
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn enzymolysis-induced energy transfer co-assembled system for spontaneously recoverable supramolecular dynamic memory†
Xuanyu Wang‡
,
Zhao Gao‡* and
Wei Tian *
*
Shaanxi Key Laboratory of Macromolecular Science and Technology, Xi'an Key Laboratory of Hybrid Luminescent Materials and Photonic Device, MOE Key Laboratory of Material Physics and Chemistry under Extraordinary Conditions, School of Chemistry and Chemical Engineering, Northwestern Polytechnical University, Xi'an 710072, P. R. China. E-mail: gaozhao@nwpu.edu.cn; happytw_3000@nwpu.edu.cn
First published on 13th June 2024
Abstract
The continuing growth of the digital world requires new ways of constructing memory devices to process and store dynamic data, because the current ones suffer from inefficiency, limited reads, and difficulty to manufacture. Here we propose a supramolecular dynamic memory (SDM) strategy based on an enzymolysis-induced energy transfer co-assembly derived from a naphthalene-based cationic monomer and organic dye sulforhodamine 101, enabling the construction of spontaneously recoverable dynamic memory devices. Benefitting from the large exciton migration rate (4.48 × 1015 L mol−1 s−1) between the monomer and sulforhodamine 101, the energy transfer process between the two is effectively achieved. Since alkaline phosphatase can selectively hydrolyze adenosine triphosphate, leading to the disruption of the co-assemblies, an enzyme-mediated time-dependent fluorochromic system is realized. On this basis, a SDM system featuring spontaneous recovery and enabling the memory of dynamic information in optical and electrical modes is successfully constructed. The current study represents a promising step in the nascent development of supramolecular materials for computational systems.
Introduction
Nowadays, information technology has become an integral part of our daily lives.1–4 Over the last few decades, there has been a tremendous development of memory devices, which form the basis of modern electronic information.5–7 Among them, dynamic random access memory (DRAM) serves as the primary memory in most computers. In DRAM, each data bit is stored in an individual capacitor. However, the minuscule charge stored in these capacitors is insufficient for direct reading and requires amplification through a sense amplifier. Moreover, the act of reading data from the bitline drains the charge from the capacitor, resulting in limited reads. To ensure that the data are correct, the readout needs to be put back into the capacitor.8 The traditional method is the top-down manufacturing process, yet according to the device dimensional scaling rule, silicon thicknesses of less than 3 nm will not only cause a significant drop in performance but also largely increase manufacturing difficulty.9–11 Hence, there is an urgent desire to find alternative materials and advanced methods to solve this issue.To address these challenges, new bottom-up approaches are continuously being presented,12–14 such as resistive random access memory,15,16 nano-random access memory,17,18 and phase-change memory.19,20 Nevertheless, these technologies still struggle with structural tunability and ease of fabrication. One promising alternative involves the use of supramolecular materials formed by non-covalent interactions.21–26 These materials offer abundant opportunities for molecular modifications and customizable stimuli-responsiveness.27–30 Not only that, but they can serve as a scaffold allowing anchoring of chromophores to realize the energy transfer process, in which the signals can be fine-tuned in a multi-mode manner.31–33 Furthermore, these materials can function at the molecular level, holding promise for molecular devices.34–36
Benefiting from the above advantages, we propose a supramolecular dynamic memory (SDM) strategy in order to mitigate the drawbacks of traditional DRAM, thereby enabling easily programmable, non-destructive, and compact dynamic memory devices. However, one challenge lies in the necessity for most supramolecular stimuli-response systems to use at least two different stimuli to switch between “on” and “off” states.37–39 This would greatly increase the complexity of the devices and introduce uncertainty to the stored data, thus hindering the production of SDM. Taking these into account, spontaneous recovery in the absence of external interference is a superior trait for realizing SDM, but is still a great challenge. Therefore, time-dependent supramolecular systems with spontaneous recovery after stimulation are greatly promising to construct SDM devices.
As a first embodiment of this design, we herein demonstrate an enzyme-controlled supramolecular co-assembly system featuring time-dependent Förster resonance energy transfer (FRET) behaviors, thus realizing spontaneously recoverable SDM. Specifically, a well-designed naphthalene-based C4-symmetric cationic monomer 1 is first synthesized, which self-assembles into nanoparticles and acts as an energy donor. When different dyes as energy acceptors are added separately into 1, FRET behaviors are observed. Furthermore, supramolecular co-assembly of 1 with adenosine triphosphate (ATP) is established, which still undergoes the FRET process with dyes (Fig. 1a). The hydrolysis process of ATP caused by alkaline phosphatase (ALP) leads to the destruction of 1/ATP, resulting in the changes in fluorescence on a time scale towards monomer 1. On this basis, time-gated logic devices are constructed, using the maximum emission band of the dyes and monomer 1 as outputs. Furthermore, an SDM device is successfully constructed (Fig. 1b). In this device matrix, each data point contains 1/dyes/ALP, and when ATP is added, they co-assemble and produce the emission of the dye due to the FRET process. On exploiting the customized photoresistor to convert the optical signals into electrical signals, digital signal “1” is obtained. Likewise, signal “0” can be obtained when the data are erased spontaneously due to the enzymolysis of ATP induced by the already present ALP.
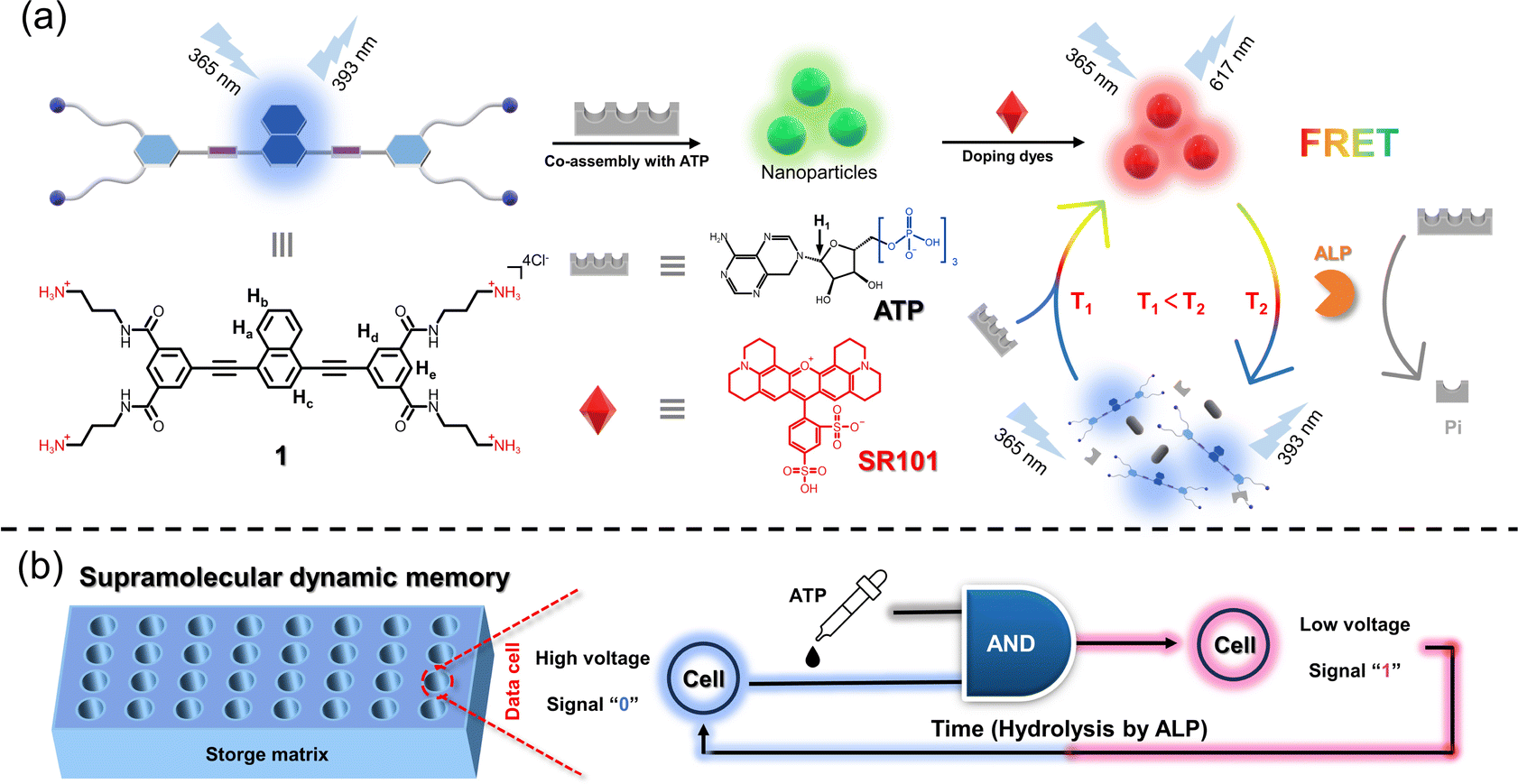 | ||
| Fig. 1 Schematic illustration of the co-assembly and dynamic memory applications of 1/ATP/dyes. (a) Supramolecular co-assembly of 1 with ATP as well as their FRET process with dyes induced by ALP. (b) Supramolecular dynamic memory applications based on the ALP-mediated co-assembly 1/ATP/dyes. The cells are in a storage matrix that contains a designed solution that can manually switch from signal 0 to 1 and spontaneously recover from signal 1 to 0. | ||
Results and discussion
Supramolecular self-assembly behaviors of monomer 1
Spectroscopic experiments were first performed for the designed monomer 1. Specifically, its UV-vis spectrum exhibited two major absorption bands in a dilute aqueous solution (365 and 380 nm, Fig. 2a, red line, ESI Fig. S1†). According to previous literature studies, the lowest energy bands with a fine structure at 398 nm were assigned to the 1Lb transition of the naphthalene unit, while the strong and broad band centered at 350 nm was assigned to the 1La band.40,41 The fluorescence spectrum showed an emission band centered at 393 nm accompanied by a vibronic peak at 412 nm (Fig. 2a, blue line, ESI Fig. S2†), which emitted bright blue light under 365 nm excitation (absolute fluorescence quantum yield, ΦF = 73.3%, ESI Fig. S3†). When the solvent changed to H2O/CH3CN solution (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :999), the absorption band exhibited a bathochromic effect, with two maximum absorption peaks at 350 nm and 400 nm (Fig. 2a, purple line). Meanwhile, the fluorescence signals dramatically red-shifted to 520 nm (Fig. 2a, green line) and the yellow-green emission was observed (ΦF = 44.1%, ESI Fig. S4†). Combining the fluorescence lifetime, density functional theory (DFT) calculations and excitation spectra (ESI Fig. S5 and S6†), we concluded that 1 self-assembled in an H-type manner. By plotting the emission band of 1 at 393 nm against concentration, the critical aggregation concentration (CAC) was measured to be 1.71 × 10−6 mol L−1 (ESI Fig. S7†). The supramolecular assembly mechanism of 1 was further elucidated by solvent-dependent UV-vis spectroscopy experiments. By varying the ratio of water, the absorption spectrum of 1 showed three isosbestic points (251, 315 and 392 nm, ESI Fig. S8†). When monitoring the aggregation species (αagg, λ = 400 nm) against the ratio of water, a sigmoidal curve was obtained (ESI Fig. S8,† inset), which indicated the involvement of an isodesmic mechanism in the supramolecular assembly process of 1.42–44
:999), the absorption band exhibited a bathochromic effect, with two maximum absorption peaks at 350 nm and 400 nm (Fig. 2a, purple line). Meanwhile, the fluorescence signals dramatically red-shifted to 520 nm (Fig. 2a, green line) and the yellow-green emission was observed (ΦF = 44.1%, ESI Fig. S4†). Combining the fluorescence lifetime, density functional theory (DFT) calculations and excitation spectra (ESI Fig. S5 and S6†), we concluded that 1 self-assembled in an H-type manner. By plotting the emission band of 1 at 393 nm against