
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFour-electron reduction of CO2: from formaldehyde and acetal synthesis to complex transformations
Sarah
Desmons†
a,
Julien
Bonin
*bc,
Marc
Robert
 *bcd and
Sébastien
Bontemps
*a
*bcd and
Sébastien
Bontemps
*a
aLCC-CNRS, Université de Toulouse, CNRS, 205 route de Narbonne, 31077 Toulouse Cedex 04, France. E-mail: sebastien.bontemps@lcc-toulouse.fr
bLaboratoire d’Electrochimie Moléculaire, Université Paris Cité, CNRS, F-75013, Paris, France. E-mail: julien.bonin@sorbonne-universite.fr; marc.robert@sorbonne-universite.fr
cInstitut Parisien de Chimie Moléculaire, Sorbonne Université, CNRS, F-75005, Paris, France
dInstitut Universitaire de France (IUF), F-75005, Paris, France
First published on 5th August 2024
Abstract
The expansive and dynamic field of the CO2 Reduction Reaction (CO2RR) seeks to harness CO2 as a sustainable carbon source or energy carrier. While significant progress has been made in two, six, and eight-electron reductions of CO2, the four-electron reduction remains understudied. This review fills this gap, comprehensively exploring CO2 reduction into formaldehyde (HCHO) or acetal-type compounds (EOCH2OE, with E = [Si], [B], [Zr], [U], [Y], [Nb], [Ta] or –R) using various CO2RR systems. These encompass (photo)electro-, bio-, and thermal reduction processes with diverse reductants. Formaldehyde, a versatile C1 product, is challenging to synthesize and isolate from the CO2RR. The review also discusses acetal compounds, emphasizing their significance as pathways to formaldehyde with distinct reactivity. Providing an overview of the state of four-electron CO2 reduction, this review highlights achievements, challenges, and the potential of the produced compounds – formaldehyde and acetals – as sustainable sources for valuable product synthesis, including chiral compounds.
 Sarah Desmons | Sarah Desmons is currently a postdoctoral researcher in the Laboratory of Inorganic Synthesis and Catalysis (LSCI) at École Polytechnique Fédérale de Lausanne (EPFL) in Switzerland. She obtained her PhD in Chemistry and Biochemistry for the chemo-enzymatic conversion of CO2 into carbohydrates from the Laboratoire de Chimie de Coordination (LCC) and the Toulouse Biotechnology Institute (TBI) in Toulouse, France. Her current research focuses on the enantioselective production of chiral amines via tailored bio-catalyzed hydroamination reactions. |
 Julien Bonin | Julien Bonin received his PhD in molecular physical chemistry from the Université Paris-Sud 11 in 2005. He then joined the Radiation Laboratory at the University of Notre Dame as a Postdoctoral Research Associate for one year. From 2006 to 2024, he has been an Associate, then a Full Professor of Chemistry at the Université Paris Cité, and he has now joined Sorbonne Université. His current research interests are related to photochemistry, redox catalysis and CO2 reduction. |
 Marc Robert | Long time Professor at Université Paris Cité, Marc Robert is now Professor at Sorbonne Université and a Senior member (Innovation Chair) of the Institut Universitaire de France (IUF). His work is dedicated to electrochemical and photochemical approaches of electron-transfer processes and the catalytic activation of small molecules (CO2, N2), in an effort to solve contemporary energy challenges. He is an International Advisory Board member at Angewandte Chemie International Edition, ChemSusChem (Wiley), Artificial Photosynthesis and ACS Electrochemistry (ACS). |
 Sébastien Bontemps | Sébastien Bontemps has received his PhD in 2006 from the University Paul Sabatier in Toulouse and carried out postdoctoral stays at the University of Chicago and McGill University. Since 2009, he has been a CNRS researcher in LCC-CNRS in Toulouse developing research studies directed at CO2 transformations, boron and hydride chemistry and at the synthesis of new first row metal complexes. |
1. Introduction
Research in CO2 transformations conducted by the Carbon Capture and Utilization (CCU) community is driven by both fundamental and applicative motivations. On the fundamental side, the goal is to find the keys to transform this thermodynamically and kinetically stable molecule. On the practical side, there is a desire to emulate nature by using CO2 either as a synthetic building block or as an energy carrier. These objectives gain urgency due to the increasing concentration of atmospheric CO2 and the inevitable depletion of fossil resources. Given its significance and broad scope, the field of CO2 transformation has been extensively covered in numerous reviews and books. Early reviews1 and comprehensive perspectives2 provide authoritative insights. Other reviews focus specifically on the use of CO2 as an energy carrier3 or as a synthetic reagent.4 From an applied perspective, the importance of Life Cycle Assessment (LCA)5 has been considered to evaluate the industrial potential of various processes.6 From a thermodynamic standpoint, this expansive field can be categorized into reactions involving a redox event at the carbon atom of CO2 (CO2 Reduction Reaction: CO2RR) or those that do not.4a Motivations for the CO2RR include using CO2 as an energy carrier, as energy is formally stored during CO2 reduction, and as a sustainable source of carbon. In the CO2RR, comprehensive reviews have focused on the type of reduction used, whether it involves thermal reduction with dihydrogen,7 hydroboranes,8 or hydrosilanes,9 (photo)electro reduction10 or bio-catalysed reduction.11 Other reviews explore the homogeneous12 or heterogeneous13 nature of the catalytic system.Within the CO2RR, the level of reduction of CO2 is arguably the most important parameter to consider. Since the carbon atom of CO2 is fully oxidized (+IV), it can be reduced by as much as 8e− giving rise to four possible even reduction states shown (Scheme 1). The +II, 0, –II and −IV carbon atom oxidation states can be reached by electroreduction or hydrogenation of CO2. The corresponding C1 reduction products are represented in Scheme 1(a): CO14 and HCOOH15 (2e−), HCHO and ROCH2OR (4e−), CH3OH15a,16 (6e−) and CH4 (ref. 17) (8e−). As shown in Scheme 1(b), the same oxidation states are reached with hydroelementation (E–H) reactions. Using mostly hydroborane (E = BR2) and hydrosilane (E = SiR3), HCOOE (2e−), EOCH2OE and HCHO (4e−), CH3OE (6e−) and CH4 (8e−) have indeed been reported. Although it does not correspond to a formal hydroelementation, the catalytic formation of CO (2e−) was reported with hydroborane18 and with diborane.19 The 4e− reduction products of CO2 – namely formaldehyde and acetal compounds – occupy a distinct position compared to other C1 reduction products (Scheme 1c). The reasons for this are explained below.
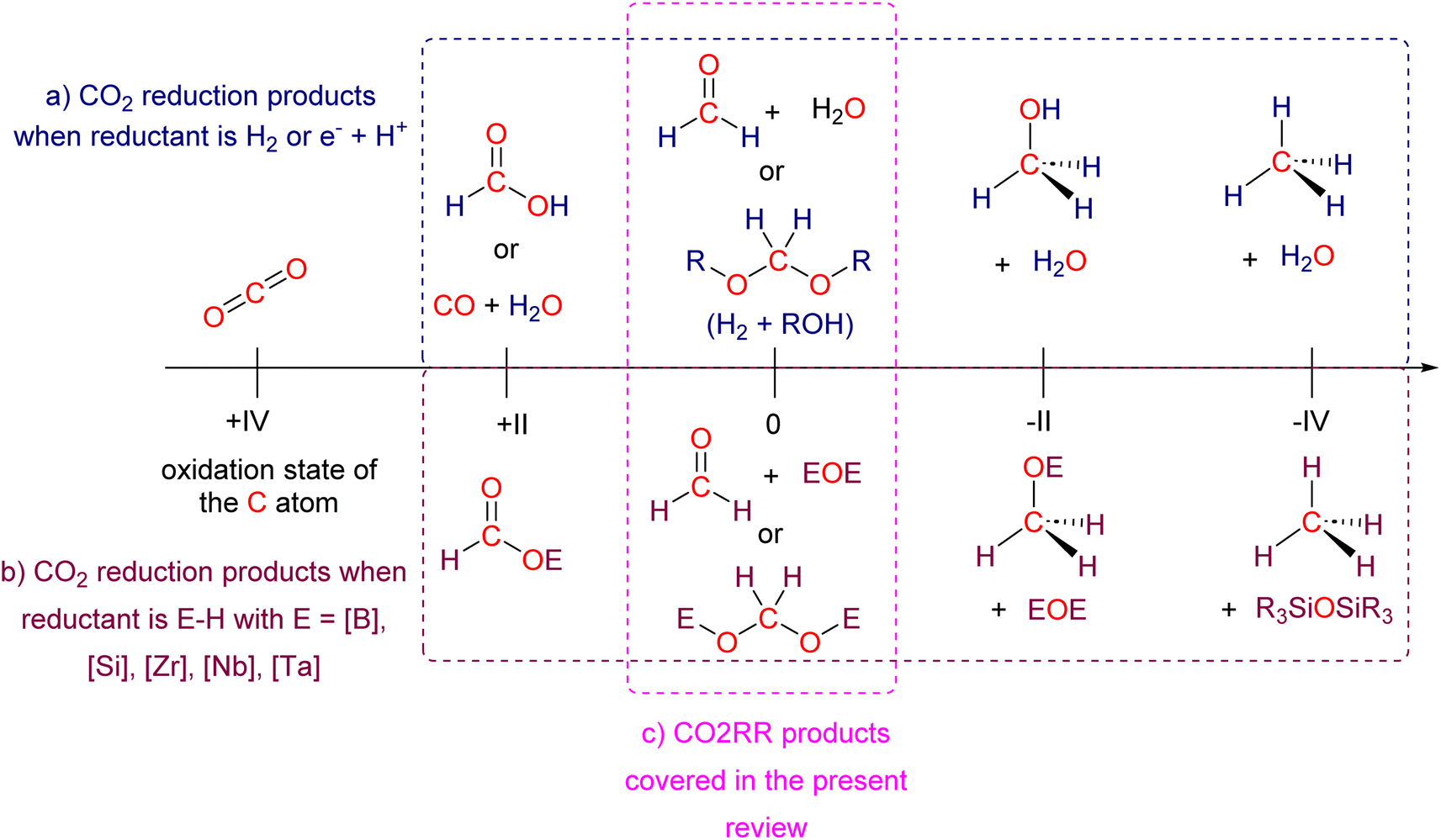 | ||
| Scheme 1 C1 products generated from CO2 reduction by (a) hydrogenation or electroreduction or (b) hydroelementation reactions. (c) Formaldehyde and acetal compounds as the products of four-electron reduction of CO2 covered in the present review. | ||
1.1. Why reducing CO2 by 4e− to formaldehyde?
In the context of an increasing demand for complex transformations of CO2 beyond the simple C1 building blocks mentioned earlier20,21 and the desire for long-term CO2 storage, formaldehyde offers unparalleled potential in terms of reactivity and versatility.22 More than 30 million tons of formaldehyde are produced and used annually by the chemical industry. This commodity compound is typically synthesized from the partial oxidation of methanol under energy-demanding conditions, representing over 35% of the total methanol transformation.22a,23 A mild and sustainable synthesis of formaldehyde from CO2 would thus represent a significant advancement for the chemical industry. Most industrial applications use formaldehyde as a reticulating agent in condensation reactions leading to various C–N, C–O, C–S, and C–C bonds.Furthermore, formaldehyde serves as a precursor to more complex molecules, including chiral ones,24 being a C1 and a Cn source in aldol25 and formose reactions,26 notably. These transformations are beyond the reach of other CO2 reduction products.
However, there is still no selective and sustainable synthesis of formaldehyde from CO2. This can be attributed to two major issues: (i) the 4e− reduction stage requires any catalytic system to overcome the energy barriers for both the 2 and 4e− reduction steps while preventing the 6e− reduction, and (ii) the detection of formaldehyde (HCHO) is challenging due to its intrinsic instability, limiting further development and optimization. Depending on the reaction media (pH, solvent, co-substrates), formaldehyde can polymerize, precipitate, or react with other chemicals present in the mixture, making its characterization difficult. Several detection methods have been used, such as optical methods, chemical derivatization for HPLC and GC analysis, or chemical conversion for colorimetric method analysis (established by Nash in the 1950s).27 Recently, a new and simple method for trapping and quantifying formaldehyde in aqueous solutions by 1H NMR was designed, otherwise allowing for isotope labelling experiments and thus unambiguous demonstration that the HCHO production arises from CO2 reduction.28
1.2. Why reducing CO2 by 4e− to acetal compounds?
The formation of acetal (EOCH2OE) from hydroelementation of CO2 is an important process linked to formaldehyde, because(i) Some of these acetal compounds were shown to quantitatively release formaldehyde, offering an efficient and selective access to it under particularly mild conditions and with isotope labelling proving the origin of formaldehyde. The generated formaldehyde was subsequently used as C1 and also Cn sources,29 and notably in the first enantioselective transformation of CO2 into a C4 carbohydrate,30 representing the highest level of complexity obtained from CO2 at that time.31
(ii) Beyond the release of formaldehyde, acetal compounds were also involved in transformations related to but distinct from formaldehyde, notably when used as a Cn source. Interestingly, in one case, boryl moieties remained on the product, demonstrating a difference in reactivity compared to formaldehyde.32 This finding should open opportunities to use the boryl or silyl functional groups as an integral part of new compounds as opposed to a waste moiety.
(iii) Finally, mild conditions and organic solvents used in hydroelementation enable in situ NMR monitoring leading to easier proofs of the origin of the acetal or formaldehyde by isotopic labelling experiments.
1.3. Objective and organization of the review
We aim at presenting the challenges and mechanistic considerations associated with formaldehyde33 and acetal synthesis from CO2 (Scheme 2a), as well as highlighting the reactions in which these reduction products have been involved (Scheme 2b). This review is organized by the type of reduction system: Hydrosilylation (Section 2), Hydroboration (Section 3), Other hydroelementation reactions (Section 4), Hydrogenation (Section 5), Electroreduction (Section 6), Photo(electro)reduction (Section 7), and Bio-catalysed reduction (Section 8). In each section, the reduction and subsequent transformations of the 4e− reduction products are presented, with special attention drawn to the mechanisms involved.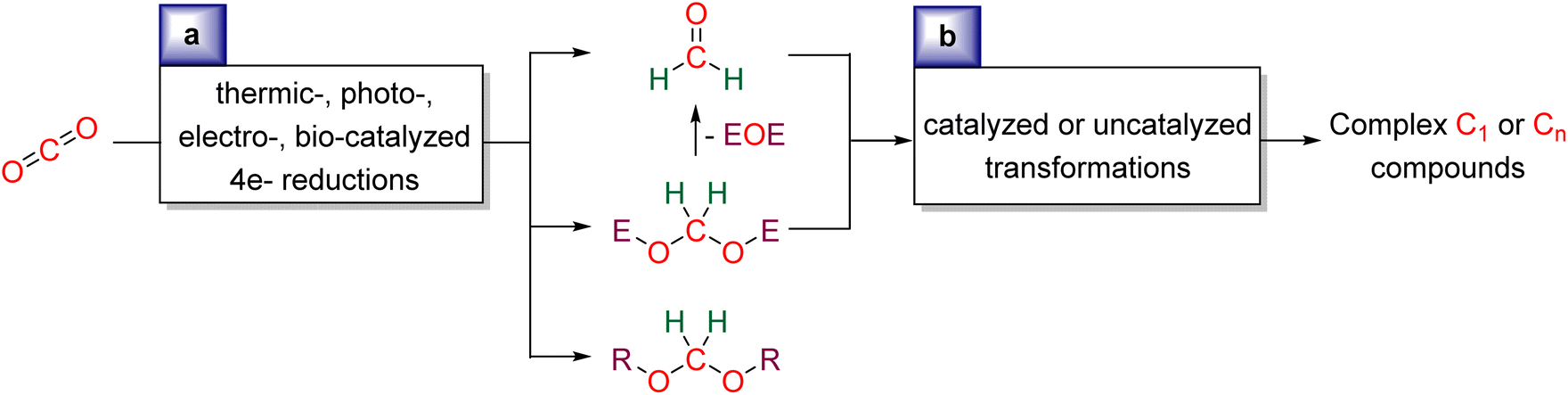 | ||
| Scheme 2 General scheme presenting the current review: (a) synthesis of formaldehyde or acetal compounds from CO2RR and (b) utilisation of formaldehyde and acetal as C1 and Cn sources. | ||
2. Hydrosilylation
Hydrosilylation of multiple C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CO and CN bonds has been intensely investigated, fuelling applications in the silicon industry as well as in synthetic chemistry, in asymmetric reductions in particular.34 Hydrosilylation of CO2 involves the addition of a Si–H bond to the CO bond, transferring the hydride to the carbon atom and forming a Si–O bond. This reaction is favoured kinetically and thermodynamically by the polarity of the Si–H bond (electronegativity (E. N.) of H = 2.2, Si = 1.9) and the formation of a strong Si–O bond (>500 kJ mol−1),35 respectively. As a consequence, CO2 hydrosilylation usually operates under mild conditions of temperature and pressure.
C, CO and CN bonds has been intensely investigated, fuelling applications in the silicon industry as well as in synthetic chemistry, in asymmetric reductions in particular.34 Hydrosilylation of CO2 involves the addition of a Si–H bond to the CO bond, transferring the hydride to the carbon atom and forming a Si–O bond. This reaction is favoured kinetically and thermodynamically by the polarity of the Si–H bond (electronegativity (E. N.) of H = 2.2, Si = 1.9) and the formation of a strong Si–O bond (>500 kJ mol−1),35 respectively. As a consequence, CO2 hydrosilylation usually operates under mild conditions of temperature and pressure.
CO2 hydrosilylation was first reported in 1981 with Ru- and Pd-based catalysts, independently.36 In both cases, the reduction process led to formoxysilane (2e− reduction product). Hydrosilylation beyond the 2e− reduction stage was hypothesized in these two initial reports based on the observation of bis(silyl)ether (R3SiOSiR3) and of a small amount of formaldehyde (<4% yield) with the Ru-based catalyst.36b In 1989, an Ir-based catalyst enabled unambiguous observation of the hydrosilylation of CO2 beyond the 2e− reduction stage. In this pioneering study, it was remarkable that the 2, 4 and 6e− reduction products – formoxysilane, bis(silyl)acetal (BSA) and methoxysilane, respectively – were subsequently observed.37 Finally, the first report of CO2 hydrosilylation into methane was published in 2006 by Matsuo and Kawaguchi with a [Zr]/B(C6F5)3 catalytic system.38
Since these initial reports, the reduction of CO2 by 4e− with hydrosilane witnessed a growing attention. The present section aims to cover in detail (i) the formation of BSA and formaldehyde in part 2.1, (ii) the mechanisms accounting for their generation in part 2.2, (iii) the proposed origin of the selectivity toward BSA in part 2.3, and (iv) their use as reactive intermediates in reductive functionalization reactions leading to more complex products in part 2.4 (Scheme 3).
 | ||
| Scheme 3 General scheme of Section 2: CO2 hydrosilylation. | ||
2.1. Hydrosilylation of CO2 into bis(silyl)acetals (BSAs) and formaldehyde
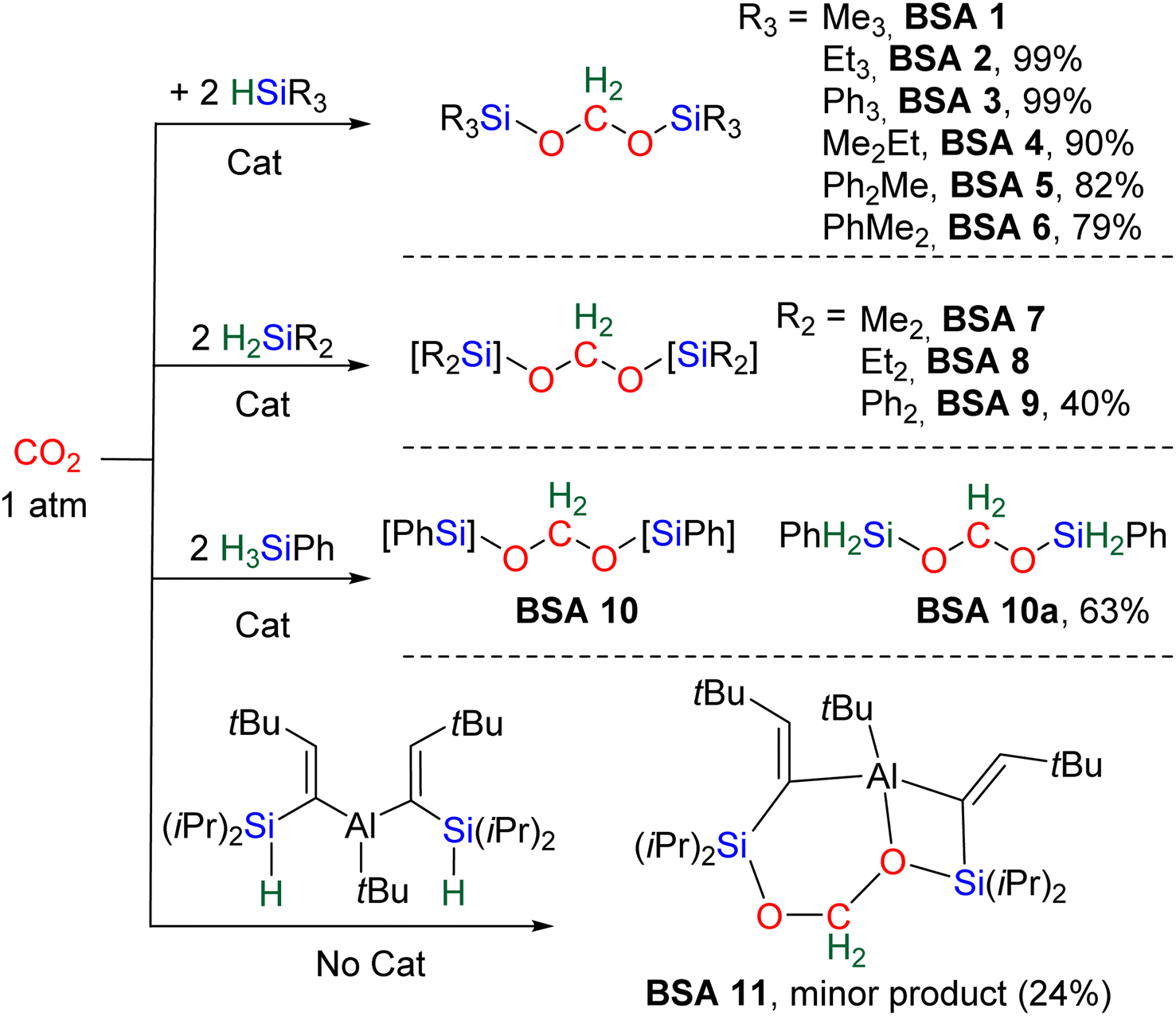 | ||
| Scheme 4 Synthesis of BSA 1–11. The yields indicated are the best reported yields. | ||
| BSA | Solvent | 13C(CH2) | δ 1H(CH2) (ppm) | δ 29Si (ppm) | Ref. | |
|---|---|---|---|---|---|---|
| δ (ppm) | 1 J CH (Hz) | |||||
| a Isolated. | ||||||
| 1 | C6D6 | — | 164 | 5.03 | — | 37 |
| 2 | C6D6 | 84.6 | — | 5.03 | 18.3 | 39 |
| 3 | C4D5Br | 85.5 | — | 5.55 | — | 38 |
| C6D6 | 85.7 | 164.4 | 5.45 | — | 38 | |
| 4 | C6D6 | 84.0 | — | 5.02 | 17.72 | 40 |
| 5 | C6D6 | 84.9 | — | 5.21 | −2.74 | 40 |
| 6 | C6D6 | 84.5 | — | 5.06 | 6.99 | 40 |
| 7 | C6D6 | 85.4 | 164 | 5.02 | — | 37 |
| 84.5 | 5.08 | |||||
| 83.8 | 5.12 | |||||
| 8 | — | — | — | — | — | 37 |
| 9 | CD3CN | 83 | — | 5.8 | — | 41 |
| 10 | C6D6 | 85.6 | — | — | — | 42 |
| 11 | C6D6 | 87.6 | — | 5.04 | 24.0 | 43 |
| 5.18 | ||||||
Most of the characterized BSA features tertiary silane moieties (BSA 1–6 and BSA 11). Three of them were isolated (BSA 2, 3 and 11) and the latter two were characterized by XRD. The higher stability of (silyl)acetals featuring tertiary silyl moieties is discussed in the mechanism section, Section 2.2. Catalysts were necessary to generate BSA but for BSA 11, which was derived from the hydrosilylation of CO2 by an aluminum complex featuring two hydrosilane pendent moieties. BSA 11 was generated as a minor product in 24% yield and features an acetal fragment stabilized by the Lewis acidic alane moiety.43
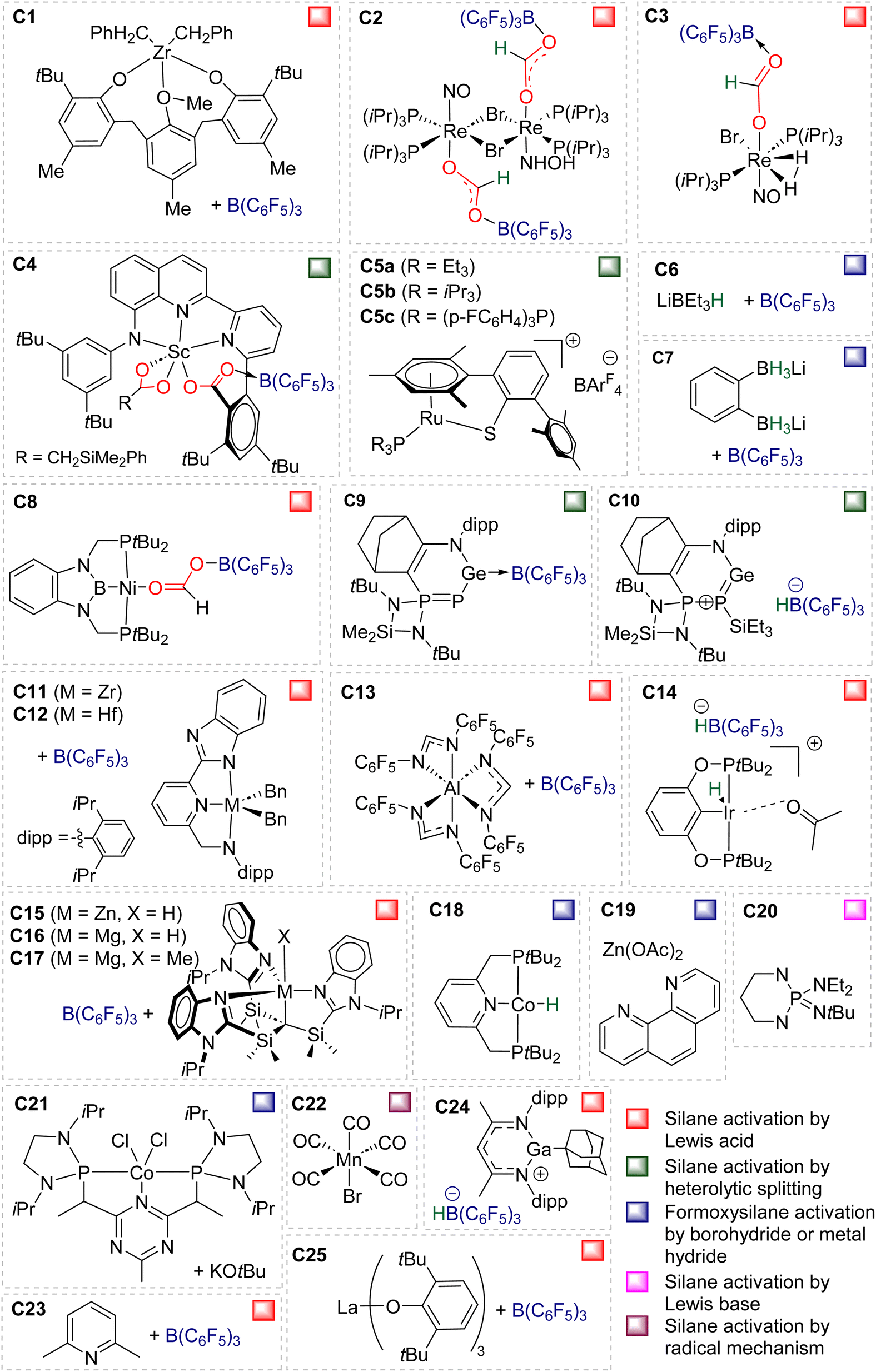 | ||
| Fig. 1 Catalysts C1–C25 leading to the selective formation of BSA. | ||
Table 2 gathers the catalytic performances (turnover number – TON, turnover frequency – TOF, yield) of catalysts C1–25. Key features are the mild conditions in terms of temperature (ranging from 25 to 80 °C) and CO2 pressure (from stoichiometric quantities to 6 atm). We have selected the conditions affording the best performances in terms of TON, TOF and/or yield for each catalytic system. TOFs are not given at the initial rate because such data were available only for C5 (Table 2). All BSA derivatives were obtained selectively with the exception of BSA 1, 7 and 8. Most of the TON values are in between 10 and 89. The catalysts C1, C8, C11, C12, C17 and C25 afford high TONs of 203, 395, 285, 620, 178 and 670, respectively. Only catalysts C4 and C8 led to exceptionally high TON values of 3400 and 1200, respectively. Moreover, C8 and C25 led to maximum TOFs of 55.8 and 52 h−1, respectively, while all the other catalysts led to TOFs between 0.1 and 10.4 h−1.
| BSA | Catalyst | TONa | TOF (h−1) | Yieldb (%) | P CO2 | T (°C) | Time (h) | [Cat] (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a TON = silane converted/catalyst loading unless otherwise stated. b In situ yield unless otherwise stated. c Initial rate measured after 1 hour. d No other product detected. e Isolated yield. f atm unless otherwise stated. g 2 eq. vs. CO2. h 4 eq. vs. CO2. i Based on hydrosilane and on CO2 conversion. j Based on CO2 conversion. | |||||||||
| 2 | C1 | 203 | 1.2 | 82 | 1 | 25 | 165 | 0.9 | 38 |
| C2 | 89 | 5.9 | 87 | 5 | 80 | 15 | 1 | 44 | |
| C3 | 95 | 7.3 | 89 | 5 | 80 | 13 | 1 | ||
| C4 | 1000 | 10.4 | 94e | 4 | 65 | 96 | 0.1 | 45 | |
| 3400 | n.d | 94e | 4 | 65 | n.d | 0.02 | |||
| C5a | 25 | 6.2/11c | 99 | 5 | 80 | 4 | 4 | 40 | |
| 49 | 1.5/8.6c | 80 | 5 | 80 | 32 | 2 | |||
| C5b | 25 | 0.51/<0.01c | 89 | 5 | 80 | 48 | 4 | ||
| C5c | 25 | 0.34/0.00c | >99 | 5 | 80 | 72 | 4 | ||
| C6 | n.d | n.d | 91 | 1 | 50 | 24 | 10 | 46 | |
| C7 | n.d | n.d | 54 | 1 | 50 | 41 | 10 | ||
| C8 | 1200 | 55.8 | 60d | 4 | 70 | 21.5 | 0.05 | 39 | |
| 910 | 26.8 | 91d | 4 | 70 | 34 | 0.1 | |||
| C9 | 20 | 0.8 | 82 | 3 | 60 | 24 | 5 | 47 | |
| C10 | |||||||||
| C11 | 395 | 6.6 | 76 | 1 | 22 | 60 | 0.24 | 48 | |
| C12 | 285 | 5.9 | 55 | 1 | 22 | 48 | 0.24 | ||
| C13 | 9 | 0.69 | 60 | 6 | 80 | 13 | 1.5 | 49 | |
| C22 | 57 | 57.3 | 86 | 4 | 50 | 1 | 1.5 | 50 | |
| C24 | 17 | 0.3 | 85 | 2 | 60 | 58 | 10 | 51 | |
| C25 | 99 | 99 | 99 | 5 | 25 | 1 | 1 | 52 | |
| 3 | C1 | 50 | 0.13 | 64 | 1 | 25 | 384 | 2 | 38 |
| C14 | 36 | 2 | 100 | 1 | 23 | 22.7 | 2.5 | 53 | |
| C15 | 12 | 0.1 | n.d | 1 | 25 | 120 | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 :10 |
54 | |
| C16 | 83 | 1.2 | n.d | 1 | 25 | 69 | 0.5:2.5 |
||
| C17 | 178 | 2.5 | n.d | 1 | 25 | 72 | 0.5:2.5 |
29a | |
| n.d | n.d | 95e | 1 | 25 | 672 | 1 | |||
| C25 | 670 | 52 | 67 | 5 | 80 | 13 | 1 | 52 | |
| 4 | C5a | 31 | 0.57/7.8c | 64 | 5 | 80 | 55 | 2 | 40 |
| 25 | 2.3/12c | 90 | 5 | 80 | 11 | 4 | |||
| 5 | C5a | 17 | 0.23/3.9c | 24 | 5 | 80 | 73 | 2 | 40 |
| 25 | 0.69/12c | 79 | 5 | 80 | 36 | 4 | |||
| C8 | 620 | 16.5 | 62d | 4 | 70 | 37.5 | 0.1 | 40 | |
| 137 | 18.3 | 82d | 4 | 70 | 7.5 | 0.6 | |||
| 6 | C5a | 32 | 0.44/6.2c | 55 | 5 | 80 | 73 | 2 | 40 |
| 24 | 0.32/9.5c | 79 | 5 | 80 | 75 | 4 | |||
| C8 | 121 | 40 | 78 | 4 | 70 | 3 | 0.6 | 39 | |
| 9 | C13 | 3.5 | 0.07 | 31 | 6 | 80 | 48 | 1 | 49 |
| C19 | 40i | 1.7i | 40i | 1 mmol | 25 | 24 | 1 | 55 | |
| C20 | 5.6 | 1.4 | 7 | 5 | 25 | 4 | 1.25 | 56 | |
| 10 | C18 | 32/126j | 1.2/4.8j | 63 | 0.4 mmol | 25 | 26 | 2 | 42 |
In addition to TON and TOF, yields are important to assess the selectivity of a given system. This is particularly important when the synthesized BSA is intended to be used as an intermediate in one-pot transformation (see reductive functionalization, Section 2.4). Most of the catalysts afforded BSA in yields higher than 73%. BSA 2 was notably isolated from C8-catalyzed CO2 hydrosilylation on the NMR scale, while larger scales of BSA 2 (570 mg, 2.1 mmol) and BSA 3 (2.67 g, 4.7 mmol) were isolated with C4- and C17-catalyzed reactions, in 94 and 95% yield respectively. This preparative scale notably facilitated the utilization of BSA 3 in subsequent transformations (see reductive functionalization, Section 2.4). The yields for hydrosilylation of CO2 are given based on the hydrosilane and not on the CO2 conversion in the vast majority of the cases. This is due to the difficulty in controlling the amount and conversion of CO2 as a gas with 13C NMR titration and to the fact that silane is considered to be the most expensive reactant. Yields based on CO2 conversion are nonetheless significant because (i) it allows for more complete description of a given catalytic system by reporting its performances based on both reactants and (ii) silane is not necessarily the most expensive substrate, notably when isotopically labelled CO2 is used. Performance data based on CO2 conversion are particularly important when 11CO2 and 14CO2 are used for radiochemistry.57
Compounds C26,58C27,37C28,59C29,60C30,61C31,41C32,62C33,49C34,49C3563 and C3664 were shown to catalyse the hydrosilylation of CO2 affording BSA as a minor product (Fig. 2). Most of these systems were optimized for the generation of methoxysilane and/or methane and not for BSA. The observation of BSA as intermediates in many cases confirms that (silyl)acetals are very often on the reaction pathway to the 6 and 8e− reduction products in hydrosilylation processes.
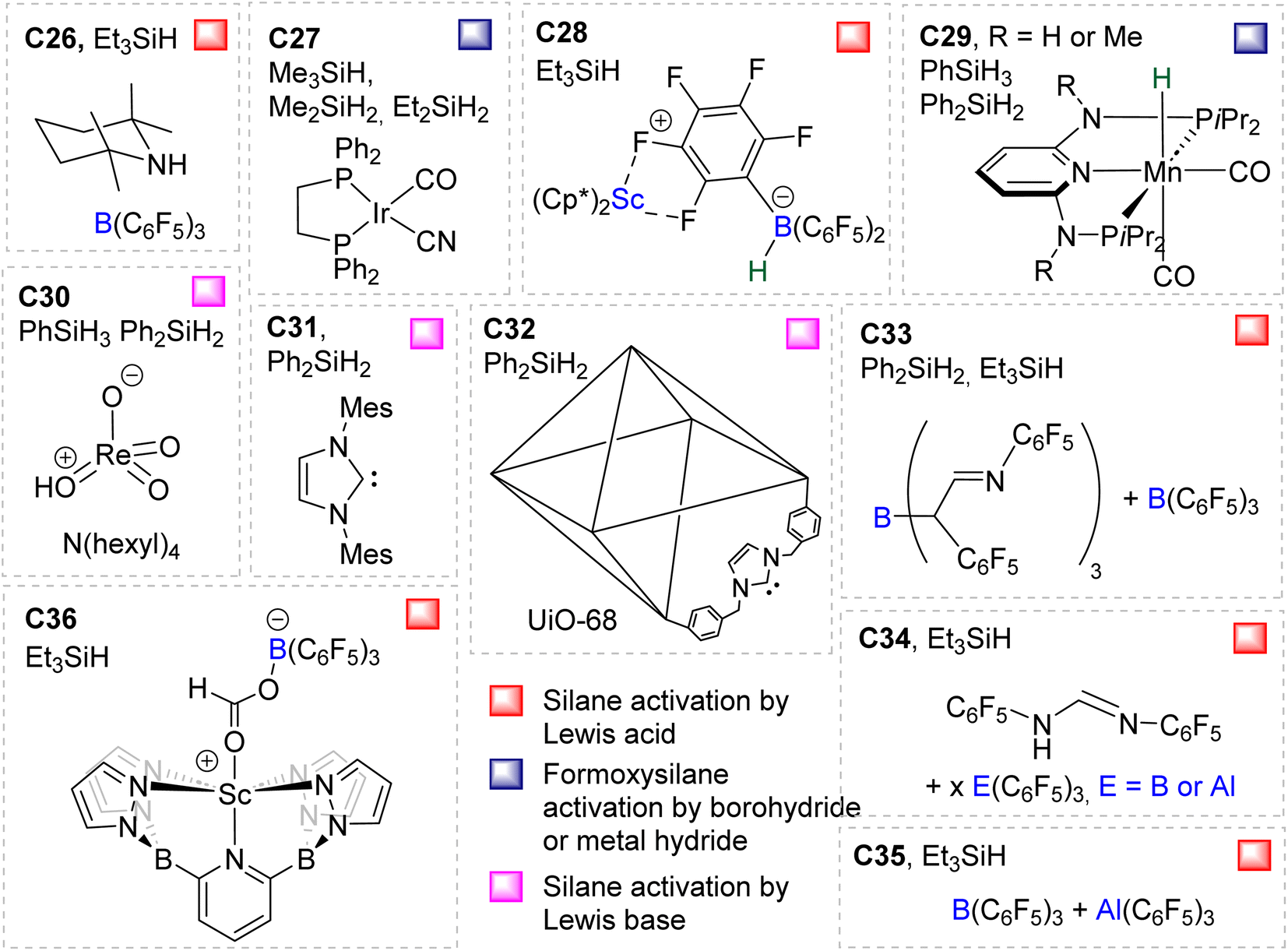 | ||
| Fig. 2 Unselective catalytic systems C26–C36 and hydrosilane employed. | ||
 | ||
| Scheme 5 Early observation of formaldehyde from CO2 hydrosilylation. | ||
More recently, BSA was utilized as an intermediate to selectively access formaldehyde (Scheme 6). The air stable BSA 3 featuring triphenylsilyl groups was shown to quantitatively deliver formaldehyde in DMSO/H2O at 100 °C or in the presence of H2SO4 at room temperature.29a Alternatively, CsF promoted the generation of formaldehyde at room temperature, presumably via the breaking of the Si–O bond after fluoride attack on the silicon centre.
