth. Catal., 2021, 363, 3380–3410 CrossRef CAS; (e) X. B. Lin, M. P Pu, X. P. Sang, S. Y. Li, X. H. Liu, Y. D. Wu and X. M. Feng, Angew. Chem., Int. Ed., 2022, 61, e202201151 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03315a |
| ‡ These two authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |