
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMild ketyl radical generation and coupling with alkynes enabled by Cr catalysis: stereoselective access to E-exocyclic allyl alcohols†
Zheng
Luo
,
Xiaoyu
Zhang
,
Zaiyang
Li
,
Meiming
Luo
 and
Xiaoming
Zeng
*
and
Xiaoming
Zeng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: zengxiaoming@scu.edu.cn
First published on 12th June 2024
Abstract
The mild catalytic generation of ketyl radicals for organic transformations remains an unsolved issue, although it facilitates the discovery of metal-catalyzed reactions with the features of high functional group tolerance. Here, we report the generation of the ketyl radicals and coupling with alkynes that was enabled by cost-effective chromium catalysis, allowing for the formation of valuable E-exocyclic allyl alcohols with high stereo- and chemoselectivity. A broad range of synthetically useful functional groups that are sensitive to strong reductants are compatible with the catalytic system, providing access to diverse substituted E-exocyclic allyl alcohols under mild conditions. Appended hydroxyl groups in products are facilely late-stage functionalized in accessing numerous derivatives, as well as the enantio-enrichment of exocyclic allyl alcohol using chiral ligands. Mechanistic studies suggest that bipyridine-ligated Cr(II) complex serves as a reactive catalyst enabling the generation of the ketyl radical for coupling, giving vinyl radical, followed by the combination of Cr and transmetalation with Cp2ZrCl moiety in affording oxazirconiumacycle. This reaction provides a new opportunity for the mild formation of transient ketyl radicals from widely accessible aliphatic aldehydes for coupling with Earth-abundant metal catalysis.
Introduction
Coupling reactions with transient radicals are one of the most powerful tools in the formation of C–C and C–heteroatom bonds in modern organic chemistry.1 In this context, carbon-centered ketyl radicals or ketyl radical anions are valuable coupling intermediates, along with cross-coupling that enables the incorporation of important carbonyl groups to construct synthetically useful aliphatic alcohols.2 However, because of the high reduction potential of carbonyl motifs,3 especially for reducing aliphatic carbonyls, stoichiometrically strong reductants, such as Na, K and Ti, are usually required for the generation of the ketyl radicals via the single electron transfer (SET) process.4 Alternatively, the thermodynamic barrier for ketyl formation can be overcome by the use of stoichiometric Kagan's reagent (SmI2),2b,5 as well as by combining strong reductants of Zn/Hg in the catalytic formation of the ketyl radicals.6 Despite the progress in developing photoredox catalysis7 and reductive electrocatalysis,8 mild strategies using metal catalysis for the reduction of carbonyls to the ketyl radicals for couplings are underdeveloped, particularly those that use earth-abundant 3d metal catalysts. In addition, to overcoming high reduction potential, chemoselectivity in metal-catalyzed reactions of the carbonyls without reducing resulting ketyl-coupled unsaturated adducts remains an issue. As a result, current studies focus on ketyl couplings with olefins to produce saturated products (Scheme 1a).9 Approaches that enable the formation of unsaturated motifs by catalytic formation of the ketyl radicals and selective couplings without over-reduction or over-addition would be value-addition to functionalized alcohol derivatives.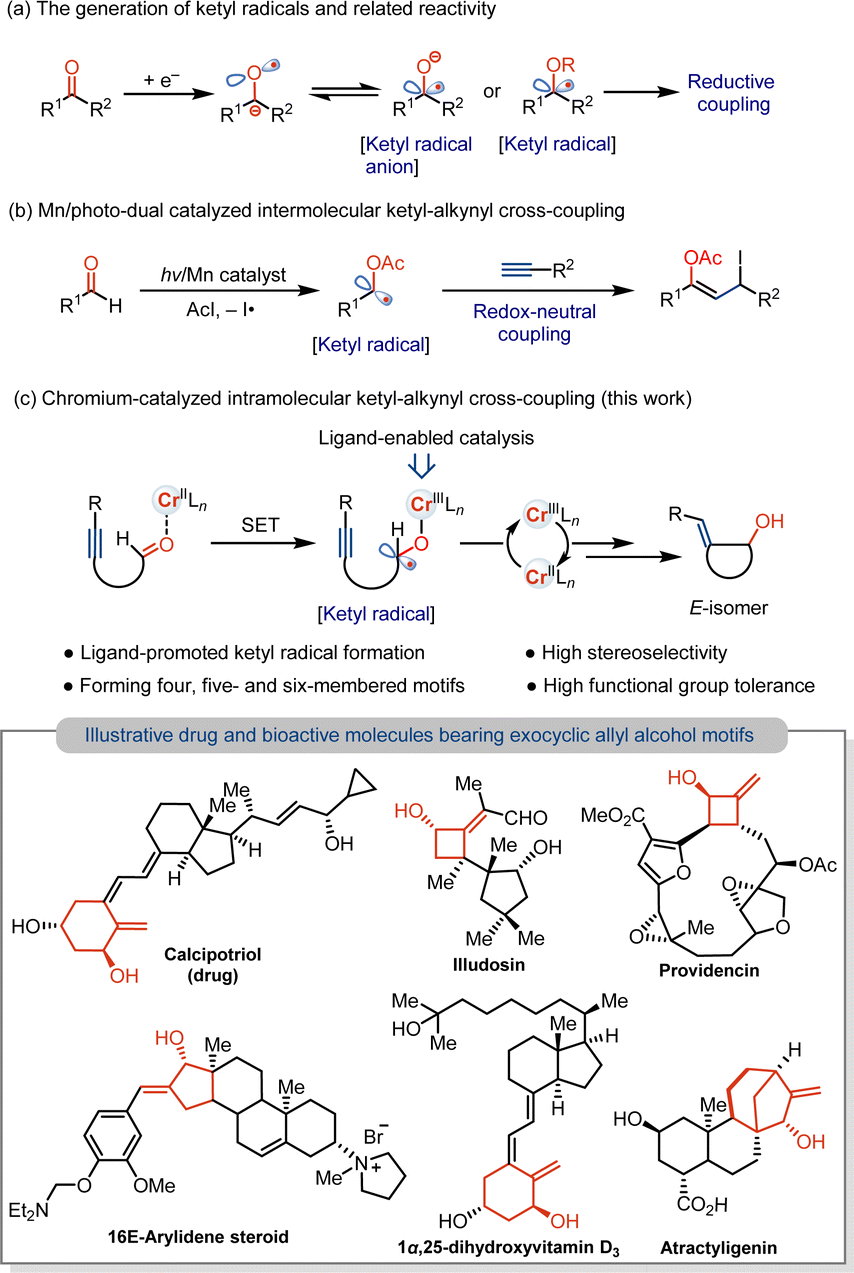 | ||
| Scheme 1 Catalytic formation of the ketyl radicals and couplings with the alkynes. | ||