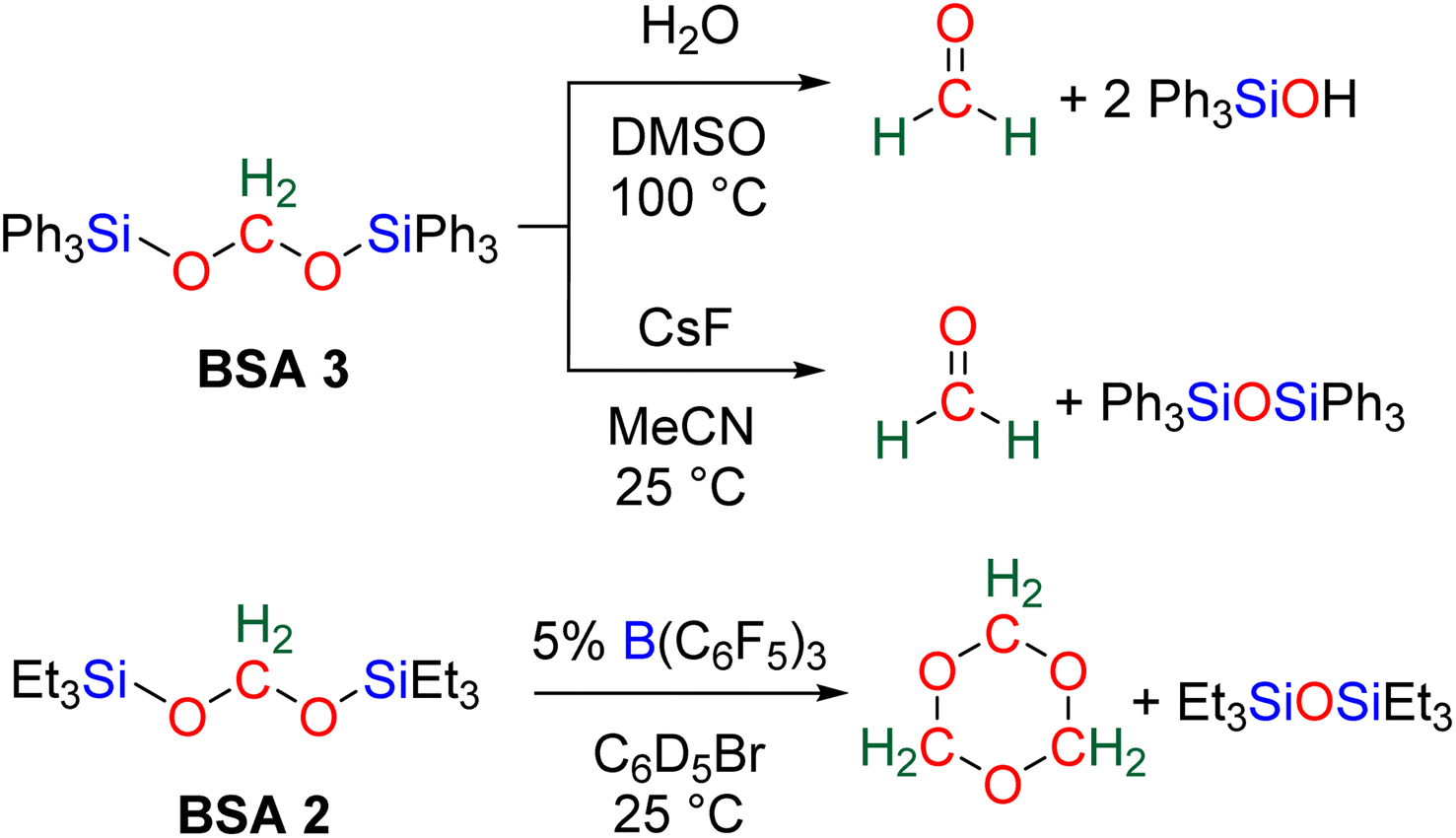 | ||
| Scheme 6 Quantitative generation of formaldehyde from BSA 2 and 3. | ||
In another study, BSA 2 featuring triethylsilyl moieties was shown to quantitatively release formaldehyde in the form of trioxane within 1 h in the presence of 5 mol% of B(C6F5)3 as the catalyst at room temperature.63 Conversely, BSA 3 did not release formaldehyde in the presence of 22 mol% of B(C6F5)3 in 10 days at room temperature.29a These contrasting reactivities between BSA 2 and BSA 3 highlight the influence of the silyl fragment on the reactivity of the BSA.
2.2. Mechanistic considerations
The 4e− reduction of CO2 into bis(silyl)acetal (BSA) or formaldehyde by hydrosilane takes place along two steps: (i) the 2e− reduction of CO2 into either formoxysilane or metal formate (Scheme 7, step (i)), followed by (ii) the hydrosilylation of the intermediate into the corresponding 4e− reduction products (Scheme 7, step (ii)). The elementary reactions accounting for step (i) have been already investigated both experimentally and theoretically in reviews focusing on CO2 hydrosilylation.9 We will focus here on the less investigated second reduction step leading to the 4e− reduction products. A thorough evaluation of the different mechanisms showed that the role of the catalyst is to activate either the hydrosilane or the formoxysilane, along different activation modes depending on the nature of the catalyst which are described hereafter for the catalysts C1–36. A summary of the different activation modes is given in Section 2.2.7 with a general scheme, Scheme 13.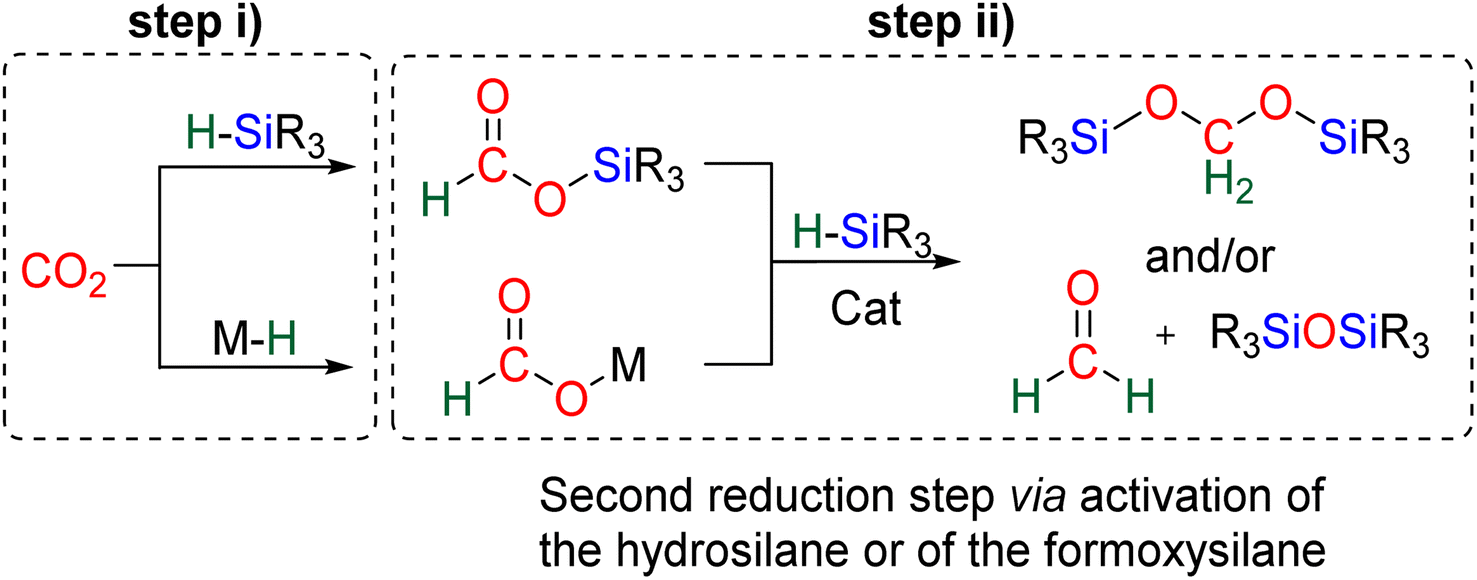 | ||
| Scheme 7 Two mechanistic pathways for the 4e− reduction of CO2 by hydrosilanes. | ||
61 and C3262 may be similar to that of C31 because of their high Lewis basicity. Theoretical investigations on the hydrosilylation of CO2 catalysed by the perrhenate anion C30 also support such views with a similar activation pathway of the hydrosilane for the first reduction step to formoxysilane.
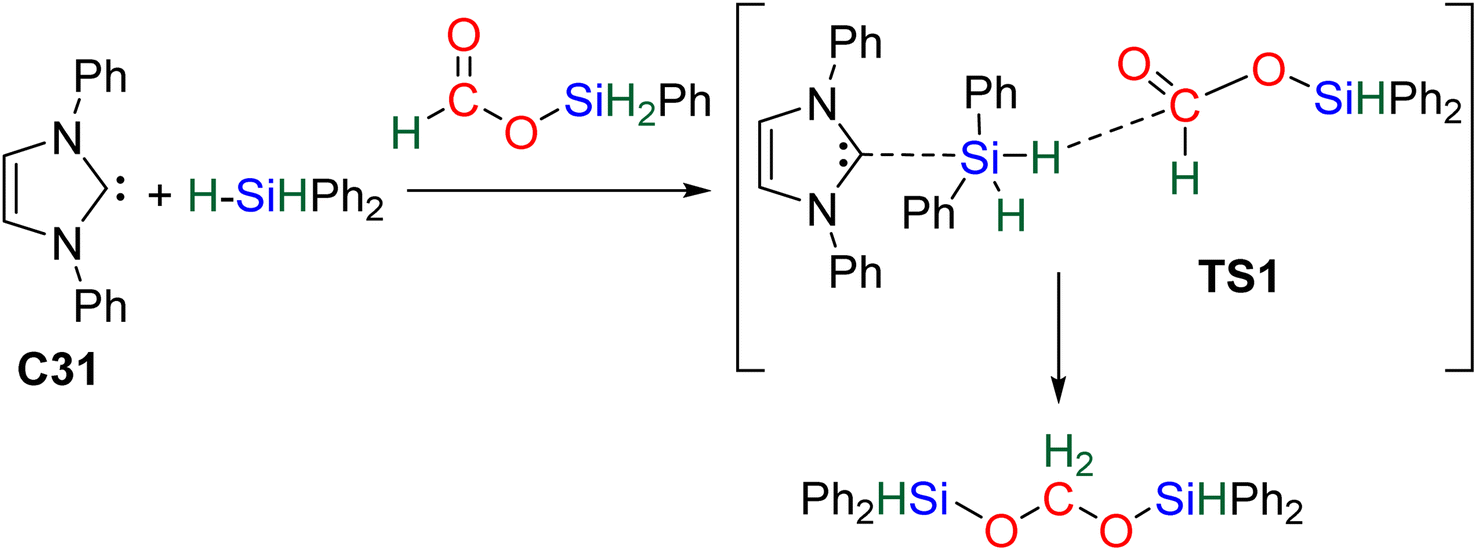 | ||
| Scheme 8 Schematic view of the computed transition state (TS1) of the Lewis basic activation of hydrosilane by catalyst C31. | ||
 | ||
| Scheme 9 Calculated Lewis acid activation of (a) silylformate or (b) metal formate intermediates. Only TS are shown for the sake of simplicity, and the movement of the electrons is schematically shown with arrows. | ||
2.2.2.1. Silyl formate intermediate. Scheme 9a describes the mechanism associated with a Lewis acidic system promoting the silylium “Si+” transfer to silyl formate species. Such a reaction occurred in an SN2-type activation going through the transition state TS1a. The resulting intermediate is then involved in a second elementary step through the transition state in which the hydride abstracted by the Lewis acid (H-LA) is transferred to the carbon atom of the carbonyl function to form the corresponding BSA derivative while regenerating the Lewis acid. Such a mechanism was computed for C14,53,67C26,58,68 and C35.63
In the cases of C14 and C26, TS1a was found to be higher in energy than TS2a, while the reverse was found with C35. The catalysts C1, C11–13, C15–17, C24–25, C28 and C36 are proposed to follow a similar pathway. The catalysts C18, C33 and C34 could also follow a similar pathway but the observed induction period points toward a more complex process.
2.2.2.2. Metal formate intermediate. Scheme 9b describes the mechanism associated with the Lewis acidic system promoting the silylium “Si+” transfer to metal formate species. In the cases of C2, C3 and C8, the second reduction step indeed occurred at a metal formate intermediate. The mechanism involving C8 has been investigated by DFT (Scheme 9b).69 Both TS1b and TS2b are similar to TS1a and TS2a respectively (Scheme 9a). The acetal obtained after these two first steps is a metal silyl acetal. After a second activation of the hydrosilane by B(C6F5)3, this metal silyl acetal intermediate is converted into the corresponding BSA derivative along with the regeneration of the active catalyst. An alternative pathway involving TS2′b was also calculated to be iso-energetic.69
40,45 and C947 are structurally different systems, the proposed mechanism of activation is similar: heterolytic cleavage of the hydrosilane prior to the reduction of the formoxysilane (Fig. 3, left). To support this assumption, catalysts C4–5 and C9 were shown to heterolytically cleave hydrosilanes to afford the three species Int-C4,45Int-C570) and C10 (Fig. 3).47 In addition, C10 was isolated and used as a catalyst in place of C9, showing similar catalytic activity toward CO2 hydrosilylation to that of C9. These compounds are proposed to transfer both silylium and hydride moieties to the formoxysilane intermediate to form the corresponding BSA derivative. Based on previous results concerning the hydrosilylation of other substrates, Oestreich et al. proposed the initial transfer of the silylium moiety followed by hydride transfer in a two elementary step mechanism with Int-C5.40 Such a two-step pathway would be very similar to the calculated pathway for the Lewis acid activation of hydrosilane described above.
 | ||
| Fig. 3 Complexes resulting from the heterolytic splitting of hydrosilane prior to CO2 hydrosilylation. | ||
 | ||
| Scheme 10 Proposed mechanism for the radical activation of Et3SiH by C22 based on experimental results. | ||
60 and C21.71 The in-depth theoretical investigation on catalyst C21 points toward the insertion of the formoxysilane into the Co–H bond leading to the formation of a metal silyl acetal intermediate.71 Following this addition, the metal centre is able to activate the hydrosilane via an oxidative addition leading to the transfer of the silyl ligand to produce BSA. Moreover, calculations enabled comparison of the three distinct catalytic cycles leading to the formoxysilane, the BSA and the methoxysilane products. The authors compared the energy gap values δE between the most stable intermediate and the highest transition state for each cycle. The smaller value (24.2 kcal mol−1) found for the formation of the formoxysilane compared to the values for the BSA (28.9 kcal mol−1) and the methoxysilane (29.9 kcal mol−1) explain why formoxysilane could be obtained selectively. The small difference between the δE of BSA vs. δE of methoxysilane explains the difficulty in selectively generating BSA in these systems. These calculations reproduced the experimental selectivity obtained under different reaction conditions of temperature and pressure. We hypothesize that the catalytic system C19 followed this pathway as well, based on the assumption that the combination of the bidentate ligand (i.e., 1,10-phenanthroline) and Zn(OAc)2 would generate a Zn–H moiety in the presence of a hydrosilane under catalytic conditions.
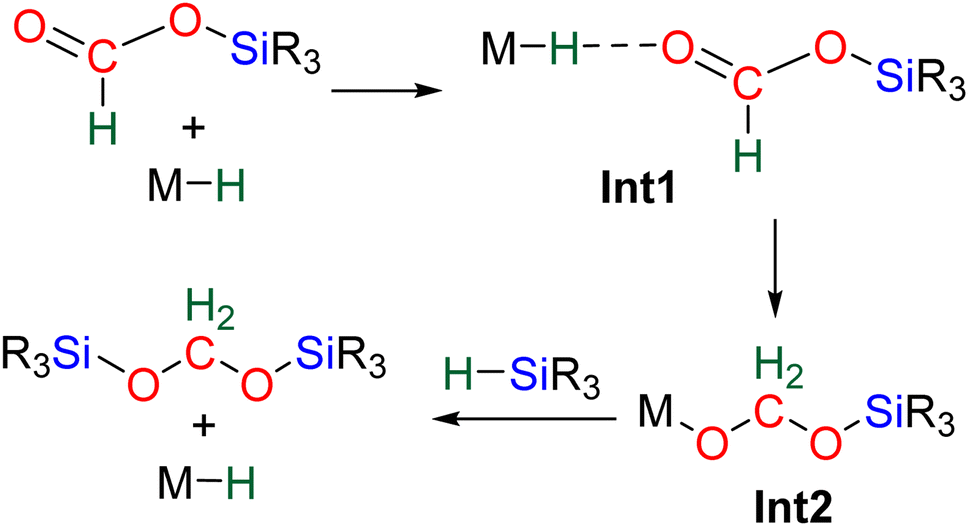 | ||
| Scheme 11 Computed pathway for the hydrosilylation of formoxysilane to BSA with M-H catalyst C29 and C21. | ||
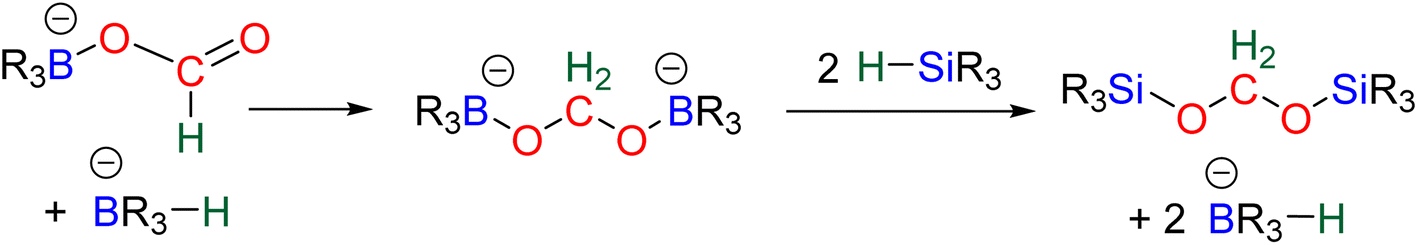 | ||
| Scheme 12 Proposed mechanism for C6 and C7 catalytic systems based on experimental results. | ||
 | ||
| Scheme 13 Activation modes of hydrosilane or formoxysilane leading to (silyl) acetals BSA (LB = Lewis base, LA = Lewis acid). | ||
2.3. Origin of the selectivity for BSA
The control or simply the understanding of the selectivity for bis(silyl)acetal (BSA) is a challenge. In this section we describe some of the main parameters which have been shown to favour BSA accumulation either upon thermodynamic stabilization or by disfavouring further reduction to methoxysilane or methane. | ||
| Scheme 14 Computational findings regarding the capability of BSA to liberate HCHO based on the type of silyl moiety. | ||
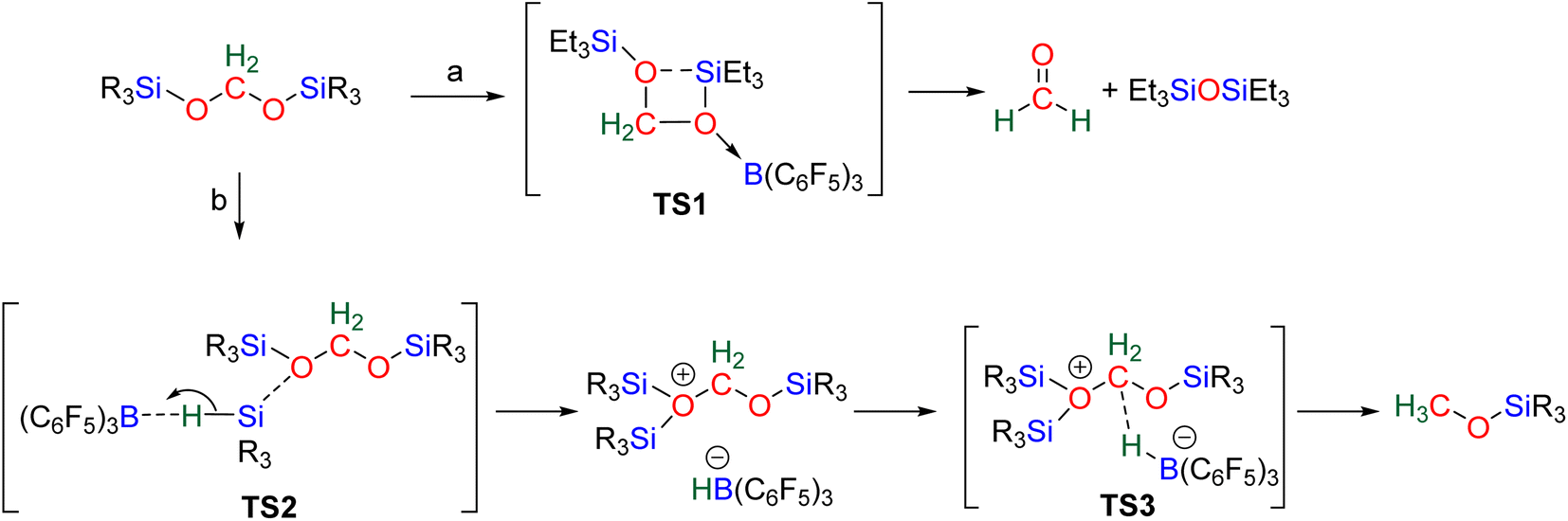 | ||
| Scheme 15 Computed B(C6F5)3-catalyzed conversion of BSA into formaldehyde or methoxysilane. | ||
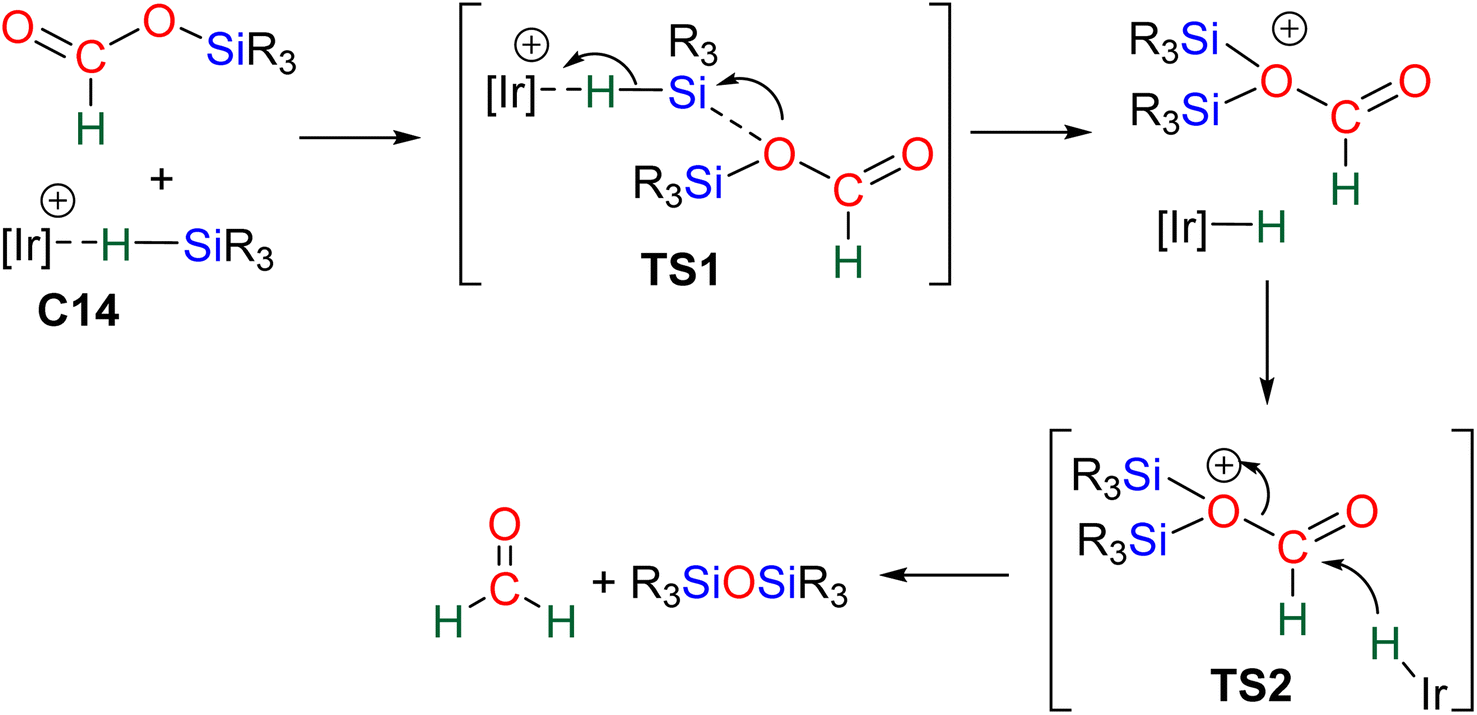 | ||
| Scheme 16 Calculated pathway for the generation of formaldehyde from formoxysilane with the Ir-based catalytic system C14. | ||
2.4. Bis(silyl)acetal as an intermediate in reductive functionalization of CO2
The reductive functionalization of CO2 refers to the reduction of CO2 combined with the functionalization of the reduced product with a co-reactant. The term was first employed in the case of the hydrosilylation of CO2 into formoxysilane, intercepted with amines to afford formamide, formamidine or methylamine derivatives.20c,74 An early reaction of this type can be traced back to a report on the preparation of formamide from CO2 reduction in the presence of amine in 1988.75 In this case, the reductant was not hydrosilane but dihydrogen. Although unexpected, the first reductive functionalization of CO2via the generation of a bis(silyl)acetal (BSA) intermediate was reported in a study devoted to the use of cationic Al-based species for catalysing the hydrosilylation of CO2 to methane.76 In the course of the reaction, the formation of deuterated diphenylmethane 1 featuring a bridged-methylene moiety was observed (Scheme 17). Experimental proofs showed that transiently generated BSA reacted with the deuterated solvent via electrophilic catalysis by the cationic Al-based compound to afford compound 1via compound 2.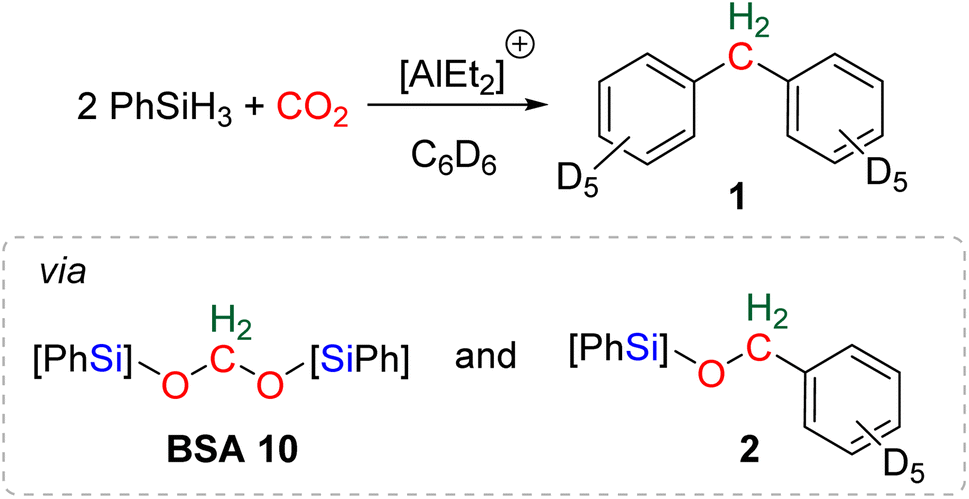 | ||
| Scheme 17 First example of reductive functionalization of CO2 into product 1, involving a BSA intermediate and compound 2. | ||
Following this preliminary report, subsequent studies reported the use of BSA as a key intermediate for the generation of more complex products in the presence of various co-reactants. Such strategies considerably enlarged the scope of compounds obtained from CO2 in using BSA as a source of methylene for the formation of C–N, C–C, C–S and C–O bonds.
2.4.1.1. TBD/PhSiH3: a pioneering reduction system in reductive functionalization. Formation of C–N and C–C bonds. The first system describing a reductive functionalization of CO2 with hydrosilane with the intermediary of BSA was reported in 2015 (Scheme 18).77 It included the use of TBD (1,5,7-triazabicyclo[4.4.0]dec-5-ene) which catalysed the hydrosilylation of CO2 with phenylsilane into the corresponding BSA, although it was never directly observed or characterized. Besides TBD, other Lewis basic catalysts including 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), Verkade's base and NHC (IPr and ItBu) were tested to catalyse the reduction step. Slightly lower yields of the final products were obtained. Various co-substrates featuring N–H (Scheme 18a) or C–H functionalities (Scheme 18b) acted as functionalizing agents in intercepting the transiently formed BSA leading to the formation of methylene-bridged “N–CH2–N” and “C–CH2–C” moieties. In total, 19 cyclic or acyclic symmetrical aminal products 3 were generated selectively with yields ranging from 14 to 99% (larger than 70% in most cases, Scheme 18a). In addition, 5 dissymmetric aminal products 4 were also generated using two different amines with yields ranging from 62 to 94% (Scheme 18a). For the synthesis of these dissymmetric aminal compounds, a key factor was to employ two amines featuring different nucleophilicities, enabling the most nucleophilic amine to first react with BSA, which affords an intermediate aminosilylacetal. Subsequently, the second (less nucleophilic) amine reacts with the aminosilylacetal intermediate yielding the corresponding aminal compound. Replacing the amine co-reactant with 2 equivalents of the nucleophilic malonate compound led to the unique formation of two C–C bonds in 58% yield (compound 5, Scheme 18b).
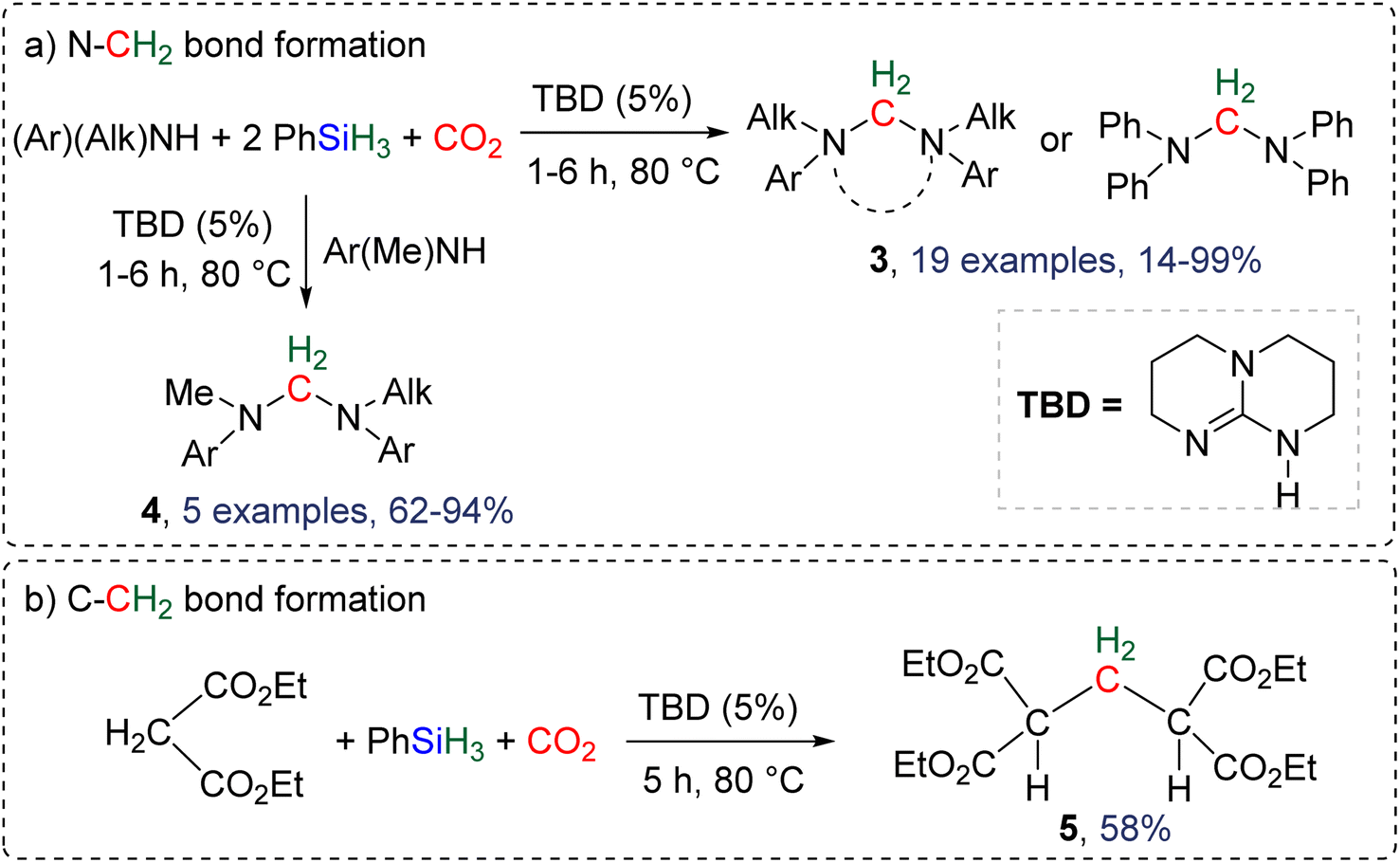 | ||
| Scheme 18 First selective reductive functionalization of CO2via the generation of a BSA intermediate leading to the formation of (a) C–N and (b) C–C bonds. | ||
2.4.1.2. Extension of the scope with the TBD/PhSiH3 reduction system. The TDB/PhSiH3 system was proven to be reliable and versatile to insert one or two methylene units in complex structures. To do so, 10–20 mol% of TBD was used at temperatures ranging from 100 to 120 °C and reaction times from 6 to 30 h (Scheme 19). Indolepyrrolidine derivatives were engaged as co-reactants leading to cyclisation reactions with the formation of C–CH2 and N–CH2 bonds. A second enantioselective reduction step of the pyrrolidine ring afforded spiro-indolepyrrolidine products 6 (Scheme 19), and 17 different compounds have been obtained in good to very good yields (55–92%).78 Reductive functionalization with amine was then used to incorporate two methylene units from the hydrosilylation of CO2 (Scheme 19, compounds 7).73 Key features of these transformations include the use of (i) a primary amine leading to the generation of an imine intermediate and (ii) a Lewis acid-catalysed cyclization reaction with various enaminones. The scope of the reaction was evaluated with 32 identified examples of tetrahydropyrimidine compounds with isolated yields (based on the enaminone) ranging from 50 to 99% (in most cases larger than 80%). The mechanism for the formation of compounds 7 was also investigated by DFT (Scheme 20). TBD was calculated to catalyse the hydrosilylation of CO2 into BSA 10 followed by the liberation of formaldehyde. The imine is then generated from the condensation of the primary amine with formaldehyde (step i). Controlled experiment proved that a stable imine can be formed when a bulky bis(diisopropyl)aniline is used. The enaminone nucleophilically adds to the intermediate imine via a Lewis acid/TBD-catalysed reaction (step ii) leading to an aza-diene compound after phenylamine elimination (step iii). The Diels–alder reaction between the aza-diene intermediate and a second equivalent of in situ generated imine finally furnishes the tetrahydropyrimidine 7 (step iv). This elegant strategy employing a primary amine as a first functionalization step transformation was further used to generate enaminones (Scheme 19, compounds 8, 22 examples, 43–94%) and 1,4-dihydropyridines (Scheme 19, compounds 9, 9 examples, 71–95%).79
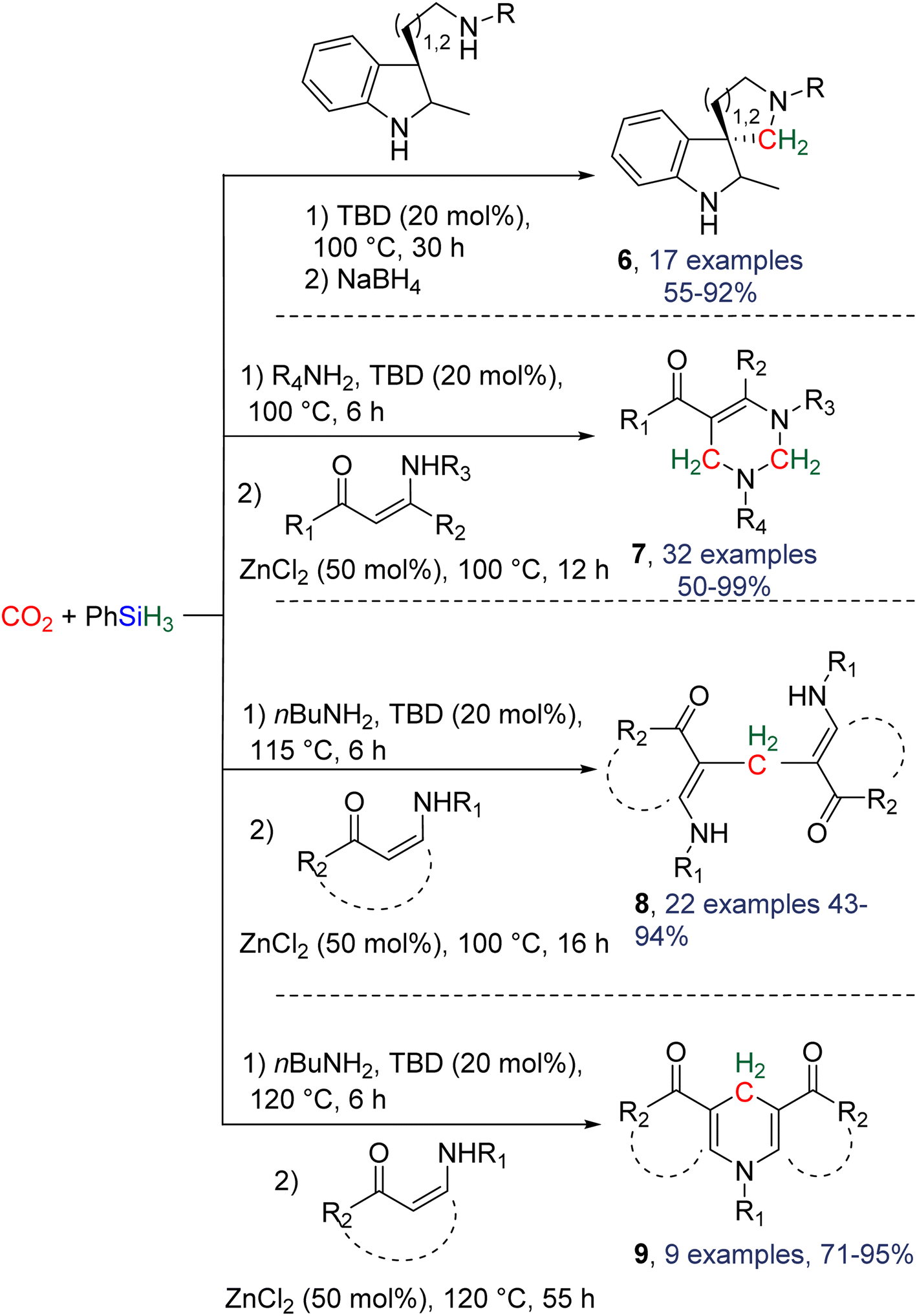 | ||
| Scheme 19 Extension of the product scope upon reductive functionalization of CO2 with hydrosilanes. | ||
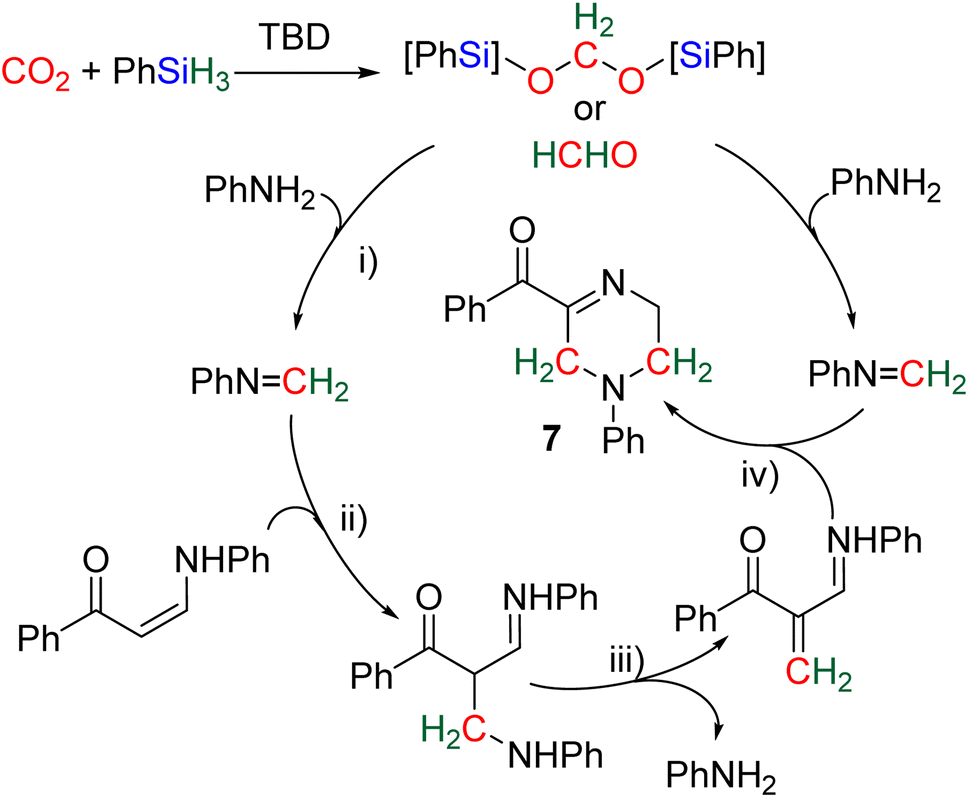 | ||
| Scheme 20 Proposed mechanism for the synthesis of tetrahydropyrimidine 7. | ||
2.4.1.3. Development of new catalysts. Besides the utilization of TBD as a catalyst, two other catalytic systems were employed for reductive functionalization with hydrosilanes. The combination of [CpFe(CO)2]2 and P(nBu)3 was shown to catalyse the hydrosilylation of CO2 with phenylsilane in the presence of tryptamine derivatives to afford a variety of tetrahydro-β-carbolines and other nitrogen-containing heterocycles 10 after 36 h at 60 °C (Scheme 21).80 In total, 19 heterocycles were obtained in yields ranging from 50 to 94%. The [CpFe(CO)2]2/P(nBu)3 system is proposed to catalyse the hydrosilylation of CO2 into BSA which is then involved in a Pictet–Spengler-type cyclization. Other Fe-based precursors did not afford any conversion, while diphenylsilane or other tertiary phosphine afforded the expected compounds although in lower yields. Another report described the use of a phosphorus ylide to intercept the reduction intermediate via a Wittig reaction (Scheme 21, compounds 11).81
 | ||
| Scheme 21 New catalysts for reductive functionalization with hydrosilanes. | ||
If the first tests were conducted with the TBD/PhSiH3 system in acetonitrile, optimization conditions revealed the beneficial role of an inorganic base and notably of sodium tert-butoxide (NaOtBu) in DMF in catalysing the hydrosilylation of CO2 with PMHS (polymethylhydrosiloxane) – a waste of the silicone industry. This process led to the synthesis of 20 olefins in yields ranging from 30 to 85%. Ionic liquids224 and covalent organic frameworks83 were also employed as catalysts in a similar synthesis of aminal by CO2 reductive functionalization.