concentration, the critical aggregation concentration (CAC) was measured to be 1.71 × 10−6 mol L−1 (ESI Fig. S7†). The supramolecular assembly mechanism of 1 was further elucidated by solvent-dependent UV-vis spectroscopy experiments. By varying the ratio of water, the absorption spectrum of 1 showed three isosbestic points (251, 315 and 392 nm, ESI Fig. S8†). When monitoring the aggregation species (αagg, λ = 400 nm) against the ratio of water, a sigmoidal curve was obtained (ESI Fig. S8,† inset), which indicated the involvement of an isodesmic mechanism in the supramolecular assembly process of 1.42–44
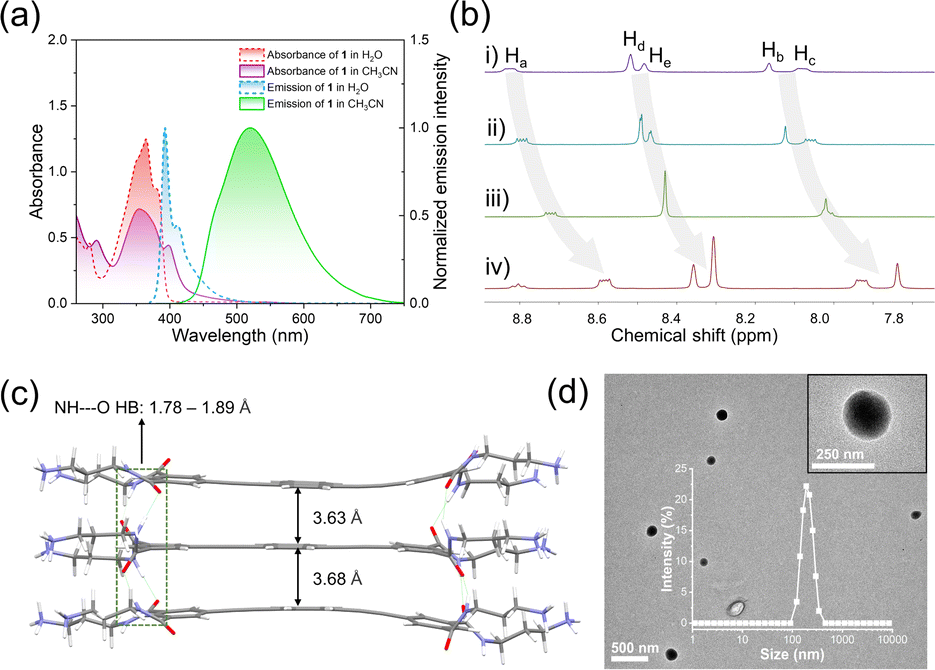 | ||
| Fig. 2 Supramolecular self-assembly behaviors and structures of 1. (a) UV-vis absorption and emission spectra of 1 (1.5 × 10−5 mol L−1) in H2O and H2O/CH3CN (v/v = 1:999). (b) Partial 1H NMR (400 MHz, 298 K) spectra of 1 in D2O/CD3CN (v/v = 7:3): (i) 1.25 mM, (ii) 2.5 mM, (iii) 5 mM, and (iv) 10 mM. (c) Optimized geometries of the trimeric structures of 1 based on DFT calculations. The BLYP functional and the def2-SVP basis set were adopted for geometry optimization calculations. (d) TEM micrograph of 1. Inset: DLS curve of the self-assembly of 1 (1.5 × 10−5 mol L−1) and partial enlargement image. | ||
To obtain deeper insight into the stacking structures of 1, concentration-dependent 1H NMR experiments were performed. Upon varying the concentration of 1 (from 1.25 to 10 mM), the aromatic protons gradually shifted upfield (Δδ = 0.24 for Ha–b, 0.25, 0.16 and 0.17 for Hc–e, Fig. 2b), revealing the existence of π–π stacking. This result was also verified by the powder X-ray diffraction peaks between 22–29° (ESI Fig. S9†). The supramolecular stacking mode of 1 was then elucidated through DFT calculations. In the optimized trimer, the monomers were bound together by two-fold N–H⋯O hydrogen bonds between neighboring amide units (bond lengths: 1.78–1.89 Å, Fig. 2c). The adjacent naphthalene adopted a face-to-face arrangement with interplanar distances of 3.63 and 3.68 Å, indicating the existence of π–π interaction. Besides, transmission electron microscopy (TEM) and dynamic light scattering (DLS) measurements revealed that small nanoparticles (∼200 nm DLS hydrodynamic diameter) were obtained for 1 (Fig. 2d and inset).