To address the selectivity problem, an impressive redox-neutral intermolecular coupling of the ketyl radicals with the alkynes has been reported by mechanisms involving atom-transfer radical addition (Scheme 1b).7i We anticipate that an intramolecular coupling of aliphatic ketyl radicals generated in situ with the alkynes could be a possibility using appropriate 3d metal catalysts. This would need designing a mild reductive strategy to achieve the chemoselective formation of cyclic allyl alcohols and retain the vinyl group. In addition to overcoming the high reduction potential of aliphatic carbonyls, combining the vinyl radical with metals and regeneration of reactive catalysts remain unresolved issues. Herein, we report that a low-cost chromium(II) catalyst supported by a bipyridine ligand enables the mild formation of the ketyl radicals from unactivated aliphatic aldehydes, allowing for the intramolecular coupling with the alkynes under the mild conditions (Scheme 1c).10 This Cr-catalyzed ketyl coupling occurs by carbonyl reduction with Cr catalyst and keeps the resulting vinyl functional group intact, achieving the stereo- and chemoselective formation of the E-exocyclic allyl alcohols that are ubiquitous structural motifs found in many pharmaceuticals and bioactive molecules.11
Results and discussion
At the outset, we commenced our studies by the exploration of the formation of the ketyl radicals and intramolecular coupling by using alkynyl aliphatic aldehyde 1a as a precursor (Fig. 1). Commercially available salt of CrCl3 was chosen as a precatalyst in combination with 4,4′-di-tert-butyl-2,2′-dipyridyl (dtbpy) as the ligand, Mn as the reductant, and Cp2ZrCl2 with LiCl as the additive. We were pleased that the ketyl coupling occurs smoothly at ambient temperature, giving exocyclic allyl alcohol compound 2a in 67% yield and excellent E/Z-selectivity (>99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The replacement of CrCl3 by CrCl2 remarkably increased the conversion to afford allylic alcohol 2a in 87% yield. The reaction using Cr(acac)3 led to the formation of 2a in low yield. Nevertheless, a wide range of metal catalysts, such as FeCl2, CoCl2, NiCl2, CuCl2, AuCl3, AgCl, VCl3, MoCl3, WCl5 and RuCl3, almost showed no efficiency in promoting the transformation under present conditions. However, relatively low stereoselectivity was obtained in these reactions. Further studies suggested that ligands play important roles in promoting the reactivity of Cr and stereochemistry of the conversion. In the absence of the dtbpy ligand, the ketyl radical coupling with the alkyne could almost not proceed with Cr catalysis. The use of bipyridine (bpy) as a ligand resulted in a relatively low conversion and E/Z-selectivity, as well as the replacement by ligands, such as 4,4′-dimethyl-2,2′-dipyridyl (dmbpy), 4,4′-dimethoxy-2,2′-dipyridyl (dmobpy) and terpyridine (tpy). In the absence of Mn or Cp2ZrCl2, the ketyl coupling reaction does not occur in almost complete recovery of 1a, indicating their important roles in promoting the transformation. The reaction by the replacement of Mn by Zn or Mg produces product 2a in low yields (see ESI†), whereas the conversion is completely inhibited using Al as a reductant.
:1). The replacement of CrCl3 by CrCl2 remarkably increased the conversion to afford allylic alcohol 2a in 87% yield. The reaction using Cr(acac)3 led to the formation of 2a in low yield. Nevertheless, a wide range of metal catalysts, such as FeCl2, CoCl2, NiCl2, CuCl2, AuCl3, AgCl, VCl3, MoCl3, WCl5 and RuCl3, almost showed no efficiency in promoting the transformation under present conditions. However, relatively low stereoselectivity was obtained in these reactions. Further studies suggested that ligands play important roles in promoting the reactivity of Cr and stereochemistry of the conversion. In the absence of the dtbpy ligand, the ketyl radical coupling with the alkyne could almost not proceed with Cr catalysis. The use of bipyridine (bpy) as a ligand resulted in a relatively low conversion and E/Z-selectivity, as well as the replacement by ligands, such as 4,4′-dimethyl-2,2′-dipyridyl (dmbpy), 4,4′-dimethoxy-2,2′-dipyridyl (dmobpy) and terpyridine (tpy). In the absence of Mn or Cp2ZrCl2, the ketyl coupling reaction does not occur in almost complete recovery of 1a, indicating their important roles in promoting the transformation. The reaction by the replacement of Mn by Zn or Mg produces product 2a in low yields (see ESI†), whereas the conversion is completely inhibited using Al as a reductant.