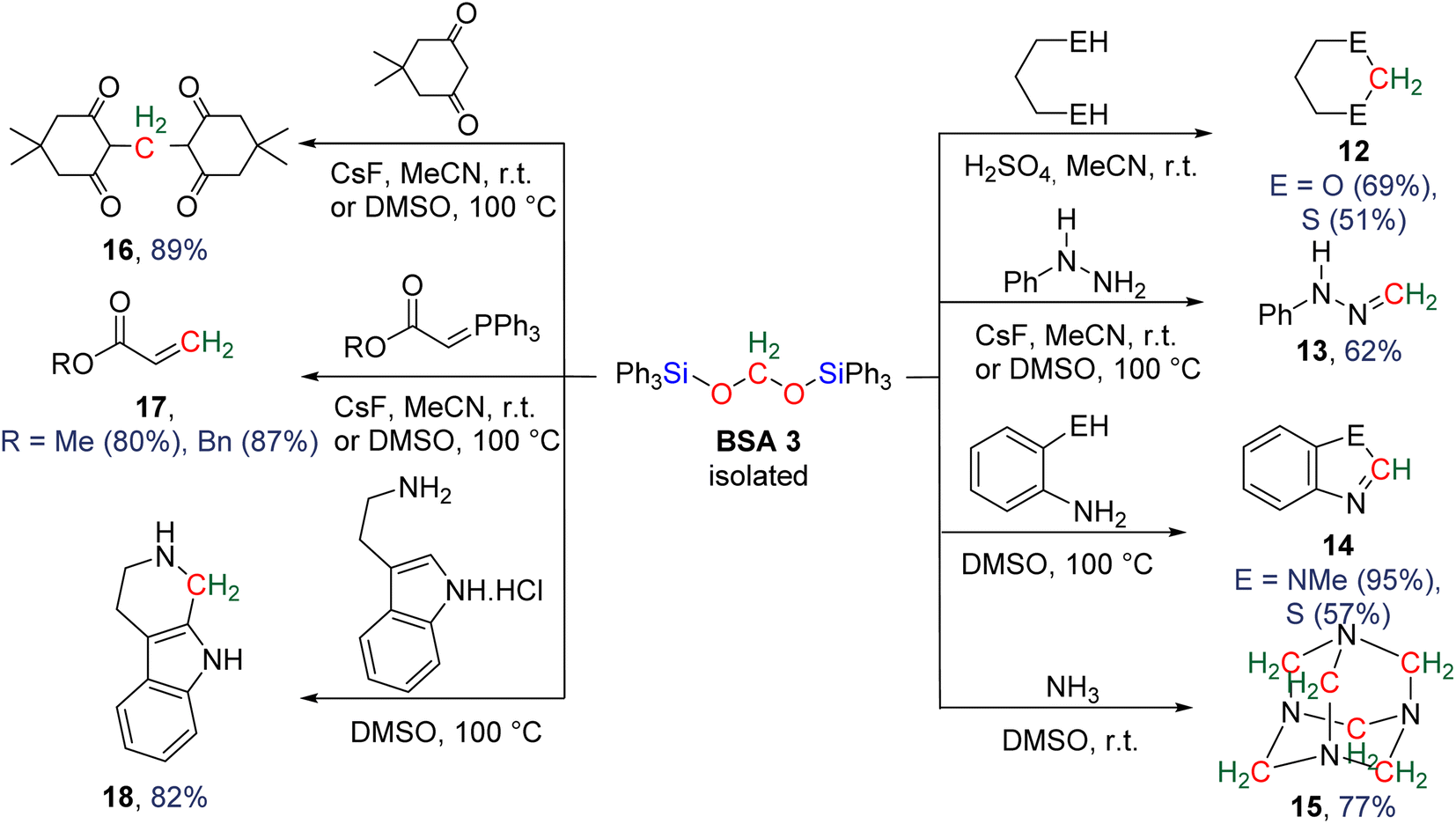 | ||
| Scheme 22 Products obtained from the functionalization of HCHO released from BSA 3. | ||
In the presence of diol or di-thiol substrates under acidic conditions, the cyclic compounds 12 were generated in good yields forming CH2–O (69% yield) and CH2–S bonds (51% yield), respectively. The reaction with hydrazine proved to be efficient to generate a CH2N bond (compound 13, 62% yield), while reactions with molecules comprising primary amine and thiol or secondary amine function led to benzimidazole and benzothiazole compounds 14 (95 and 57% yields), respectively. Finally, the reaction with ammonia afforded hexamethylene tetra-amine 15 in 77% yield. In addition, C–C bonds were also generated (i) with malonate leading to 5,5-dimethylcyclohexane-1,3-dione compound 16 (89% yield), (ii) through a Wittig reaction leading to the acrylate compounds 17 (80 and 87% yields) and finally (iii) through a Pictet–Spengler-type cyclization reaction leading to compound 18 (82% yield). It must be noted that 13C-labelled BSA 3 could also be efficiently generated and isolated from the catalytic hydrosilylation of 13CO2 and was used as a dry source of 13C-labeled HCHO.
2.5. Conclusion and perspectives
In conclusion, the four-electron reduction of CO2 with hydrosilane is noteworthy for several reasons. Firstly, it distinguishes itself with a considerable number and variety of selective catalytic systems (C1–24), primarily based on the combination of transition metal-based complexes and B(C6F5)3. Comprehensive experimental and theoretical studies have revealed five distinct activation modes, elucidating the 2 to 4 electron reduction stage. Despite the highest reported TON of 3400 and TOF of 10.4 h−1, there is room for further enhancements. Secondly, it stands out due to the diverse array of characterized and isolated bis(silyl)acetal (BSA) compounds, with a current total of 11 reported. These isolated BSAs were predominantly obtained from tertiary silanes, demonstrating high selectivity (79 to 99% yields) under mild conditions. Finally, the process is notable for numerous transformations involving BSA in reductive functionalization, ranging from simple condensation to multi-step reactions with co-reactants. Interestingly, when primary silanes were employed, BSA formation was never observed, likely due to its high reactivity. In contrast, tertiary silanes led to the isolation of BSA before engaging in subsequent functionalization through formaldehyde release.Perspectives in the field of 4e− CO2 hydrosilylation could involve rationalizing the stability of the generated bis(silyl)acetal (BSA) based on the choice of hydrosilane. Generating BSAs with intermediate stability holds particular interest, offering potential for expanding the reactivity scope of BSA compounds. A significant breakthrough would be utilizing BSAs as a Cn source for producing high-value added products, an area yet to be explored compared to their borylated counterparts discussed in the next section. However, the primary challenge remains the treatment of the siloxane by-product produced during CO2 hydrosilylation, currently considered as waste. Exploring potential uses for these siloxane moieties or designing a regeneration system for the sustainable recycling of hydrosilane presents a promising avenue for further research.
3. Hydroboration
In the realm of hydroelementation reactions, hydroboration holds an important and specific place. Since the discovery of hydroboration of olefins,84 this reaction has been largely developed for the reduction of unsaturated bonds.8b,85 Beyond reduction, the formal addition of B–H bonds is used as a privileged access to organoboron compounds which are then used as reagents in coupling reactions.86 Hydroboration of CO2 involves the addition of a B–H bond to the CO bond, transferring the hydride to the carbon atom and forming a B–O bond. It is favoured both kinetically and thermodynamically by the polarity of the B–H bond (E. N. of H = 2.2, B = 2.0) and the strength of the formed B–O bond (BDE(B–O) >500 kJ mol−1),87 respectively. As a consequence, CO2 hydroboration reactions, like CO2 hydrosilylation ones, operate under much milder conditions – usually room temperature and 1 atm of CO2 – than CO2 hydrogenation.8a However, it is noticeable that the catalytic systems used for the hydroboration or the hydrosilylation of CO2 are different in most cases.
The first catalytic hydroboration of CO2 was reported in 2010 with a Ni-based catalytic system and was shown to afford the 6e− reduction product, methoxyborane, readily hydrolysed to methanol.88 While the 2e− reduction product – formoxyborane – was observed, no 4e− reduction product was detected, although formaldehyde was proposed as an elusive intermediate.88,89 The Ni-based pincer system used as the catalyst remains one of the most efficient catalyst of CO2 hydroboration into methoxyborane with TOF and TON of 495 and 495 h−1, respectively. It is worth noticing that an earlier report described the use of borane in the reduction of CO2. However, it was not a hydroboration reaction, but the reduction of CO2 into CO by B2pin2 and a (NHC)Cu-based catalyst.19b It was only in 2012 and 2014 that bis(boryl)acetal (BBA)90 and formaldehyde91 were respectively characterized from the hydroboration of CO2, although not in a selective manner. Later on, the efficient and selective generation of BBA derivatives was reported using different hydroboranes as reductants. These transformations (Scheme 23, step a) will be presented in the first part 3.1 and the mechanism will be discussed in part 3.2. Finally, the use of these 4e− reduced products as reactants in further transformations (Scheme 23, step b) will be described in part 3.3 and a general conclusion will be drawn in part 3.4.
 | ||
| Scheme 23 General scheme for Section 3: CO2 hydroboration. | ||
3.1. Hydroboration of CO2 into bis(boryl)acetals (BBAs) and formaldehyde
In total, 7 BBA derivatives have been synthesized by the hydroboration of CO2.92BBA 1–4 were obtained from the hydroboration of CO2 (Scheme 24) catalysed by compounds C36–C45 (selective catalysts, Fig. 4) and C46–C63 (unselective catalysts, Fig. 4). BBA 5–7 were obtained in the absence of a catalytic system (Scheme 25). BBA 2 and 4 were shown to be key intermediates leading to formaldehyde in high yields. These transformations are described hereafter.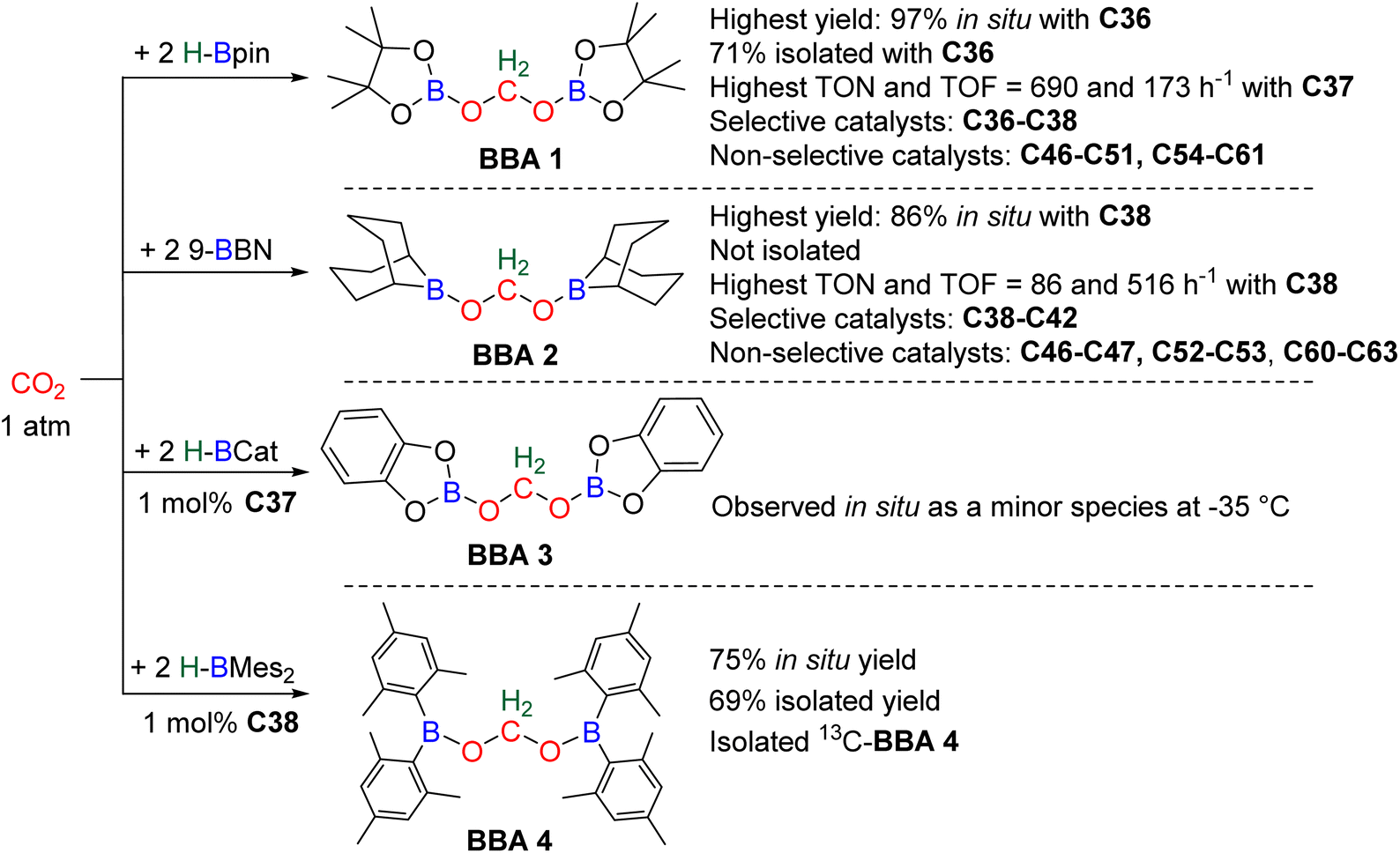 | ||
| Scheme 24 Catalytic generation of BBA 1–4 with C36–C63 catalysts. | ||
 | ||
| Fig. 4 Catalysts C36–C63 for the generation of BBA 1–7 and formaldehyde: (a) selective catalysts C36–C45 and (b) unselective catalysts C46–C63. | ||
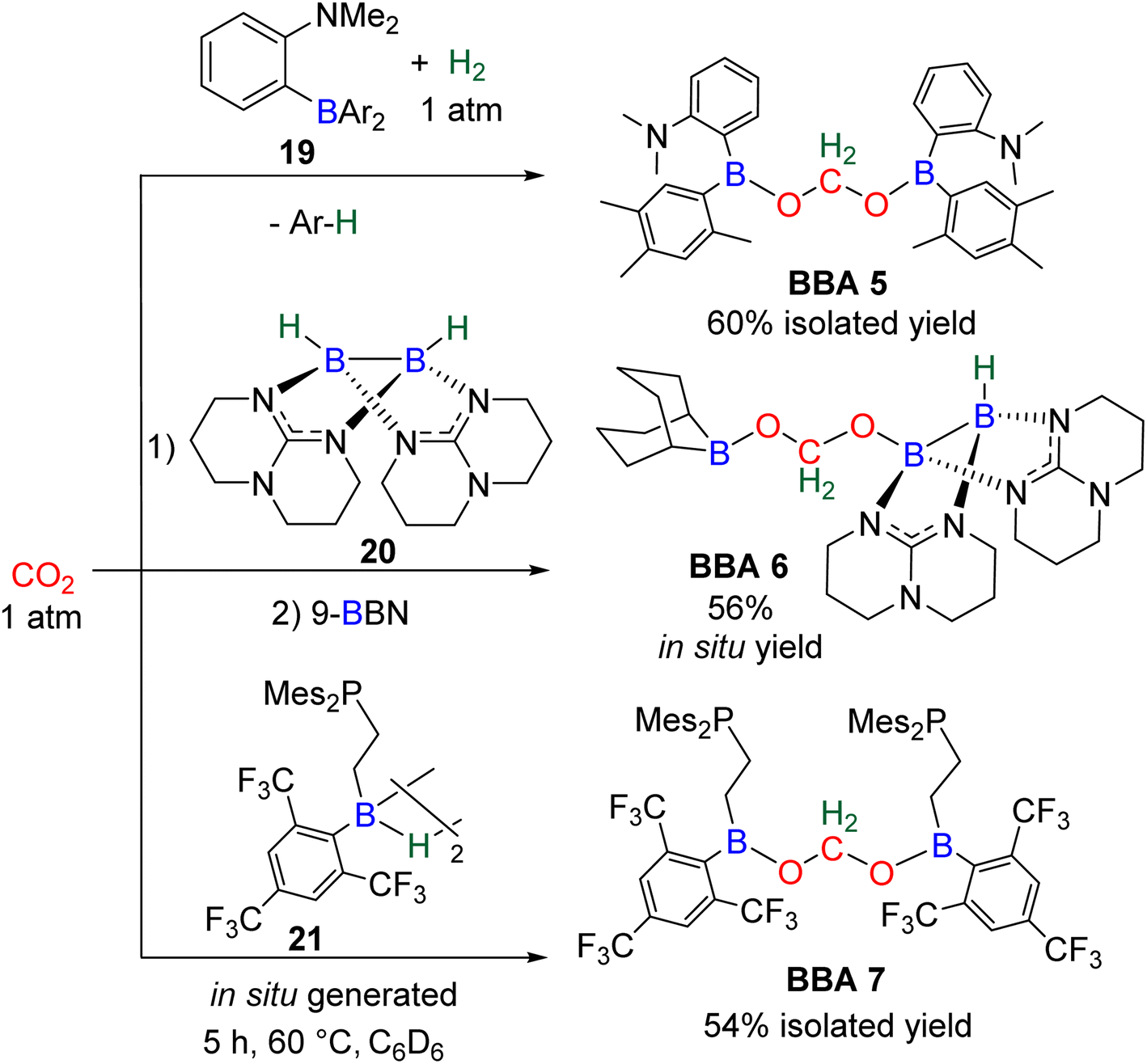 | ||
| Scheme 25 Catalyst free generation of BBA 5–7. | ||
95 (15 min at room temperature with 2% of the Ni-based precatalyst) and C4496 (2 h at 90 °C with 1% of the Mo-based catalyst) were shown to afford BBA 1 as the major product. It must be noted that BBA 1 was observed as a minor product in hydroboration reactions catalysed by compounds C46–C47,90C48–C50,94,97C51,98 and C54–C61.99,100
BBA 2 was reported for the first time in 2014 in CO2 hydroboration catalysed by the nitrogen-based Lewis base TBD (C45).101 This study focused on the reduction of CO2 to the methoxyborane compound with various metal-free catalysts and hydroboranes. Using 9-BBN and 2.5 mol% of catalyst C45, BBA 2 was detected as the major intermediate after 1 h (85% ratio) before decaying, leading to the final methoxyborane compound. The selective generation of BBA 2 and yield quantification (instead of ratio) were then described with C37–C39.
In 2015, the well-defined iron dihydride bis-diphosphine catalyst C38 was used at room temperature and under 1 atm of CO2 to afford BBA 2 in 85% yield (TOF = 109 h−1) with a catalyst loading of 1 mol%.93 The importance of the THF solvent was identified since such selectivity was not observed with benzene or toluene. Moreover, a high TOF of 516 h−1 was measured upon increasing the temperature to 60 °C with the generation of BBA 2 in 86% yield in only 10 min. In 2016, the well-defined [Co]- and [Cu]-precatalysts C41 and C42 were shown to generate BBA 2 as the major product after 24 h at room temperature and 60 °C, respectively.102 Beyond these selective systems, catalysts C46–C47,90C52–C53,103C60–C61104 and C62–C63105 were also reported to catalyze the generation of BBA 2 as a minor product under mild conditions. BBA 3 has only been partially characterized once as a minor compound, despite numerous studies using HBCat for CO2 hydroboration. This is in marked contrast with the selective generation of BBA 1 and BBA 2, and might be explained by the ability of HBCat to readily reduce CO2 to the methoxyborane stage (vide infra in mechanistic consideration).
In this context, BBA 3 was characterized in situ as a minor intermediate, during methoxyborane preparation with catalyst C37.97 Following the reaction, a minor signal was observed by 1H NMR in the characteristic area of acetals. Its broadness suggested the reversible formation of BBA 3. This assumption was confirmed by its sharpening at −35 °C leading to the first characterization of the acetal moiety of BBA 3. When 13CO2 was used, this acetal signal appeared as a doublet, which finally disappeared upon further reduction to the methoxyborane product. BBA 4 was obtained from the CO2 hydroboration with HBMes2 catalysed by the iron hydride catalyst C38 with an in situ yield of 75%.30 This BBA exhibits higher stability as compared to BBA 1–3, as shown by an isolated yield of 69% and its characterization by X-ray diffraction analysis.30 Moreover, BBA 4 featuring a 13C-labelled methylene fragment was also isolated in similar yield from 13CO2 hydroboration. The stability of BBA 4 was used to perform enzymatic transformations (vide infra). It is noteworthy that catalyst C38 is able to catalyse the selective generation of three BBA derivatives, namely, BBA 1, 2 and 4.
The performances of catalysts C36–C42 for the selective generation of BBA 1–2 and BBA 4 are reported in Table 3. The performances of catalysts C43–C45 are not indicated because only the ratio between the detected products were measured and not the quantitative yields. The performances of catalysts C46–C63 are not given since BBA 1–3 were only observed as minor compounds. Among the seven selective catalytic systems (C36–C42), one can notice that all of them are transition metal (TM)-based catalysts and five based on 3d-TM (C36–C39, C41). The active centre is a metal hydride fragment either present in the initial catalyst or presumably generated in situ during the reaction with the hydroborane. The best performances in terms of TON and TOF were obtained with C37 (487 and 122 h−1) and C38 (85, 109 h−1 at room temperature and 86, 516 h−1 at 60 °C) for the generation of BBA 1 and BBA 2, respectively. The best yields were obtained with catalysts C36 (97% yield) and C38 (86% yield) for the generation of BBA 1 and 2, respectively. The selectivity is an important factor to be considered when the generated BBA is further engaged in additional reactions (see Section 3.3). If a catalytic system exhibits high TON and TOF but poor selectivity, the presence of other products or of the remaining hydroborane may be detrimental for the subsequent reactions.
| BBA | Catalyst | TON | TOF (h−1) | Yielda (%) | P CO2 (atm) | T (°C) | Timeb | Cat loading (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a In situ yield unless otherwise stated. b Hour unless otherwise stated. c Isolated yield. | |||||||||
| 1 | C36 | 487 | 122 | 97 | 1 | rt | 4 | 0.2 | 94 |
| 71c | |||||||||
| C37 | 690 | 173 | 69 | 1 | rt | 4 | 0.1 | 97 | |
| C38 | 6 | 1.2 | 30 | 1 | 25 | 5 | 5 | 93 | |
| 2 | C38 | 85 | 109 | 85 | 1 | rt | 47 min | 1 | 93 |
| 86 | 516 | 86 | 1 | 60 | 10 min | 1 | |||
| C39 | 32 | 2 | 64 | 1 | rt | 16 | 1 | 97 | |
| C40 | 15 | 3.8 | 75 | 1 | rt | 4 | 5 | 106 | |
| C41 | 25 | 1 | 38 | 1 | 60 | 24 | 1.5 | 102 | |
| C42 | 53 | 2 | 79 | 1 | rt | 24 | 1.5 | ||
| 4 | C38 | 75 | 75 | 75 | 1 | 25 | 1 | 1 | 30 |
| 69c | |||||||||
| BBA | Isolated | XRD | Solvent | δ 13C(CH2) | δ 1H(CH2) | δ 11B | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | Yes | No | C6D6 | 85.7 | 5.51 | 22.8 | 94 |
| 85.4 | 5.49 | — | 90 | ||||
| 1 J HC = 167.2 Hz | |||||||
| 2 | No | No | THF-d8 | 87.0 | 5.54 | 58.6 (ref. 100c) | 93 |
| 1 J HC = 165.2 Hz | |||||||
| 3 | No | No | C6D6 | 81.9 | 5.27 | n.d | 97 |
| 4 | Yes | Yes | THF-d8 | 89.6 | 5.49 | 48.5 | 30 |
| 5 | Yes | Yes | C6D6 | 90.9 | 5.72 | 46.1 | 110 |
| 6 | No | No | CD2Cl2 | 91.3 | 5.20 | −5.4 | 107 |
| 6.8 | |||||||
| 55.5 | |||||||
| 7 | Yes | Yes | C6D6 | 88.5 | 4.90 | 51.0 | 32 |
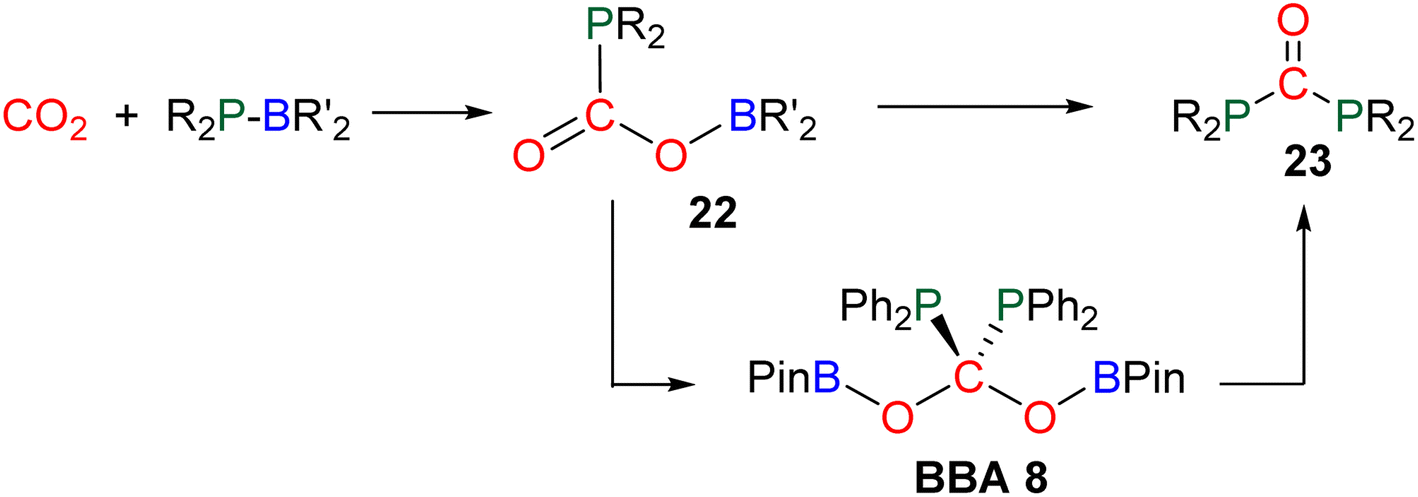 | ||
| Scheme 26 Synthesis of BBA 8 by the phosphoborylation of CO2. | ||
 | ||
| Scheme 27 Hydroboration of CO2 into formaldehyde: (a) generation and (b) selective trapping of HCHO. | ||
Observation of formaldehyde from CO2 hydroboration was also mentioned in two other studies.95,112 In both cases, it appears to result from the evolution of BBA 1. Using the selective formation of BBA as a tool to access formaldehyde was further developed recently. BBA 2 and BBA 4 were selectively generated with catalyst C38, and subsequently hydrolysed with 10 equivalents of D2O. This led to their complete hydrolysis into formaldehyde at room temperature within 30 min and less than 2 h for BBA 2 and BBA 4, respectively (Scheme 28). The generated formaldehyde was then further transformed in organo-29b or enzymatic30-catalyzed reactions (see Section 3.3). It is remarkable that BBA 2 and BBA 4 were stable enough to be generated selectively – and even isolated in the case of BBA 4 – and yet efficiently released formaldehyde under mild hydrolytic conditions.
 | ||
| Scheme 28 Generation of formaldehyde from BBA 2 and BBA 4 hydrolysis. | ||
3.2. Mechanistic considerations
This section is dedicated to mechanistic considerations for the formation of bis(boryl)acetal (BBA) or formaldehyde. It is divided into three parts: (i) metal-hydride catalysis (C36–C44, C46–C51), (ii) organic catalysis (C45, C52–C63) and (iii) uncatalyzed generation of BBA 5–7.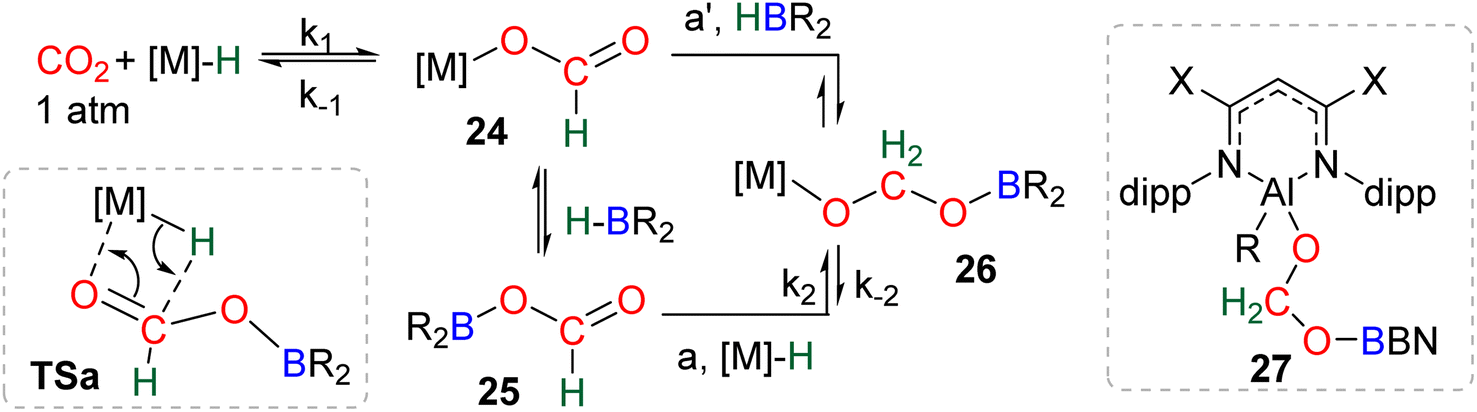 | ||
| Scheme 29 Calculated elementary steps (a and a′) leading to the generation of the elusive intermediate 26 and structure of the only metal boryl acetal (27) so far characterized. | ||
Numerous experimental proofs demonstrated that formate complex 24 obtained from CO2 insertion into M–H bond could be used in hydroboration reactions.8a,90,91 However, although the metal boryl acetal species 26 has been calculated to be central for the 4e− reduction of CO2, identification and or isolation of these species are still lacking. The aluminum compound 27 is the only metal boryl acetal compound characterized without ambiguity (Scheme 29).114 It was obtained from the subsequent reaction of the corresponding [Al]–H with CO2 and then with 9-BBN and 27 was fully analysed by NMR and X-ray diffraction (XRD) studies. It is noticeable that only 9-BBN gave access to this metal boryl acetal structure. Analogous structures could indeed neither be observed with HBpin because of the lack of reactivity nor with HBCat or BH3 because of subsequent reduction processes. It is also important to notice that like boron, aluminium is a group 13 element and that despite mimicking important steps of CO2 hydroboration, neither the starting Al hydride complex nor complex 27 catalyses the CO2 hydroboration reaction because the initial Al–H species is not regenerated under the reaction conditions. Step a′ (Scheme 29) is an alternative step to generate 26 from the direct hydroboration of the formate complex 24. To the best of our knowledge, it has not been reported in DFT investigations but we believe that it should not be discarded ipso facto. Compound 26 is a key intermediate because it serves as a bifurcation point either toward BBA (Scheme 30, step b) or formaldehyde (Scheme 30, step c). BBA is formed from the hydroboration of the metallic acetal intermediate 26. TSb represents the type of transition state calculated to account for this step. It formally corresponds to an M–O metathesis. Step c illustrates the route leading to formaldehyde and the boryl ether 28 which eventually leads to the 6e− reduction hydroboration product 29 through step d. This last step was experimentally identified with the Ru–H complex C46 that was shown to catalyse the hydroboration of formaldehyde into methoxyborane with HBpin. The first transition state TSc-1 does not involve HBR2 and is simply a rearrangement of the metal boryl acetal 26. The aliphatic chain rearranges to enable the interaction of the second oxygen atom with the metal centre. This reorganization weakens the M–O bond of the first oxygen atom and the O–CH2 bond of the second oxygen atom. This leads to the release of formaldehyde and the formation of an M–OBR2 intermediate which is subsequently hydroborated to regenerate the metal hydride and affords the ether compound 28viaTSc-2.
 | ||
| Scheme 30 Calculated elementary steps (b, c, c′, d) and transition states (TSb, TSc-1 and TSc-2) from metallic borylacetal. | ||
The factors influencing the selectivity have been explored in a thorough study involving the use of various hydride pincers of Ni- and Pd-based catalysts and three different hydroboranes, namely, HBpin, HBCat and 9-BBN.97 The origin of the selectivity between the 2e− and 4e− reduction products was proposed to be dependent on the relative rate of reaction between the metal hydride fragment (M–H) and either (i) CO2 to afford the formoxyborane 25 (k1) or (ii) the formoxyborane intermediate 25 to afford the metal boryl acetal 26 (k2) (Scheme 29). It was indeed shown that a lower pressure of CO2 led to a higher selectivity toward the 4e− reduction product presumably by disfavouring k1. It was also proposed that k2 could be favoured by (i) increasing the concentration of the hydroborane, (ii) decreasing the steric hindrance of the boryl unit, (iii) increasing its electrophilicity and (iv) using a Lewis acid to co-activate the formoxyborane.
The origin of the selectivity between the 4e− and the 6e− reduction products appears to be the preference between the formation of BBA and the formation of formaldehyde, since the latter readily leads to further reduction. However, the factors influencing the preference toward BBA or formaldehyde remain to be disclosed. In addition, the formation of BBA was shown to be reversible which eventually led to the final generation of the more stable methoxyborane in various systems.90,97 This feature explains the key role that the boryl moieties may play in the kinetic and/or thermodynamic stabilization of the corresponding BBA. Step c′ is an alternative path generating formaldehyde directly from BBA (Scheme 30). Such evolution has not been proposed as a plausible path for M–H catalysis but for organic catalysis (vide infra).96,98 The direct generation of a methoxyborane compound 29 from the metal boryl acetal 26 has not been identified so far.
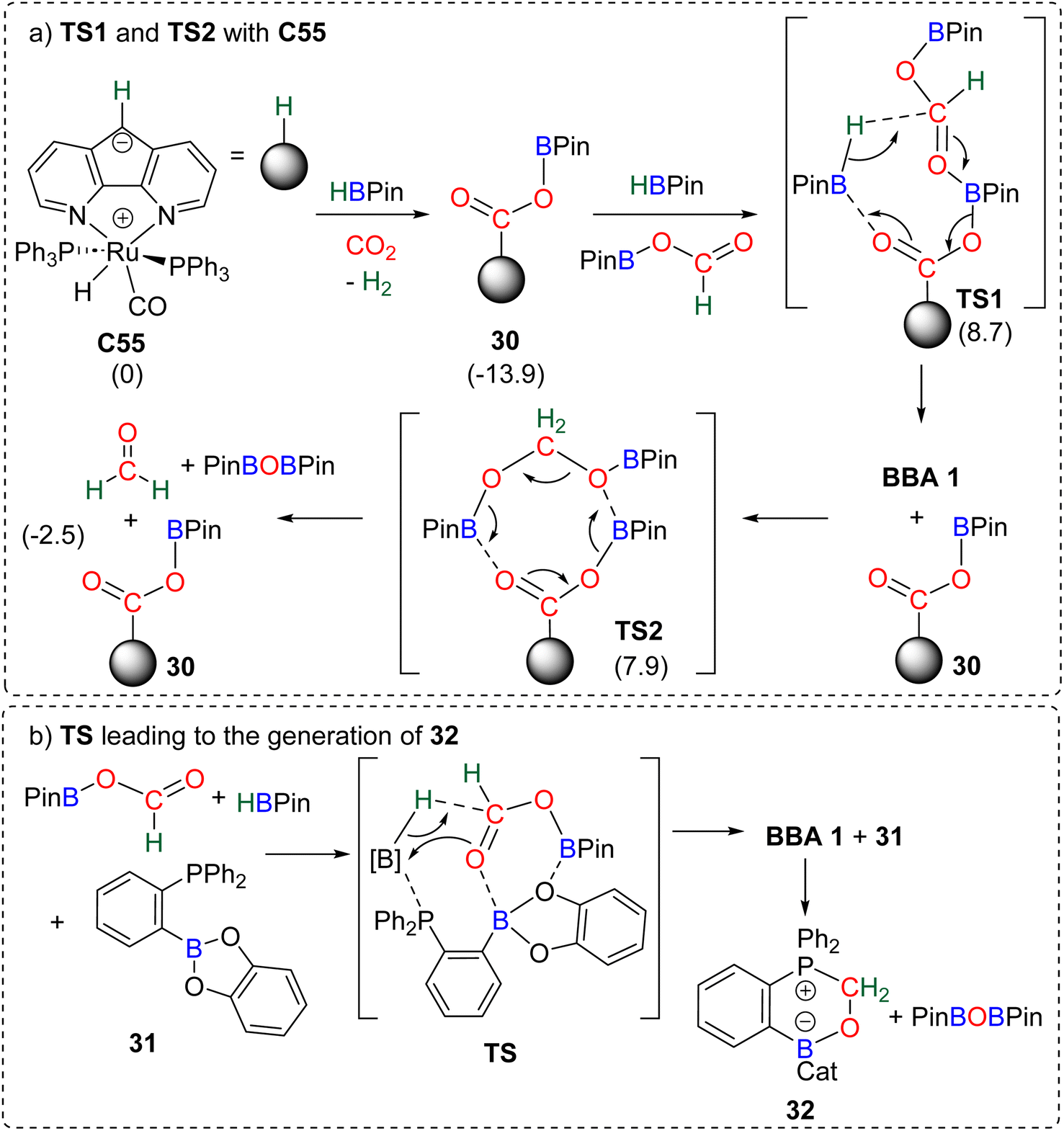 | ||
| Scheme 31 Calculated elementary steps for the 4e− reduction mechanism of CO2 with (a) C55 (energies indicated in parenthesis are ΔG values in kcal mol−1) and (b) compound 31. | ||
The carbanion located on the backbone of the ligand of the complex was found to react both with HBPin and CO2 to produce the catalytic species 30. Compound 30 was proposed to activate the formoxyborane intermediate 25 and HBPin concomitantly through TS1 to afford BBA 1. The regenerated catalyst 30 could then be involved in the transformation of BBA 1 into formaldehyde via an 8-membered-ring (TS2). In this reaction, formaldehyde was not experimentally observed but was proposed to be a transient intermediate en route to the observed methoxyborane structure. The ambiphilic phosphine borane 31 was shown to catalyse CO2 hydroboration into methoxyborane with different hydroboranes (HBCat, HBPin, 9-BBN and BH3) (Scheme 31b).118 Although no free formaldehyde nor BBA was observed, a pathway, similar on various aspects to the previous one (Scheme 31a), was proposed. Theoretical investigations notably concluded on the co-activation of HBPin and formoxyborane (TS, Scheme 31b) to afford BBA 1. BBA 1 then evolves to formaldehyde which ultimately reacts with compound 31 to form the formaldehyde adduct 32. Interestingly, labelling experiments proved that the formaldehyde moiety of compound 32 was not reduced further to afford the methoxyborane.109a Instead, the adduct 32 acts as a more effective catalyst in the hydroboration of CO2 into methoxyborane with the various boranes tested. This feature is responsible for an observed induction period, which was also noticed (i) in the CO2 hydroboration reactions with catalyst C52 leading to the generation of C53,103,119 (ii) in the hydroboration of CO2 with PtBu3 in which a formaldehyde derived intermediate (compound 36, Scheme 33) was also observed,120 and (iii) when using catalyst C60 leading to C61 which was proven to be a more active catalyst.104 The similar pathway identified in these two studies implies (i) the reduction of CO2 into formoxyborane as the 2e− reduction intermediates are observed with metal hydride catalysts and (ii) the subsequent generation of BBA (Scheme 32, step a) and then formaldehyde (Scheme 32, step b) before further reduction to the 6e− reduction product.121 This latter feature stands in contrast with the metal hydride systems in which BBA and formaldehyde are not generated along the same pathway (Scheme 30). This common pathway remains to be confirmed by further studies on additional organo-catalysed systems. Generalizing such a mechanism remains highly hypothetical because of (i) the very small number of reported theoretical investigations, (ii) the large variety of organic catalysts C45 and C52–C63 and (iii) the difficulty to study very reactive intermediates experimentally. A general trend is the ability of these main group organic catalysts to co-activate several substrates and/or intermediates (CO2, hydroborane, formoxyborane). This feature highlights the powerful synergy observed between moderately reactive centres (moderate Lewis acid and Lewis base fragments) for the transformation of very stable molecules such as carbon dioxide.
 | ||
| Scheme 32 Elementary steps a and b for the reduction of formoxyborane 25 to methoxyborane 29 calculated with compound 32. | ||
3.3. Use of formaldehyde and bis(boryl)acetal as C1 and Cn sources
The known high reactivity of formaldehyde was first evidenced in four side reactions involving CO2 reduction. In these reactions, in situ generated formaldehyde reacted with other products or catalysts present in solution (Scheme 33a).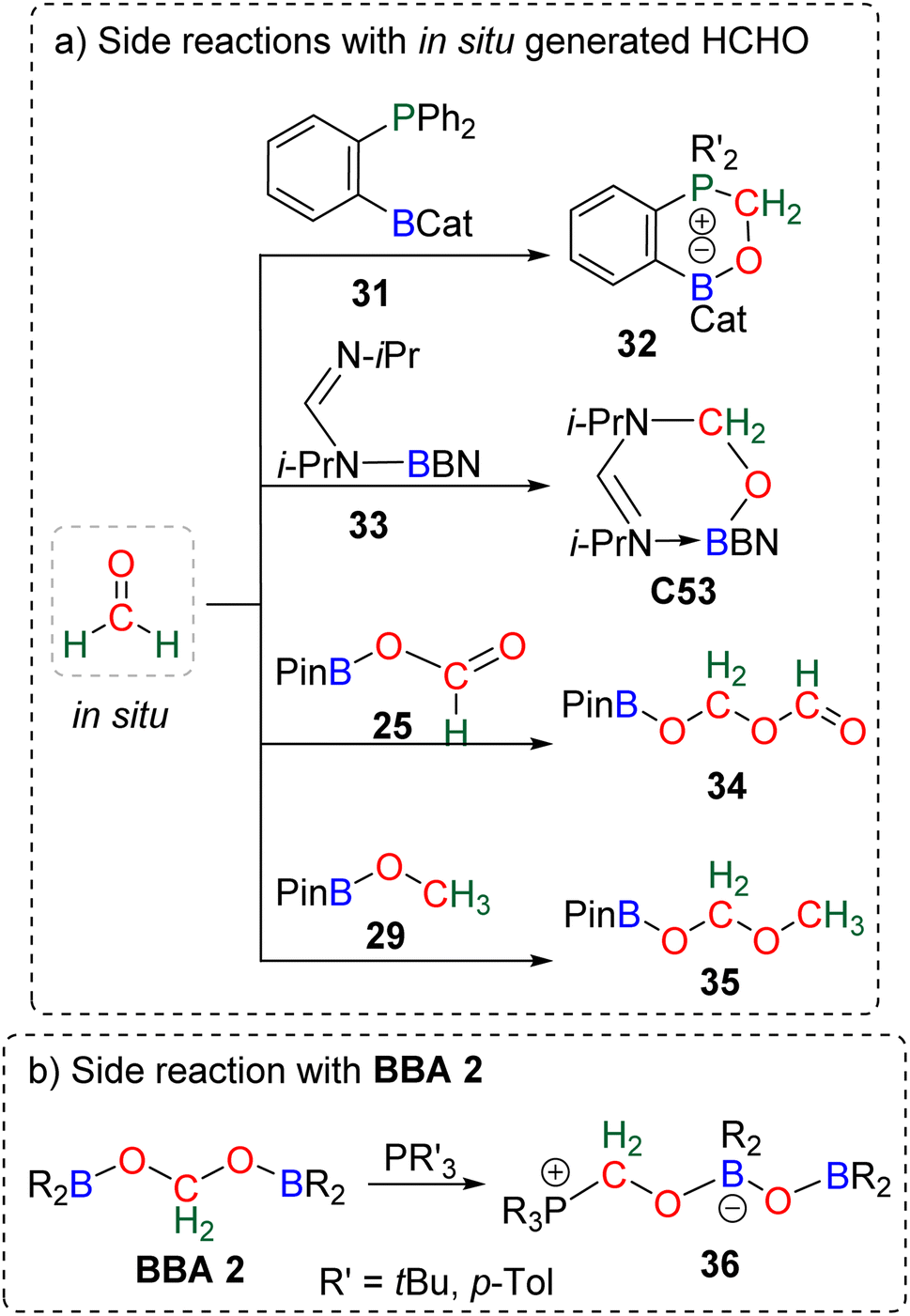 | ||
| Scheme 33 Side reactions obtained with in situ generated HCHO (a) and BBA 2 (b). | ||
The formation of compounds 32109a,118 and C53103 was postulated from calculation to arise from the reaction between transiently generated formaldehyde and the ambiphilic compounds 31 and 33, respectively. Compounds 3490,112,123 and 3591 were shown experimentally to arise from the reaction of formaldehyde with 25 and 29, respectively, which was confirmed by labelling experiments. BBA compounds were also shown to readily react under mild conditions. We believe that the characterization of compound 36 was the first observation of such BBA reactivity (Scheme 33b).120 Although the authors did not elaborate on the mechanism of its formation, we propose that 36 could be formed from the reaction of BBA 2 with the phosphine used as the catalyst, on the basis of a similar reaction later observed between BBA 2 and an NHC leading to compound 48 (Scheme 37) analogous to compound 36. These examples showed that BBA intermediates or formaldehyde is prone to react. Such “undesired” yet attractive reactivities may pave the way to more controlled transformations. Along this idea, we present hereafter different examples using BBA or formaldehyde in subsequent reactions either (i) as a C1 source for the formation of C–O, C–N, C–P and C–C bonds, or (ii) as a Cn source in oligomerization reactions leading to the formation of carbon chains and the synthesis of chiral carbon centres.