FRET properties of supramolecular self-assembly of 1 with dyes
Taking advantage of the broad emission band of the self-assembly of 1, we believe that it can be used as a universal donor matrix for transferring energy to a variety of acceptors. To confirm this hypothesis, we chose four organic dyes, sulforhodamine 101 (SR101), eosin Y (EY), phloxine B (PB), and rhodamine B (RhB) as acceptors. The FRET properties of 1/SR101 were first investigated. Here, we used the same solvent conditions as for the self-assembly, i.e., a mixture of H2O/CH3CN solution (v/v = 1:999), for the FRET property studies. The spectral overlap integral between the emission band of 1 and the absorption band of SR101 was calculated to be 1.96 × 1015 M−1 cm−1 nm4 (ESI Fig. S10a and b†), which facilitates the FRET of 1/SR101 at the excited state. Upon gradually adding SR101 into 1, the emission band of 1 at 520 nm was reduced, while the SR101 emission band centred at 617 nm was enhanced (Fig. 3a). Meanwhile, the CIE coordinate diagram showed that the emission color gradually changed from blue-green to red (Fig. 3b). Surprisingly, almost white light emission (0.31, 0.34) was observed with the addition of 0.09 mol% SR101 (Fig. 3b, inset). The FRET behaviors from 1 to SR101 were further demonstrated by the two-dimensional excitation spectra (Fig. 3c). Upon excitation at 300–400 nm, the emission signal of 1 at 400–530 nm significantly decayed, and 1/SR101 was almost completely dominated by the acceptor's emission bands at 600–640 nm. In this FRET process, the addition of SR101 has no effect on the original supramolecular assembly structures (ESI Fig. S11–S14†).
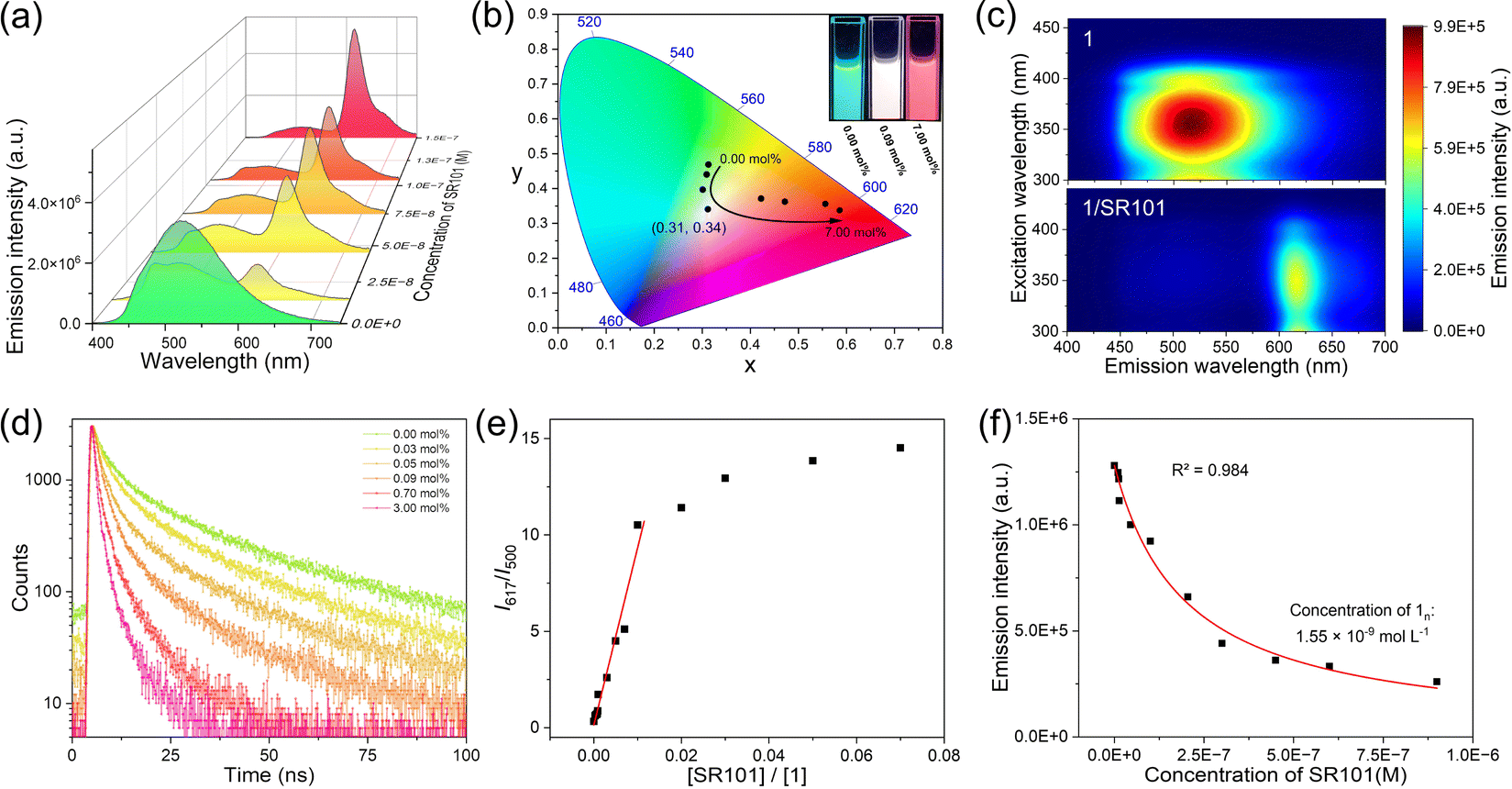 | ||