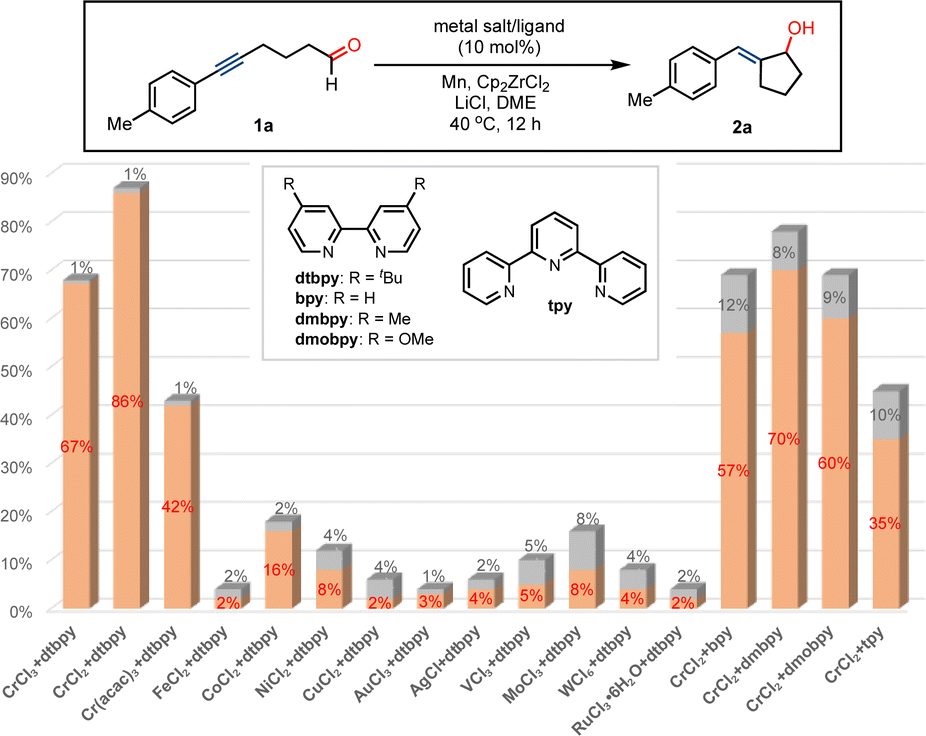 | ||
| Fig. 1 Studying the effect of metal salts and ligands on ketyl coupling to produce E-exocyclic allyl alcohol. | ||
Having optimal reaction conditions in hand, we examined the scope of the radical coupling using diverse substituted alkynyl aliphatic aldehydes. As shown in Scheme 2, the treatment of 5-hexynals that contain electron-donating substituents, such as tert-butyl, methoxy, cyclopropylmethoxy, benzyloxy, phenoxy and tert-butyldimethylsilyloxy on aryls by dtbpy-ligated Cr(II) catalysis enables the formation of annulated (E)-2-benzylidenecyclopentanol motifs 2c–h in 81–91% yields and 94:6–99:1 stereoselectivities. The incorporation of electron-withdrawing groups of phenyl and naphthyl into the alkynyl does not affect the coupling reaction and provides access to (E)-(biphenyl-4-ylmethylene)cyclopentanol 2i and (E)-(naphthylmethylene)cyclopentanols (2j and 2k) in good yields and high stereoselectivities. It was noted that the steric hindrance caused by the mesityl substituent around the alkynyl does not impede annulated coupling and produces the sterically congested compound 2l. Due to the mild conditions with Cr(II) catalyst and manganese reductant, halogen substituents, such as fluoride, polyfluoride, chloride, bromide, and iodide can be effectively compatible with the coupling system, providing a valuable strategy in the preparation of various halide-bearing 2-methylenecyclopentanols (2m–2s). It was noted that the thio group on the aryl of 5-hexynal does not hinder the catalysis, offering a strategy in the synthesis of product 2t. Using this methodology, 2-methylenecyclopentanols containing a range of synthetically useful groups, such as cyano, alkoxycarbonyl, trifluoromethyl, trifluoromethoxy, trimethylsilyl, boronate ester, alkenyl and alkynyl groups can be facilely accessible (2u–2ac). In addition, the reaction can be employed in the synthesis of methylenecyclopentanol derivatives that contain a range of heterocycle functional groups, such as benzofuranyl, dibenzo[b,d]furanyl, benzo[d][1,3]dioxol, 2,2-difluorobenzo[d][1,3]dioxolyl, 9H-carbazolyl, 1H-indazolyl, 1H-pyrazolyl and thiophenyl groups (2ad–2al).