N bonds. The reactivity was further extended to the generation of the hemiaminal 39 (67% yield, 1 h) and both cyclic and acyclic aminal compounds 40 (77% yield, 3 h) and 41 (84% yield, 1 h), respectively. Finally, the reaction with ammonia afforded the cage compound hexamethylenetetramine 15 in 84% yield after 16 h. In the latter example, it is remarkable that the six fully deoxygenated carbon atoms are issued from BBA 2 and thus from CO2. BBA 1 was also used as a source of methylene in reductive functionalization reactions (Scheme 35).94 Being selectively generated (71% isolated yield), its reactivity was explored first with (diisopropyl)aniline giving rise to the corresponding imine 38 (86% yield, 24 h). It is interesting to note that similar yields were obtained starting from both BBA 1 and BBA 2 (i.e., 83% and 86% yields, respectively) but with longer reaction time with BBA 1 (24 h) than with BBA 2 (20 min). The reaction with phenylmethylamine afforded the aminal 42 (79% yield, 24 h). Finally, the reaction with diphenylphosphine gave rise to a single product assigned to compound 43. Subsequent reaction with phenylamine led to the isolation of compound 44 (78% yield, 48 h) featuring for the first time a C–P bond.
 | ||
| Scheme 34 Formation of CH2–N, CH2N and CH2–O bonds from BBA 2: generation of compounds 15, 37–41. | ||
 | ||
| Scheme 35 Generation of CH2–N and CH2–P bonds from BBA 1. | ||
C bonds could be successfully achieved from CO2.93
 | ||
| Scheme 36 Generation of C–C and CC bonds from BBA 2. | ||
 | ||
| Scheme 37 Homocoupling of BBA 2 resulting in the formation of compounds 49 and 50. | ||
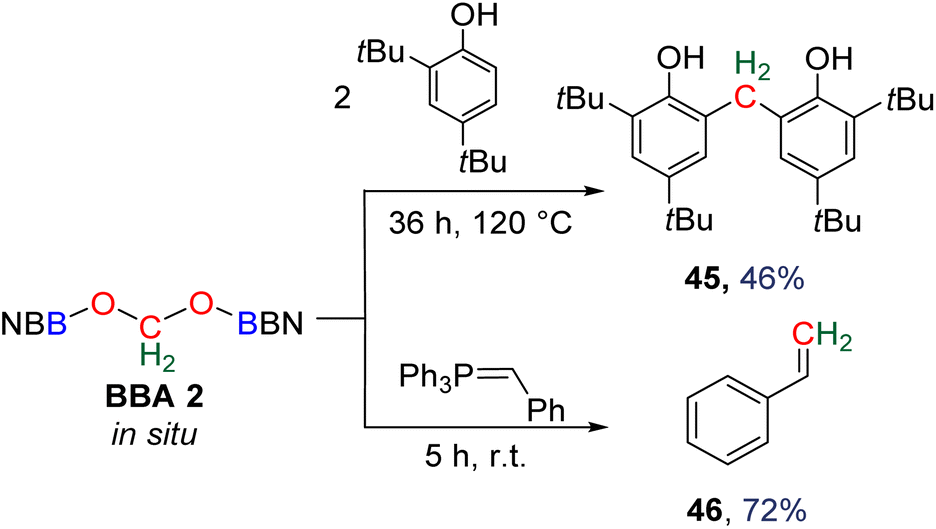
Compound 49 features a new C–C bond between two carbon atoms arising from two CO2 molecules. In this reaction, the new C–C bond was formed thanks to the nucleophilic character of the carbon atom linked to the NHC. This Umpolung process – classical of organocatalyzed C–C bond formation with such a carbene – was further used to generate compound 50 from the homocoupling of three BBA 2 molecules in the presence of 33 mol% of NHC, generating a chain of three carbon atoms (C1, C2, C3) arising from three CO2 molecules. It must be pointed out that two of the three carbon atoms (C2, C3) of the chain are asymmetric carbons and were generated with a high diastereocontrol, thanks to the presence of the boryl moiety. DFT investigations were conducted to get insights on the elementary steps accounting for these transformations,32 which stand as the first reported example of the generation of asymmetric carbon centres from CO2. In the process leading to 50, it should be noticed that formaldehyde is never released from BBA 2. The oligomerization reaction involves BBA without the intermediary of formaldehyde, as shown from the presence of boryl moieties in the final product 50. The possibility that formaldehyde could be released from BBA – in the same vein as what was explored with BSA 3 – before its engagement in the oligomerization process, was recently explored with BBA 2 and BBA 4 (Scheme 38). Despite its powerful nature, the formose reaction remains a challenging reaction due to the difficulty in controlling both the length of the carbon chain and the chirality of the generated carbohydrates.26,126,128
 | ||
| Scheme 38 Chemo- and enzyme-catalysed oligomerization of BBA 2 and 4. | ||
As presented earlier, BBA 2 was shown to quantitatively release formaldehyde after 1 h at room temperature in the presence of 10 equivalents of D2O. Optimization of both the hydrolysis and the formose steps led to the synthesis of the C2 carbohydrate glycolaldehyde 51 in 62% yield after 30 min at 80 °C using 2.5 mol% of NHC 47 as the catalyst (Scheme 37).29b Glycolaldehyde was further engaged in one-pot reactions leading to the formation of compound 52 (37% isolated yield) via a Petasis–Borono–Mannich reaction or to the formation of the C4 carbohydrates erythrose (53, 40% yield) and threose (54, 24% yield) generated from Zn(pro)-catalysed dimerization reactions (pro = proline).
In a parallel study, BBA 4 was identified as a promising candidate to conduct enzymatic transformations.30 Its isolation, in contrast to BBA 2, was seen as an important feature toward this aim. If it was stable enough for isolation, BBA 4 was nonetheless shown to readily release formaldehyde upon hydrolysis under relatively similar conditions to those with BBA 2. Enzymatic transformations were then thought about in order to control the length of the generated carbon chain and the chirality of the product. While formaldehyde is usually responsible for enzyme inhibition and even denaturation,129D-fructose-6-phosphate aldolase (FSA) and formolase (FLS) were shown to catalyse aldolization and formose transformations of formaldehyde, respectively.24,130 These enzymes were thus selected to perform the transformation of formaldehyde released from BBA 4. The enzymatic transformations were conducted at 30 °C for 48 h in a triethanolamine (TEA) buffered solution (25 mM, pH 7.0 with 1 mM MgSO4 and 0.1 mM thiamine pyrophosphate (TPP)) with THF. The presence of 10% THF was found optimal to ensure the solubility of BBA 4, while limiting the detrimental effect of this organic solvent on FLS and FSA activities (Scheme 38).30 Under these conditions, BBA 4 was oligomerized by FLS into dihydroxyacetone (55) in 86% yield. BBA 4 was then reacted with commercial dihydroxyacetone in the presence of the mono-mutant FSA A129S131 to afford the corresponding C4 carbohydrate, L-erythrulose (56) in 53% yield with a perfect enantiocontrolled aldol reaction. In this case, one carbon atom of the carbon chain arose from BBA 4. Following recently reported cascade reactions with formaldehyde,132 these two enzymes were then combined in a one-pot cascade reaction to achieve the transformation of BBA 4 into L-erythrulose 57 in 35% yield. All four carbon atoms of the carbohydrate arose from CO2, including the chiral one. This reaction corresponds to the first cell-free enantioselective transformation of CO2.
3.4. Conclusion and perspectives
In conclusion, the 4e− hydroboration of CO2 stands out for several reasons: (i) the uncatalyzed formation of three bis(boryl)acetal (BBA) compounds derived from specific hydroboranes; (ii) the highest reported turnover frequency (TOF) at 516 h−1 among hydroelementation reactions; (iii) the reactive nature of BBA at room temperature, facilitating condensation reactions or formaldehyde release; (iv) the utilization of BBA as a Cn source with or without formaldehyde release, wherein the reaction without formaldehyde release results in the formation of an O-borylated C3 compound; (v) the use of BBA in the pioneering enantioselective generation of a carbon centre from CO2.30–32 This significant breakthrough was achieved through formaldehyde release and subsequent enzymatic cascade reactions.As perspectives in CO2 hydroboration, the primary challenge lies in the stoichiometric utilization of hydroborane and thus the stoichiometric amount of borinic acid (R2BOH) generated as a co-product following the hydrolysis of BBA compounds. Since there is currently no sustainable process for regenerating hydroborane from this co-product, CO2 hydroboration is scarcely applicable unless the product synthesized from BBA holds a very high value. For example, carbohydrates, particularly rare and non-natural ones, are of significant interest to the pharmaceutical industry. Their synthesis from CO2, especially with isotope labelling, could be an avenue worth exploring for generating chiral complex Cn products from CO2. On the other hand, the prospect of generating O-borylated Cn products could open new avenues to retain boryl moieties in CO2 transformation products, instead of treating them as waste after B–O bond hydrolysis. This would allow for further functionalization of the generated compounds. To achieve these objectives, future efforts could focus on exploring a broader variety of hydroboranes with diverse properties. Such a range of hydroboranes could lead to the formation of a diverse array of BBA compounds with varying stability and reactivity, suitable for various applications. Additionally, other CO2 hydroelementation reactions, beyond hydrosilylation and hydroboration, may provide an even greater variety of acetal compounds with unique properties. Specific CO2 hydroelementation reactions involving early transition-metal hydride species are discussed in the subsequent section of this review.
4. Other hydroelementation reactions
The CO2 hydroelementation involving metal hydrides and phosphines has seen limited development. We have compiled these results in this concise standalone section, separated from the broader and more extensive sections.4.1. Hydroelementation with actinide, early TM and rare earth metal hydride species to acetal complexes
Hydride complexes of U, Zr, Ta, Nb and Y were shown to react with CO2 under mild conditions to afford the bi-metallic acetal or methylene diolate complexes shown in Scheme 39. The reaction of the U(IV) hydride complex (Cp′)3U(H) (Cp′ = C5H4(SiMe3)) with 1 atm CO2 led to the expected formoxy uranium (Cp′)3U(OCHO). However, when only 0.5 equiv. of CO2 was introduced in an NMR tube, the bis(uranyl)acetal complex (BUA) was generated.131 This complex was also isolated by crystallization from the stoichiometric reaction of (Cp′)3U(OCHO) with (Cp′)3U(H) in 75% yield after 4 h at 70 °C. The methylene fragment was characterized at 73.83 ppm in 1H NMR due to the proximity with the paramagnetic uranium centre. The reaction of the Schwartz's reagent, Cp2Zr(H)Cl, was investigated with 13CO2.134 A frozen THF-d8 solution of the Zr complex was prepared and reaction with 13CO2 was investigated at an early stage. After initiating the reaction by a gentle shaking of the frozen solution, the acetal BZrA 1 could be observed by 1H NMR analysis for only 10 min. To further characterize the acetal species, 1H and 13C{1H} NMR analyses were conducted at 235 K.134b The methylene signal appeared at δ1H = 5.36 ppm and δ13C = 101.2 ppm. Low temperature diffusion ordered NMR spectroscopy (DOSY) confirmed the dimeric nature of BZrA 1 in solution.134a The reduction of CO2 was also investigated with Ta–H and Nb–H complexes. The acetal complexes BTaA 1, BTaA 2 and BNbA 1 were isolated and characterized, notably by XRD in the case of the two former ones (Scheme 39).135 Two diastereoisomers with diastereotopic methylene protons, the rac and meso forms, were observed for BTaA 1 and BNbA 1, being the result of a C2 and Cs symmetry, respectively. The [Ta]- and [Nb]-based hydride complexes featured a peculiar aziridine-metallacycle which was shown to generate a 5-membered cycle leading to BTaA 2. Another Ta acetal complex BTaA 3 was later reported from the reaction of a dinuclear Ta tetrahydride complex with 0.9 equivalent of CO2 at low temperature.134 The methylene signal was characterized in solution at δ1H = 6.11 ppm and δ13C = 110.6 ppm and the complex was structurally characterized by X-ray diffraction analysis. Finally, a tetranuclear yttrium tetrahydride complex was also shown to readily react with 1 atm of CO2 to afford BYA featuring two methenediolate bridge fragments maintaining the tetranuclear structure. The complex was analysed in the solid state by X-ray diffraction and in solution by NMR (CH2 fragment: δ1H = 6.27 ppm and δ13C = 94.3 ppm).137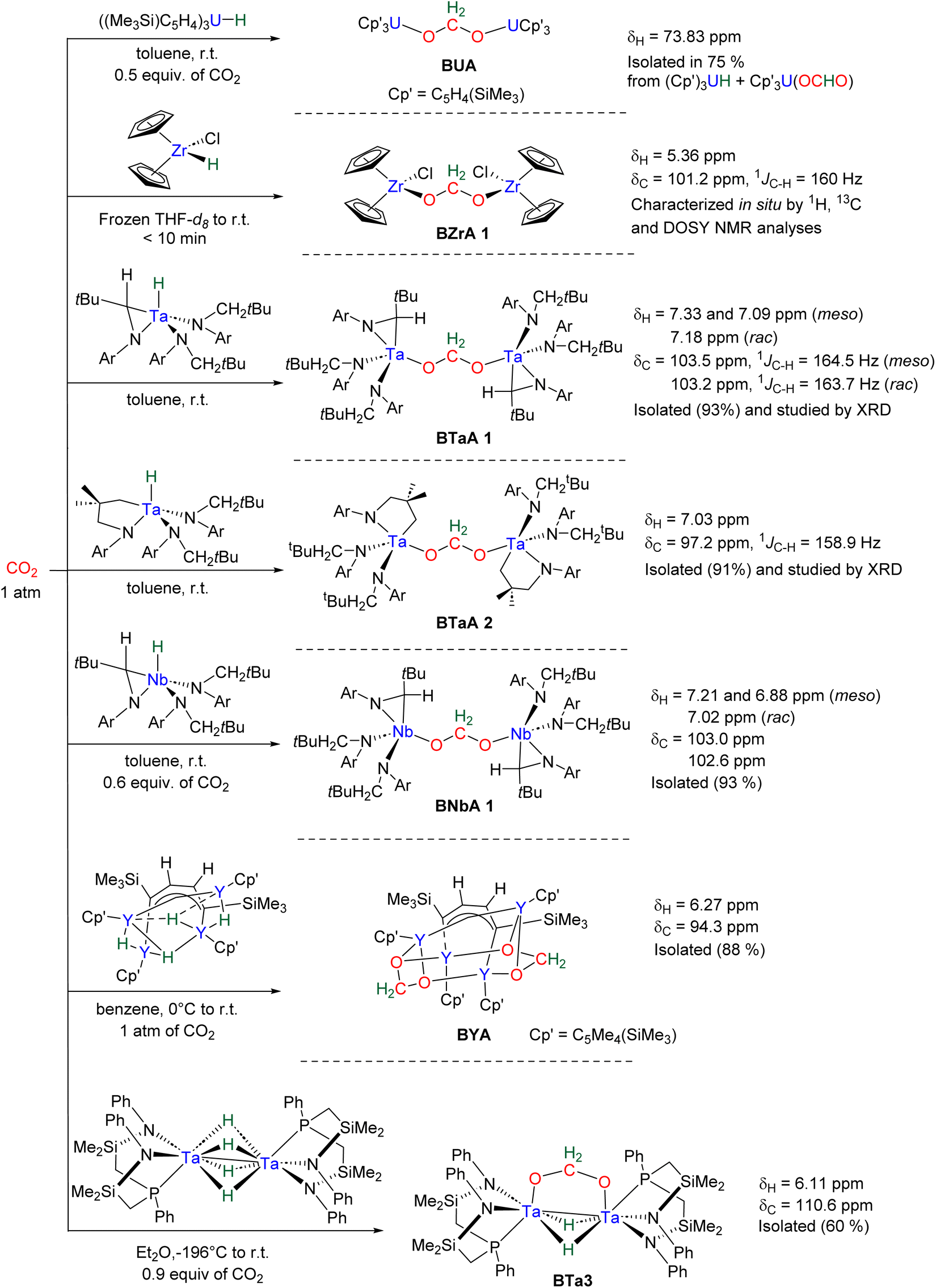 | ||
| Scheme 39 Generation of BZrA 1, BTaA 1, BTaA 2 and BNbA 1. | ||
The methylene signal of these acetal products appeared at a lower field in 1H NMR (6.11 ppm < δ1H < 7.33 ppm) as compared to BBA, BSA and BZrA 1 structures while the 13C{1H} NMR chemical shifts of the early-TM acetals are in a slightly lower field area as compared to the ones obtained from BBA and BSA derivatives (101.2 ppm < δ13C < 103.5 ppm).
DFT investigations89 were conducted to compare the reactivity of CO2 toward the Schwartz's reagent [Zr]–H complexes134vs. [Ni]–H complexes used as catalysts in CO2 hydroboration,88 regarding the formation of the corresponding metallic acetal species (Scheme 40). This study disclosed a rare comparison between the reactivities of early and late TM complexes toward CO2.138 The formation of the acetal from two equivalents of [M]–H and CO2 (steps (i) + (ii), Scheme 36) is exergonic with ΔG0 = −19.0 kcal mol−1 for the Zr complex, while it is endergonic for the Ni analogue by +42.8 kcal mol−1. Replacing the bulky tBu group of the pincer system on Ni led to a more accessible, yet still endergonic reaction by +26.9 kcal mol−1. The thermodynamic barriers (ΔG#) for step (ii) were calculated to equal 23.6 and 62.7 kcal mol−1 for the Zr–H and Ni–H, respectively. These calculations explained why the bis-nickel acetal was never observed in contrast to the Zr analogue.89
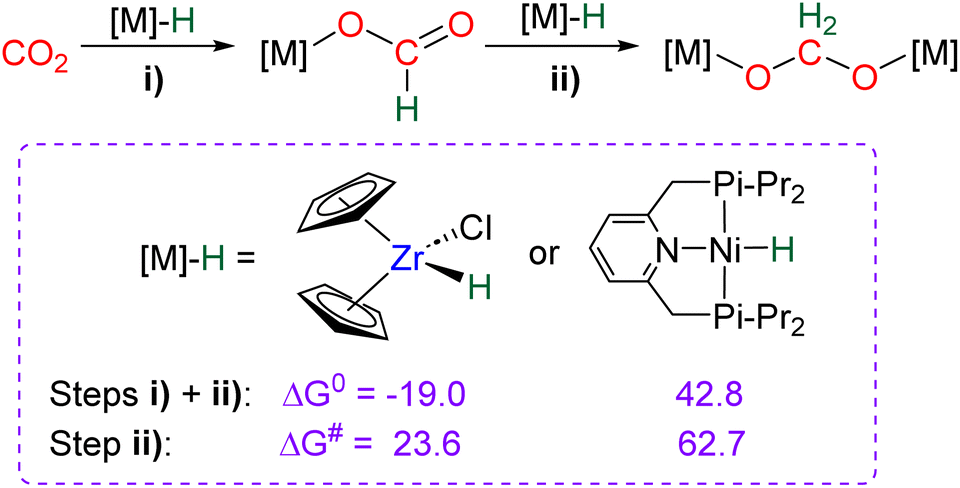 | ||
| Scheme 40 DFT investigations for the formation of metallic acetal upon reaction of [Ni]–H and [Zr]–H hydrides with CO2 (free enthalpies in kcal mol−1). | ||
4.2. Hydroelementation to formaldehyde and reductive functionalization
While the formation of BZrA 1 is favourable as shown in experiments and upon DFT calculations, it is not stable at room temperature, in contrast to BTaA 1, BTaA 2 and BNbA 1. The reaction of group IV elements toward CO2 has been studied for a long time, with an early report in the 1970s.139 Using Schwartz's reagent as the reductant, CO2 was readily reduced to formaldehyde at room temperature (Scheme 41).139a,b Besides the formation of a [ZrOZr] complex, the proof for the formation of formaldehyde was obtained by in situ condensation with 5,5-dimethylcyclohexane-1,3-dione to afford the corresponding maledone compound 16 which displays a methylene-bridged structure. This study reported the actual first example of reductive CO2 functionalization.139a,140 | ||
| Scheme 41 Condensation of a cyclohexanedione with formaldehyde generated from CO2 hydrozirconation. | ||
4.3. Hydrophosphination
CO2 hydrophosphination was reported to afford the corresponding formate compound.141 Despite similar electronegativity values for P and H atoms, 1,3,2-diazaphospholene (NHP–H) exhibits zwitterionic properties142 leading to the 2e− reduction of CO2 into a phosphorus formate moiety in high yield (93% isolated yield). However, no further reduction to the 4, 6 or 8e− reduction levels was evidenced. One can imagine that further structural modification would be necessary to achieve further reduction.4.4. Conclusion
In conclusion, the 4e− hydroelementation with early TM presented in this section stands out by the fact that (i) only four examples were reported so far; (ii) formaldehyde is spontaneously released at low temperature from BZrA 1; and (iii) the insertion of CO2 into the early TM hydride bond is uncatalyzed.As perspectives, hydroelementation of CO2 to the 4e− reduction level might offer a wide range of reactivity via the modification of the coordination sphere of the metal fragment.
5. Hydrogenation
The hydrogenation of CO2 into HCOOH, HCHO, CH3OH and CH4 are reactions of utmost importance in the actual momentum of producing green H2 from water electrolysis.13f,15a,16a,143 As opposed to hydroelementation, hydrogenation of CO2 is a 100% atom economical reaction making CO2 perfectly fitted to store energy and/or to afford key C1 building blocks for the chemical industry. As a consequence, these transformations have been intensively investigated but for formaldehyde synthesis. There are indeed currently only five catalytic systems, including heterogeneous and homogeneous processes, which have reported the generation and detection of formaldehyde in CO2 hydrogenation (Scheme 42a). These systems are presented in Section 5.1. Three of these examples generated formaldehyde as the major product and none disclosed its isolation. Reasons are because of (i) the disfavoured thermodynamic parameters in gas and in liquid phases, (ii) the highly reactive nature of formaldehyde and (iii) its volatility as a gas under atmospheric conditions. | ||
| Scheme 42 CO2 hydrogenation into 4e− reduction products: (a) formaldehyde (Section 5.1) and (b) acetals (Section 5.2). | ||
To circumvent such hurdles, the hydrogenation reaction conducted in the presence of alcohol as the solvent and co-reactant led to the stabilization of the 4e− reduction stage in the form of acetals (Scheme 42b) as described in Section 5.2, which is in line with the formation of BSA and BBA presented in the previous sections. The specific roles of H2 and alcohol are thoroughly explained in the mechanism described in Section 5.2.3.
5.1. Hydrogenation of CO2 into formaldehyde
Catalysts C64–C68 were reported to generate formaldehyde from CO2 hydrogenation (Scheme 43). The catalytic conditions and performances are presented in Table 5. It is worth mentioning that the origin of the detected HCHO was not confirmed with 13CO2 labelling experiments in any of these cases.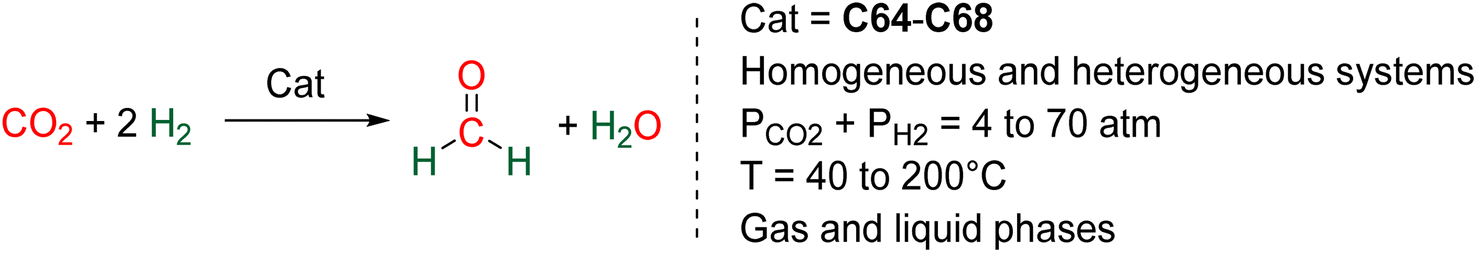 | ||
| Scheme 43 CO2 hydrogenation into formaldehyde catalysed by C64–C68. | ||
| Catalytic systems | K[Ru(EDTA-H)Cl]·H2O | Pt–Cu/SiO3 | Ni–Co NPs | Pt–Ni and Pt–Cu/Al2O3 | Ru/LDH |
|---|---|---|---|---|---|
|
C64144 |
C65145 |
C66146 |
C67147 |
C68148 |
|
| Performances | Rate = 2.79 × 10−2 M h−1 | 5.22 × 10−3 M gcat−1 h−1 | CO2 conv. = 0.24% | 0.41 × 10−5 M gcat−1 h−1 | 0.037 M gcat−1 h−1 |
| HCHO ratio = 30% | |||||
| T (°C) | 40 | 150 | 200 | 25 | 50 |
| P CO2 (atm) | 17 | 0.3 | 1 | 4 | 10 |
| P H2 (atm) | 17 | 5.7 | 3 | 66 | 10 |
| Medium | H2O | CO2 gas | CO2 gas | CH3OH | H2O |
| HCHO as the major product? | No | Yes | No | Yes | Yes |
C65,145C67147 and C68148 gave rise to the generation of formaldehyde as the major product. C64144 is the only reported molecular homogeneous system and operates in liquid media similarly to C67 and C68, while C65 and C66146 operate in the gas phase. Reaction temperatures range between 40 and 200 °C, while the total gas pressure (PCO2 + PH2) ranges between 4 and 70 atm. The positive impact of a high PH2/PCO2 ratio on the selectivity toward HCHO formation is a common feature observed for C64, C65 and C67. It can also be noticed that both selective systems, C65 and C67, relied on a Pt-based catalyst system. The small number of catalytic systems and their disparities preclude further general considerations. C64–C68 systems are described separately hereafter in a chronological order.
The first report of formaldehyde generation dates back to 1989 with the use of the homogeneous Ru-based catalyst C64.144 The reaction was performed in water at 40 °C with a total gas pressure (PCO2 + PH2) of 34 atm. Under these catalytic conditions, CO2 hydrogenation led to the generation of HCOOH and HCHO which then decomposed into CO and H2O. The generation of CO was monitored by GC analysis while formation of HCOOH and HCHO was followed spectrophotometrically upon reaction with Nash's reagent.149 Kinetic investigation showed that the decrease of PCO2 while keeping PH2 constant led to a rate increase in HCHO formation. The initial rate of formation of formaldehyde was measured at 2.79 × 10−2 M h−1 using 1 mmol of catalyst loading with an estimated activation energy of 8.3 kcal mol−1. The rate of formation of formaldehyde was found to be lower than its rate of decomposition, explaining the lack of formaldehyde build-up within the system. In addition, the rate of formation of formic acid was always higher than that of formaldehyde explaining the favoured selectivity towards formic acid. In 2001, an impregnated Pt/Cu (ratio = 0.03) on SiO2 surface (C65) was shown to afford formaldehyde and methanol with a total gas pressure (PCO2 + PH2) of 6 atm in the gas phase.145 The reaction was monitored by temperature programmed surface reaction spectroscopy indicating an optimum temperature of 150 °C for the formation of formaldehyde. While methanol was favoured at a PH2/PCO2 ratio of 3, the selectivity was reversed when this ratio was increased to 20. At this ratio, the rate of formation reached 0.12 gcat−1 h−1 for methanol and 5.22 × 10−3 gcat−1 h−1 for formaldehyde, respectively. In the absence of Pt, the analogous Cu-based catalyst afforded only methanol. The importance of Pt in the formation of formaldehyde was attributed to the known ability of Pt to adsorb H2. Indeed, the Pt activation of H2 along with a higher pressure of H2 was proposed to favour a higher concentration of H2 in the proximity of the Cu centres, responsible for shifting the product formation from methanol to formaldehyde. These observations suggest that the formation of formaldehyde and methanol follows two distinct competitive pathways and that the accessibility of activated H2 is part of the rate determining step for formaldehyde but not for methanol. In 2015, bimetallic Ni–Co nanoparticles (NPs) (C66) were produced in a bottom-up approach and their ability to catalyse CO2 hydrogenation was studied.144 Their catalytic activity was evaluated after deposition of the Ni–Co NPs on a mesoporous silica support and exposition to a CO2:H2:He (6.6:20.7:14.7) gas mixture at a total pressure of 6 atm. At 200 °C, a low conversion of CO2 (0.24%) was observed by GC analysis with 30% selectivity for formaldehyde and 70% selectivity for CO generated as the major product. Only a trace amount of formaldehyde was detected upon increasing the temperature to 350 °C. Pt–Ni and Pt–Cu (C67) catalysts selectively afforded formaldehyde in methanol medium at 25 °C and a total gas pressure (PCO2 + PH2) of 70 atm.147 The catalysts were prepared by a wet impregnation method using γ-alumina as the support. The final Pt–Ni and Pt–Cu supported catalysts contained 10 wt% Ni or Cu content and 1 wt% Pt content. Once again, a high PH2/PCO2 ratio (16) led to the generation of formaldehyde with a molar yield of 0.9 × 10−4 M gcat−1 in 22 h corresponding to 0.41 × 10−5 M gcat−1 h−1. This activity is still lower than the one reported with the C65 catalyst (5.22 × 10−3 M gcat−1 h−1).
Compound C68 is a heterogeneous catalyst consisting of highly dispersed Ru on layered double hydroxide (LDH). The use of 0.5 wt% Ru was recently shown to catalyse the hydrogenation of CO2 (PCO2 = 10 atm, PH2 = 10 atm) into 0.446 M gcat−1 of formaldehyde in water as the solvent in 12 h, corresponding to 0.037 M gcat−1 h−1.148 The detection and quantification of formaldehyde was performed using the colorimetric method with 3-methyl-2-benzothiazolinone hydrazone (MBTH) and high-performance liquid chromatography (HPLC), respectively. In the same study, dihydrogen was replaced by glycerol as a transfer hydrogenation reagent. In a 0.2 M aqueous glycerol solution, catalytic transfer hydrogenation (CTH) of CO2 afforded formaldehyde in a slightly better yield at 0.1 and 3 wt% Ru under otherwise the same conditions. It is worth mentioning that in the proposed mechanism, initiation of the reaction via the dehydrogenation of glycerol into the C3 carbohydrate dihydroxyacetone (DHA) or glyceraldehyde is proposed. However, the possibility that glycerol – notably via the generation of these carbohydrates – could be a source of formaldehyde prior to the CTH of CO2 was not investigated.150
5.2. Hydrogenation of CO2 into acetals
As presented above, the selective 4e− hydrogenation of CO2 into HCHO is still a widely open challenge. In contrast, the selective 4e− hydrogenation of CO2 into acetal was pioneered rather recently in 2016 and 2017 with homogeneous Ru- and Co-based catalytic systems.151 The formation of an acetal from CO2 hydrogenation consists in the addition of alcohol to stop at the 4e− reduction stage with the formation of acetal species (Scheme 42b). Mechanistic investigations have shown that formaldehyde is never formed during the process which alternates between reduction (hydrogenation) and functionalization (with the alcohol) steps giving rise to the acetal. The interest in the formation of dialkoxymethane derivatives is dual. First, it provides a selective access to formaldehyde since some of them were found to release HCHO in strong acidic media,153 although to our knowledge, the proof of concept from CO2 has never been reported. Second, dialkoxymethane derivatives are compounds of interest as fuel additives to lower the formation of soot because of their high oxygen content and the absence of C–C bonds.151,153 The catalytic performances of the reported systems, the in-depth optimization of the catalytic conditions, the proposed mechanism and the extent of the scope of alcohols employed are presented hereafter. | ||
| Fig. 5 Catalytic systems C69–C78 leading to the hydrogenation of CO2 into DMM. | ||
| Entry | Cat, n (μmol) | Acid, n (μmol) | P H2 (atm) | P CO2 (atm) | V CH3OH (mL) | Time (h) | T (°C) | TON | TOF (h−1) | Sela (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Selectivity = TON(DMM)/[TON(MF) + TON(DMM)]. | |||||||||||
| 1 | C69, 12.5 | Al(OTf)3, 25 | 60 | 20 | 2 | 18 | 80 | 214 | 12 | 67 | 151 |
| 2 | C70, 15 | HN(Tf)2, 45 | 60 | 20 | 2 | 22 | 100 | 92 | 4 | 74 | 151b |
| 3 | C71, 15 | HN(Tf)2, 45 | 60 | 20 | 2 | 22 | 100 | 120 | 5 | 79 | 151b |
| 4 | C72, 15 | HN(Tf)2, 45 | 60 | 20 | 2 | 22 | 100 | 157 | 7 | 81 | 151b |
| 5 | C73, 1.5 | Al(OTf)3, 6.25 | 60 | 20 | 0.5 | 18 | 80 | 32 | 2 | 28 | 154 |
| 6 | C74, 1.5 | Al(OTf)3, 6.25 | 60 | 20 | 0.5 | 18 | 80 | 221 | 12 | 71 | 154 |
| 7 | C75, 1.5 | Al(OTf)3, 6.25 | 60 | 20 | 0.5 | 18 | 80 | 292 | 16 | 72 | 154 |
| 8 | C75, 1.5 | Al(OTf)3, 6.25 | 90 | 5 | 0.5 | 18 | 90 | 97 | 5 | 92 | 154 |
| 9 | C75, 0.38 | Al(OTf)3, 1.56 | 90 | 20 | 0.5 | 18 | 90 | 786 | 44 | 60 | 154 |
| 10 | C75, 0.075 | Al(OTf)3, 3.576 | 105 | 12.5 | 0.55 | 21 | 105 | 2761 | 131 | 61 | 155 |
| 11 | C75, 0.75 | Al(OTf)3, 35.76 | 100 | 15 | 0.5 | 20 | 100 | 3874 | 194 | 73 | 155 |
| 12 | C76, 0.38 | Al(OTf)3, 3.13 | 90 | 20 | 0.5 | 18 | 90 | 685 | 38 | 60 | 156 |
| 13 | C77, 0.38 | Al(OTf)3, 3.13 | 90 | 20 | 0.5 | 18 | 90 | 439 | 24 | 46 | 156 |
| 14 | C78, 3.1 | Al(OTf)3, 25 | 20 | 100 | 5 | 20 | 100 | 1176 | 59 | 70 | 157 |
The characteristic methylene signal of DMM was observed at 4.39 ppm in the 1H NMR analysis in CD3OD. With the Ru-based precatalyst C69 featuring a triphosphine ligand (Scheme 44) and a Lewis acid co-catalyst (Al(OTf)3), the hydrogenation of CO2 in methanol was shown to afford the 2e− reduction product methoxyformate (MF) and the 4e− reduction product dimethoxymethane (DMM) (Scheme 5).151a Among the different conditions tested (variation of the Ru-based precatalyst, temperature, pressure, nature of the acid and time), the best TON of 214 was found with C69 under the following conditions: PCO2 = 20 atm, PH2 = 60 atm, 18 h, 80 °C, 2 mL of CH3OH, 12.5 μmol of Ru-based precatalyst and 25 μmol of Al(OTf)3 (Table 6, entry 1). The selectivity toward DMM vs. MF was 67%. Following this initial report, catalytic systems C70–C78 were investigated in the same reaction (Fig. 5).