| Fig. 3 FRET properties of 1/SR101. (a) Steady-state fluorescence spectra changes of 1 (1.5 × 10−5 mol L−1) upon increasing the molar ratio of SR101. (b) CIE coordinate changes upon increasing the molar ratio of SR101. Inset: photographs of the self-assembly of 1 doped with different molar ratios of SR101 under 365 nm excitation. (c) Two-dimensional excitation spectra of the assemblies of 1 and 1/SR101 (3 mol% of SR101). (d) Fluorescence lifetime decay profiles of self-assembly of 1 upon increasing the molar ratio of SR101. (e) Ratiometric plot of SR101 in 1/SR101 upon direct excitation of 1. I617 and I500 denote the emission intensity of SR101 at 617 nm and 1 at 500 nm, respectively. (f) Non-linear fitting of the emission intensity of 1 versus the concentration of SR101. n is the ratio between the concentration of 1 and the concentration of donor matrix 1n. The concentrations of 1 for these experiments were all fixed at 1.5 × 10−5 mol L−1. | ||
The FRET mechanism was then elucidated. Upon increasing the molar ratio of SR101, the fluorescence lifetime (τ) of 1 exhibited a significant decrease (from 20.82 ns to 2.43 ns, Fig. 3d). Besides, the ratio plots of 1/SR101 (I617 nm/I500 nm) increased linearly on increasing the molar ratio of SR101 at the initial state from 0 mol% to 1.0 mol% (Fig. 3e), indicating the individual dispersion of the acceptors into the self-assembly matrix of 1. Furthermore, the I617 nm/I500 nm values reached a plateau and the emission of SR101 in 1/SR101 decreased a little (Fig. 3e). These results confirmed the tendency of acceptor SR101 to form clusters in 1/SR101.45 Importantly, the energy transfer efficiency (ΦET) and antenna effect were calculated to be 94.7% and 161 according to the Fröster mechanism (ESI Fig. S10c†).