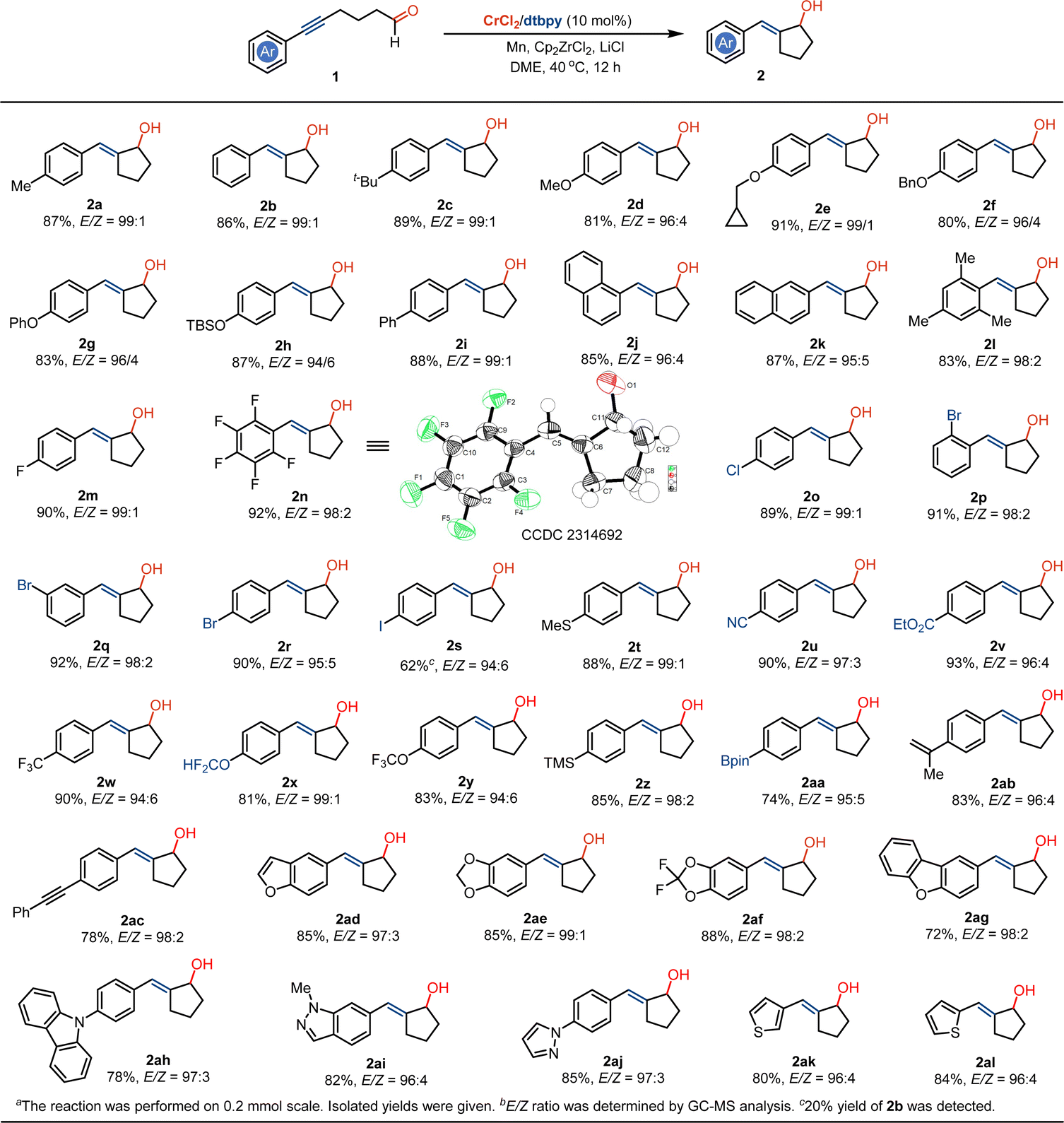 | ||
| Scheme 2 Cr(II)-catalyzed ketyl radical coupling reactions with alkynyl aliphatic aldehydes.a,b | ||
Inspired by these results, we examined whether the radical coupling could be applied in the construction of four- and six-membered ring-containing cyclitols. Using 4-pentynal as a precursor, we were pleased that the four-member ring-based exocyclic butanol 4a was obtained in moderate yields and high E-selectivity (Scheme 3). The treatment of 6-heptynal with Cr catalysis delivers six-membered (benzylidene)cyclohexanol in 73% yield and 99:1 E/Z selectivity (4b). The use of oxygen moiety as a linker in the radical coupling of alkynyl alcohol does not affect the conversion, offering a strategy in the formation of exocyclic tetrahydro-2H-pyranol 4c. Moreover, benzylidenyl-based 2,3-dihydro-1H-indenol and 1,2,3,4-tetrahydronaphthol motifs 4d and 4e can be prepared at ambient temperatures. In addition to the coupling with the alkyne, we wondered whether the ketyl radical coupling reaction with olefin could occur with Cr catalysis. By the use of alkenyl-substituted aliphatic aldehydes as precursors, we found that corresponding saturated five-membered cyclopentanols 4f–4i were formed in moderate-to-good diastereoselectivity.