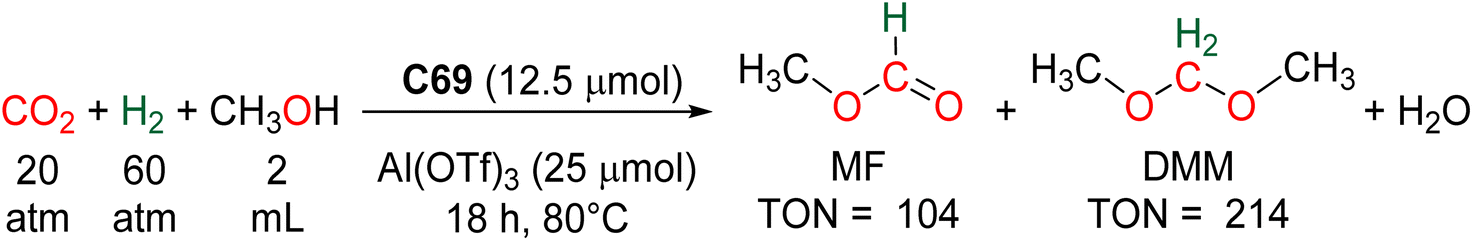 | ||
| Scheme 44 DMM synthesis with C69. | ||
Pre-catalysts C70–C72 are Co-based in contrast to all the other catalysts which are Ru-based.151bC70–C72 were found to generate DMM with slightly lower TON of 92 to 157 than C69 (Table 6, entries 2–4 vs. entry 1, resp.), with an interesting maximal selectivity of 81% for DMM. Better TONs were observed by replacing Al(OTf)3 with HN(Tf)2 as the acid, longer reaction time (22 h) and a higher temperature (100 °C). The impact of the triphosphine ligand was also probed. The use of triphosphine ligands featuring P(Xyl)2 and P(Tol)2 moieties increased the TON to 120 and 157 for C71 and C72, respectively. Optimization of the Ru-based system was further explored in detail by another group.154–156C73–C75 were tested in a first study.154 While the phosphinite moieties in C73 gave lower TONs than the other reported catalytic systems (Table 6, entry 5), replacement of the central C atom of the backbone of the ligand by a N atom in C74 and C75 enabled slight increase of the TON to 221 and 292, respectively (Table 6, entries 6 and 7). With C75, the optimization conditions led to a net increase of either the selectivity for DMM to 92% but with a TON of 97 (Table 6, entry 8) or of the TON to 786 but with a selectivity for DMM reduced to 60% (Table 6, entry 9). The quantities of the catalyst and co-catalyst were shown to be the main factor enabling this TON increase. In this optimization study for C75, all possible parameters were carefully optimized individually (univariate optimization) to improve the TON from 214 to 786 (Table 6, entries 1 and 9, resp.) which represents a 367% increase in TON. A subsequent study using a multivariate optimization led to a larger improvement of TON. The pre-catalyst C75 indeed provided TONs of 2761 and 3874 with a 10 fold increase of the scale of the reaction leading to the formation of 2.88 mmol of DMM (Table 6, entries 10 and 11, resp.).155 This corresponds to a significant improvement of further 493% from the univariate optimization (Table 6, entry 9). These impressive results were made possible mainly by acting on the quantities of the catalyst and co-catalyst. In another report, the replacement of the heteroelement of the backbone by Si or P atoms (C76 and C77, respectively) did not enable any further increase of the catalytic performances or selectivity (Table 6, entries 12 and 13).156 In order to have a closer look at the active catalyst, dihydride compound C78 was synthesized and tested (Table 6, entry 14).157 Pre-catalysts C69–77 are indeed sought to generate M–H species which act as the active catalyst in the reaction. This study showed the beneficial aspect of adding tertiary phosphine or phosphite in order to prevent catalyst deactivation by the formation of a carbonyl ligand from the CO2 decarbonylation. Overall, optimization strategies presented with catalysts C69–C78 were dedicated to the improvement of TON as the most important parameter. Although interesting selectivities of DMM vs. MM were obtained, the lesser attention to this parameter can be explained by the possibility of hydrogenating MF into DMM in a second step.154,156 However, the TOF parameter was not optimized. As indicated in Table 6, it ranges between 2 and 194 h−1, leaving room for improvement in these systems.
151a and C70 (Fig. 5),151b 11 examples were obtained and characterized with TON ranging from 16 to 118 (Scheme 45a). Aliphatic alcohols used included linear chain (containing 2 to 10 carbon atoms), branched (tBuOH and iPrOH) and benzyl alcohol. In a subsequent study, the authors used bio-sourced diols to increase the sustainability of the process and the product scope.158 These diols were shown to afford either a cyclic acetal (C-DAM) or a linear acetal (L-DAM) (Scheme 45b). In total, eight products were characterized and selectivity toward either C-DAM or L-DAM was reached depending on the nature of the diol substrate (n = 1–6). A selectivity of 92% was notably observed toward the formation of the C-DAM, 4-methyl-1,3-dioxane, while reversed selectivity of 99% to L-DAM was obtained with the diol featuring a 5-carbon linker.
 | ||
| Scheme 45 Synthesis of (a) linear DAM with C69 or C70 and (b) cyclic or linear C-DAM or L-DAM, respectively, with C69. | ||
 | ||
| Scheme 46 Proposed mechanism for the formation of DMM. | ||
Two recent in-depth studies published concomitantly in 2021 proved to be particularly complementary since one is an experimental investigation of the most used C75/Al(OTf)3 catalytic system,161 while the other is a DFT investigation on the Co-based C70/HN(Tf)2 system.153b It is noteworthy that these studies confirmed the elementary steps proposed initially in 2016. The C75/Al(OTf)3 catalytic system was investigated by in situ infrared (IR) spectroscopy at three relevant temperatures: 333 K, 353 K and 373 K enabling the determination of rate constants and activation parameters for the three steps: CO2 to MF, MF to MM and MM to DMM.161 These key steps were included in a reaction network analysis. The rate determining step (rds) was shown to be the first CO2 to MF step with an activation parameters ΔG# (333 K) of 119.0 kJ mol−1 and the lowest rate constant of 1.41 × 10−6 s−1 M−2 at the same temperature. In accord with the experiment, the competitive formation of CH3OH from the hydrogenation of MM was shown to be accessible at higher temperature and the formation of DMM was favoured by the large excess of CH3OH used as the solvent. Furthermore, this reaction network analysis was also used as a predictive tool to propose new experimental conditions that would favour one product over the other. The positive effect of H2O removal on the formation of DMM was notably proposed.
The DFT study of the C70/HN(Tf)2 system proposed a detailed kinetic and thermodynamic analysis of each elementary step accounting for the formation of DMM but also for the over-reduction to CH3OH.153b The thermodynamic values ΔG0 for the subsequent formation of formic acid, MF, MM and DMM, indicated in Scheme 47, have been calculated to be 34, 19, 53 and 41 kJ mol−1, respectively. A major input of this study was to define the exact role of the TM-based catalyst and the acid co-catalyst. The active Co-based species were calculated to be the monohydride cationic Co complex ([P3Co–H]+) depicted in Scheme 47 (purple box) and was calculated to be the catalyst responsible for the hydrogenation of CO2 into formic acid and of MF into MM. The two steps (i) and (iii) have been calculated with ΔG# of 79 and 98 kJ mol−1, respectively. On the other hand, the esterification of formic acid into MM (step ii) and the etherification of MM into DMM (step iv) are catalysed by the acid catalyst HN(Tf)2. It was found that the methylating agent responsible for the etherification of MM to DMM was not directly methanol, but CH3O-NTf2in situ generated from the reaction of CH3OH with HN(Tf)2. Nonetheless, the calculated Gibbs free energy barrier from the most stable Co-formate species is 179 kJ mol−1 for the last step (iv), making it the rate determining step (rds) of the whole process. As in the previous study, the access to DMM via formaldehyde was calculated to be higher in energy and thus discarded. The formation of formaldehyde as an intermediate to DMM was proposed in a related Ru-based system using ethanol instead of methanol, but not with the same level of kinetic or thermodynamic investigations.162
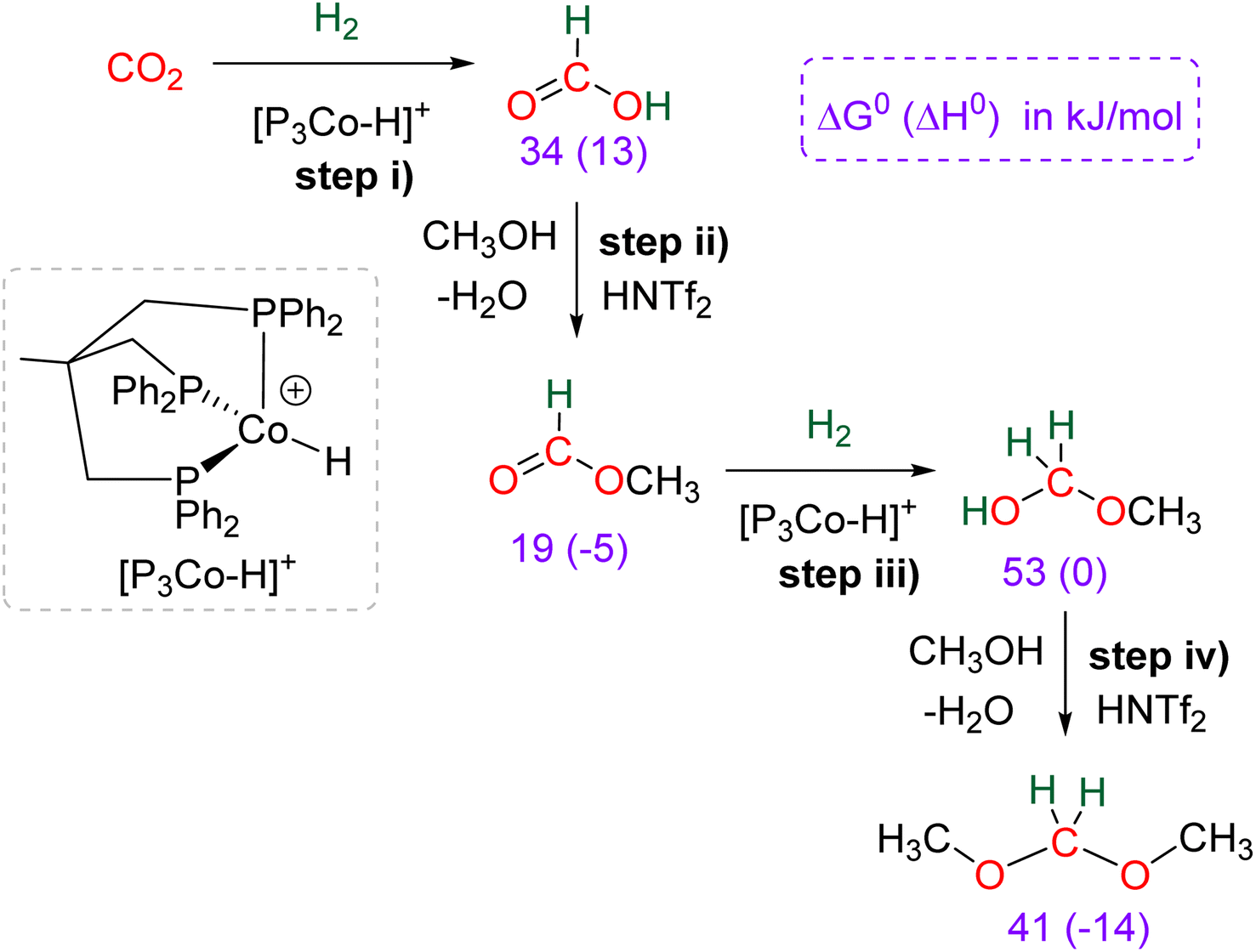 | ||
| Scheme 47 Mechanism of formation of DMM based on calculations. | ||
5.3. Conclusion
In conclusion, the selective 4e− reduction of CO2 by hydrogenation stands out for several reasons: (i) the development of highly active catalytic systems resulting in the reduction of CO2 into acetals, achieving the highest reported turnover number (TON) of 3874 for this reduction stage, following extensive optimization studies; (ii) the synthesis of acetals, which are of interest as fuel additives, in contrast to bis(silyl)acetal (BSA) and bis(boryl)acetal (BBA) compounds that lack known applications. Consequently, a variety of DAM derivatives have been synthesized, utilizing various alcohols, particularly those derived from biomass.As perspectives, future endeavours could focus on broadening the applicability of DAM, encouraging additional investigations into acetal syntheses via CO2 hydrogenation, and exploring diverse catalytic processes beyond [P3Ru] and [P3Co] systems.163 It would be intriguing to explore the potential use of the generated acetal as both C1 and Cn sources, similar to what has been described with BSA and BBA. However, the hydrogenation of CO2 into formaldehyde is still in its early stages, with only three selective systems described so far, operating in gas or liquid phases, involving catalysts C65, C67, and C68. Moreover, no isolation or utilization of the generated formaldehyde has been reported to date. Given its perfect atom efficiency and the reactive nature of formaldehyde, the primary challenge lies in developing an efficient system for hydrogenating CO2 into formaldehyde, enabling its isolation and/or utilization.
Finally, although the processes of 4e− reduction of CO2 into formaldehyde and into acetal are at very different stages of maturity, it is noteworthy that the stabilization of the 4e− reduction stage in the form of acetal parallels the stabilization of monomeric formaldehyde (HCHO) in the form of acetals of various lengths in formalin solution – the common commercial source of formaldehyde for chemical or biochemical transformations.24–26,128a–130,132,164 It is indeed noticeable that the two examples described in liquid medium for the hydrogenation of CO2 with C64 and C67 were conducted in H2O, CH3OH or glycerol. Presumably, these solvents stabilize the reactive in situ generated monomeric formaldehyde in the form of methanediol or acetal derivatives while avoiding its polymerization as paraformaldehyde.
6. Electroreduction
Electrochemical CO2 reduction reaction (eCO2RR) offers a green and sustainable approach for converting this gas into various products, under mild conditions (room temperature and pressure) and both in organic or aqueous media. Regarding C1 products, while the formation of 2e− reduction products has been performed with several catalysts with high selectivity and rate,10a the formation of 4, 6 and 8e− reduction products has been much more rarely observed (Scheme 48).165 If methanol and methane have been produced in a few cases, formaldehyde remains an elusive product to date. | ||
| Scheme 48 Electrochemical reduction of CO2 into C1 products. | ||
6.1. Electrocatalytic systems
One of the early examples of CO2 electrochemical reduction to formaldehyde concerned the use of a Ru based complex with a terpyridine–bipyridine ligand (Fig. 6) which was shown to produce a small amount of HCHO, through the formation of a formyl complex, in a mixture of water and ethanol, at low temperature (−20 °C) upon electrolysis at an Hg pool working electrode (E = −1.70 V vs. Ag/AgCl).166 A faradaic efficiency (FE) of ca. 5% was obtained for HCHO, while methanol and remarkably C–C coupling products were also identified (H(O)CCOOH, HOCH2COOH). Unexpectedly, a metal-free organic catalyst, 6,7-dimethyl-4-hydroxy-2-mercaptopteridine (a natural cofactor, Fig. 6) was reported to electrocatalyze CO2 reduction to various products, including not only formate, but also methanol and formaldehyde (not quantified).167 The reaction was conducted in a phosphate buffered solution at a reticulated vitreous carbon electrode (E = −0.65 V vs. Ag/AgCl), and it was suggested that CO2 was first reduced to formate, which was subsequently reduced to formaldehyde and methanol (the latter product was obtained with FE 10–23%) via a carbamate compound. Recently, a transition metal based polyoxometalate (TOA)6[a-SiW11O39Co(_)] (TOA: tetraoctyl ammonium, _ = vacant position at the Co atom) was shown to electroreduce CO2 under homogeneous conditions in dichloromethane as the solvent, and a potential of −1.5 V vs. Hg2Cl2/Hg at a Hg electrode.168 In the presence of an acid as a co-reactant (water and H2SO4), the products were identified as CO, with about 13% FE, and formaldehyde with a maximum efficiency (FE) of ca. 25%. The use of an organic and reducible solvent (which is shown to generate HCHO under an Ar atmosphere), limits the development of the process but this example is an interesting case of molecular electrochemical approach for CO2-to-HCHO conversion. Another molecular cobalt complex, a Co corrole (triphenylphosphine 5,10,15-tris(2,3,5,6-tetrafluoro-4-(MeO-PEG(7)thiophenyl)), Fig. 6) modified at the para positions with thiol bound poly(ethylene glycol) chains terminated by methoxy groups was adsorbed at the carbon paper electrode and then investigated for the eCO2RR into methanol and ethanol.169 In 0.1 M phosphate buffer solutions with 0.1 M NaClO4 at potentials ranging from −0.51 V to −0.8 V vs. NHE, low formaldehyde efficiencies from 10 to 1%, respectively, were obtained among other liquid phase products (formate, acetate, glyoxal), corresponding to low current densities (from 0.2 to 0.02 mA cm−2 respectively).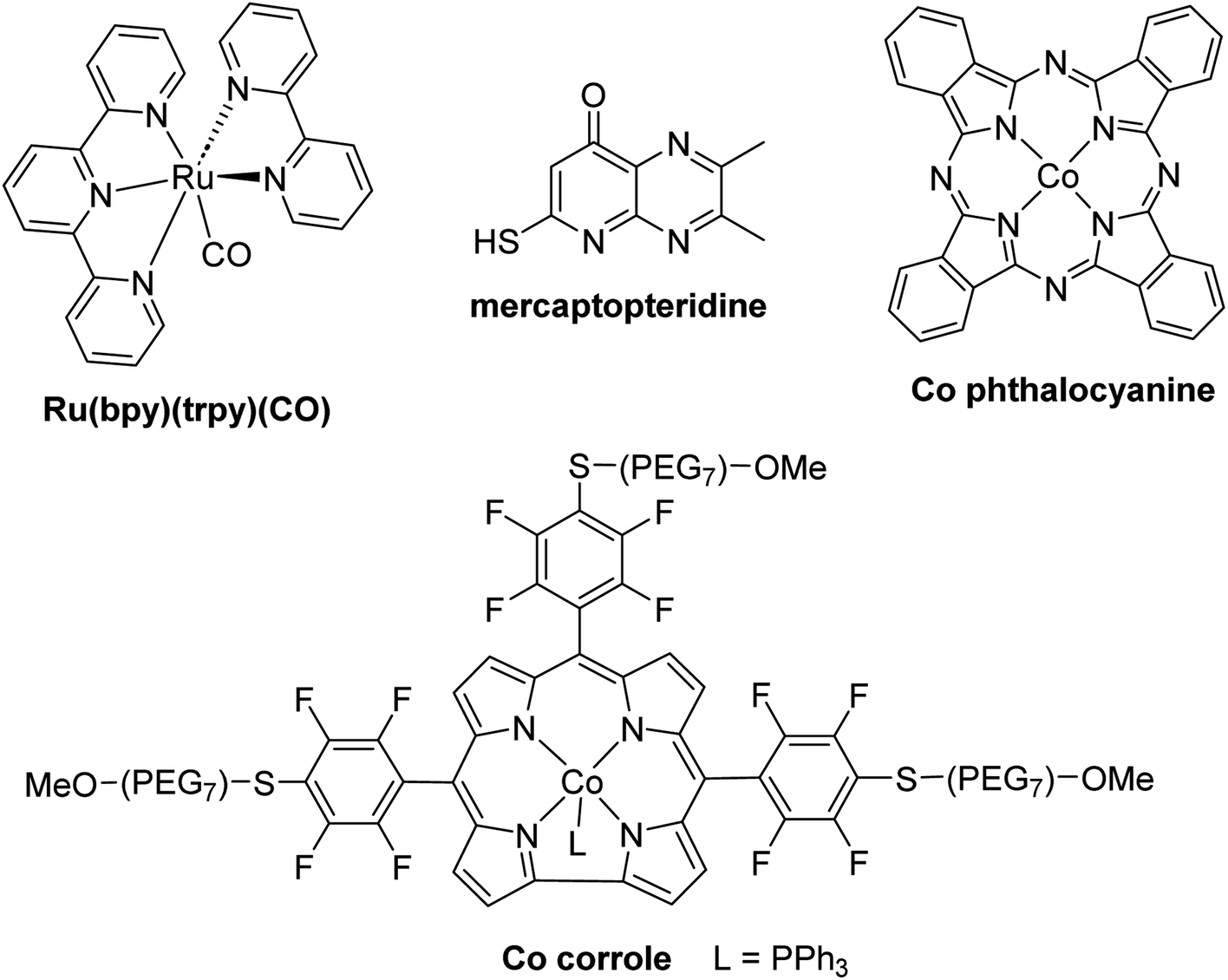 | ||
| Fig. 6 Examples of molecular catalysts capable of generating formaldehyde from CO2 electroreduction. | ||
Another strategy was developed upon polymerizing electrode films of metal complexes at the surface of conductive carbon. Cr, Co and Fe bis-terpyridine complexes bearing the vinyl group were used to grow electroactive films upon oxidation, which were further employed as catalysts in aqueous solutions with 0.1 M NaClO4.170 At potentials close to −1.1 V vs. Ag/AgCl, faradaic yields of 28% (Fe), 39% (Co) and 87% (Cr) were reported for formaldehyde, thus showing very high activity for first row transition metals while second and third row transition metals (Ru and Os respectively) gave barely any reactivity, which was assigned to the presence of metal-based redox processes in the former cases compared to ligand-based redox processes in the latter. The CO2 to CO conversion being now a well-controlled reaction, and CO being a possible intermediate towards formaldehyde production, a parallel approach was developed using CO directly as the reactant and supramolecular catalysts: thin films of Co phthalocyanine and 4,4′,4′′,4′′′-tetracarboxyphthalocyanine made at the carbon fibre were investigated and shown to produce mainly formaldehyde as the CO reduction product with very little methanol, at a potential of −0.7 V vs. SCE (Saturated Calomel Electrode) in sulfuric acid solutions.171 Recently, it was further shown that CO reduction with a phthalocyanine monomer complex also led to formaldehyde with unambiguous confirmation from labelled studies.172 These results opened the door to understanding the mechanism of formaldehyde pathway from both CO2 and CO as reactants, with possibilities of tuning the quantity of formaldehyde being produced and the ratio between HCHO and methanol. It also opens the possibility of setting a cascade approach in which the CO produced from CO2 reduction could further be used to generate HCHO. A very recent study explored such electroreduction of CO to HCHO with Co phthalocyanine deposited at a carbon paper electrode. Upon optimization of the pH (12) and of the electrode potential, a partial current density of 0.64 mA cm−2 (17.5% faradaic efficiency) was achieved for HCHO, with a maximum ratio between HCHO and CH3OH of ca. 7.5. Mechanistic insights illustrate that the binding of CO to the metal cobalt site is strong enough for further reducing it, while controlling the amount of protons and the electrode potential is key to favouring formaldehyde formation and release at the expense of methanol production.173 A puzzling discovery was made upon direct electroreduction of CO2 in various solutions (methanol, water and even sea water) at boron doped diamond (BDD) electrodes without any additional catalyst.174 These materials have several advantages, including a large potential window, high stability and furthermore the presence of sp3-bonded carbon atoms on BDD which are believed to be active sites for triggering chemical transformations. Upon 1 h electrolysis at E = −1.7 V vs. Ag/AgCl, a maximum yield (FE) of 74% was obtained for formaldehyde, along with formic acid and a very small amount of dihydrogen as a by-product. The formaldehyde was deemed to be formed from the 2e− reduction of formic acid initially obtained from CO2 conversion, while current densities were low, typically in the range of 0.1 to 0.2 mA cm−2. Finally, an elegant indirect approach has been proposed with methanol as the solvent. While methanol itself was oxidized to formaldehyde in the anodic compartment, electroreduction of CO2 at the Sn electrode led to formic acid, which reacted with the solvent to give methyl formate. Finally, the latter was reduced to formaldehyde, with faradaic efficiencies for HCHO from 10 to 50% depending on the electrolyte conditions.175
6.2. Conclusion and perspectives
In conclusion, the electrochemical reduction of CO2 to formaldehyde poses a formidable challenge, with only a limited number of examples identified thus far. These cases exhibit low yields, low current density, and, in many instances, uncertainties regarding the origin of the produced formaldehyde. The challenge of obtaining formaldehyde also arises from its susceptibility to further transformation at the electrode into more reduced products, such as methanol. Additionally, the precise catalytic mechanisms involved in these processes remain elusive with only very few mechanistic studies.173As perspectives, the prospect of in situ generating a substantial quantity of formaldehyde under mild conditions across a range of precisely controlled potentials is enticing. This potential development could pave the way for utilizing formaldehyde in subsequent chemical reactions, particularly in coupling reactions. As an early illustrative example, co-reduction scenarios involving CO2 (or CO) with nitrogen-containing species like nitrite (NO2−)176 or nitrate (NO3−)176b,177 could facilitate C–N bond formation, yielding a diverse array of products, including amines and urea. The exploration of these processes, along with related ones, under the controlled conditions afforded by electrochemistry is poised to yield new and stimulating results.
7. Photo(electro)reduction
The photochemical (PC) and photoelectrochemical (PEC) reduction of CO2 (Scheme 49), mostly using semiconducting materials, have been extensively studied in the last four decades.178 However, if compared to other reduction products such as CO or formate, only a limited number of studies so far reported the formation of formaldehyde as a major product and even less studies report a selective HCHO production, mainly because of its high reactivity.179 Moreover, very few of these studies conducted proper isotope labelled measurements to confirm the origin of the carbon atom, which constitutes a severe drawback.180 | ||
| Scheme 49 Photochemical and photoelectrochemical reduction of CO2. | ||
A very early report questioned the possible formation of formaldehyde from water and CO2 as the first stage of the photosynthetic process forming carbohydrates.181 This study showed that an aqueous solution, free of catalyst, containing carbon dioxide gives formaldehyde when irradiated with UV-light (λ = 200 nm) in a first stage, and that subsequent irradiation with lower energy light (λ = 290 nm) induced the polymerization of HCHO to sugars. It also showed that the formation of HCHO can be photocatalyzed under visible light through the use of “coloured basic substances” such as malachite-green or methyl-orange. Remarkably, the role of HCHO in the course of the photochemically induced formation of highly reduced compounds from CO2 was thus already questioned a century ago. Later, a pioneer work reported that using suspensions of various semiconductors (TiO2, ZnO, CdS, GaP and SiC, Table 7, entries 1–5) in CO2-saturated purified water, the generation of formic acid, formaldehyde and methanol was observed up to the millimolar concentration range after 7 h of visible light illumination (500 W Xe or Hg lamp).182 Trace of methane was also detected. The reduction products were quantified by gas chromatography although no labelled experiment was conducted to assert the origin of the products. The yield in methanol was observed to increase as the conduction band (CB) of the material was more negative compared to the reduction potential of the CO2-to-CH3OH reaction. A quantum yield of ca. 5 × 10−4 was reported for the formaldehyde production with both TiO2 and SiC photocatalysts. Using semiconductor electrodes polarized at −1.5 V vs. SCE, CO2 reduction to formic acid, formaldehyde and methanol was also observed with non-illuminated TiO2 and illuminated GaP. Since this seminal paper, the majority of studies published so far have explored various semiconductors,198 based on metal and metal oxides, doped or not, eventually in combination with co-catalysts,3c,199 in the form of suspensions, bulk materials or thin films. Some other examples, albeit quite rare, employed either a fully molecular, a heterogeneous material or even a photo-enzymatic approach.
| Suspensions | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Loading | Conditions | Light source | Products | Yield (μmol h−1 gcat−1) | Ref. |
| a n.d: not determined. b Catalyst deposited on a wet paper filter at the bottom of the reaction vessel. c In μmol L−1 h−1 cm−2 (reactor volume not specified). | |||||||
| 1 | TiO2 | 1 g/100 mL | Water | 500 W Xe or h.-p. Hg arc lamp | HCOOH | n.d | 182 |
| CH3OH | 3.3 | ||||||
| HCHO | 15.7 | ||||||
| 2 | ZnO | 1 g/100 mL | Water | 500 W Xe or h.-p. Hg arc lamp | HCOOH | n.d | 182 |
| CH3OH | 5 | ||||||
| HCHO | 17.1 | ||||||
| 3 | CdS | 1 g/100 mL | Water | 500 W Xe or h.-p. Hg arc lamp | HCOOH | n.d | 182 |
| CH3OH | 16.7 | ||||||
| HCHO | 28.6 | ||||||
| 4 | GaP | 1 g/100 mL | Water | 500 W Xe or h.-p. Hg arc lamp | HCOOH | n.d | 182 |
| CH3OH | 15.7 | ||||||
| HCHO | 13.3 | ||||||
| 5 | SiC | 1 g/100 mL | Water | 500 W Xe or h.-p. Hg arc lamp | HCOOH | n.d | 182 |
| CH3OH | 15.7 | ||||||
| HCHO | 13.3 | ||||||
| 6 | SrTiO3 | 4–10 g L−1 | Water | 70 W h.-p. Hg lamp (>300 nm) | CH3OH | 1.79 | 183 |
| 67 °C | HCHO | 0.01 | |||||
| 7 | WO3 | 4–10 g L−1 | Water | 70 W h.-p. Hg lamp (>300 nm) | CH3OH | 1.26 | 183 |
| 67 °C | HCHO | 0.05 | |||||
| 8 | TiO2 | 4–10 g L−1 | Water | 70 W h.-p. Hg lamp (>300 nm) | CH3OH | 0.51 | 183 |
| 34 °C | HCHO | 0.08 | |||||
| 9 | SrTiO3 + 0.5 mole % LaCrO3 | 1 g/160 mL | Water | 70 W h.-p. Hg lamp | HCOOH | 0.98 | 184 |
| 60 °C | CH3OH | 0.065 | |||||
| HCHO | 0.042 | ||||||
| 10 | CdS | 0.33 g L−1 | Water | 100 W quartz-iodine lamp (>350 nm) | HCOOH | 1.22 | 185 |
| H2S 0.097 M | HCHO | 1.95 | |||||
| 11 | Bi2S3 | 0.5 g L−1 | Water | 100 W quartz-iodine lamp (>350 nm) | HCOOH | 1.0 | 185 |
| H2S 0.097 M | HCHO | 1.58 | |||||
| 12 | TiO2 (MCB) | 0.3 g | Water | 500 W h.-p. Xe lamp | HCOOH | 0.94 | 186 |
| 60 °C | HCHO | 1.98 | |||||
| 13 | TiO2 + 0.1 mole % WO3 | 0.3 g | Water | 500 W h.-p. Xe lamp | HCHO | 1.18 | 186 |
| 60 °C | |||||||
| 14 | TiO2 + 2 mole % WO3 | 0.3 g | Water | 500 W h.-p. Xe lamp | HCHO | 1.4 | 186 |
| 60 °C | |||||||
| 15 | 1 mole % Rh/TiO2 | 0.3 g | Water | 500 W h.-p. Xe lamp | HCOOH | 0.69 | 186 |
| 60 °C | CH3OH | 0.53 | |||||
| HCHO | 0.37 | ||||||
| 16 | 0.1 wt% Au/TiO2 | 0.6 g/1.2 L | Water, Na2SO3 0.85 g L−1 | med.-p. Hg lamp (254–364 nm) | HCOOH | 148 | 187 |
| 80 °C | CH3OH | 721 | |||||
| 7 bar CO2 | HCHO | 270 | |||||
| 17 | Au/TiO2 NT photonic crystals | 85 μg Au per TiO2 wafer | Water vapor | 50 W white-cold LED (>400 nm) | CO | 323 | 188 |
| HCHO | 420 | ||||||
| 18 | TiO2 | 0.5 g L−1 | Water | 125 W med.-p. Hg lamp (254–364 nm) | H2 | 102 | 189 |
| 80 °C | HCOOH | 2954 | |||||
| 7 bar CO2 | HCHO | 16537 |
|||||
| 19 | TiO2/ZrO2 (ZrO2 40 weight%) | 0.2 g spread on a Cu plate | “Wet” (70% humidity) | Real sunlight | CH3OH | 732 | 190 |
| HCHO | 1392 | ||||||
| 20 | NaTaO3–C (annealed at 650 °C) | 100 mg/200 mL | Water | Pen ray lamp (254 nm) | HCHO | 19.5 | 191 |
| 21 | Pd/TiO2–Fe2O3 | 0.05 g/50 mL | Water pH 8.5 | 500 W Xe lamp | HCHO | 33.5 | 192 |
| Na2CO3 | C2H5OH | 101.7 | |||||
| CH3CO2H | 51 | ||||||
| 22 | CoPc/TiO2 (1% mass ratio, calcined at 400 °C) | 10 g/100 mL | 0.1 N NaOH aq. sln | 500 W tungsten–halogen lamp | HCOOH | 15.7 | 193 |
| CO | 1 | ||||||
| CH3OH | 0.5 | ||||||
| CH4 | 0.8 | ||||||
| HCHO | 2.5 | ||||||
| 23 | RuL1OH/TiO2 (0.125 μM Ru complex per gcat) | 10 mg/10 mL | 0.8 mM TEOA aq. sln | 250 W lamp (>420 nm) | CO | 2.1 | 194 |
| CH4 | 1 | ||||||
| HCHO | 11.3 | ||||||
| 24 | Sn75–Cu25/C3N4 | 1.5 mg/3.5 mL | 0.1 M KHCO3 | 300 W Xe lamp | HCOOH | 6.9 | 195 |
| 10v vol% TEA aq. sln | (>420 nm) | HCHO | 10.8 | ||||
| 25b | K2Ti6O13 + 0.3 wt% Pt | 0.3 g/4 mL | Water | 150 W Hg-lamp | H2 | 68 | 196 |
| r.t | HCOOH | 0.23 | |||||
| CH4 | 0.2 | ||||||
| HCHO | 0.476 | ||||||
| 26b | Cu/ZnO/K2Ti6O13 + 0.3 wt% Pt | 0.3 g/4 mL | Water | 150 W Hg-lamp | H2 | 17 | 196 |
| r.t | HCOOH | 3.04 | |||||
| CH4 | 0.27 | ||||||
| HCHO | 0.57 | ||||||
| 27b | K2Ti6O13 + 0.3 wt% Pt | 0.3 g/4 mL | Water | Concentrated sunlight | H2 | 32.8 | 196 |
| 274 °C | HCOOH | 20.7 | |||||
| CH4 | 0.23 | ||||||
| HCHO | 5.59 | ||||||
| 28 | TiO2 | 30 mg/20 mL | Aq. sln pH 6.5, NADH 5 mM, [Cp*Rh(bpy) (H2O)]2+ 0.25 mM | 23 W UV lamps (max 365 nm) | HCHO | 27.6 | 197 |
| (PB buff.) | |||||||
| HCHO | |||||||
| (NaOH-EDTA buff.) | 548 | ||||||
| 29 | TiO2 coupled to FDH:FADH (ratio 1:03) loaded membrane |
30 mg/20 mL | Aq. sln pH 5.5, NADH 5 mM, [Cp*Rh(bpy) (H2O)]2+ 0.25 mM | 23 W UV lamps (max 365 nm) | HCHO | 13.5 | 197 |
| (PB buff.) | |||||||
| HCHO | 345 | ||||||
| (NaOH-EDTA buff.) | |||||||
| Photoelectrodes | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Bias (V) | Conditions | Light source | Products | Yield (μmol h−1 cm−2) | Ref. |
| 30 | Polyaniline coated p-Si | −1.9 V vs. SCE | 0.1 M LiClO4 aq. sln | Xe lamp (150 mW cm−2) | HCOOH | 60.5 | 200 |
| HCHO | 15 | ||||||
| 31 | p-GaAs | −0.5 V vs. SCE | 4 M HCl aq. sln with 0.32 M V(II)/V(III) couple, 50 °C | 150 W tungsten or Xe lamp | HCOOH | n.d | 201 |
| CH3OH | |||||||
| HCHO | |||||||
| 32 | Bifunctional TiO2 with Ru N719 dye | No bias | 2 M NaOH aq. sln pH 10 | 300 W Xe lamp (cutoff > 420 nm) | HCOOH | 16.7 | 202 |
| CH3OH | 35.6 | ||||||
| HCHO | 25.8 | ||||||
| 33b | Cu2O film on Cu | No bias | 0.1 M Na2CO3/NaHCO3 aq. sln pH 9.0 | 125 W h.-p. Hg lamp | CH3OH | 9.5 | 203 |
| HCHO | 125 | ||||||
| 34 | 040-BiVO4|Cu | +0.9 V vs. RHE | 0.5 M NaCl aq. sln | 300 W Xe lamp with an AM 1.5G filter | HCHO | 108c | 204 |
| CH3OH | n.d | ||||||
| C2H5OH | n.d | ||||||
| 35 | Cu/rGO/PVP/Nafion | +0.68 V vs. RHE | 0.1 M NaOH aq. sln | 300 W Xe lamp with a KG3 filter | HCHO | 1.18 | 205 |
7.1. Suspension of a semiconductor
Aqueous suspensions of semiconducting SiTiO3, WO3 and TiO2 under both Hg lamp or natural sunlight were reported to generate a mixture of methanol (a few μmol per hour) and formaldehyde (a few tens of μmol per hour) from dissolved carbon dioxide, as quantified by gas chromatography (CH3OH) and colorimetric (HCHO) measurements (Table 7, entries 6–8).183 Traces of methane were also detected. In a follow-up study,184 aqueous suspensions of strontium titanate (SrTiO3) treated with various transition metal–TiO2 deposits, namely Rh, Pt, In and Au, and saturated with CO2 were irradiated with a 75 W Hg-lamp or a solar concentrator. When using Ir oxide as an additive (4.55 mol%), formic acid was the main product with a rate of ca. 1 μmol h−1. With Ru oxide (4.55 mol%) as the additive, methanol was the main product. The highest rate for formaldehyde formation was obtained with SrTiO3 with 0.5 mole % of lanthanum chromite (LaCrO3) as an additive (Table 7, entry 9). However, the change in selectivity with the nature of the additive was not discussed. Aqueous suspensions of n-type semiconductors such as bismuth (Bi2S3) and cadmium (CdS) sulphides were reported to generate formic acid and formaldehyde at ca. 10−5 M concentration after 1 h under wide spectrum irradiation (55 mW cm−2) when H2S was present in the system (Table 7, entries 10 and 11).185 This was explained by a sequential process producing first formic acid by 2e−/2H+ transfers then formaldehyde by additional electron transfers. The catalysis, which was shown to be proportional to the light intensity, stopped due to the poisoning deposition of elemental sulphur at the surface of the particles. The formation of HCHO as the only CO2 reduction product was also observed at a rate up to 0.25 μmol h−1 with a CO2 saturated aqueous suspension of TiO2 at 333 K under 500 W high pressure Xe lamp irradiation (Table 7, entry 12), but the origin of such selectivity is unknown.186 TiO2 doping with 0.1 mol% WO3 further increased this rate up to 0.42 μmol h−1 (Table 7, entries 13 and 14). The deposition of Rh on TiO2 led to the formation of HCOOH (major), CH3OH (secondary) and HCHO (minor) up to a total rate of 0.67 μmol h−1 (Table 7, entry 15). The doping of TiO2 with Au was more thoroughly studied. At 7 bars and 80 °C, a reactor containing 0.6 g of 0.1–0.5 wt% loading of Au/TiO2 composites led to the highest yield of formaldehyde (270 mmol kgcat−1 h−1, Table 7, entry 16).187 Another study reported the use of a gold nanoparticle decorated TiO2 photonic crystal as the photocatalyst (Au-PMTiNTs).188 When irradiated with an AM1.5 G source, methane was the main reduction product (302 μmol gcat−1 h−1, 89.3% selectivity), whereas, under the irradiation of a UV-poor white lamp, formaldehyde (420 μmol gcat−1 h−1) and CO (323 μmol gcat−1 h−1) were the major products (Table 7, entry 17). This so-called optical control of selectivity was explained by the plasmonic effects of the gold surface involving hot electrons but no discussion was made on the hydrogen atom source. Another example of TiO2 based photocatalysis was reported in which sodium sulfite was used as the reducing agent in order to fill in photogenerated holes. Under 7 bars of CO2, at 80 °C and at a TiO2 loading of 0.5 g−1 in basic (pH 14) aqueous solutions, the formation of H2 (102 μmol h−1 gcat−1), formaldehyde (16537 μmol h−1 gcat−1) and formic acid (2954 μmol h−1 gcat−1) were measured depending on experimental conditions (time and pH) under UV irradiation from a medium pressure Hg vapor 125 W lamp introduced into the photoreactor (Table 7, entry 18).189 The higher formation rate for formaldehyde was observed at the beginning of the reaction (few hours), which was explained by a branched mechanism, with on one hand a CO2-to-formic acid reduction later generating gaseous products (only detected after several hours of irradiation once the hole scavenger is consumed), and on the other hand the direct reduction of carbonates into HCHO. Composites made of nanometre-sized TiO2 particles and micrometre-sized zirconium oxide (ZrO2) particles have been reported to generate high yields of formaldehyde and methanol (maximum 300 μmol gcat−1 total after only 300 s) under real solar light illumination (Table 7, entry 19).190 For that, the composite was scattered on a copper plate and allowed to humidify in a fridge to form a thin layer of water on the composite surface. The assembly was then placed in a gas-barrier plastic bag (allowing CO2 diffusion only) containing ambient air and then was illuminated with solar light. It was proposed that reduction products originated from the reduction of bicarbonate ions resulting from the dissolution of CO2 contained (from ambient air) in the thin layer of water. A highly crystalline carbon-doped NaTaO3 perovskite was also reported to reduce CO2 to formaldehyde in neutral aqueous solutions at room temperature and under 254 nm irradiation (4.4 mW cm−2), at the optimal rate of 39 μmol gcat−1 after 2 h (Table 7, entry 20).191 This rate, determined through complexometric measurements, was shown to be dependent on the annealing temperature used to prepare the material as well as the carbon content since the undoped material showed very low formaldehyde yield. Three hematite-based materials, namely Fe2O3, TiO2–Fe2O3 and Pd/TiO2–Fe2O3 were suspended as nanoparticles in an aqueous sodium carbonate solution (pH 8.5) and illuminated with a 500 W xenon lamp. After 2 hours, the highest rate for formaldehyde was obtained with Pd/TiO2–Fe2O3 (1338 μmol L−1 gcat−1, Table 7, entry 21), when compared with the simple Fe2O3 (878 μmol L−1 gcat−1) and TiO2–Fe2O3 (399 μmol L−1 gcat−1) and was explained by the hydrogenation of formic acid with hydrogen atoms on the photocatalyst surface.192 An example of a system combining a semiconducting material and a molecule consisted of cobalt phthalocyanine loaded TiO2 nanoparticles (CoPc/TiO2) of ca. 11 nm diameter prepared by a sol–gel method and dispersed in alkaline (NaOH) aqueous solutions under a CO2 atmosphere.193 When irradiated with a tungsten–halogen 500 W lamp, a mixture of products containing CO, HCOOH, CH3OH, CH4 and HCHO was produced at a maximum yield of 407 μmol gcat−1 after 20 h under optimized conditions (CoPc/TiO2 mass ratio, calcination temperature), the latter accounting for ca. 50 μmol gcat−1 (Table 7, entry 22). The CO2 reduction to formaldehyde utilizing a heteroleptic ruthenium metal complex covalently linked to TiO2 nanoparticles was also reported to occur in aqueous medium at pH 7 under visible light (λ > 420 nm) illumination.194 When the hybrid catalyst was dispersed in neutral water, in the presence of TEOA (triethanolamine) as a sacrificial donor agent, the formation of formaldehyde as the major product (maximum TON of 720 after 5 h, corresponding to a formation rate of 18 μmol h−1 gcat−1, Table 7, entry 23) was observed in the liquid phase as determined by HPLC, with CO and CH4 as minor (TON 136 and 64, respectively), gaseous, products. Moreover, recyclability tests showed that the loss of activity was limited to 10–15% after the sixth cycle, due to some material loss during washing steps. In addition, this system used the in situ generated formaldehyde in a condensation reaction with amine, generating new H2C–N bonds (Scheme 50). To the best
of our knowledge, this represents the only example of reductive functionalization of CO2via formaldehyde in (photo)electroreduction systems.
 | ||
| Scheme 50 Visible light reduction of CO2 into formaldehyde followed by in situ condensation with an amine.194 | ||
Very recently,195 a single-atom catalyst with dual-atom sites (DAS) composed of neighbouring Sn(II) and Cu(I) centres embedded in a C3N4 framework was reported to generate HCHO from CO2 with a selectivity of 61% and a productivity of 259.1 μmol g−1 (Table 7, entry 24) after 24 h irradiation. The catalyst was dispersed in a CO2-saturated 0.1 M aqueous KHCO3 containing 10 vol% triethylamine (TEA) as the sacrificial electron donor and was irradiated using a 300 W Xe lamp equipped with a 420 nm cut-off filter. The best productivity was obtained with the Sn75–Cu25/C3N4 composition, and the production of HCHO from CO2, asserted by 13C labelled experiments and FTIR and NMR characterization, was attributed to the 2e− reduction of HCHO through the synergetic action of the two metals.
An original strategy was also tested with a Pt-loaded potassium hexatitanate (Pt–K2Ti6O13) photocatalyst (0.3 mg) deposited on a filter at the bottom of a flask containing 4 mL of water under a CO2 atmosphere.196 Upon illumination with a 150 W Hg-lamp, formaldehyde was detected as a secondary reduction product (H2 as the main one) by GC at a rate of 0.48 μmol h−1 gcat−1, together with CH4 and HCOOH (Table 7, entry 25). When combined with a Cu/ZnO catalyst, the resulting composite produced HCHO as the secondary product (H2 – major) at a rate of 0.57 μmol h−1 gcat−1 along with CH4 and HCOOH (Table 7, entries 26 and 27).
Finally, semiconductors can also be associated with an enzymatic process. For example, a very recent study reported the immobilization of two kinds of dehydrogenases, namely formate dehydrogenase (FDH) and formaldehyde dehydrogenase (FADH) (see Section 8 for more details on these enzymes), on the surface of polyethylene hollow fibre membranes, coupled with UV induced, TiO2-based photocatalytic coenzyme regeneration.197 In this system, the excitation of TiO2 causes charge separation and electrons are transferred to the NAD+ formed during the enzymatic cascade CO2 reduction, for coenzyme NADH regeneration, using EDTA as the electron donor and pentamethylcyclopentadienyl rhodium bipyridine ([Cp*Rh(bpy)(H2O)]2+) as the redox mediator. In the absence of an enzyme, HCHO was formed at a maximum rate of 0.207 mM (with H2O as the electron donor) and 4.11 mM (with EDTA as the electron donor), at pH 6.5 and after 5 h UV irradiation (365 nm max). With the photo-enzyme assembly, the best results were obtained at pH 5.5 with EDTA as the electron donor, reaching 2.59 mM of formaldehyde after 5 h of reaction (Table 7, entries 28 and 29). The production of HCHO proceeded for 48 h with limited loss of activity.
7.2. Photoelectrodes
A p-type Si photoelectrode was coated with a polyaniline film of ca. 50 nm thickness upon electro polymerization. Under illumination with a Xe lamp (150 mW cm−2) at a bias potential of −1.9 V vs. SCE, the photocathode immersed in a CO2-saturated 0.1 M LiClO4 aqueous solution generated several micromoles of formic acid and formaldehyde as determined by a colorimetric method (Table 7, entry 30).200 The bare photoelectrode did not show any activity towards CO2 reduction. The total faradaic yield reached 20 to 28% even though no methane or methanol could be detected by gas chromatography. In the presence of the V(II)–V(III) chloride redox couple presumably acting as an electron relay, a p-type GaAs photoelectrode was reported to generate formic acid, formaldehyde and methanol in low yields (from 1 to 3% faradaic yields) in an acidic (4 M HCl) aqueous solution under visible light illumination (tungsten–halogen lamp, 980 mW cm−2) at a bias potential of −0.5 V vs. SCE and at 50 °C (Table 7, entry 31).201The sequential formation of formic acid (detected by HPLC), formaldehyde (UV-Vis) and methanol (GC) was reported with a bifunctionalized TiO2 film under visible-light irradiation (300 W Xe lamp with a cut-off filter) in acetonitrile solution containing DMPImI electrolyte and the iodide/triiodide couple as a redox mediator.202 The film was separated into two zones, a sensitized area containing a ruthenium complex (N719) as the dye and a catalytic area composed of a pure TiO2 zone. Under illumination, electrons generated in the sensitized zone migrate to the catalytic one through TiO2 particles. This film was used as the reduction half-cell in a H-type reactor with Pt as the anode material, in order to prevent re-oxidation of CO2 reduction products. Formic acid was generated first, then formaldehyde and methanol, with a respective yield of ca. 0.05 mmol cm−2 after 5 h (Table 7, entry 32). This yield was enhanced when an external voltage was applied between the reference electrode and the anode thus enabling the regeneration of the redox mediator and consequently of the dye. The effect of the bias potential and the pH on the product distribution was studied with a PEC assembly composed of the Cu/Cu2O electrode irradiated with a 125 W high pressure mercury lamp.203 In 0.1 M Na2CO3/NaHCO3 electrolyte (pH 9.0) saturated with CO2, a high rate for HCHO was observed at 0 V bias (Table 7, entry 33), whereas methanol was the main product at +0.2 V bias. At the positive potential bias, some HCHO was also observed at pH 8 to 11 as a minor product.
A PEC assembly consisting of a (040)-facet engineered BiVO4 plate as the photoanode (040-BVO) and a Cu plate immersed in NaCl electrolyte as the cathode has been reported to generate various C1 products from CO2 when illuminated with solar light under an external bias potential.204 Depending on the applied potential, the selectivity could be tuned towards a specific product, with a general trend showing that at low potentials, kinetically favourable products are dominant, whereas at high potentials, thermodynamically stable products are dominant. Formaldehyde is produced with 85% faradaic efficiency at an applied potential of 0.90 V vs. RHE, corresponding to a yield of 143 μM h−1 (Table 7, entry 34). Very recently, it has been shown that a system combining calcium and iron co-doped TiO2 films as the photoanode and multilayered Cu based dark electrode as the cathode was able to selectively generate formaldehyde and/or acetaldehyde at the cathode surface by tuning the anode bias potential.205 Both electrodes were immersed in a 0.1 NaOH aqueous electrolyte saturated with CO2, and formaldehyde was detected as the only product with a reduced graphene oxide (rGO) coated copper electrode (Table 7, entry 35). The highest yield was 470 μM cm−2 after 8 h (25% faradaic efficiency) under solar simulated irradiation with an applied bias of 0.68 V vs. RHE. It is important to note that in the last example 13C labelled experiments were conducted to confirm the origin of the products.
7.3. Solid material approaches
Some studies were also conducted with catalytic solid materials. Two Mo-based heterogeneous catalysts, namely a mixed-valence polyoxomolybdate cluster and composite consisting of a molybdenum keplerate cluster embedded in reduced graphene oxide (RGO), were shown to reduce CO2 and oxidize water at the same time, generating sub-mmol amounts of formic acid as the main product and formaldehyde as the secondary product, as quantified by HPLC.206 Experiments were conducted in pure water with no sensitizer and light irradiation was set at 373 nm (19 mW cm−2). The turnover number and frequency for formic acid (not calculated for formaldehyde) were claimed to reach ca. 106 and ca. 6 × 102 s−1, respectively. This study shows the possibility of using cheap metal oxides such as oxometallates as catalytic materials for both CO2 reduction and water oxidation.7.4. Molecular approaches
To the best of our knowledge, only two studies employing a fully molecular approach have been reported. A homogeneous system employing pyridine as a catalyst in aqueous solution in the presence of Ru(phen)3 photosensitizer, ascorbic acid as an electron donor and KCl as an electrostatic stabilizing agent reported the formation, under 470 nm monochromatic illumination, of formate (NMR quantified) as the main product with methanol (GC-MS) as the minor product and traces of formaldehyde detected by NMR.207 Two ruthenium(II) polypyridyl complexes covalently modified with a pendant pyridyl function have also been shown to be active towards CO2 reduction under both electrochemical and photochemical conditions in a DMF:water mixture. In the presence of TEA (triethylamine) as a sacrificial donor agent and under 470 nm irradiation, the formation of formate (main product), formaldehyde and methanol (minor) was observed after 1 h irradiation albeit with a very low TON of 16, 7 and 1, respectively.208 The rapid loss of activity was attributed to ligand photolabilization leading to complex degradation. In neither of these examples has the mechanism been elucidated.
7.5. Conclusion and perspectives
In conclusion, achieving the selective conversion of CO2 into formaldehyde (HCHO) using light as the primary, if not the sole, energy source is far from realized, as outlined in Table 7 with key examples. The predominant outcome in the majority of studies involves a product mixture, lacking control over selectivity, with low yields and demonstrating limited mechanistic understanding. The inherent high reactivity of HCHO poses a clear challenge to its detection. Therefore, improvements in analytical detection methodologies are imperative. The use of visible (solar) light still remains scarce, with most systems requiring UV light.As perspectives, functionalized materials, in particular associating a photoactive material and molecular tuneable unit, open new possibilities to finely control the local environment of the catalytic site and thus to favour the formaldehyde pathway.
8. Enzyme-catalysed CO2 reduction into formaldehyde
Biological fixation of CO2 occurs through six major pathways including the Calvin–Benson–Bassham (CBB) cycle which represents the most common pathway.209 Interestingly, the transformation of CO2 into carbohydrates in the CBB cycle was initially hypothesized to occur via a formaldehyde pathway.181,210 However, it was later demonstrated that the CBB cycle does not involve the intermediate formation of formaldehyde, but the CO2 fixation to form a carboxylate which is then further reduced in subsequent steps.211 It is only with isolated enzymes that HCHO and CH3OH were obtained from CO2. The dehydrogenases, formate dehydrogenases (FDHs, EC 1.2.1.2),212 formaldehyde dehydrogenase (FaldDH, EC 1.2.1.46)211 and alcohol dehydrogenase (ADH, EC 1.1.1.1)213 are able to catalyse the reduction of CO2, HCOOH and HCHO into HCOOH, HCHO and CH3OH, respectively.11b,214 Although most of the reports focused on the selective generation of formate, notably because of its implication as an intermediate in the Wood–Ljungdah pathway in acetogens215 or methanol,216 only a handful of studies focused on the selective generation of formaldehyde. The two-step enzyme cascade combining formate dehydrogenase (FDH) and formaldehyde dehydrogenase (FaldDH) is currently the only reported process (Scheme 51) for the enzymatic conversion of CO2 into HCHO. Both enzymes are commercially available and have similar pH and temperature optima. Each step of the cascade is endowed with a 2e− reduction of CO2 and requires one equivalent of NADH (nicotinamide adenine dinucleotide hydride) as a co-factor, serving as the reductant. Two molecules of NADH are thus oxidized into the corresponding NAD+ during the biocatalyzed reduction of CO2 to formaldehyde. | ||
| Scheme 51 Enzymatic sequential generation of formate and formaldehyde with FDH (formate dehydrogenase) and FaldDH (formaldehyde dehydrogenase). | ||
The main parameters and performances of these selective catalytic systems are summarized in Table 8. The results are described in three sections: (i) optimized reaction conditions with free enzyme systems, (ii) immobilized and compartmentalized enzymes and, finally, (iii) systems coupled to the NADH regeneration process.
| Entry | Immobilization | Substrate | P or [C] | NADH regeneration | Time (h) | HCHO generation (mmol L−1) | Ref. |
|---|---|---|---|---|---|---|---|
| a HDMMCs = hybrid double membrane microcapsules, NPs = nanoparticles, NPSCSs = nanoparticle-stabilized capsules, MCF-MP = siliceous mesostructured cellular foams functionalized with mercaptopropyl groups, TCPP = 5,10,15,20-tetrakis(4-carboxyphenyl)porphyrin, ZIF = zeolite-like imidazole framework material, PE-HFMs = polyethylene hollow fibre membranes. | |||||||
| 1 | Free enzymes | KHCO3 | 100 mM | No | 72 | 0.060 | 217 |
| 2 | Free enzymes | CO2(g) | 0.5 MPa | No | 12 | 0.901 | 218 |
| 3 | HDMMC-based system | CO2(g) | 0.3 MPa | No | 8 | 0.022 | 219 |
| 4 | TiO2-NPs | CO2(g) | 0.3 MPa | No | 4 | — | 220 |
| 5 | NPSCs | CO2(g) | 0.3 MPa | No | 4 | 0.125 | 221 |
| 6 | MCF-MP | KHCO3 | 200 mM | No | — | 0.037 | 222 |
| 7 | 3% TCPP@ZIF-8 | NaHCO3 | 10 μM | Yes | 8 | 0.008 | 223 |
| 8 | PE-HFMs | CO2(g) | 3 mL min−1 | Yes | 4 | 0.034 | 197 |
8.1. Optimization of the reaction conditions with free enzyme systems
Studies have shown that FDH/FaldDH ratio lower than 1 is beneficial for the production of formaldehyde over formate since the formate is a good substrate for the FDH (Table 8, entry 1).217 In the same vein, an excess of NADH (NADH/NAD+ molar ratio > 2000) was also found necessary to drive the reaction. Under optimized conditions, 60 μM of formaldehyde was produced from KHCO3 instead of CO2 in 72 h with 15 g L−1 of FaldDH and 1 g L−1 of FDH. The formaldehyde concentration was quantified using a colorimetric test (Nash reagent)149 as it was found that indirect detection through UV quantification of NADH disappearance could be erroneous due to interaction with CO2. Following this study, the same cascade was explored with gaseous CO2 (Table 8, entry 2).218 Higher amount of FaldDH as compared to FDH was again necessary and the study disclosed the positive impact of a high CO2 pressure (from 0.2 to 0.5 MPa) to generate a high amount of formaldehyde. Under optimized conditions, 901 μM of formaldehyde was produced (0.5 MPa of CO2) after 12 h. A plateau was found at 10–12 h before a net decrease of formaldehyde, tentatively explained by the consumption of formaldehyde via the reverse reaction (i.e., oxidation of HCHO into CO2) and/or the instability of formaldehyde in the reaction medium.8.2. Enzyme immobilization and compartmentalization
Immobilized systems were explored to improve the performances of the FDH/FaldDH cascade with the aim to increase enzyme stability, activity224 and reusability compared to the enzymes freely diffusing systems. FDH and FaldDH were immobilized and spatially separated at organic–inorganic hybrid double membrane microcapsules (HDMMCs) (Table 8, entry 3).219 FDH was encapsulated in the lumen of the HDMMCs while FaldDH was encapsulated in the intermembrane space. The study showed an improved activity with formaldehyde production of 22 μmol compared to the control experiment with free enzymes (9 μmol). The same group encapsulated FDH in Ti nanoparticles (NPs) while FaldDH was immobilized at the surface of the particles (Table 8, entry 4).220 The size of the NPs (from 75 to 375 nm) was shown to have a significant impact on the reaction. The best performances were obtained with NP size of 75 nm. The recycling stability of the enzyme-containing NPs was evaluated and displayed a loss of half of the initial performance after 10 cycles. It was improved in a following report describing the preparation of nanoparticle-stabilized capsules (NPSCs) containing FDH entrapped in the capsule and FaldDH conjugated on the capsule surface (Table 8, entry 5).221 Furthermore, the distance between both enzymes was evaluated using Förster resonance energy transfer (FRET) analysis on the enzyme labelled with carbocyanine dyes (Table 8, entry 6).222 While no detectable FRET was observed free in the enzyme system with an estimated distance of 40 nm between the two enzymes, co-immobilization of the enzymes in the silica materials displayed an increased transfer efficiency >50% with an estimated distance of less than 10 nm. This feature was proposed to be responsible for the enhanced activity in the particles compared to the enzyme free system, due to probability increase of direct substrate transfer between the two enzymes.8.3. NADH regeneration
NADH is a co-factor that is regenerated in biological systems. When implemented in vitro, its stoichiometric use negatively impacts the atom-efficiency of any given transformation. The regeneration of NADH from NAD+ has thus been the subject of intense investigation with the development of bio-, electro- and photo-regeneration systems.225 In the case of the FDH/FaldDH cascade reaction transforming CO2 into formaldehyde, only two reports described the combination of the cascade with a regeneration process. FDH and FaldDH were co-immobilized in the cavities of the zeolitic imidazolate framework-8 (ZIF-8) combined with 5,10,15,20-tetrakis(4-carboxyphenyl) porphyrin (TCPP) as a photosensitizer fixed to the surface of the zeolite (Table 8, entry 7).223 Triethanolamine (TEOA) was used as an electron donor, Cp*Rh(bpy)Cl (M, Cp* = pentamethylcyclopentadienyl, bpy = bipyridine) as an electron mediator and water as a proton donor. Under optimum conditions (1 mg mL−1 3% TCPP@ZIF-8, pH = 8.0) the NADH regeneration reached 75% yield after 3 h under visible-light exposure. Combining this regeneration system with FDH/FaldDH (2:1 ratio) afforded the conversion of 10 μM of NaHCO3 into 7.74 μM of formaldehyde (conversion rate 77%) after 8 h under visible-light exposure. The use of titanium dioxide (TiO2) in a photocatalytic system182 was also probed with immobilized FDH/FaldDH on the surface of polyethylene (PE) hollow fibre membranes (HFMs) (Table 8, entry 8).197 Despite side reactions due to the known ability of the UV/TiO2 system to photoreduce CO2 into various products (e.g., CO, HCOOH, HCHO, CH3OH, CH4),182 NADH regeneration was observed and 34 μM of formaldehyde was generated under the best conditions (ratio FDH:FaldDH = 1:0.3, CO2 3 mL min−1, H2O as the electron donor, PB buffer, pH 7.0, [NADH] = 1 mmol L−1, exposure to two 23 W UV lamps at λ = 365 nm).
8.4. Conclusion and perspectives
In conclusion, the enzymatic conversion of CO2 into formaldehyde is still at an early stage of development. Only a few reports have highlighted the selective formation of formaldehyde in cascade reactions involving FDH and FaldDH. Strategies for enzyme immobilization were identified to enhance both the reusability and activities of FDH and FaldDH when compared to free enzyme systems.As perspectives, discovering more active and/or robust enzymes from diverse organisms or engineering existing enzymes for improved stability and activity is crucial for advancing current strategies. Additionally, the development of efficient co-factor regeneration systems is essential to establish an energetically sustainable and cost-effective process. The design of an effective co-immobilized system for enzymes coupled with a co-factor regeneration process remains undisclosed. An attractive approach involves substituting the natural co-factor NADH with a more sustainable electron and proton supplier system, as previously demonstrated in the biocatalyzed generation of formate or methanol from CO2.11b,213 Similarly, electrochemically driven enzymatic systems represent a promising avenue. Finally, the biocatalyzed four-electron reduction of CO2 to formaldehyde has yet to be explored in subsequent transformations. While such processes are enticing, the undefined form taken by the generated formaldehyde and its instability in the enzymatic reaction medium have so far hindered its isolation or utilization in subsequent transformations for the synthesis of more complex products.
9. General conclusions
In this comprehensive review, we have examined the reduction of CO2 with four electrons, leading to the production of formaldehyde and acetal derivatives through thermal (hydrogenation or hydroelementation reactions), electrochemical, photoelectrochemical, and bio-driven processes. Formaldehyde holds considerable appeal for both energy carrier applications and use as a chemical feedstock. Particularly noteworthy is its versatility for the latter application compared to formic acid, methanol, or methane obtained from the CO2 reduction reaction (CO2RR). Despite its promising attributes, synthesizing and isolating formaldehyde from the CO2RR remains significantly more challenging than the aforementioned C1 compounds. To date, only a limited number of systems have been reported to generate formaldehyde from CO2 as the major product, and in many cases, conclusive evidence regarding the origin of formaldehyde is lacking. The most notable performances of these systems are summarized in Table 9.|
|
|||
|---|---|---|---|
| Reduction system | Hydrogenation | Electroreduction | Photoelectroreduction |
| Yield | 0.037 M gcat−1 h−1 | 400–500 μmol h−1 gcat−1 (1 bar CO2) | |
| 16500 μmol h−1 gcat−1 (7 bars CO2) |
|||
| TON | — | 104 | |
| Selectivity (%) | — | 90 | 60–70 |
| Faradaic efficiency (%) | n.a | 85–90 | NA |
| Current density (mA cm−2) | n.a | 0.1–0.2 | NA |
| C1 source | ✗ | ✗ | CH2–N |
| Cn source | ✗ | ✗ | ✗ |
| Asymmetric C center | ✗ | ✗ | ✗ |

In hydrogenation reactions, the highest reported yield of formaldehyde (HCHO) was 0.037 M gcat−1 h−1 achieved with a Ru-based heterogeneous catalyst in an aqueous medium.148 Another notable instance involved electroreduction, where a film of electropolymerized chromium vinylterpyridine complex exhibited 90% faradaic efficiency (FE) and a turnover number (TON) of 104.170 In photoelectroreduction, optimal performances were observed at 16500 μmol h−1 gcat−1 under 7 bars of CO2 and 80 °C with a TiO2 catalyst, achieving 70% selectivity.189 Remarkably, in all these systems, the generated formaldehyde was utilized only once as a substrate in a cascade reaction to form a C–N bond, leaving significant untapped potential in this area for future exploration.194 Further investigations are thus imperative, both for fundamental understanding and catalyst development, and it is crucial to gather solid evidence regarding the origin of the generated formaldehyde in each case. In addition to the challenge of generating formaldehyde, detecting and isolating it proves to be a formidable task due to its reactive nature.
On the other hand, the reduction of CO2 into acetal products has seen successful developments in thermic processes, specifically in (i) the hydrogenation of CO2 in the presence of alcohol and (ii) hydroelementation reactions involving hydrosilanes, hydroboranes, and early transition metal hydride species. In contrast to the synthesis of formaldehyde, numerous acetal compounds were selectively and efficiently generated, and eventually isolated. Notably, the formation of acetals from the CO2RR currently stands as the sole pathway to formaldehyde, enabling its consistent use not only as a C1 source but also as a Cn source. Bis(silyl)acetal (BSA) and bis(boryl)acetal (BBA) were indeed shown to release formaldehyde, which was then employed in subsequent C1 and Cn transformations. It was also demonstrated that these acetals could serve as a formaldehyde surrogate without releasing formaldehyde. The primary drawback of the acetal approach is its lack of atom efficiency. While the regeneration of hydroborane or hydrosilane in a sustainable reduction process would be an ideal solution, the formation of high value-added compounds via complex transformations may compensate for this issue to some extent. Table 10 compiles the highest reported performances (TON, TOF, yield, selectivity) in each of the reductive processes leading to acetal compounds. While the chemistry of formaldehyde is well-established and developed, the acetal compounds obtained from the four-electron reduction of CO2 are, for the most part, novel molecules, and their reactivities are still under investigation. Pioneering studies on their reactivity have led to the utilization of several BSA, BBA, and BZrA 1 as a C1 source in multicomponent reactions. This facilitated the incorporation of one carbon atom from CO2 into very diverse molecules in the form of methylene. Table 10 also summarizes the types of C–E bonds (E = N, O, S, P) formed from BSA, BBA, and BZrA 1. Moreover, two BBA compounds (BBA 2 and 4) were employed as a Cn source, generating C2, C3, and C4 carbohydrates. In the case of the latter two, asymmetric carbon centres were even generated in a diastereo- and enantioselective manner, respectively. Formaldehyde was directly observed only once in 4% yield with hydrosilanes and only once among a mixture of other products with hydroboranes. The intermediary of acetals (BSA 3, BBA 2 and 4, BZrA 1) was essential for the selective generation of formaldehyde, as it was quantitatively released either spontaneously, from hydrolysis, or from CsF activation depending on the acetal properties.
|
|
||||
|---|---|---|---|---|
| Reduction system | Hydrogenation | Hydrosilylation | Hydroboration | Hydrozirconation |
| TON | 3874 | 3400 | 690 | — |
| TOF (h−1) | 194 | 10.4 | 516 | — |
| Yield (%) | — | 99 | 97 | — |
| Scale | 2.88 mmol | 4.70 mmol | 0.55 mmol | |
| C1 source | ✗ | CH2–N, –O, –S, –C | CH2–N, –O, -P, –C | CH2–C |
| Cn source | ✗ | ✗ | C2, C3, C4 | ✗ |
| Asymmetric C center | ✗ | ✗ | Diastereo- and enantio-selective transformations | ✗ |
To conclude, the 4e− reduction of CO2 is a two-face journey. On one hand, the generation of acetal compounds witnessed important advances both to control this challenging reduction stage and to use it in subsequent complex transformations but at the expense of low atom-efficiency. On the other hand, the generation of formaldehyde from hydrogenation or photo(electro)reduction is more sustainable, but still in its infancy compared to other C1 reduction compounds such as CO or methanol.
We believe that intertwined processes from the different fields involved in the CO2RR (single vs. cascade processes, coupling between electro- and organo-catalysis) may be instrumental in addressing the challenging and sustainability issues associated with the 4e− reduction of CO2.
Data availability
Correspondence and requests should be addressed to E-mail: julien.bonin@sorbonne-universite.fr; , sebastien.bontemps@lcc-toulouse.fr; or marc.robert@sorbonne-universite.fr.Author contributions
S. B. conceptualized the review. S. B, S. D., J. B. and M. R. wrote and edited the review.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Partial financial support to M. R. from the Institut Universitaire de France (IUF) is warmly thanked. S. D. thanks Région Midi-Pyrénées and Université Fédérale de Toulouse for a doctoral fellowship. S. B. thanks the CNRS for financial and technical support.References
- (a) H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Catalysis Research of Relevance to Carbon Management: Progress, Challenges, and Opportunities, Chem. Rev., 2001, 101, 953–996 CrossRef; (b) E. Fujita, Photochemical carbon dioxide reduction with metal complexes, Coord. Chem. Rev., 1999, 185–186, 373–384 CrossRef; (c) W. Leitner, The coordination chemistry of carbon dioxide and its relevance for catalysis: A critical study, Coord. Chem. Rev., 1996, 155, 247 CrossRef; (d) D. H. Gibson, The organometallic chemistry of carbon dioxide, Chem. Rev., 1996, 96, 2063–2095 CrossRef; (e) P. Braunstein, D. Matt and D. Nobel, Reactions of carbon dioxide with carbon-carbon bond formation catalyzed by transition-metal complexes, Chem. Rev., 1988, 88, 747–764 CrossRef; (f) D. B. Dell'Amico, F. Calderazzo, L. Labella, F. Marchetti and G. Pampaloni, Converting Carbon Dioxide into Carbamato Derivatives, Chem. Rev., 2003, 103, 3857–3898 CrossRef PubMed.
- (a) A. Goeppert, M. Czaun, J.-P. Jones, G. K. Surya Prakash and G. A. Olah, Recycling of carbon dioxide to methanol and derived products - closing the loop, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC; (b) M. Aresta, A. Dibenedetto and A. Angelini, Catalysis for the Valorization of Exhaust Carbon: from CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2, Chem. Rev., 2014, 114, 1709–1742 CrossRef; (c) C. Costentin, M. Robert and J.-M. Saveant, Catalysis of the electrochemical reduction of carbon dioxide, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC; (d) A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation, Chem. Rev., 2013, 113, 6621–6658 CrossRef PubMed; (e) G. Centi and S. Perathoner, Opportunities and prospects in the chemical recycling of carbon dioxide to fuels, Catal. Today, 2009, 148, 191–205 CrossRef; (f) E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels, Chem. Soc. Rev., 2009, 38, 89–99 RSC; (g) T. Sakakura, J.-C. Choi and H. Yasuda, Transformation of Carbon Dioxide, Chem. Rev., 2007, 107, 2365–2387 CrossRef PubMed; (h) M. Aresta and A. Dibenedetto, Utilisation of CO2 as a chemical feedstock: opportunities and challenges, Dalton Trans., 2007, 2975–2992 RSC; (i) M. Aresta, in Activation of small molecules, ed. W. B. Tolman, Wiley, Weiheim, Germany, 2006, pp. 1–42 Search PubMed; (j) M. D. Burkart, N. Hazari, C. L. Tway and E. L. Zeitler, Opportunities and Challenges for Catalysis in Carbon Dioxide Utilization, ACS Catal., 2019, 9, 7937–7956 CrossRef CAS.
- (a) A. Modak, P. Bhanja, S. Dutta, B. Chowdhury and A. Bhaumik, Catalytic reduction of CO2 into fuels and fine chemicals, Green Chem., 2020, 22, 4002–4033 RSC; (b) F. Marques Mota and D. H. Kim, From CO2 methanation to ambitious long-chain hydrocarbons: alternative fuels paving the path to sustainability, Chem. Soc. Rev., 2019, 48, 205–259 RSC; (c) X. Li, J. Yu, M. Jaroniec and X. Chen, Cocatalysts for Selective Photoreduction of CO2 into Solar Fuels, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed; (d) K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols, Chem. Rev., 2018, 118, 372–433 CrossRef CAS; (e) S. Remiro-Buenamañana and H. García, Photoassisted CO2 Conversion to Fuels, ChemCatChem, 2018, 11, 342–356 CrossRef; (f) J. Qiao, Y. Liu, F. Hong and J. Zhang, A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels, Chem. Soc. Rev., 2014, 43, 631–675 RSC; (g) A. J. Morris, G. J. Meyer and E. Fujita, Molecular Approaches to the Photocatalytic Reduction of Carbon Dioxide for Solar Fuels, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef PubMed.
- (a) F. Della Monica and A. W. Kleij, Mechanistic guidelines in nonreductive conversion of CO2: the case of cyclic carbonates, Catal. Sci. Technol., 2020, 10, 3483–3501 RSC; (b) Y. Yang and J.-W. Lee, Toward ideal carbon dioxide functionalization, Chem. Sci., 2019, 10, 3905–3926 RSC; (c) S. Wang and C. Xi, Recent advances in nucleophile-triggered CO2-incorporated cyclization leading to heterocycles, Chem. Soc. Rev., 2019, 48, 382–404 RSC; (d) A. Cherubini-Celli, J. Mateos, M. Bonchio, L. Dell'Amico and X. Companyó, Transition Metal-Free CO2 Fixation into New Carbon–Carbon Bonds, ChemSusChem, 2018, 11, 3056–3070 CrossRef PubMed; (e) J. Luo and I. Larrosa, C−H Carboxylation of Aromatic Compounds through CO2 Fixation, ChemSusChem, 2017, 10, 3317–3332 CrossRef PubMed; (f) T. Janes, Y. Yang and D. Song, Chemical reduction of CO2 facilitated by C-nucleophiles, Chem. Commun., 2017, 53, 11390–11398 RSC; (g) F.-G. Fontaine, M.-A. Courtemanche, M.-A. Légaré and É. Rochette, Design principles in frustrated Lewis pair catalysis for the functionalization of carbon dioxide and heterocycles, Coord. Chem. Rev., 2017, 334, 124–135 CrossRef CAS; (h) Y. Li, X. Cui, K. Dong, K. Junge and M. Beller, Utilization of CO2 as a C1 Building Block for Catalytic Methylation Reactions, ACS Catal., 2016, 1077–1086, DOI:10.1021/acscatal.6b02715; (i) D. Yu, S. P. Teong and Y. Zhang, Transition metal complex catalyzed carboxylation reactions with CO2, Coord. Chem. Rev., 2015, 293–294, 279–291 CrossRef CAS; (j) Q. Liu, L. Wu, R. Jackstell and M. Beller, Using carbon dioxide as a building block in organic synthesis, Nat. Commun., 2015, 6, 1–15 Search PubMed; (k) L. Wu, Q. Liu, R. Jackstell and M. Beller, Carbonylations of Alkenes with CO Surrogates, Angew. Chem., Int. Ed., 2014, 53, 6310–6320 CrossRef CAS; (l) I. Omae, Recent developments in carbon dioxide utilization for the production of organic chemicals, Coord. Chem. Rev., 2012, 256, 1384–1405 CrossRef CAS; (m) K. Huang, C.-L. Sun and Z.-J. Shi, Transition-metal-catalyzed C-C bond formation through the fixation of carbon dioxide, Chem. Soc. Rev., 2011, 40, 2435–2452 RSC; (n) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Transformation of Carbon Dioxide with Homogeneous Transition-Metal Catalysts: A Molecular Solution to a Global Challenge?, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS; (o) S. N. Riduan and Y. Zhang, Recent developments in carbon dioxide utilization under mild conditions, Dalton Trans., 2010, 39, 3347–3357 RSC.
- (a) G. Garcia-Garcia, M. C. Fernandez, K. Armstrong, S. Woolass and P. Styring, Analytical Review of Life-Cycle Environmental Impacts of Carbon Capture and Utilization Technologies, ChemSusChem, 2021, 14, 995–1015 CrossRef CAS; (b) A. Sternberg, C. M. Jens and A. Bardow, Life cycle assessment of CO2-based C1-chemicals, Green Chem., 2017, 19, 2244–2259 RSC; (c) J. A. Martens, A. Bogaerts, N. De Kimpe, P. A. Jacobs, G. B. Marin, K. Rabaey, M. Saeys and S. Verhelst, The Chemical Route to a Carbon Dioxide Neutral World, ChemSusChem, 2017, 10, 1039–1055 CrossRef CAS PubMed; (d) N. von der Assen, P. Voll, M. Peters and A. Bardow, Life cycle assessment of CO2 capture and utilization: a tutorial review, Chem. Soc. Rev., 2014, 43, 7982–7994 RSC.
- R. I. Masel, Z. Liu, H. Yang, J. J. Kaczur, D. Carrillo, S. Ren, D. Salvatore and C. P. Berlinguette, An industrial perspective on catalysts for low-temperature CO2 electrolysis, Nat. Nanotechnol., 2021, 16, 118–128 CrossRef CAS.
- (a) J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Selective Catalytic Synthesis Using the Combination of Carbon Dioxide and Hydrogen: Catalytic Chess at the Interface of Energy and Chemistry, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS; (b) W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS; (c) W. Wang, S. Wang, X. Ma and J. Gong, Recent advances in catalytic hydrogenation of carbon dioxide, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC; (d) P. G. Jessop, F. Joo and C.-C. Tai, Recent advances in the homogeneous hydrogenation of carbon dioxide, Coord. Chem. Rev., 2004, 248, 2425–2442 CrossRef CAS; (e) T. Schaub, CO2-based hydrogen storage: CO2 hydrogenation to formic acid, formaldehyde and methanol, Phys. Sci. Rev., 2018, 3, 20170015 Search PubMed.
- (a) S. Bontemps, Boron-Mediated Activation of Carbon Dioxide, Coord. Chem. Rev., 2016, 308(Part 2), 117–130 CrossRef CAS; (b) C. C. Chong and R. Kinjo, Catalytic Hydroboration of Carbonyl Derivatives, Imines, and Carbon Dioxide, ACS Catal., 2015, 5, 3238–3259 CrossRef CAS.
- (a) F. J. Fernández-Alvarez and L. A. Oro, Homogeneous Catalytic Reduction of CO2 with Silicon-Hydrides, State of the Art, ChemCatChem, 2018, 10, 4783–4796 CrossRef; (b) F. J. Fernandez-Alvarez, A. M. Aitani and L. A. Oro, Homogeneous catalytic reduction of CO2 with hydrosilanes, Catal. Sci. Technol., 2014, 4, 611–624 RSC.
- (a) E. Boutin, L. Merakeb, B. Ma, B. Boudy, M. Wang, J. Bonin, E. Anxolabéhère-Mallart and M. Robert, Molecular catalysis of CO2 reduction: recent advances and perspectives in electrochemical and light-driven processes with selected Fe, Ni and Co aza macrocyclic and polypyridine complexes, Chem. Soc. Rev., 2020, 49, 5772–5809 RSC; (b) S. Fukuzumi, Y.-M. Lee, H. S. Ahn and W. Nam, Mechanisms of catalytic reduction of CO2 with heme and nonheme metal complexes, Chem. Sci., 2018, 9, 6017–6034 RSC; (c) K. M. Waldie, A. L. Ostericher, M. H. Reineke, A. F. Sasayama and C. P. Kubiak, Hydricity of Transition-Metal Hydrides: Thermodynamic Considerations for CO2 Reduction, ACS Catal., 2017, 8, 1313–1324 CrossRef; (d) N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Molecular polypyridine-based metal complexes as catalysts for the reduction of CO2, Chem. Soc. Rev., 2017, 46, 761–796 RSC; (e) H. Takeda, C. Cometto, O. Ishitani and M. Robert, Electrons, Photons, Protons and Earth-Abundant Metal Complexes for Molecular Catalysis of CO2 Reduction, ACS Catal., 2016, 7, 70–88 CrossRef; (f) A. M. Appel and M. L. Helm, Determining the Overpotential for a Molecular Electrocatalyst, ACS Catal., 2014, 4, 630–633 CrossRef CAS; (g) Y. Dong, P. Duchesne, A. Mohan, K. K. Ghuman, P. Kant, L. Hurtado, U. Ulmer, J. Y. Y. Loh, A. A. Tountas, L. Wang, A. Jelle, M. Xia, R. Dittmeyer and G. A. Ozin, Shining light on CO2: from materials discovery to photocatalyst, photoreactor and process engineering, Chem. Soc. Rev., 2020, 49, 5648–5663 RSC; (h) C. S. Yeung, Photoredox Catalysis as a Strategy for CO2 Incorporation: Direct Access to Carboxylic Acids from a Renewable Feedstock, Angew. Chem., Int. Ed., 2019, 58, 5492–5502 CrossRef CAS; (i) L. j. Guo, Y. j. Wang and T. He, Photocatalytic Reduction of CO2 over Heterostructure Semiconductors into Value-Added Chemicals, Chem. Rec., 2016, 16, 1918–1933 CrossRef CAS; (j) X. Chang, T. Wang and J. Gong, CO2 photo-reduction: insights into CO2 activation and reaction on surfaces of photocatalysts, Energy Environ. Sci., 2016, 9, 2177–2196 RSC; (k) J. L. White, M. F. Baruch, J. E. Pander Iii, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Light-Driven Heterogeneous Reduction of Carbon Dioxide: Photocatalysts and Photoelectrodes, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS; (l) S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- (a) M. Yuan, M. J. Kummer and S. D. Minteer, Strategies for Bioelectrochemical CO2 Reduction, Chem.–Eur. J., 2019, 25, 14258–14266 CrossRef CAS PubMed; (b) S. Schlager, A. Dibenedetto, M. Aresta, D. H. Apaydin, L. M. Dumitru, H. Neugebauer and N. S. Sariciftci, Biocatalytic and Bioelectrocatalytic Approaches for the Reduction of Carbon Dioxide using Enzymes, Energy Technol., 2017, 5, 812–821 CrossRef CAS.
- (a) K. A. Grice, Carbon dioxide reduction with homogenous early transition metal complexes: Opportunities and challenges for developing CO2 catalysis, Coord. Chem. Rev., 2017, 336, 78–95 CrossRef CAS; (b) A. Pinaka and G. C. Vougioukalakis, Using sustainable metals to carry out “green” transformations: Fe- and Cu-catalyzed CO2 monetization, Coord. Chem. Rev., 2015, 288, 69–97 CrossRef CAS; (c) L. Zhang and Z. Hou, N-Heterocyclic carbene (NHC)-copper-catalysed transformations of carbon dioxide, Chem. Sci., 2013, 4, 3395–3403 RSC; (d) S. P and S. K. Mandal, From CO2 activation to catalytic reduction: a metal-free approach, Chem. Sci., 2020, 11, 10571–10593 RSC.
- (a) K. Liu, P. Cao, W. Chen, C. I. Ezeh, Z. Chen, Y. Luo, Q. Liu, H. Zhao, Z. Rui, S. Gao, Z. Yin, X. Sun and X. Yu, Electrocatalysis enabled transformation of earth-abundant water, nitrogen and carbon dioxide for a sustainable future, Mater. Adv., 2022, 3, 1359–1400 RSC; (b) H. Lu, J. Tournet, K. Dastafkan, Y. Liu, Y. H. Ng, S. K. Karuturi, C. Zhao and Z. Yin, Noble-Metal-Free Multicomponent Nanointegration for Sustainable Energy Conversion, Chem. Rev., 2021, 121, 10271–10366 CrossRef CAS PubMed; (c) F. Naseem, P. Lu, J. Zeng, Z. Lu, Y. H. Ng, H. Zhao, Y. Du and Z. Yin, Solid Nanoporosity Governs Catalytic CO2 and N2 Reduction, ACS Nano, 2020, 14, 7734–7759 CrossRef CAS PubMed; (d) L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Surface strategies for catalytic CO2 reduction: from two-dimensional materials to nanoclusters to single atoms, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC; (e) H. Yin, K. Xing, Y. Zhang, D. M. A. S. Dissanayake, Z. Lu, H. Zhao, Z. Zeng, J.-H. Yun, D.-C. Qi and Z. Yin, Periodic nanostructures: preparation, properties and applications, Chem. Soc. Rev., 2021, 50, 6423–6482 RSC; (f) A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes, Chem. Rev., 2017, 117, 9804–9838 CrossRef; (g) L. Zhang, Z.-J. Zhao, T. Wang and J. Gong, Nano-designed semiconductors for electro- and photoelectro-catalytic conversion of carbon dioxide, Chem. Soc. Rev., 2018, 47, 5423–5443 RSC; (h) J. Zhong, X. Yang, Z. Wu, B. Liang, Y. Huang and T. Zhang, State of the art and perspectives in heterogeneous catalysis of CO2 hydrogenation to methanol, Chem. Soc. Rev., 2020, 49, 1385–1413 RSC.