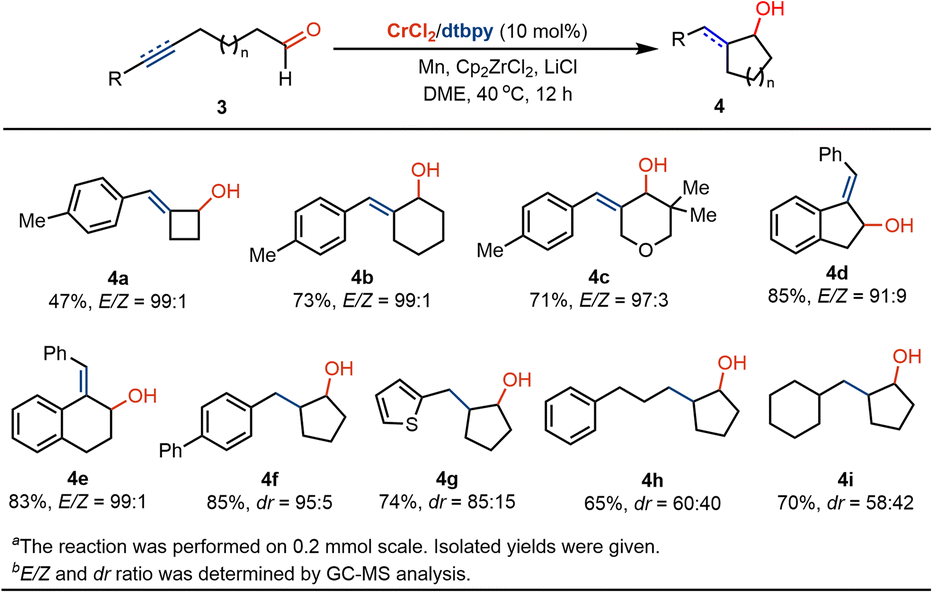 | ||
| Scheme 3 Application in the formation of cyclobutanols, cyclopentanols and cyclohexanol derivatives.a,b | ||
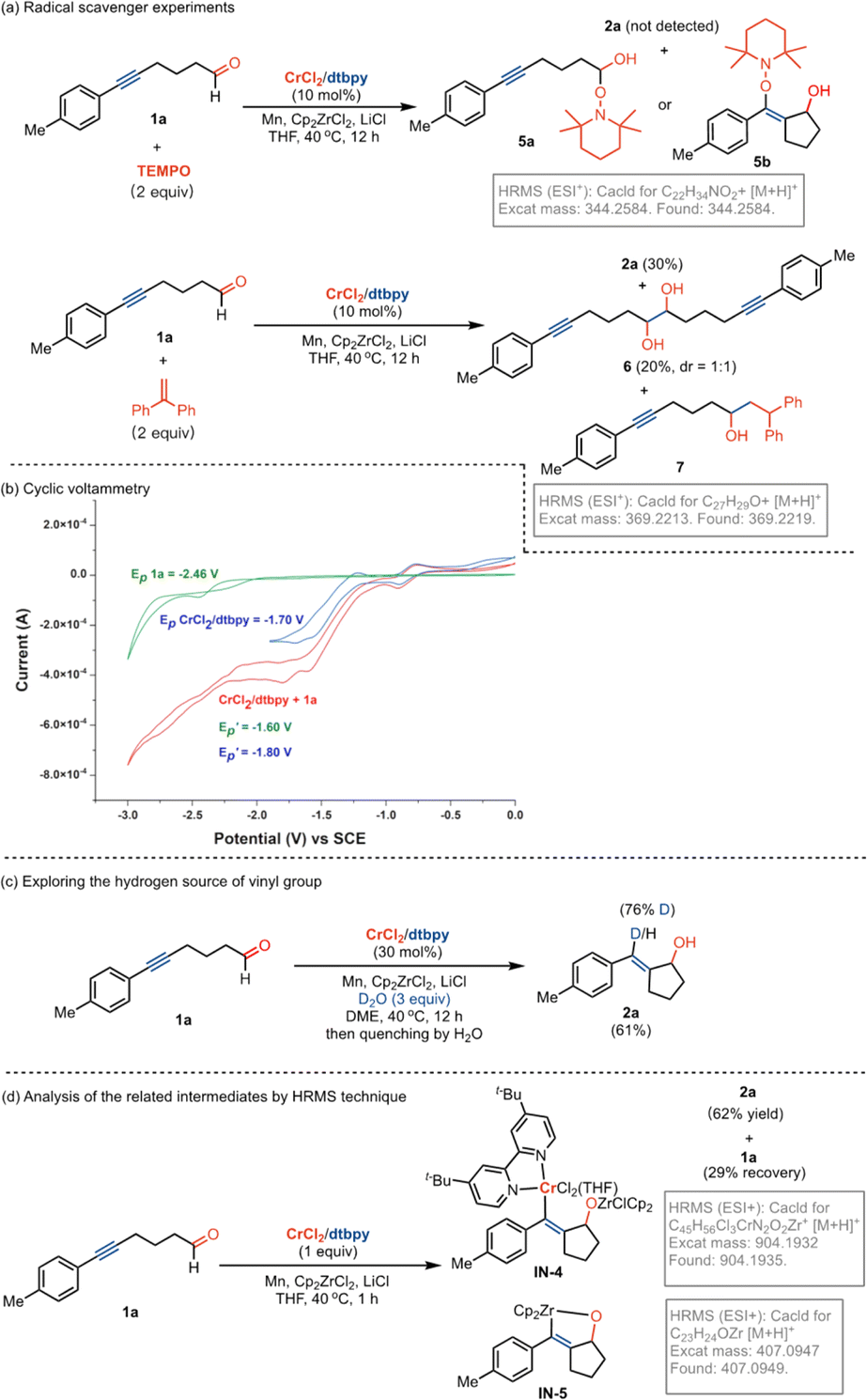
To gain insight into the reaction pathway, mechanistic experiments were performed. By the addition of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) as a radical scavenger into the reaction system, we found that the coupling was completely inhibited without the production of related compound 2a (Fig. 2a). The analysis of the residue by the high-resolution mass spectrum (HRMS) technique suggested that the TEMPO-trapped compound 5a or 5b might be formed. Interestingly, the replacement of TEMPO by 1,1-diphenylethylene in the reaction led to the formation of dialkynyl-containing diol 6 in 20% yield, combined with a small amount of adduct 7 by the addition of aldehyde to olefin. These results indicate that the formation of the ketyl radicals could be considered in the reaction. The experiments of cyclic voltammetry (CV) were carried out to study the activation of carbonyl by Cr catalysis. A high reduction potential for 5-hexynal 1a (Ep = −2.46 V) was observed (Fig. 2b). The coordination with the dtbpy ligand leads to the promotion of the reduction potential of CrCl2 (CrCl2/dtbpy: Ep = −1.70 V). Interestingly, the combination of the CrCl2/dtbpy complex with 1a leads to a great decrease in the reduction potential of 1a with the increased potential of the complex
with the increased potential of the complex 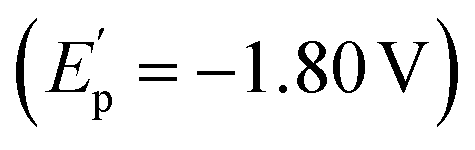 . It indicates that the coordination of CrCl2/dtbpy with 1a might promote the reduction process in giving the related ketyl radical. Subsequently, we explored the source of hydrogen in the vinyl scaffold of 2a. By the addition of 3.0 equivalent D2O in the reaction, we found that the coupling occurs smoothly to afford the compound 2a in 61% yield, with the incorporation of around 76% of deuterium into the vinyl group (Fig. 2c). The related metalated vinyl intermediate may be formed during the process. We explored related intermediates in the stoichiometric reaction