- (a) S. Liang, L. Huang, Y. Gao, Q. Wang and B. Liu, Electrochemical Reduction of CO2 to CO over Transition Metal/N-Doped Carbon Catalysts: The Active Sites and Reaction Mechanism, Adv. Sci., 2021, 8, 2102886 CrossRef CAS; (b) S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Advances and Challenges for the Electrochemical Reduction of CO2 to CO: From Fundamentals to Industrialization, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS; (c) R. Küngas, Electrochemical CO2 Reduction for CO Production: Comparison of Low- and High-Temperature Electrolysis Technologies, J. Electrochem. Soc., 2020, 167, 044508 CrossRef; (d) F.-Y. Gao, R.-C. Bao, M.-R. Gao and S.-H. Yu, Electrochemical CO2-to-CO conversion: electrocatalysts, electrolytes, and electrolyzers, J. Mater. Chem. A, 2020, 8, 15458–15478 RSC.
- (a) R. Sun, Y. Liao, S.-T. Bai, M. Zheng, C. Zhou, T. Zhang and B. F. Sels, Heterogeneous catalysts for CO2 hydrogenation to formic acid/formate: from nanoscale to single atom, Energy Environ. Sci., 2021, 14, 1247–1285 RSC; (b) P. Duarah, D. Haldar, V. S. K. Yadav and M. K. Purkait, Progress in the electrochemical reduction of CO2 to formic acid: A review on current trends and future prospects, J. Environ. Chem. Eng., 2021, 9, 106394 CrossRef CAS; (c) M. F. Philips, G.-J. M. Gruter, M. T. M. Koper and K. J. P. Schouten, Optimizing the Electrochemical Reduction of CO2 to Formate: A State-of-the-Art Analysis, ACS Sustain. Chem. Eng., 2020, 8, 15430–15444 CrossRef CAS; (d) P. Ding, H. Zhao, T. Li, Y. Luo, G. Fan, G. Chen, S. Gao, X. Shi, S. Lu and X. Sun, Metal-based electrocatalytic conversion of CO2 to formic acid/formate, J. Mater. Chem. A, 2020, 8, 21947–21960 RSC; (e) W. Leitner, Carbon-Dioxide as a Raw-Material - the Synthesis of Formic-Acid and Its Derivatives from CO2, Angew. Chem., Int. Ed., 1995, 34, 2207–2221 CrossRef CAS.
- (a) X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Recent Advances in Carbon Dioxide Hydrogenation to Methanol via Heterogeneous Catalysis, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed; (b) E. Alberico and M. Nielsen, Towards a methanol economy based on homogeneous catalysis: methanol to H2 and CO2 to methanol, Chem. Commun., 2015, 51, 6714–6725 RSC; (c) Y.-N. Li, R. Ma, L.-N. He and Z.-F. Diao, Homogeneous hydrogenation of carbon dioxide to methanol, Catal. Sci. Technol., 2014, 4, 1498–1512 RSC; (d) G. A. Olah, Towards Oil Independence Through Renewable Methanol Chemistry, Angew. Chem., Int. Ed., 2012, 52, 104–107 CrossRef PubMed.
- (a) S. W. Sheehan, Electrochemical methane production from CO2 for orbital and interplanetary refueling, iScience, 2021, 24, 102230 CrossRef CAS; (b) C. Vogt, M. Monai, G. J. Kramer and B. M. Weckhuysen, The renaissance of the Sabatier reaction and its applications on Earth and in space, Nat. Catal., 2019, 2, 188–197 CrossRef CAS; (c) P. Melo Bravo and D. P. Debecker, Combining CO2 capture and catalytic conversion to methane, Waste Dispos. Sustain. Energy., 2019, 1, 53–65 CrossRef.
- S. Bagherzadeh and N. P. Mankad, Catalyst Control of Selectivity in CO2 Reduction Using a Tunable Heterobimetallic Effect, J. Am. Chem. Soc., 2015, 137(34), 10898–10901 CrossRef CAS.
- (a) H. Zhao, Z. Lin and T. B. Marder, Density Functional Theory Studies on the Mechanism of the Reduction of CO2 to CO Catalyzed by Copper(I) Boryl Complexes, J. Am. Chem. Soc., 2006, 128, 15637–15643 CrossRef CAS PubMed; (b) D. S. Laitar, P. Mueller and J. P. Sadighi, Efficient Homogeneous Catalysis in the Reduction of CO2 to CO, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef CAS PubMed.
- (a) Y. Zhang, T. Zhang and S. Das, Catalytic transformation of CO2 into C1 chemicals using hydrosilanes as a reducing agent, Green Chem., 2020, 22, 1800–1820 RSC; (b) M. Hulla and P. J. Dyson, Pivotal Role of the Basic Character of Organic and Salt Catalysts in C−N Bond Forming Reactions of Amines with CO2, Angew. Chem., Int. Ed., 2020, 59, 1002–1017 CrossRef CAS PubMed; (c) A. Tlili, E. Blondiaux, X. Frogneux and T. Cantat, Reductive functionalization of CO2 with amines: an entry to formamide, formamidine and methylamine derivatives, Green Chem., 2015, 17, 157–168 RSC.
- (a) T. K. Todorova, M. W. Schreiber and M. Fontecave, Mechanistic Understanding of CO2 Reduction Reaction (CO2RR) Toward Multicarbon Products by Heterogeneous Copper-Based Catalysts, ACS Catal., 2020, 10, 1754–1768 CrossRef CAS; (b) G. Prieto, Carbon Dioxide Hydrogenation into Higher Hydrocarbons and Oxygenates: Thermodynamic and Kinetic Bounds and Progress with Heterogeneous and Homogeneous Catalysis, ChemSusChem, 2017, 10, 1056–1070 CrossRef CAS PubMed; (c) M. D. Porosoff, B. Yan and J. G. Chen, Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: challenges and opportunities, Energy Environ. Sci., 2016, 9, 62–73 RSC; (d) S. A. Fors and C. A. Malapit, Homogeneous Catalysis for the Conversion of CO2, CO, CH3OH, and CH4 to C2+ Chemicals via C–C Bond Formation, ACS Catal., 2023, 13, 4231–4249 CrossRef CAS.
- (a) L. E. Heim, H. Konnerth and M. H. G. Prechtl, Future Perspectives for Formaldehyde: Pathways for Reductive Synthesis and Energy Storage, Green Chem., 2017, 19, 2347–2355 RSC; (b) G. Reuss, W. Disteldorf, A. O. Gamer and A. Hilt, Formaldehyde in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, Weinheim, 2003 Search PubMed; (c) M. Trincado, H. Grützmacher and M. H. G. Prechtl, CO2-based hydrogen storage – Hydrogen generation from formaldehyde/water, Phys. Sci. Rev., 2018, 3, 20170013 Search PubMed.
- A. W. Franz, H. Kronemayer, D. Pfeiffer, R. D. Pilz, G. Reuss, W. Disteldorf, A. O. Gamer and A. Hilt, in Ullmann's Encyclopedia of Industrial Chemistry, 2016, pp. 1–34, DOI:10.1002/14356007.a11_619.pub2.
- S. Desmons, R. Fauré and S. Bontemps, Formaldehyde as a Promising C1 Source: The Instrumental Role of Biocatalysis for Stereocontrolled Reactions, ACS Catal., 2019, 9, 9575–9588 CrossRef CAS.
- S. Meninno and A. Lattanzi, Asymmetric Aldol Reaction with Formaldehyde: a Challenging Process, Chem. Rec., 2016, 16, 2016–2030 CrossRef CAS.
- (a) I. Zafar and N. Senad, The Formose Reaction: A Tool to Produce Synthetic Carbohydrates Within a Regenerative Life Support System, Curr. Org. Chem., 2012, 16, 769–788 CrossRef; (b) I. V. Delidovich, A. N. Simonov, O. P. Taran and V. N. Parmon, Catalytic Formation of Monosaccharides: From the Formose Reaction towards Selective Synthesis, ChemSusChem, 2014, 7, 1833–1846 CrossRef CAS.
- T. Nash, The colorimetric estimation of formaldehyde by means of the Hantzsch reaction, Biochem. J., 1953, 55, 416–421 CrossRef CAS PubMed.
- T. Chatterjee, E. Boutin and M. Robert, Manifesto for the routine use of NMR for the liquid product analysis of aqueous CO2 reduction: from comprehensive chemical shift data to formaldehyde quantification in water, Dalton Trans., 2020, 49, 4257–4265 RSC.
- (a) M. Rauch, Z. Strater and G. Parkin, Selective Conversion of Carbon Dioxide to Formaldehyde via a Bis(silyl)acetal: Incorporation of Isotopically Labeled C1 Moieties Derived from Carbon Dioxide into Organic Molecules, J. Am. Chem. Soc., 2019, 141, 17754–17762 CrossRef CAS; (b) D. Zhang, C. Jarava-Barrera and S. Bontemps, Selective Reductive Dimerization of CO2 into Glycolaldehyde, ACS Catal., 2021, 11, 4568–4575 CrossRef CAS.
- S. Desmons, K. Grayson-Steel, N. Nuñez-Dallos, L. Vendier, J. Hurtado, P. Clapés, R. Fauré, C. Dumon and S. Bontemps, Enantioselective Reductive Oligomerization of Carbon Dioxide into L-Erythrulose via a Chemoenzymatic Catalysis, J. Am. Chem. Soc., 2021, 143, 16274–16283 CrossRef CAS PubMed.
- T. Cai, H. Sun, J. Qiao, L. Zhu, F. Zhang, J. Zhang, Z. Tang, X. Wei, J. Yang, Q. Yuan, W. Wang, X. Yang, H. Chu, Q. Wang, C. You, H. Ma, Y. Sun, Y. Li, C. Li, H. Jiang, Q. Wang and Y. Ma, Cell-free chemoenzymatic starch synthesis from carbon dioxide, Science, 2021, 373, 1523–1527 CrossRef CAS PubMed.
- S. Zhao, H.-Q. Liang, X.-M. Hu, S. Li and K. Daasbjerg, Challenges and Prospects in the Catalytic Conversion of Carbon Dioxide to Formaldehyde, Angew. Chem., Int. Ed., 2022, 61, e202204008 CrossRef CAS.
- A. Béthegnies, Y. Escudié, N. Nuñez-Dallos, L. Vendier, J. Hurtado, I. del Rosal, L. Maron and S. Bontemps, Reductive CO2 Homocoupling: Synthesis of a Borylated C3 Carbohydrate, ChemCatChem, 2019, 11, 760–765 CrossRef.
- (a) H. Maciejewski, C. Pietraszuk, P. Pawluć and B. Marciniec, Hydrosilylation - A Comprehensive Review on Recent Advances, Springer, Berlin, 2010 Search PubMed; (b) B. Marciniec, Catalysis by transition metal complexes of alkene silylation–recent progress and mechanistic implications, Coord. Chem. Rev., 2005, 249, 2374–2390 CrossRef CAS.
- C. Hühn, A. Erlebach, D. Mey, L. Wondraczek and M. Sierka, Ab Initio energetics of Si-O bond cleavage, J. Comput. Chem., 2017, 38, 2349–2353 CrossRef.
- (a) G. Sussfink and J. Reiner, Anionic Ruthenium Clusters as Catalysts in the Hydrosilylation of Carbon-Dioxide, J. Organomet. Chem., 1981, 221, C36–C38 CrossRef; (b) H. Koinuma, F. Kawakami, H. Kato and H. Hirai, Hydrosilylation of carbon dioxide catalysed by ruthenium complexes, J. Chem. Soc., Chem. Commun., 1981, 213–214 RSC.
- T. C. Eisenschmid and R. Eisenberg, The iridium complex catalyzed reduction of carbon dioxide to methoxide by alkylsilanes, Organometallics, 1989, 8, 1822–1824 CrossRef CAS.
- T. Matsuo and H. Kawaguchi, From Carbon Dioxide to Methane: Homogeneous Reduction of Carbon Dioxide with Hydrosilanes Catalyzed by Zirconium−Borane Complexes, J. Am. Chem. Soc., 2006, 128, 12362–12363 CrossRef CAS PubMed.
- P. Rios, N. Curado, J. Lopez-Serrano and A. Rodriguez, Selective reduction of carbon dioxide to bis(silyl)acetal catalyzed by a PBP-supported nickel complex, Chem. Commun., 2016, 52, 2114–2117 RSC.
- T. T. Metsänen and M. Oestreich, Temperature-Dependent Chemoselective Hydrosilylation of Carbon Dioxide to Formaldehyde or Methanol Oxidation State, Organometallics, 2015, 34, 543–546 CrossRef.
- S. N. Riduan, Y. Zhang and J. Y. Ying, Conversion of Carbon Dioxide into Methanol with Silanes over N-Heterocyclic Carbene Catalysts, Angew. Chem., Int. Ed., 2009, 48, 3322–3325 CrossRef CAS.
- M. L. Scheuermann, S. P. Semproni, I. Pappas and P. J. Chirik, Carbon Dioxide Hydrosilylation Promoted by Cobalt Pincer Complexes, Inorg. Chem., 2014, 53, 9463–9465 CrossRef CAS.
- M. Tolzmann, L. Schürmann, A. Hepp, W. Uhl and M. Layh, Hydrosilylation and Hydrogermylation of CO2 and CS2 by Al and Ga Functionalized Silanes and Germanes – Cooperative Reactivity with Formation of Silyl Formates and Disilylacetals, Eur. J. Inorg. Chem., 2020, 2020, 4024–4036 CrossRef CAS.
- Y. Jiang, O. Blacque, T. Fox and H. Berke, Catalytic CO2 Activation Assisted by Rhenium Hydride/B(C6F5)3 Frustrated Lewis Pairs—Metal Hydrides Functioning as FLP Bases, J. Am. Chem. Soc., 2013, 135, 7751–7760 CrossRef CAS PubMed.
- F. A. LeBlanc, W. E. Piers and M. Parvez, Selective Hydrosilation of CO2 to a Bis(silylacetal) Using an Anilido Bipyridyl-Ligated Organoscandium Catalyst, Angew. Chem., Int. Ed., 2014, 53, 789–792 CrossRef CAS PubMed.
- Z. Lu, H. Hausmann, S. Becker and H. A. Wegner, Aromaticity as Stabilizing Element in the Bidentate Activation for the Catalytic Reduction of Carbon Dioxide, J. Am. Chem. Soc., 2015, 137, 5332–5335 CrossRef CAS PubMed.
- N. Del Rio, M. Lopez-Reyes, A. Baceiredo, N. Saffon-Merceron, D. Lutters, T. Müller and T. Kato, N,P-Heterocyclic Germylene/B(C6F5)3 Adducts: A Lewis Pair with Multi-reactive Sites, Angew. Chem., Int. Ed., 2017, 56, 1365–1370 CrossRef CAS.
- L. Luconi, A. Rossin, G. Tuci, Z. Gafurov, D. M. Lyubov, A. A. Trifonov, S. Cicchi, H. Ba, C. Pham-Huu, D. Yakhvarov and G. Giambastiani, Benzoimidazole-Pyridylamido Zirconium and Hafnium Alkyl Complexes as Homogeneous Catalysts for Tandem Carbon Dioxide Hydrosilylation to Methane, ChemCatChem, 2019, 11, 495–510 CrossRef CAS.
- W. Huang, T. Roisnel, V. Dorcet, C. Orione and E. Kirillov, Reduction of CO2 by Hydrosilanes in the Presence of Formamidinates of Group 13 and 12 Elements, Organometallics, 2020, 39, 698–710 CrossRef CAS.
- T. González and J. J. García, Catalytic CO2 hydrosilylation with [Mn(CO)5Br] under mild reaction conditions, Polyhedron, 2021, 203, 115242 CrossRef.
- A. Caise, J. Hicks, M. Ángeles Fuentes, J. M. Goicoechea and S. Aldridge, Partnering a Three-Coordinate Gallium Cation with a Hydroborate Counter-Ion for the Catalytic Hydrosilylation of CO2, Chem.–Eur. J., 2021, 27, 2138–2148 CrossRef CAS.
- K. Chang, I. del Rosal, X. Zheng, L. Maron and X. Xu, Hydrosilylative reduction of carbon dioxide by a homoleptic lanthanum aryloxide catalyst with high activity and selectivity, Dalton Trans., 2021, 50, 7804–7809 RSC.
- S. Park, D. Bézier and M. Brookhart, An Efficient Iridium Catalyst for Reduction of Carbon Dioxide to Methane with Trialkylsilanes, J. Am. Chem. Soc., 2012, 134, 11404–11407 CrossRef CAS.
- M. Rauch and G. Parkin, Zinc and Magnesium Catalysts for the Hydrosilylation of Carbon Dioxide, J. Am. Chem. Soc., 2017, 139, 18162–18165 CrossRef CAS PubMed.
- Q. Zhang, N. Fukaya, T. Fujitani and J.-C. Choi, Carbon Dioxide Hydrosilylation to Methane Catalyzed by Zinc and Other First-Row Transition Metal Salts, Bull. Chem. Soc. Jpn., 2019, 92, 1945–1949 CrossRef CAS.
- M.-A. Courtemanche, M.-A. Legare, E. Rochette and F.-G. Fontaine, Phosphazenes: efficient organocatalysts for the catalytic hydrosilylation of carbon dioxide, Chem. Commun., 2015, 51, 6858–6861 RSC.
- (a) B. H. Rotstein, S. H. Liang, M. S. Placzek, J. M. Hooker, A. D. Gee, F. Dolle, A. A. Wilson and N. Vasdev, 11C=O bonds made easily for positron emission tomography radiopharmaceuticals, Chem. Soc. Rev., 2016, 45, 4708–4726 RSC; (b) B. H. Rotstein, S. H. Liang, J. P. Holland, T. L. Collier, J. M. Hooker, A. A. Wilson and N. Vasdev, 11CO2 fixation: a renaissance in PET radiochemistry, Chem. Commun., 2013, 49, 5621–5629 RSC; (c) M. Ahamed, J. Verbeek, U. Funke, J. Lecina, A. Verbruggen and G. Bormans, Recent Progress in Metal Catalyzed Direct Carboxylation of Aryl Halides and Pseudo Halides Employing CO2: Opportunities for 11C Radiochemistry, ChemCatChem, 2016, 8, 3692–3700 CrossRef CAS; (d) S. Monticelli, A. Talbot, P. Gotico, F. Caillé, O. Loreau, A. Del Vecchio, A. Malandain, A. Sallustrau, W. Leibl, A. Aukauloo, F. Taran, Z. Halime and D. Audisio, Unlocking full and fast conversion in photocatalytic carbon dioxide reduction for applications in radio-carbonylation, Nat. Commun., 2023, 14, 4451 CrossRef CAS PubMed; (e) A. Malandain, M. Molins, A. Hauwelle, A. Talbot, O. Loreau, T. D'Anfray, S. Goutal, N. Tournier, F. Taran, F. Caillé and D. Audisio, Carbon Dioxide Radical Anion by Photoinduced Equilibration between Formate Salts and [11C, 13C, 14C]CO2: Application to Carbon Isotope Radiolabeling, J. Am. Chem. Soc., 2023, 145, 16760–16770 CrossRef CAS PubMed; (f) G. Destro, K. Horkka, O. Loreau, D.-A. Buisson, L. Kingston, A. Del Vecchio, M. Schou, C. S. Elmore, F. Taran, T. Cantat and D. Audisio, Transition-Metal-Free Carbon Isotope Exchange of Phenyl Acetic Acids, Angew. Chem., Int. Ed., 2020, 59, 13490–13495 CrossRef CAS PubMed; (g) G. Destro, O. Loreau, E. Marcon, F. Taran, T. Cantat and D. Audisio, Dynamic Carbon Isotope Exchange of Pharmaceuticals with Labeled CO2, J. Am. Chem. Soc., 2019, 141, 780–784 CrossRef CAS; (h) P. Gotico, A. Del Vecchio, D. Audisio, A. Quaranta, Z. Halime, W. Leibl and A. Aukauloo, Visible-Light-Driven Reduction of CO2 to CO and Its Subsequent Valorization in Carbonylation Chemistry and 13C Isotope Labeling, ChemPhotoChem, 2018, 2, 715–719 CrossRef CAS.
- A. Berkefeld, W. E. Piers and M. Parvez, Tandem Frustrated Lewis Pair/Tris(pentafluorophenyl)borane-Catalyzed Deoxygenative Hydrosilylation of Carbon Dioxide, J. Am. Chem. Soc., 2010, 132, 10660–10661 CrossRef CAS PubMed.
- A. Berkefeld, W. E. Piers, M. Parvez, L. Castro, L. Maron and O. Eisenstein, Decamethylscandocinium-hydrido-(perfluorophenyl)borate: fixation and tandem tris(perfluorophenyl)borane catalysed deoxygenative hydrosilation of carbon dioxide, Chem. Sci., 2013, 4, 2152–2162 RSC.
- F. Bertini, M. Glatz, B. Stöger, M. Peruzzini, L. F. Veiros, K. Kirchner and L. Gonsalvi, Carbon Dioxide Reduction to Methanol Catalyzed by Mn(I) PNP Pincer Complexes under Mild Reaction Conditions, ACS Catal., 2019, 9, 632–639 CrossRef CAS.
- D. S. Morris, C. Weetman, J. T. C. Wennmacher, M. Cokoja, M. Drees, F. E. Kühn and J. B. Love, Reduction of carbon dioxide and organic carbonyls by hydrosilanes catalysed by the perrhenate anion, Catal. Sci. Technol., 2017, 7, 2838–2845 RSC.
- X. Zhang, J. Sun, G. Wei, Z. Liu, H. Yang, K. Wang and H. Fei, In Situ Generation of an N-Heterocyclic Carbene Functionalized Metal–Organic Framework by Postsynthetic Ligand Exchange: Efficient and Selective Hydrosilylation of CO2, Angew. Chem., Int. Ed., 2019, 58, 2844–2849 CrossRef CAS PubMed.
- J. Chen, L. Falivene, L. Caporaso, L. Cavallo and E. Y. X. Chen, Selective Reduction of CO2 to CH4 by Tandem Hydrosilylation with Mixed Al/B Catalysts, J. Am. Chem. Soc., 2016, 138, 5321–5333 CrossRef CAS PubMed.
- D. W. Beh, W. E. Piers, B. S. Gelfand and J.-B. Lin, Tandem deoxygenative hydrosilation of carbon dioxide with a cationic scandium hydridoborate and B(C6F5)3, Dalton Trans., 2020, 49, 95–101 RSC.
- F. Huang, G. Lu, L. Zhao, H. Li and Z.-X. Wang, The Catalytic Role of N-Heterocyclic Carbene in a Metal-Free Conversion of Carbon Dioxide into Methanol: A Computational Mechanism Study, J. Am. Chem. Soc., 2010, 132, 12388–12396 CrossRef CAS.
- (a) T. Robert and M. Oestreich, Si-H Bond Activation: Bridging Lewis Acid Catalysis with Brookhart's Iridium(III) Pincer Complex and B(C6F5)3, Angew. Chem., Int. Ed., 2013, 52, 5216–5218 CrossRef CAS PubMed; (b) D. J. Parks, J. M. Blackwell and W. E. Piers, Studies on the Mechanism of B(C6F5)3-Catalyzed Hydrosilation of Carbonyl Functions, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS; (c) V. Gevorgyan, M. Rubin, S. Benson, J.-X. Liu and Y. Yamamoto, A Novel B(C6F5)3-Catalyzed Reduction of Alcohols and Cleavage of Aryl and Alkyl Ethers with Hydrosilanes, J. Org. Chem., 2000, 65, 6179–6186 CrossRef CAS; (d) D. J. Parks and W. E. Piers, Tris(pentafluorophenyl)boron-Catalyzed Hydrosilation of Aromatic Aldehydes, Ketones, and Esters, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- S. Fang, H. Chen and H. Wei, Insight into catalytic reduction of CO2 to methane with silanes using Brookhart's cationic Ir(III) pincer complex, RSC Adv., 2018, 8, 9232–9242 RSC.
- M. Wen, F. Huang, G. Lu and Z.-X. Wang, Density Functional Theory Mechanistic Study of the Reduction of CO2 to CH4 Catalyzed by an Ammonium Hydridoborate Ion Pair: CO2 Activation via Formation of a Formic Acid Entity, Inorg. Chem., 2013, 52, 12098–12107 CrossRef CAS.
- P. Ríos, A. Rodríguez and J. López-Serrano, Mechanistic Studies on the Selective Reduction of CO2 to the Aldehyde Level by a Bis(phosphino)boryl (PBP)-Supported Nickel Complex, ACS Catal., 2016, 6, 5715–5723 CrossRef.
- T. Stahl, H. F. T. Klare and M. Oestreich, C(sp3)–F Bond Activation of CF3-Substituted Anilines with Catalytically Generated Silicon Cations: Spectroscopic Evidence for a Hydride-Bridged Ru–S Dimer in the Catalytic Cycle, J. Am. Chem. Soc., 2013, 135, 1248–1251 CrossRef CAS PubMed.
- H. H. Cramer, S. Ye, F. Neese, C. Werlé and W. Leitner, Cobalt-Catalyzed Hydrosilylation of Carbon Dioxide to the Formic Acid, Formaldehyde, and Methanol Level—How to Control the Catalytic Network?, JACS Au, 2021, 1, 2058–2069 CrossRef CAS PubMed.
- (a) K. Fujiwara, S. Yasuda and T. Mizuta, Reduction of CO2 to Trimethoxyboroxine with BH3 in THF, Organometallics, 2014, 33, 6692–6695 CrossRef CAS; (b) I. Knopf and C. C. Cummins, Revisiting CO2 Reduction with NaBH4 under Aprotic Conditions: Synthesis and Characterization of Sodium Triformatoborohydride, Organometallics, 2015, 34, 1601–1603 CrossRef CAS.
- Y. Zhao, X. Liu, L. Zheng, Y. Du, X. Shi, Y. Liu, Z. Yan, J. You and Y. Jiang, One-Pot Methylenation–Cyclization Employing Two Molecules of CO2 with Arylamines and Enaminones, J. Org. Chem., 2020, 85, 912–923 CrossRef CAS PubMed.
- (a) O. Jacquet, C. Das Neves Gomes, M. Ephritikhine and T. Cantat, Recycling of Carbon and Silicon Wastes: Room Temperature Formylation of N–H Bonds Using Carbon Dioxide and Polymethylhydrosiloxane, J. Am. Chem. Soc., 2012, 134, 2934–2937 CrossRef CAS; (b) C. Das Neves Gomes, O. Jacquet, C. Villiers, P. Thuéry, M. Ephritikhine and T. Cantat, A Diagonal Approach to Chemical Recycling of Carbon Dioxide: Organocatalytic Transformation for the Reductive Functionalization of CO2, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef CAS.
- S. Schreiner, J. Y. Yu and L. Vaska, Reversible homogeneous catalysis of carbon dioxide hydrogenation/reduction at room temperature and low pressures, J. Chem. Soc., Chem. Commun., 1988, 602–603 RSC.
- M. Khandelwal and R. J. Wehmschulte, Deoxygenative Reduction of Carbon Dioxide to Methane, Toluene, and Diphenylmethane with [Et2Al]+ as Catalyst, Angew. Chem., Int. Ed., 2012, 51, 7323–7326 CrossRef CAS.
- X. Frogneux, E. Blondiaux, P. Thuéry and T. Cantat, Bridging Amines with CO2: Organocatalyzed Reduction of CO2 to Aminals, ACS Catal., 2015, 5, 3983–3987 CrossRef CAS.
- D.-Y. Zhu, L. Fang, H. Han, Y. Wang and J.-B. Xia, Reductive CO2 Fixation via Tandem C–C and C–N Bond Formation: Synthesis of Spiro-indolepyrrolidines, Org. Lett., 2017, 19, 4259–4262 CrossRef CAS PubMed.
- Y. Zhao, X. Guo, X. Ding, Z. Zhou, M. Li, N. Feng, B. Gao, X. Lu, Y. Liu and J. You, Reductive CO2 Fixation via the Selective Formation of C–C Bonds: Bridging Enaminones and Synthesis of 1,4-Dihydropyridines, Org. Lett., 2020, 22, 8326–8331 CrossRef CAS.
- W.-D. Li, J. Chen, D.-Y. Zhu and J.-B. Xia, Fe-Catalyzed Pictet-Spengler-Type Cyclization via Selective Four-Electron Reductive Functionalization of CO2, Chin. J. Chem., 2021, 39, 614–620 CrossRef CAS.
- D.-Y. Zhu, W.-D. Li, C. Yang, J. Chen and J.-B. Xia, Transition-Metal-Free Reductive Deoxygenative Olefination with CO2, Org. Lett., 2018, 20, 3282–3285 CrossRef CAS PubMed.
- X.-Y. Li, S.-S. Zheng, X.-F. Liu, Z.-W. Yang, T.-Y. Tan, A. Yu and L.-N. He, Waste Recycling: Ionic Liquid-Catalyzed 4-Electron Reduction of CO2 with Amines and Polymethylhydrosiloxane Combining Experimental and Theoretical Study, ACS Sustain. Chem. Eng., 2018, 6, 8130–8135 CrossRef CAS.
- Z.-J. Mu, X. Ding, Z.-Y. Chen and B.-H. Han, Zwitterionic Covalent Organic Frameworks as Catalysts for Hierarchical Reduction of CO2 with Amine and Hydrosilane, ACS Appl. Mater. Interfaces, 2018, 10, 41350–41358 CrossRef CAS.
- (a) R. Köster and I. Borverbindungen, Darstellung von Bortrialkylen und ihre Reaktionen mit Olefinen, Justus Liebigs Ann. Chem., 1958, 618, 31–43 CrossRef; (b) H. C. Brown and G. Zweifel, Hydroboration. VII. Directive Effects in the Hydroboration of Olefins, J. Am. Chem. Soc., 1960, 82, 4708–4712 CrossRef CAS; (c) H. C. Brown and B. C. S. Rao, A new technique for the conversion of olefins into organoboranes and related alcohols, J. Am. Chem. Soc., 1956, 78, 5694–5695 CrossRef CAS.
- (a) E. J. Corey, R. K. Bakshi and S. Shibata, Highly enantioselective borane reduction of ketones catalyzed by chiral oxazaborolidines. Mechanism and synthetic implications, J. Am. Chem. Soc., 1987, 109, 5551–5553 CrossRef CAS; (b) E. J. Corey and C. J. Helal, Reduction of Carbonyl Compounds with Chiral Oxazaborolidine Catalysts: A New Paradigm for Enantioselective Catalysis and a Powerful New Synthetic Method, Angew. Chem., Int. Ed., 1998, 37, 1986–2012 CrossRef CAS.
- A. Suzuki, Cross-Coupling Reactions Of Organoboranes: An Easy Way To Construct C-C Bonds (Nobel Lecture), Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS.
- Y.-R. Luo and Editor, Comprehensive Handbook of Chemical Bond Energies, CRC Press, 2007 Search PubMed.
- S. Chakraborty, J. Zhang, J. A. Krause and H. Guan, An Efficient Nickel Catalyst for the Reduction of Carbon Dioxide with a Borane, J. Am. Chem. Soc., 2010, 132, 8872–8873 CrossRef CAS PubMed.
- F. Huang, C. Zhang, J. Jiang, Z.-X. Wang and H. Guan, How Does the Nickel Pincer Complex Catalyze the Conversion of CO2 to a Methanol Derivative? A Computational Mechanistic Study, Inorg. Chem., 2011, 50, 3816–3825 CrossRef CAS.
- S. Bontemps, L. Vendier and S. Sabo-Etienne, Borane-Mediated Carbon Dioxide Reduction at Ruthenium: Formation of C1 and C2 Compounds, Angew. Chem., Int. Ed., 2012, 51, 1671–1674 CrossRef CAS.
- S. Bontemps, L. Vendier and S. Sabo-Etienne, Ruthenium-Catalyzed Reduction of Carbon Dioxide to Formaldehyde, J. Am. Chem. Soc., 2014, 136, 4419–4425 CrossRef CAS.
- S. Desmons, Y. Zhou, D. Zhang, C. Jarava-Barrera, A. Coffinet, A. Simonneau, L. Vendier, G. Luo and S. Bontemps, CO2 Hydroboration: Impact of the Boryl Moieties on the Reactivity of Four Bis(boryl)acetal Compounds toward 2,6-Diisopropylaniline, Eur. J. Org Chem., 2023, 26, e202300525 CrossRef CAS.
- G. Jin, C. G. Werncke, Y. Escudié, S. Sabo-Etienne and S. Bontemps, Iron-Catalyzed Reduction of CO2 into Methylene: Formation of C–N, C–O, and C–C Bonds, J. Am. Chem. Soc., 2015, 137, 9563–9566 CrossRef CAS.
- L. J. Murphy, H. Hollenhorst, R. McDonald, M. Ferguson, M. D. Lumsden and L. Turculet, Selective Ni-Catalyzed Hydroboration of CO2 to the Formaldehyde Level Enabled by New PSiP Ligation, Organometallics, 2017, 36, 3709–3720 CrossRef CAS.
- H.-W. Suh, L. M. Guard and N. Hazari, Synthesis and reactivity of a masked PSiP pincer supported nickel hydride, Polyhedron, 2014, 84, 37–43 CrossRef CAS.
- R. Pal, T. L. Groy and R. J. Trovitch, Conversion of Carbon Dioxide to Methanol Using a C–H Activated Bis(imino)pyridine Molybdenum Hydroboration Catalyst, Inorg. Chem., 2015, 54, 7506–7515 CrossRef CAS PubMed.
- M. R. Espinosa, D. J. Charboneau, A. Garcia de Oliveira and N. Hazari, Controlling Selectivity in the Hydroboration of Carbon Dioxide to the Formic Acid, Formaldehyde, and Methanol Oxidation Levels, ACS Catal., 2019, 9, 301–314 CrossRef CAS.
- M. D. Anker, M. Arrowsmith, P. Bellham, M. S. Hill, G. Kociok-Kohn, D. J. Liptrot, M. F. Mahon and C. Weetman, Selective reduction of CO2 to a methanol equivalent by B(C6F5)3-activated alkaline earth catalysis, Chem. Sci., 2014, 5, 2826–2830 RSC.
- L. Li, H. Zhu, L. Liu, D. Song and M. Lei, A Hydride-Shuttle Mechanism for the Catalytic Hydroboration of CO2, Inorg. Chem., 2018, 57, 3054–3060 CrossRef CAS PubMed.
- (a) Y. Yang, L. Yan, Q. Xie, Q. Liang and D. Song, Zwitterionic indenylammonium with carbon-centred reactivity towards reversible CO2 binding and catalytic reduction, Org. Biomol. Chem., 2017, 15, 2240–2245 RSC; (b) T. Janes, K. M. Osten, A. Pantaleo, E. Yan, Y. Yang and D. Song, Insertion of CO2 into the carbon-boron bond of a boronic ester ligand, Chem. Commun., 2016, 52, 4148–4151 RSC; (c) Y. Yang, M. Xu and D. Song, Organocatalysts with carbon-centered activity for CO2 reduction with boranes, Chem. Commun., 2015, 51, 11293–11296 RSC; (d) C. Weetman, A. Porzelt, P. Bag, F. Hanusch and S. Inoue, Dialumenes – aryl vs. silyl stabilisation for small molecule activation and catalysis, Chem. Sci., 2020, 11, 4817–4827 RSC; (e) J. Fan, J.-Q. Mah, M.-C. Yang, M.-D. Su and C.-W. So, A N-Phosphinoamidinato NHC-Diborene Catalyst for Hydroboration, J. Am. Chem. Soc., 2021, 143, 4993–5002 CrossRef PubMed.
- C. Das Neves Gomes, E. Blondiaux, P. Thuéry and T. Cantat, Metal-Free Reduction of CO2 with Hydroboranes: Two Efficient Pathways at Play for the Reduction of CO2 to Methanol, Chem.–Eur. J., 2014, 20, 7098–7106 CrossRef CAS PubMed.
- A. Aloisi, J.-C. Berthet, C. Genre, P. Thuery and T. Cantat, Complexes of the tripodal phosphine ligands PhSi(XPPh2)3 (X = CH2, O): synthesis, structure and catalytic activity in the hydroboration of CO2, Dalton Trans., 2016, 45, 14774–14788 RSC.
- A. Ramos, A. Antinolo, F. Carrillo-Hermosilla, R. Fernandez-Galan, A. Rodriguez-Dieguez and D. Garcia-Vivo, Carbodiimides as catalysts for the reduction of CO2 with boranes, Chem. Commun., 2018, 54, 4700–4703 RSC.
- A. Ramos, A. Antiñolo, F. Carrillo-Hermosilla and R. Fernández-Galán, Ph2PCH2CH2B(C8H14) and Its Formaldehyde Adduct as Catalysts for the Reduction of CO2 with Hydroboranes, Inorg. Chem., 2020, 59, 9998–10012 CrossRef CAS.
- S. C. Sau, R. Bhattacharjee, P. K. Vardhanapu, G. Vijaykumar, A. Datta and S. K. Mandal, Metal-Free Reduction of CO2 to Methoxyborane under Ambient Conditions through Borondiformate Formation, Angew. Chem., Int. Ed., 2016, 55, 15147–15151 CrossRef CAS PubMed.
- X. Wang, K. Chang and X. Xu, Hydroboration of carbon dioxide enabled by molecular zinc dihydrides, Dalton Trans., 2020, 49, 7324–7327 RSC.
- M. Frick, J. Horn, H. Wadepohl, E. Kaifer and H. J. Himmel, Catalyst-Free Hydroboration of CO2 With a Nucleophilic Diborane(4), Chem.–Eur. J., 2018, 24, 16983–16986 CrossRef CAS.
- J. Li, C. G. Daniliuc, G. Kehr and G. Erker, Preparation of the Borane (Fmes)BH2 and its Utilization in the FLP Reduction of Carbon Monoxide and Carbon Dioxide, Angew. Chem. Int. Ed., 2019, 58, 6737–6741 CrossRef CAS.
- (a) R. Declercq, G. Bouhadir, D. Bourissou, M.-A. Légaré, M.-A. Courtemanche, K. S. Nahi, N. Bouchard, F.-G. Fontaine and L. Maron, Hydroboration of Carbon Dioxide using Ambiphilic Phosphine-Borane Catalysts: On the Role of the Formaldehyde Adduct, ACS Catal., 2015, 5, 2513–2520 CrossRef CAS; (b) S. Y. F. Ho, C.-W. So, N. Saffon-Merceron and N. Mezailles, Formation of a zwitterionic boronium species from the reaction of a stable carbenoid with borane: CO2 reduction, Chem. Commun., 2015, 51, 2107–2110 RSC.
- M.-A. Courtemanche, A. P. Pulis, E. Rochette, M.-A. Legare, D. W. Stephan and F.-G. Fontaine, Intramolecular B/N frustrated Lewis pairs and the hydrogenation of carbon dioxide, Chem. Commun., 2015, 51, 9797–9800 RSC.
- J. H. W. LaFortune, Z. W. Qu, K. L. Bamford, A. Trofimova, S. A. Westcott and D. W. Stephan, Double Phosphinoboration of CO2: A Facile Route to Diphospha-Ureas, Chem.–Eur. J., 2019, 25, 12063–12067 CrossRef CAS.
- S. Bontemps and S. Sabo-Etienne, Trapping Formaldehyde in the Homogeneous Catalytic Reduction of Carbon Dioxide, Angew. Chem., Int. Ed., 2013, 52, 10253–10255 CrossRef CAS PubMed.
- (a) C. Dong, M. Ji, X. Yang, J. Yao and H. Chen, Reaction Mechanisms of CO2 Reduction to Formaldehyde Catalyzed by Hourglass Ru, Fe, and Os Complexes: A Density Functional Theory Study, Catalysts, 2017, 7, 5 CrossRef; (b) F. Huang, Q. Wang, J. Guo, M. Wen and Z.-X. Wang, Computational mechanistic study of Ru-catalyzed CO2 reduction by pinacolborane revealing the σ-π coupling mechanism for CO2 decarbonylation, Dalton Trans., 2018, 47, 4804–4819 RSC.
- A. Caise, D. Jones, E. L. Kolychev, J. Hicks, J. M. Goicoechea and S. Aldridge, On the Viability of Catalytic Turnover via Al−O/B−H Metathesis: The Reactivity of β-Diketiminate Aluminium Hydrides towards CO2 and Boranes, Chem.–Eur. J., 2018, 24, 13624–13635 CrossRef CAS.
- N. Ma, C. Tu, Q. Xu, W. Guo, J. Zhang and G. Zhang, Computational study on the mechanism of hydroboration of CO2 catalysed by POCOP pincer nickel thiolate complexes: concerted catalysis and hydride transfer by a shuttle, Dalton Trans., 2021, 50, 2903–2914 RSC.
- (a) X. Wan, M. Li and R.-Z. Liao, Ligand-assisted Hydride Transfer: A Pivotal Step for CO2 Hydroboration Catalyzed by a Mononuclear Mn(I) PNP Complex, Chem.–Asian J., 2021, 16, 2529–2537 CrossRef CAS; (b) C. Erken, A. Kaithal, S. Sen, T. Weyhermüller, M. Hölscher, C. Werlé and W. Leitner, Manganese-catalyzed hydroboration of carbon dioxide and other challenging carbonyl groups, Nat. Commun., 2018, 9, 4521 CrossRef.
- H. Sabet-Sarvestani, M. Izadyar and H. Eshghi, Theoretical evaluation of the organocatalytic behavior of the negatively charged carbon atom in a fused five-member ring in carbon dioxide transformation to methanol, Energy, 2017, 134, 493–503 CrossRef CAS.
- (a) M.-A. Courtemanche, M.-A. Légaré, L. Maron and F.-G. Fontaine, Reducing CO2 to Methanol Using Frustrated Lewis Pairs: On the Mechanism of Phosphine–Borane-Mediated Hydroboration of CO2, J. Am. Chem. Soc., 2014, 136, 10708–10717 CrossRef CAS PubMed; (b) M.-A. Courtemanche, M.-A. Légaré, L. Maron and F.-G. Fontaine, A Highly Active Phosphine–Borane Organocatalyst for the Reduction of CO2 to Methanol Using Hydroboranes, J. Am. Chem. Soc., 2013, 135, 9326–9329 CrossRef CAS.
- Z.-W. Qu, H. Zhu and S. Grimme, Frustrated Lewis Pair Catalyzed Reduction of Carbon Dioxide Using Hydroboranes: New DFT Mechanistic Insights, ChemCatChem, 2020, 12, 3656–3660 CrossRef CAS.
- T. Wang and D. W. Stephan, Phosphine catalyzed reduction of CO2 with boranes, Chem. Commun., 2014, 50, 7007–7010 RSC.
- Y. C. A. Sokolovicz, O. Nieto Faza, D. Specklin, B. Jacques, C. S. López, J. H. Z. dos Santos, H. S. Schrekker and S. Dagorne, Acetate-catalyzed hydroboration of CO2 for the selective formation of methanol-equivalent products, Catal. Sci. Technol., 2020, 10, 2407–2414 RSC.
- Z. M. Heiden and A. P. Lathem, Establishing the Hydride Donor Abilities of Main Group Hydrides, Organometallics, 2015, 34, 1818–1827 CrossRef CAS.
- H. Gao, J. Jia, C.-H. Tung and W. Wang, Iron-Catalyzed Selective Hydroboration of CO2 by Cooperative B–H Bond Activation, Organometallics, 2023, 42, 944–951 CrossRef CAS.
- W. Huadsai, M. Westerhausen and S. Bontemps, Alkaline Earth Catalyzed CO2 Hydroboration into Acetal Derivatives Leading to C–S Bond Formation, Organometallics, 2023, 42, 2921–2926 CrossRef CAS.
- A. Butlerow, Formation synthétique d'une substance sucrée, Cr. Acad. Sci., 1861, 53, 145–147 Search PubMed.
- J. Henrique Teles, J.-P. Melder, K. Ebel, R. Schneider, E. Gehrer, W. Harder, S. Brode, D. Enders, K. Breuer and G. Raabe, The Chemistry of Stable Carbenes. Part 2. Benzoin-type condensations of formaldehyde catalyzed by stable carbenes, Helv. Chim. Acta, 1996, 79, 61–83 CrossRef.
- A. M. Fajardo, N. Queyriaux, A. Camy, L. Vendier, M. Grellier, I. del Rosal, L. Maron and S. Bontemps, A Masked Form of an O-Borylated Breslow Intermediate for the Diastereoselective FLP-Type Activation of Aldehydes, Chem.–Eur. J., 2022, 28, e202104122 CrossRef CAS.
- (a) I. V. Delidovich, A. N. Simonov, O. P. Pestunova and V. N. Parmon, Catalytic condensation of glycolaldehyde and glyceraldehyde with formaldehyde in neutral and weakly alkaline aqueous media: Kinetics and mechanism, Kinet. Catal., 2009, 50, 297–303 CrossRef CAS; (b) J. Castells, F. Geijo and F. López-Calahorra, The “formoin reaction” : A promising entry to carbohydrates from formaldehyde, Tetrahedron Lett., 1980, 21, 4517–4520 CrossRef CAS.
- J. J. A. G. Kamps, R. J. Hopkinson, C. J. Schofield and T. D. W. Claridge, How formaldehyde reacts with amino acids, Commun. Chem., 2019, 2, 126 CrossRef.
- (a) T. Zhou, J. J. Vallooran, S. Assenza, A. Szekrenyi, P. Clapés and R. Mezzenga, Efficient Asymmetric Synthesis of Carbohydrates by Aldolase Nano-Confined in Lipidic Cubic Mesophases, ACS Catal., 2018, 8, 5810–5815 CrossRef CAS; (b) A. Szekrenyi, X. Garrabou, T. Parella, J. Joglar, J. Bujons and P. Clapés, Asymmetric Assembly of Aldose Carbohydrates from Formaldehyde and Glycolaldehyde by Tandem Biocatalytic Aldol Reactions, Nat. Chem., 2015, 7, 724–729 CrossRef CAS PubMed; (c) J. A. Castillo, C. Guérard-Hélaine, M. Gutiérrez, X. Garrabou, M. Sancelme, M. Schürmann, T. Inoue, V. Hélaine, F. Charmantray, T. Gefflaut, L. Hecquet, J. Joglar, P. Clapés, G. A. Sprenger and M. Lemaire, A Mutant D-Fructose-6-Phosphate Aldolase (Ala129Ser) with Improved Affinity towards Dihydroxyacetone for the Synthesis of Polyhydroxylated Compounds, Adv. Synth. Catal., 2010, 352, 1039–1046 CrossRef CAS; (d) S. Güner, V. Wegat, A. Pick and V. Sieber, Design of a synthetic enzyme cascade for the in vitro fixation of a C1 carbon source to a functional C4 sugar, Green Chem., 2021, 23, 6583–6590 RSC; (e) T. Li, Z. Tang, H. Wei, Z. Tan, P. Liu, J. Li, Y. Zheng, J. Lin, W. Liu, H. Jiang, H. Liu, L. Zhu and Y. Ma, Totally atom-economical synthesis of lactic acid from formaldehyde: combined bio-carboligation and chemo-rearrangement without the isolation of intermediates, Green Chem., 2020, 22, 6809–6814 RSC; (f) J. B. Siegel, A. L. Smith, S. Poust, A. J. Wargacki, A. Bar-Even, C. Louw, B. W. Shen, C. B. Eiben, H. M. Tran, E. Noor, J. L. Gallaher, J. Bale, Y. Yoshikuni, M. H. Gelb, J. D. Keasling, B. L. Stoddard, M. E. Lidstrom and D. Baker, Computational Protein Design Enables a Novel one-Carbon Assimilation Pathway, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3704–3709 CrossRef CAS PubMed; (g) S. Poust, J. Piety, A. Bar-Even, C. Louw, D. Baker, J. D. Keasling and J. B. Siegel, Mechanistic Analysis of an Engineered Enzyme that Catalyzes the Formose Reaction, ChemBioChem, 2015, 16, 1950–1954 CrossRef CAS PubMed.
- X. Garrabou, J. A. Castillo, C. Guérard-Hélaine, T. Parella, J. Joglar, M. Lemaire and P. Clapés, Asymmetric Self- and Cross-Aldol Reactions of Glycolaldehyde Catalyzed by D-Fructose-6-phosphate Aldolase, Angew. Chem., Int. Ed., 2009, 121, 5629–5633 CrossRef.
- J. Yang, S. Sun, Y. Men, Y. Zeng, Y. Zhu, Y. Sun and Y. Ma, Transformation of Formaldehyde into Functional Sugars via Multi-Enzyme Stepwise Cascade Catalysis, Catal. Sci. Technol., 2017, 7, 3459–3463 RSC.
- J.-C. Berthet and M. Ephritikhine, Reactions of the uranium (IV) hydride (C5H4SiMe3)3UH including the first transformation:2 [M]-H + CO2 -> [M]-O-CH2-O-[M], New J. Chem., 1992, 16, 767–768 CAS.
- (a) N. E. Schlörer, E. J. Cabrita and S. Berger, Characterization of Reactive Intermediates by Diffusion-Ordered NMR Spectroscopy: A Snapshot of the Reaction of 13CO2 with [Cp2Zr(Cl)H], Angew. Chem., Int. Ed., 2002, 41, 107–109 CrossRef; (b) N. E. Schlörer and S. Berger, First Spectroscopical Evidence of a Dioxomethylene Intermediate in the Reaction of CO2 with Cp2Zr(H)Cl: A 13C NMR Study, Organometallics, 2001, 20, 1703–1704 CrossRef.
- (a) A. Mendiratta, J. S. Figueroa and C. C. Cummins, Synthesis of a four-coordinate titanium(iv) oxoanion via deprotonation and decarbonylation of complexed formate, Chem. Commun., 2005, 3403–3405 RSC; (b) M. A. Rankin and C. C. Cummins, Carbon Dioxide Reduction by Terminal Tantalum Hydrides: Formation and Isolation of Bridging Methylene Diolate Complexes, J. Am. Chem. Soc., 2010, 132, 10021–10023 CrossRef CAS.
- J. Ballmann, F. Pick, L. Castro, M. D. Fryzuk and L. Maron, Reduction of Carbon Dioxide Promoted by a Dinuclear Tantalum Tetrahydride Complex, Inorg. Chem., 2013, 52, 1685–1687 CrossRef CAS.
- O. Tardif, D. Hashizume and Z. Hou, Hydrogenation of Carbon Dioxide and Aryl Isocyanates by a Tetranuclear Tetrahydrido Yttrium Complex. Isolation, Structures, and CO2 Insertion Reactions of Methylene Diolate and μ3-Oxo Yttrium Complexes, J. Am. Chem. Soc., 2004, 126, 8080–8081 CrossRef CAS.
- (a) K.-C. Lau, B. J. Petro, S. Bontemps and R. F. Jordan, Comparative Reactivity of Zr– and Pd–Alkyl Complexes with Carbon Dioxide, Organometallics, 2013, 32, 6895–6898 CrossRef CAS; (b) M. Hill and O. F. Wendt, Reactivity of Carbon Dioxide toward Zirconocene Cations, Organometallics, 2005, 24, 5772–5775 CrossRef CAS; (c) N. Hazari and J. E. Heimann, Carbon Dioxide Insertion into Group 9 and 10 Metal–Element σ Bonds, Inorg. Chem., 2017, 56(22), 13655–13678 CrossRef CAS; (d) A. P. Deziel, M. R. Espinosa, L. Pavlovic, D. J. Charboneau, N. Hazari, K. H. Hopmann and B. Q. Mercado, Ligand and solvent effects on CO2 insertion into group 10 metal alkyl bonds, Chem. Sci., 2022, 13, 2391–2404 RSC.
- (a) S. Gambarotta, S. Strologo, C. Floriani, A. Chiesi-Villa and C. Guastini, Stepwise reduction of carbon dioxide to formaldehyde and methanol: reactions of carbon dioxide and carbon dioxide like molecules with hydridochlorobis(cyclopentadienyl)zirconium(IV), J. Am. Chem. Soc., 1985, 107, 6278–6282 CrossRef CAS; (b) G. Fachinetti, C. Floriani and S. Pucci, Stoicheiometric reduction of CO and CO2 to methanol: evidence for carbon monoxide insertion into zirconium–hydrogen bond, J. Chem. Soc., Chem. Commun., 1978, 269–270 RSC; (c) J. A. Labinger, Titanium, zirconium and hafnium: Annual survey covering the year 1976, J. Organomet. Chem., 1977, 138, 185–210 CrossRef CAS; (d) G. Fachinetti, C. Floriani, A. Chiesi-Villa and C. Guastini, Carbon dioxide activation. Deoxygenation and disproportionation of carbon dioxide promoted by bis(cyclopentadienyl)titanium and -zirconium derivatives. A novel bonding mode of the carbonato and a trimer of the zirconyl unit, J. Am. Chem. Soc., 1979, 101, 1767–1775 CrossRef CAS.
- D. Spencer and T. Henshall, The Kinetics and Mechanism of the Reaction of Formaldehyde with Dimedone. Part I, J. Am. Chem. Soc., 1955, 77, 1943–1948 CrossRef CAS.
- C. C. Chong and R. Kinjo, Hydrophosphination of CO2 and Subsequent Formate Transfer in the 1,3,2-Diazaphospholene-Catalyzed N-Formylation of Amines, Angew. Chem., Int. Ed., 2015, 54, 12116–12120 CrossRef CAS PubMed.
- D. Gudat, in Encyclopedia of Inorganic and Bioinorganic Chemistry, 2018, pp. 1–23, DOI:10.1002/9781119951438.eibc2642.
- J. Wei, R. Yao, Y. Han, Q. Ge and J. Sun, Towards the development of the emerging process of CO2 heterogenous hydrogenation into high-value unsaturated heavy hydrocarbons, Chem. Soc. Rev., 2021, 50, 10764–10805 RSC.
- M. M. T. Khan, S. B. Halligudi and S. Shukla, Reduction of CO2 by molecular hydrogen to formic acid and formaldehyde and their decomposition to CO and H2O, J. Mol. Catal., 1989, 57, 47–60 CrossRef CAS.
- D.-K. Lee, D.-S. Kim and S.-W. Kim, Selective formation of formaldehyde from carbon dioxide and hydrogen over PtCu/SiO2, Appl. Organomet. Chem., 2001, 15, 148–150 CrossRef CAS.
- S. Carenco, C.-H. Wu, A. Shavorskiy, S. Alayoglu, G. A. Somorjai, H. Bluhm and M. Salmeron, Synthesis and Structural Evolution of Nickel–Cobalt Nanoparticles Under H2 and CO2, Small, 2015, 11, 3045–3053 CrossRef CAS PubMed.
- F. L. Chan, G. Altinkaya, N. Fung and A. Tanksale, Low temperature hydrogenation of carbon dioxide into formaldehyde in liquid media, Catal. Today, 2018, 309, 242–247 CrossRef CAS.
- L. Deng, X. Liu, J. Xu, Z. Zhou, S. Feng, Z. Wang and M. Xu, Transfer hydrogenation of CO2 into formaldehyde from aqueous glycerol heterogeneously catalyzed by Ru bound to LDH, Chem. Commun., 2021, 57, 5167–5170 RSC.
- U. Kleeberg and W. Klinger, Sensitive formaldehyde determination with NASH's reagent and a ‘tryptophan reaction’, J. Pharmacol. Methods, 1982, 8, 19–31 CrossRef CAS PubMed.
- J. Zhang, C. Xing, B. Tiwari and Y. R. Chi, Catalytic Activation of Carbohydrates as Formaldehyde Equivalents for Stetter Reaction with Enones, J. Am. Chem. Soc., 2013, 135, 8113–8116 CrossRef CAS PubMed.
- (a) K. Thenert, K. Beydoun, J. Wiesenthal, W. Leitner and J. Klankermayer, Ruthenium-Catalyzed Synthesis of Dialkoxymethane Ethers Utilizing Carbon Dioxide and Molecular Hydrogen, Angew. Chem., Int. Ed., 2016, 55, 12266–12269 CrossRef CAS PubMed; (b) B. G. Schieweck and J. Klankermayer, Tailor-made Molecular Cobalt Catalyst System for the Selective Transformation of Carbon Dioxide to Dialkoxymethane Ethers, Angew. Chem., Int. Ed., 2017, 56, 10854–10857 CrossRef CAS.
- (a) F. A. Long and D. McIntyre, Acid-catalyzed Hydrolysis of Methylal. II. Kinetic and Equilibrium Salt Effects and Correlation with H0, J. Am. Chem. Soc., 1954, 76, 3243–3247 CrossRef CAS; (b) D. McIntyre and F. A. Long, Acid-catalyzed Hydrolysis of Methylal. I. Influence of Strong Acids and Correlation with Hammett Acidity Function, J. Am. Chem. Soc., 1954, 76, 3240–3242 CrossRef CAS.
- (a) S. Dabral and T. Schaub, The Use of Carbon Dioxide (CO2) as a Building Block in Organic Synthesis from an Industrial Perspective, Adv. Synth. Catal., 2019, 361, 223–246 CrossRef CAS; (b) Z. Wei, X. Tian, M. Bender, M. Beller and H. Jiao, Mechanisms of CoII and Acid Jointly Catalyzed Domino Conversion of CO2, H2, and CH3OH to Dialkoxymethane: A DFT Study, ACS Catal., 2021, 6908–6919 CrossRef CAS.
- M. Siebert, M. Seibicke, A. F. Siegle, S. Kräh and O. Trapp, Selective Ruthenium-Catalyzed Transformation of Carbon Dioxide: An Alternative Approach toward Formaldehyde, J. Am. Chem. Soc., 2019, 141, 334–341 CrossRef CAS.
- M. Siebert, G. Krennrich, M. Seibicke, A. F. Siegle and O. Trapp, Identifying high-performance catalytic conditions for carbon dioxide reduction to dimethoxymethane by multivariate modelling, Chem. Sci., 2019, 10, 10466–10474 RSC.
- M. Seibicke, M. Siebert, A. F. Siegle, S. M. Gutenthaler and O. Trapp, Application of Hetero-Triphos Ligands in the Selective Ruthenium-Catalyzed Transformation of Carbon Dioxide to the Formaldehyde Oxidation State, Organometallics, 2019, 38, 1809–1814 CrossRef CAS.
- R. Konrath, K. Sekine, I. Jevtovikj, R. A. Paciello, A. S. K. Hashmi and T. Schaub, Performance enhancing additives for reusable ruthenium-triphos catalysts in the reduction of CO2 to dimethoxymethane, Green Chem., 2020, 22, 6464–6470 RSC.
- K. Beydoun and J. Klankermayer, Ruthenium-Catalyzed Synthesis of Cyclic and Linear Acetals by the Combined Utilization of CO2, H2, and Biomass Derived Diols, Chem.–Eur. J., 2019, 25, 11412–11415 CrossRef CAS PubMed.
- C. A. Huff and M. S. Sanford, Cascade Catalysis for the Homogeneous Hydrogenation of CO2 to Methanol, J. Am. Chem. Soc., 2011, 133, 18122–18125 CrossRef CAS.
- N. M. Rezayee, C. A. Huff and M. S. Sanford, Tandem Amine and Ruthenium-Catalyzed Hydrogenation of CO2 to Methanol, J. Am. Chem. Soc., 2015, 137, 1028–1031 CrossRef CAS PubMed.
- M. Leopold, M. Siebert, A. F. Siegle and O. Trapp, Reaction Network Analysis of the Ruthenium-Catalyzed Reduction of Carbon Dioxide to Dimethoxymethane, ChemCatChem, 2021, 13, 2807–2814 CrossRef CAS.
- J. D. Erickson, A. Z. Preston, J. C. Linehan and E. S. Wiedner, Enhanced Hydrogenation of Carbon Dioxide to Methanol by a Ruthenium Complex with a Charged Outer-Coordination Sphere, ACS Catal., 2020, 7419–7423 CrossRef CAS.
- (a) S. Wesselbaum, V. Moha, M. Meuresch, S. Brosinski, K. M. Thenert, J. Kothe, T. vom Stein, U. Englert, M. Holscher, J. Klankermayer and W. Leitner, Hydrogenation of carbon dioxide to methanol using a homogeneous ruthenium-Triphos catalyst: from mechanistic investigations to multiphase catalysis, Chem. Sci., 2015, 6, 693–704 RSC; (b) S. Wesselbaum, T. vom Stein, J. Klankermayer and W. Leitner, Hydrogenation of Carbon Dioxide to Methanol by Using a Homogeneous Ruthenium–Phosphine Catalyst, Angew. Chem., Int. Ed., 2012, 51, 7499–7502 CrossRef CAS PubMed; (c) K. Beydoun, G. Ghattas, K. Thenert, J. Klankermayer and W. Leitner, Ruthenium-Catalyzed Reductive Methylation of Imines Using Carbon Dioxide and Molecular Hydrogen, Angew. Chem., Int. Ed., 2014, 53, 11010–11014 CrossRef CAS; (d) K. Beydoun, T. vom Stein, J. Klankermayer and W. Leitner, Ruthenium-Catalyzed Direct Methylation of Primary and Secondary Aromatic Amines Using Carbon Dioxide and Molecular Hydrogen, Angew. Chem., Int. Ed., 2013, 52, 9554–9557 CrossRef CAS; (e) Y. Li, T. Yan, K. Junge and M. Beller, Catalytic Methylation of C-H Bonds Using CO2 and H2, Angew. Chem., Int. Ed., 2014, 126, 10644–10648 CrossRef; (f) Y. Li, I. Sorribes, T. Yan, K. Junge and M. Beller, Selective Methylation of Amines with Carbon Dioxide and H2, Angew. Chem., Int. Ed., 2013, 52, 12156–12160 CrossRef CAS.
- A. N. Simonov, L. G. Matvienko, O. P. Pestunova, V. N. Parmon, N. A. Komandrova, V. A. Denisenko and V. E. Vas’kovskii, Selective synthesis of erythrulose and 3-pentulose from formaldehyde and dihydroxyacetone catalyzed by phosphates in a neutral aqueous medium, Kinet. Catal., 2007, 48, 550–555 CrossRef CAS.
- E. Boutin and M. Robert, Molecular Electrochemical Reduction of CO2 beyond Two Electrons, Trends Chem., 2021, 3, 359–372 CrossRef CAS.
- (a) H. Nagao, T. Mizukawa and K. Tanaka, Carbon-Carbon Bond Formation in the Electrochemical Reduction of Carbon Dioxide Catalyzed by a Ruthenium Complex, Inorg. Chem., 1994, 33, 3415–3420 CrossRef CAS; (b) N. Hirotaka, M. Tetsunori and T. Koji, Carbon-Carbon Bond Formation in Multi-electron Reduction of Carbon Dioxide Catalyzed by [Ru(bpy)(trpy)(CO)]2+ (bpy = 2,2′-bipyridine; trpy = 2,2′:6′,2′′-terpyridine), Chem. Lett., 1993, 22, 955–958 CrossRef.
- D. Xiang, D. Magana and R. B. Dyer, CO2 Reduction Catalyzed by Mercaptopteridine on Glassy Carbon, J. Am. Chem. Soc., 2014, 136, 14007–14010 CrossRef CAS.
- M. Girardi, S. Blanchard, S. Griveau, P. Simon, M. Fontecave, F. Bedioui and A. Proust, Electro-Assisted Reduction of CO2 to CO and Formaldehyde by (TOA)6[α-SiW11O39Co(_)] Polyoxometalate, Eur. J. Inorg. Chem., 2015, 2015, 3642–3648 CrossRef CAS.
- S. Gonglach, S. Paul, M. Haas, F. Pillwein, S. S. Sreejith, S. Barman, R. De, S. Müllegger, P. Gerschel, U.-P. Apfel, H. Coskun, A. Aljabour, P. Stadler, W. Schöfberger and S. Roy, Molecular cobalt corrole complex for the heterogeneous electrocatalytic reduction of carbon dioxide, Nat. Commun., 2019, 10, 3864 CrossRef PubMed.
- J. A. Ramos Sende, C. R. Arana, L. Hernandez, K. T. Potts, M. Keshevarz-K and H. D. Abruna, Electrocatalysis of CO2 Reduction in Aqueous Media at Electrodes Modified with Electropolymerized Films of Vinylterpyridine Complexes of Transition Metals, Inorg. Chem., 1995, 34, 3339–3348 CrossRef CAS.
- K. Kusuda, R. Ishihara, H. Yamaguchi and I. Izumi, Electrochemical investigation of thin films of cobalt phthalocyanine and cobalt-4,4′,4′′,4′′′-tetracarboxyphthalocyanine and the reduction of carbon monoxide, formic acid and formaldehyde mediated by the Co(I) complexes, Electrochim. Acta, 1986, 31, 657–663 CrossRef CAS.
- E. Boutin, A. Salamé, L. Merakeb, T. Chatterjee and M. Robert, On the Existence and Role of Formaldehyde During Aqueous Electrochemical Reduction of Carbon Monoxide to Methanol by Cobalt Phthalocyanine, Chem.–Eur. J., 2022, 28, e202200697 CrossRef CAS.
- A. Singh, A. Zamader, R. Khakpour, K. Laasonen, M. Busch and M. Robert, Molecular electrochemical catalysis of CO-to-formaldehyde conversion with a Cobalt complex, J. Am. Chem. Soc., 2024, 146(32), 22129–22133 CrossRef CAS PubMed.
- (a) K. Nakata, T. Ozaki, C. Terashima, A. Fujishima and Y. Einaga, High-Yield Electrochemical Production of Formaldehyde from CO2 and Seawater, Angew. Chem., Int. Ed., 2014, 53, 871–874 CrossRef CAS; (b) D. Luo, S. Liu, K. Nakata and A. Fujishima, Electrochemical reduction of CO2 and degradation of KHP on boron-doped diamond electrodes in a simultaneous and enhanced process, Chin. Chem. Lett., 2019, 30, 509–512 CrossRef CAS.
- J. Bin Yeo, J. Ho Jang, Y. In Jo, J. Woo Koo and K. Tae Nam, Paired Electrosynthesis of Formaldehyde Derivatives from CO2 Reduction and Methanol Oxidation, Angew. Chem., Int. Ed., 2024, 63, e202316020 CrossRef CAS.
- (a) M. Shibata and N. Furuya, Electrochemical synthesis of urea at gas-diffusion electrodes: Part VI. Simultaneous reduction of carbon dioxide and nitrite ions with various metallophthalocyanine catalysts, J. Electroanal. Chem., 2001, 507, 177–184 CrossRef CAS; (b) M. Shibata, K. Yoshida and N. Furuya, Electrochemical synthesis of urea on reduction of carbon dioxide with nitrate and nitrite ions using Cu-loaded gas-diffusion electrode, J. Electroanal. Chem., 1995, 387, 143–145 CrossRef.
- Y. Wu, Z. Jiang, Z. Lin, Y. Liang and H. Wang, Direct electrosynthesis of methylamine from carbon dioxide and nitrate, Nat. Sustain., 2021, 4, 725–730 CrossRef.
- (a) B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Photochemical and Photoelectrochemical Reduction of CO2, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed; (b) J. Wu, Y. Huang, W. Ye and Y. Li, CO2 Reduction: From the Electrochemical to Photochemical Approach, Adv. Sci., 2017, 4, 1700194 CrossRef PubMed; (c) A. U. Pawar, C. W. Kim, M.-T. Nguyen-Le and Y. S. Kang, General Review on the Components and Parameters of Photoelectrochemical System for CO2 Reduction with in Situ Analysis, ACS Sustain. Chem. Eng., 2019, 7, 7431–7455 CrossRef CAS; (d) P. R. Yaashikaa, P. Senthil Kumar, S. J. Varjani and A. Saravanan, A review on photochemical, biochemical and electrochemical transformation of CO2 into value-added products, J. CO2 Util., 2019, 33, 131–147 CrossRef CAS.
- T. D. Nguyen, T. Van Tran, S. Singh, P. T. T. Phuong, L. G. Bach, S. Nanda and D.-V. N. Vo, in Conversion of Carbon Dioxide into Hydrocarbons Vol. 2 Technology, ed. Inamuddin, A. M. Asiri and E. Lichtfouse, Springer International Publishing, Cham, 2020, ch. 6, pp. 159–183, DOI:10.1007/978-3-030-28638-5_6.
- M. Bonchio, J. Bonin, O. Ishitani, T.-B. Lu, T. Morikawa, A. J. Morris, E. Reisner, D. Sarkar, F. M. Toma and M. Robert, Best practices for experiments and reporting in photocatalytic CO2 reduction, Nat. Catal., 2023, 6, 657–665 CrossRef.
- E. C. C. Baly, I. M. Heilbron and W. F. Barker, CX.—Photocatalysis. Part I. The synthesis of formaldehyde and carbohydrates from carbon dioxide and water, J. Chem. Soc., Trans., 1921, 119, 1025–1035 RSC.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders, Nature, 1979, 277, 637–638 CrossRef CAS.
- B. Aurian-Blajeni, M. Halmann and J. Manassen, Photoreduction of carbon dioxide and water into formaldehyde and methanol on semiconductor materials, Sol. Energy, 1980, 25, 165–170 CrossRef CAS.
- M. Ulman, A. H. A. Tinnemans, A. Mackor, B. Aurian-Blajeni and M. Halmann, Photoreduction of Carbon Dioxide to Formic Acid, Formaldehyde, Methanol, Acetaldehyde and Ethanol Using Aqueous Suspensions of Strontium Titanate with Transition Metal Additives, Int. J. Sol. Energy, 1982, 1, 213–222 CrossRef CAS.
- S. M. Aliwi and K. F. Al-Jubori, Photoreduction of CO2 by metal sulphide semiconductors in presence of H2S, Sol. Energy Mater., 1989, 18, 223–229 CrossRef CAS.
- F. Solymosi and I. Tombácz, Photocatalytic reaction of H2O+CO2 over pure and doped Rh/TiO2, Catal. Lett., 1994, 27, 61–65 CrossRef CAS.
- E. Bahadori, A. Tripodi, A. Villa, C. Pirola, L. Prati, G. Ramis, N. Dimitratos, D. Wang and I. Rossetti, High pressure CO2 photoreduction using Au/TiO2: unravelling the effect of co-catalysts and of titania polymorphs, Catal. Sci. Technol., 2019, 9, 2253–2265 RSC.
- S. Zeng, E. Vahidzadeh, C. G. VanEssen, P. Kar, R. Kisslinger, A. Goswami, Y. Zhang, N. Mahdi, S. Riddell, A. E. Kobryn, S. Gusarov, P. Kumar and K. Shankar, Optical control of selectivity of high rate CO2 photoreduction via interband- or hot electron Z-scheme reaction pathways in Au-TiO2 plasmonic photonic crystal photocatalyst, Appl. Catal., B, 2020, 267, 118644 CrossRef CAS.
- F. Galli, M. Compagnoni, D. Vitali, C. Pirola, C. L. Bianchi, A. Villa, L. Prati and I. Rossetti, CO2 photoreduction at high pressure to both gas and liquid products over titanium dioxide, Appl. Catal., B, 2017, 200, 386–391 CrossRef CAS.
- I. Moriya, Converting of CO2 into low-molecular-weight organic compounds with the TiO2/ZrO2 composites under solar irradiation, Sci. Rep., 2017, 7, 14446 CrossRef PubMed.
- J. M. Mora-Hernandez, A. M. Huerta-Flores and L. M. Torres-Martínez, Photoelectrocatalytic characterization of carbon-doped NaTaO3 applied in the photoreduction of CO2 towards the formaldehyde production, J. CO2 Util., 2018, 27, 179–187 CrossRef CAS.
- M. Lashgari and S. Soodi, Photocatalytic Conversion of CO2 into Oxygenate Fuels/Chemicals Using Efficient, Eco-Friendly, Titania/Hematite-Based Nanostructured Solar-Energy Materials, J. Nanosci. Nanotechnol., 2019, 19, 3237–3243 CrossRef CAS PubMed.
- Z. Zhao, J. Fan, S. Liu and Z. Wang, Optimal design and preparation of titania-supported CoPc using sol–gel for the photo-reduction of CO2, Chem. Eng. J., 2009, 151, 134–140 CrossRef CAS.
- A. Kumar and R. Ananthakrishnan, Visible light-assisted reduction of CO2 into formaldehyde by heteroleptic ruthenium metal complex–TiO2 hybrids in an aqueous medium, Green Chem., 2020, 22, 1650–1661 RSC.
- B. Kim, D. Kwon, J.-O. Baeg, M. Austeria P, G. H. Gu, J.-H. Lee, J. Jeong, W. Kim and W. Choi, Dual-Atom-Site Sn-Cu/C3N4 Photocatalyst Selectively Produces Formaldehyde from CO2 Reduction, Adv. Funct. Mater., 2023, 33, 2212453 CrossRef CAS.
- G. Guan, T. Kida, T. Harada, M. Isayama and A. Yoshida, Photoreduction of carbon dioxide with water over K2Ti6O13 photocatalyst combined with Cu/ZnO catalyst under concentrated sunlight, Appl. Catal., A, 2003, 249, 11–18 CrossRef CAS.
- M. Guo, F. Gu, L. Meng, Q. Liao, Z. Meng and W. Liu, Synthesis of formaldehyde from CO2 catalyzed by the coupled photo-enzyme system, Sep. Purif. Technol., 2022, 286, 120480 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Photocatalytic Reduction of CO2 on TiO2 and Other Semiconductors, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- X. Chang, T. Wang, P. Yang, G. Zhang and J. Gong, The Development of Cocatalysts for Photoelectrochemical CO2 Reduction, Adv. Mater., 2019, 31, 1804710 CrossRef.
- B. Aurian-Blajeni, I. Taniguchi and J. O. M. Bockris, Photoelectrochemical reduction of carbon dioxide using polyaniline-coated silicon, J. Electroanal. Chem., 1983, 149, 291–293 CAS.
- M. Zafrir, M. Ulman, Y. Zuckerman and M. Halmann, Photoelectrochemical reduction of carbon dioxide to formic acid, formaldehyde and methanol on p-gallium arsenide in an aqueous V(II)-V(III) chloride redox system, J. Electroanal. Chem., 1983, 159, 373–389 CrossRef CAS.
- G. Qin, Y. Zhang, X. Ke, X. Tong, Z. Sun, M. Liang and S. Xue, Photocatalytic reduction of carbon dioxide to formic acid, formaldehyde, and methanol using dye-sensitized TiO2 film, Appl. Catal., B, 2013, 129, 599–605 CrossRef CAS.
- J. F. de Brito, A. R. Araujo, K. Rajeshwar and M. V. B. Zanoni, Photoelectrochemical reduction of CO2 on Cu/Cu2O films: Product distribution and pH effects, Chem. Eng. J., 2015, 264, 302–309 CrossRef.
- C. W. Kim, M. J. Kang, S. Ji and Y. S. Kang, Artificial Photosynthesis for Formaldehyde Production with 85% of Faradaic Efficiency by Tuning the Reduction Potential, ACS Catal., 2018, 8, 968–974 CrossRef CAS.
- A. U. Pawar, U. Pal, J. Y. Zheng, C. W. Kim and Y. S. Kang, Thermodynamically controlled photo-electrochemical CO2 reduction at Cu/rGO/PVP/Nafion multi-layered dark cathode for selective production of formaldehyde and acetaldehyde, Appl. Catal., B, 2022, 303, 120921 CrossRef CAS.
- S. Das, S. Biswas, T. Balaraju, S. Barman, R. Pochamoni and S. Roy, Photochemical reduction of carbon dioxide coupled with water oxidation using various soft-oxometalate (SOM) based catalytic systems, J. Mater. Chem. A, 2016, 4, 8875–8887 RSC.
- D. J. Boston, C. Xu, D. W. Armstrong and F. M. MacDonnell, Photochemical Reduction of Carbon Dioxide to Methanol and Formate in a Homogeneous System with Pyridinium Catalysts, J. Am. Chem. Soc., 2013, 135, 16252–16255 CrossRef CAS.
- D. J. Boston, Y. M. F. Pachón, R. O. Lezna, N. R. de Tacconi and F. M. MacDonnell, Electrocatalytic and Photocatalytic Conversion of CO2 to Methanol using Ruthenium Complexes with Internal Pyridyl Cocatalysts, Inorg. Chem., 2014, 53, 6544–6553 CrossRef CAS.
- A. Alissandratos and C. J. Easton, Biocatalysis for the application of CO2 as a chemical feedstock, Beilstein J. Org. Chem., 2015, 11, 2370–2387 CrossRef CAS.
- E. C. C. Baly, I. M. Heilbron and D. P. Hudson, CXXX.—Photocatalysis. Part II. The photosynthesis of nitrogen compounds from nitrates and carbon dioxide, J. Chem. Soc., Trans., 1922, 121, 1078–1088 RSC.
- J. A. Bassham, A. A. Benson, L. D. Kay, A. Z. Harris, A. T. Wilson and M. Calvin, The Path of Carbon in Photosynthesis. XXI. The Cyclic Regeneration of Carbon Dioxide Acceptor1, J. Am. Chem. Soc., 1954, 76, 1760–1770 CrossRef CAS.
- (a) L. F. Li, L. Ljungdahl and H. G. Wood, Properties of Nicotinamide Adenine Dinucleotide Phosphate-Dependent Formate Dehydrogenase from Clostridium thermoaceticum, J. Bacteriol., 1966, 92, 405–412 CrossRef CAS; (b) L. Calzadiaz-Ramirez and A. S. Meyer, Formate dehydrogenases for CO2 utilization, Curr. Opin. Biotechnol., 2022, 73, 95–100 CrossRef CAS.
- K. Murashima, H. Yoneda, H. Sumi and Y. Amao, Electrocatalytic production of formaldehyde with formaldehyde dehydrogenase using a viologen redox mediator, New J. Chem., 2022, 46, 10004–10011 RSC.
- J. Shi, Y. Jiang, Z. Jiang, X. Wang, X. Wang, S. Zhang, P. Han and C. Yang, Enzymatic conversion of carbon dioxide, Chem. Soc. Rev., 2015, 44, 5981–6000 RSC.
- (a) M. Miller, W. E. Robinson, A. R. Oliveira, N. Heidary, N. Kornienko, J. Warnan, I. A. C. Pereira and E. Reisner, Interfacing Formate Dehydrogenase with Metal Oxides for the Reversible Electrocatalysis and Solar-Driven Reduction of Carbon Dioxide, Angew. Chem., Int. Ed., 2019, 58, 4601–4605 CrossRef CAS; (b) B. S. Jayathilake, S. Bhattacharya, N. Vaidehi and S. R. Narayanan, Efficient and Selective Electrochemically Driven Enzyme-Catalyzed Reduction of Carbon Dioxide to Formate using Formate Dehydrogenase and an Artificial Cofactor, Acc. Chem. Res., 2019, 52, 676–685 CrossRef CAS PubMed.
- (a) S. K. Kuk, R. K. Singh, D. H. Nam, R. Singh, J.-K. Lee and C. B. Park, Photoelectrochemical Reduction of Carbon Dioxide to Methanol through a Highly Efficient Enzyme Cascade, Angew. Chem., Int. Ed., 2017, 56, 3827–3832 CrossRef CAS PubMed; (b) K. Ma, O. Yehezkeli, E. Park and J. N. Cha, Enzyme Mediated Increase in Methanol Production from Photoelectrochemical Cells and CO2, ACS Catal., 2016, 6, 6982–6986 CrossRef CAS; (c) R. K. Yadav, G. H. Oh, N.-J. Park, A. Kumar, K.-j. Kong and J.-O. Baeg, Highly Selective Solar-Driven Methanol from CO2 by a Photocatalyst/Biocatalyst Integrated System, J. Am. Chem. Soc., 2014, 136, 16728–16731 CrossRef CAS.
- R. Cazelles, J. Drone, F. Fajula, O. Ersen, S. Moldovan and A. Galarneau, Reduction of CO2 to methanol by a polyenzymatic system encapsulated in phospholipids–silica nanocapsules, New J. Chem., 2013, 37, 3721–3730 RSC.
- W. Liu, Y. Hou, B. Hou and Z. Zhao, Enzyme-catalyzed Sequential Reduction of Carbon Dioxide to Formaldehyde, Chin. J. Chem. Eng., 2014, 22, 1328–1332 CrossRef CAS.
- J. Shi, L. Zhang and Z. Jiang, Facile Construction of Multicompartment Multienzyme System through Layer-by-Layer Self-Assembly and Biomimetic Mineralization, ACS Appl. Mater. Interfaces, 2011, 3, 881–889 CrossRef CAS.
- J. Shi, X. Wang, Z. Jiang, Y. Liang, Y. Zhu and C. Zhang, Constructing spatially separated multienzyme system through bioadhesion-assisted bio-inspired mineralization for efficient carbon dioxide conversion, Bioresour. Technol., 2012, 118, 359–366 CrossRef CAS PubMed.
- J. Shi, X. Wang, W. Zhang, Z. Jiang, Y. Liang, Y. Zhu and C. Zhang, Synergy of Pickering Emulsion and Sol-Gel Process for the Construction of an Efficient, Recyclable Enzyme Cascade System, Adv. Funct. Mater., 2013, 23, 1450–1458 CrossRef CAS.
- P. S. Nabavi Zadeh, M. Zezzi do Valle Gomes, B. Åkerman and A. E. C. Palmqvist, Förster Resonance Energy Transfer Study of the Improved Biocatalytic Conversion of CO2 to Formaldehyde by Coimmobilization of Enzymes in Siliceous Mesostructured Cellular Foams, ACS Catal., 2018, 8, 7251–7260 CrossRef CAS.
- J. Zhou, S. Yu, H. Kang, R. He, Y. Ning, Y. Yu, M. Wang and B. Chen, Construction of multi-enzyme cascade biomimetic carbon sequestration system based on photocatalytic coenzyme NADH regeneration, Renewable Energy, 2020, 156, 107–116 CrossRef CAS.
- P. Bauler, G. Huber, T. Leyh and J. A. McCammon, Channeling by Proximity: The Catalytic Advantages of Active Site Colocalization Using Brownian Dynamics, J. Phys. Chem. Lett., 2010, 1, 1332–1335 CrossRef CAS.
- (a) J. Liu and M. Antonietti, Bio-inspired NADH regeneration by carbon nitride photocatalysis using diatom templates, Energy Environ. Sci., 2013, 6, 1486–1493 RSC; (b) A. Weckbecker, H. Gröger and W. Hummel, in Biosystems Engineering I: Creating Superior Biocatalysts, ed. C. Wittmann and R. Krull, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, pp. 195–242 Search PubMed.
Footnote |
| † Present address: Laboratory of Inorganic Synthesis and Catalysis, Institute of Chemical Sciences and Engineering, Ecole Polytechnique Fédérale de Lausanne (EPFL), EPFL-ISIC-LSCI, BCH 3305, Lausanne, CH 1015 Switzerland. |
| This journal is © The Royal Society of Chemistry 2024 |