by HRMS analysis (Fig. 2d). Both the vinylated chromate species IN-4 that contained the Cp2ZrCl moiety and oxazirconiumacycle IN-5 were detected, indicating that the ketyl radical coupling with the alkynyl, followed by combination with dtbpy-Cr(II), might be considered, which may undergo a transmetalation process by a further reaction with Cp2ZrCl2 in forming oxazirconiumacycle.
. It indicates that the coordination of CrCl2/dtbpy with 1a might promote the reduction process in giving the related ketyl radical. Subsequently, we explored the source of hydrogen in the vinyl scaffold of 2a. By the addition of 3.0 equivalent D2O in the reaction, we found that the coupling occurs smoothly to afford the compound 2a in 61% yield, with the incorporation of around 76% of deuterium into the vinyl group (Fig. 2c). The related metalated vinyl intermediate may be formed during the process. We explored related intermediates in the stoichiometric reaction by HRMS analysis (Fig. 2d). Both the vinylated chromate species IN-4 that contained the Cp2ZrCl moiety and oxazirconiumacycle IN-5 were detected, indicating that the ketyl radical coupling with the alkynyl, followed by combination with dtbpy-Cr(II), might be considered, which may undergo a transmetalation process by a further reaction with Cp2ZrCl2 in forming oxazirconiumacycle.
 | ||
| Fig. 2 Mechanistic studies. | ||
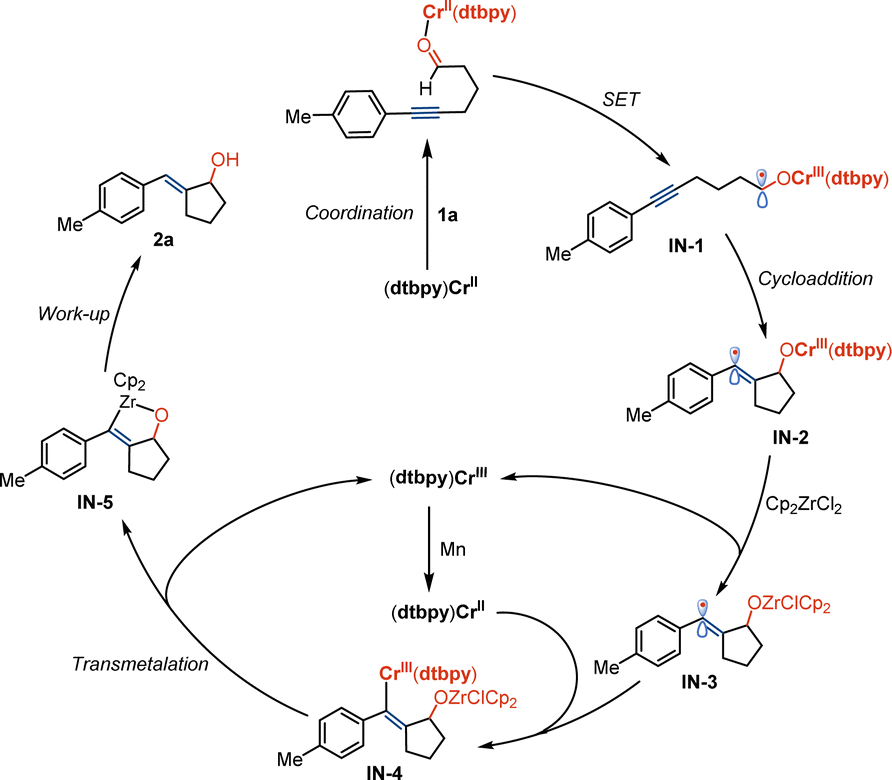
Based on the preliminary results and studies of Cr/Zr-catalyzed organic transformations,10e–k we hypothesized that the reaction is initiated by a single electron transfer process between dtbpy–Cr complex and 1a, resulting in the formation of ketyl radical IN-1 (Scheme 4). It may deliver the related vinyl radical IN-2 by the process of annulation and transmetalation with Cp2ZrCl2 to produce IN-3 and the (dtbpy)Cr(III) complex. The reactive Cr(II) catalyst can be regenerated by reduction with manganese and combination with vinyl radical by transmetalation between (dtbpy)Cr(III) and Zr species to produce oxazirconiumacycle IN-5. Exocyclic E-allyl alcohol compound 2a is formed by following the work-up process.
 | ||
| Scheme 4 Presumed reaction pathway. | ||
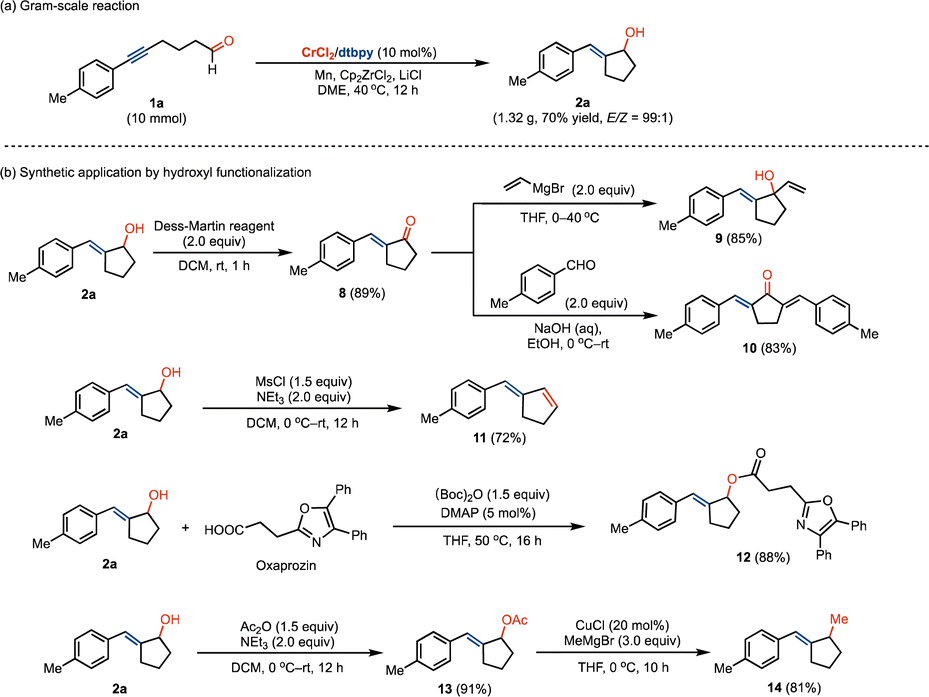
The Cr-catalyzed ketyl radical coupling reaction can be conducted on a gram-scale without loss of efficiency, giving exocyclic E-allyl alcohol products with high stereoselectivity (Scheme 5a). We investigated the late-stage functionalization of the resulting exocyclic allyl alcohol in the preparation of valuable motifs. By oxidation using the Dess–Martin reagent, α,β-unsaturated ketone 8 can be easily accessed, which reacts with vinyl Grignard reagent to afford vinylcyclopentanol 9 in good yields (Scheme 5b). The reaction of 8 with benzaldehyde led to the formation of functionalized bis(benzylidene)cyclopentanone motif 10. By the elimination of the hydroxyl group, the related 1,3-diene compound 11 could be obtained in 72% yield. The hydroxyl scaffold can be facilely functionalized in accessing the pharmaceutical compound of oxaprozin (12) by esterification. In addition, the transformation of the hydroxyl group by sequential esterification and methylation allowed us to prepare the methylated derivative 14.
 | ||
| Scheme 5 Gram-scale reaction and late-stage functionalization. | ||
Chiral exocyclic pentanols are synthetically intriguing motifs that serve as core structures and are widely found in numerous biologically active molecules and pharmaceuticals.11 As a result, the development of asymmetric catalysis in the stereoselective construction of these motifs is of significant interest.12 To probe the possibility of developing the Cr-catalyzed asymmetrical ketyl radical coupling, we studied the effect of chiral ligands on the transformation of enantioselective formation of chiral exocyclic allyl alcohols. As shown in Scheme 6, the use of chiral tert-butyl-substituted bis(oxazoline) ligand of L1, we found that the ketyl radical coupling occurs smoothly, leading to the formation of the product (R)-2a in 62% yield and 48% value of enantiomeric excess (ee). The replacement of dimethyl substituents by cyclopropanyl or methylbenzyl in the linker does not strongly promote enantioselectivity with Cr catalysis (L2 and L3). The reaction used phenyl-substituent bis(oxazoline) L4 to form the product (R)-2a with a good yield and ee value. Further studies suggested that the change of substituents of the linker and oxazolinyl groups did not greatly improve the transformation (L5–L13). While the incorporation of quinolinyl or pyridinyl as coordination groups into oxazoline (L14 and L15) led to relatively low conversions and enantioselectivity.
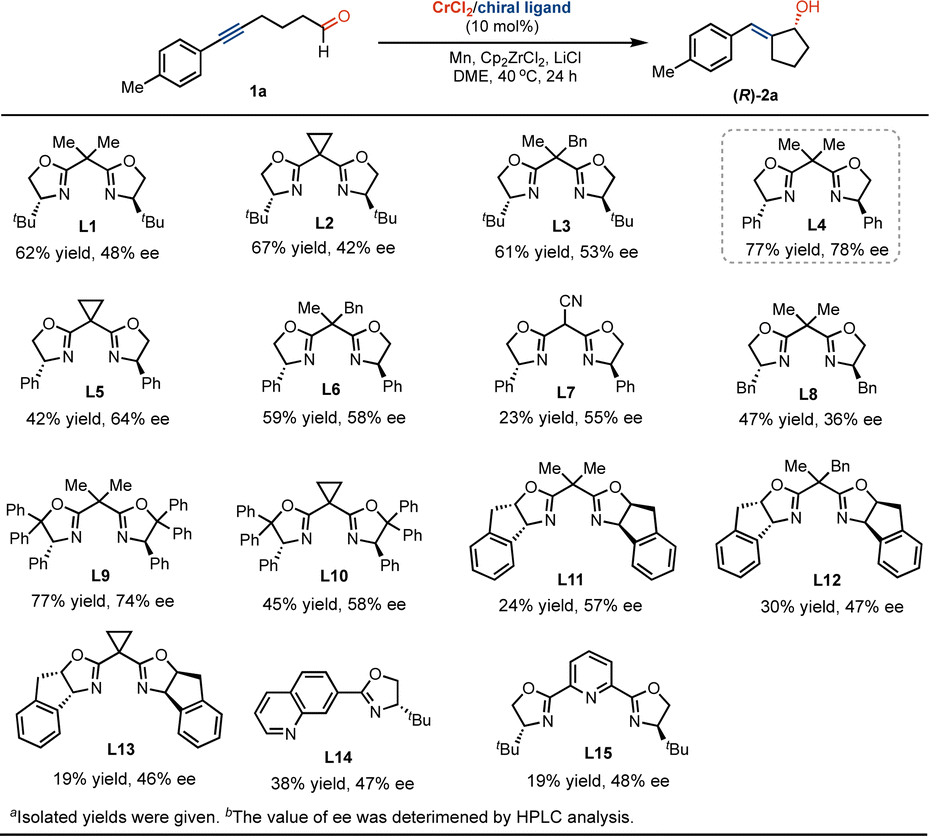 | ||
| Scheme 6 Exploration of Cr-catalyzed asymmetric ketyl radical coupling reaction.a,b | ||
Conclusions
In summary, we have developed a chromium-catalyzed ligand-enabled ketyl radical formation in coupling with the alkynes. This reaction was initiated by a bipyridine-supported Cr(II) catalyst for the generation of the ketyl radicals for alkyne hydrofunctionalization, offering a stereo- and chemoselective strategy in the synthesis of valuable E-exocyclic allyl alcohols. Because of mild catalytic conditions, a range of synthetically useful functionalities, including iodide, bromide, chloride, fluoride, cyano, amino, alkoxycarbonyl, alkynyl, and olefin groups, can be compatible with reaction systems. Compared with previous reports using strong reductants, this reaction employs (dtbpy)Cr(II) catalyst as a mild reagent to reduce aldehydes to the ketyl radicals, by the annulation and transmetalation with Cp2ZrCl motif in giving oxazirconiumacycle species. Formed exocyclic allyl alcohol derivatives can be modified by hydroxyl functionalization in the construction of various synthetically useful molecules. Further improvement is underway in the enantioselectivity for the development of asymmetric ketyl radical coupling reaction by Cr catalysis.Data availability
Experimental details and characterization of the products are available in the ESI.†Author contributions
X. Zeng conceived and directed the overall research project. Z. Luo, X. Zhang, and Z. Li designed and conducted the experimental studies. Z. Luo, M. Luo, and X. Zeng analyzed the data and contributed to the preparation of the manuscript. X. Zeng wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (grants 22125107 and 21971168) and Fundamental Research Funds for the Central Universities (20826041D4117) for financial support. The authors thank the Analytical & Testing Center, Dr D. Deng from the College of Chemistry, and the public Platform of the Analytical & Testing Center at Sichuan University for NMR testing.References
-
(a) U. Wille, Chem. Rev., 2013, 113, 813–853 CrossRef CAS PubMed
; (b) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed
-
(a) Á. Péter, S. Agasti, O. Knowles, E. Pye and D. J. Procter, Chem. Soc. Rev., 2021, 50, 5349–5365 RSC
- H. G. Roth, Synlett, 2016, 27, 714–723 CAS
-
(a) B. E. Kahn and R. D. Rieke, Chem. Rev., 1988, 88, 733–745 CrossRef CAS
-
(a) G. A. Molander and C. R. Harris, Chem. Rev., 1996, 96, 307–338 CrossRef CAS PubMed
- E. J. Corey and G. Z. Zheng, Tetrahedron Lett., 1997, 38, 2045–2048 CrossRef CAS
-
(a) K. T. Tarantino, P. Liu and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 10022–10025 CrossRef CAS PubMed
-
(a) P. Hu, B. K. Peters, C. A. Malapit, J. C. Vantourout, P. Wang, J. Li, L. Mele, P.-G. Echeverria, S. D. Minteer and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 20979–20986 CrossRef CAS PubMed
-
(a) P. Bichovski, T. M. Haas, D. Kratzer and J. Streuff, Chem.–Eur. J., 2015, 21, 2339–2342 CrossRef CAS PubMed
-
(a) M. Inoue, T. Suzuki and M. Nakada, J. Am. Chem. Soc., 2003, 125, 1140–1141 CrossRef CAS PubMed
-
(a) A. Kumar, M. I. Siddiqi and S. Miertus, J. Mol. Model., 2010, 16, 693–712 CrossRef CAS PubMed
-
(a) K. Zhang, Q. Liu, R. He, D. Chen, Z. Deng, N. Huang and H. Zhou, Green Chem., 2021, 23, 1628–1632 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2314692. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02967d |
| This journal is © The Royal Society of Chemistry 2024 |