Furthermore, Stern–Volmer plots of the average fluorescence lifetime and the steady-state fluorescence intensity of 1 did not overlap (ESI Fig. S10d†), which was characteristic of the fluorescence quenching of donor 1 that contained both dynamic and static quenching.46 However, the calculated dynamic quenching efficiency (ηdyn) was much higher than the static quenching efficiency (ηstat) (ESI Tables S7 and S8†), indicating that the dynamic quenching of the donor dominated in the FRET process. We then employed a mathematical model to determine the number of donors (n) that were quenched by a single acceptor.47–50 Plotting the donor emission intensity against the acceptor concentration, a non-linear curve fitting yielded an n value of 9677 (Fig. 3f), demonstrating a very strong quenching effect of the acceptor towards the donor. Coulombic homo-transfer was used here to illustrate excitation energy delocalization in 1/SR101, which occurred as exciton diffusion.51–53 By plotting 1/τ of 1 against the concentration of SR101, the second-order exciton migration rate for SR101 was calculated to be 4.48× 1015 L mol−1 s−1 (ESI Fig. S10f†), which represents a large value of the FRET from 1 to SR101.
Similarly, for the other organic dyes, EY, PB and RhB could also serve as energy acceptors for an excellent FRET process with 1 due to their large spectral overlap (ESI Fig. S15a–f†). ΦET and antenna effects were calculated to be 90.7% and 62 for 1/EY, 81.0% and 50 for 1/PB and 74.1% and 185 for 1/RhB, respectively (ESI Fig. S15g–i†). Like 1/SR101, the ηdyn value of these dyes was separately higher than their corresponding ηstat (ESI Fig. S16a–c and S17a, Tables S1–S6†). Outstandingly, 1/PB has the highest second-order exciton migration rate of 8.59 × 1015 compared to the others (ESI Fig. S16d–f and S17b†). This tendency was reflected by the efficient quenching of 1 by PB (n value = 12820 for PB, 7042 for EY, and 1056 for RhB, ESI Fig. S18†).
Spontaneous fluorochromism in co-assembly 1/ATP
After elucidating the FRET features between the self-assembly of 1 and dyes, we then sought to investigate the spontaneous fluorochromism behaviors of the co-assembly 1/ATP with dyes on the participation of ALP. The co-assembly of 1 with ATP was first clarified. With the gradual addition of ATP into the aqueous solution of 1 (1.5 × 10−5 mol L−1), the original emission band of monomer 1 centred at 393 nm gradually decreased, while a new broad emission band centred at 540 nm appeared (Fig. 4a). Simultaneously, the absorption band located between 325 and 375 nm decreased on the addition of ATP, while a set of bands centred at 406 nm appeared with an isosbestic point at 390 nm (Fig. 4a, inset). The upfield shifted resonances (Δδ = 0.55 for Ha–e and 0.72 for H1) also confirmed the co-assembly of 1/ATP (Fig. 4b). The CAC value of 1/ATP in aqueous solution was obtained to be 1.61 × 10−5 mol L−1 (ESI Fig. S19†). Furthermore, the co-assembly mechanism of 1/ATP was elaborated by temperature-dependent UV-vis measurements.54–56 When monitoring αagg (λ = 406 nm) versus temperature, the obtained sigmoidal melting curve indicated the participation of the isodesmic mechanism in the co-assembly process of 1/ATP (ESI Fig. S20†). In addition, scanning electron microscopy (SEM) demonstrated the larger nanoparticles of 1/ATP than that self-assembly of 1 in acetonitrile (DLS hydrodynamic diameter of ∼350 nm, ESI Fig. S21†). These phenomena confirmed that ATP can induce 1 to form the co-assembly 1/ATP in an aqueous medium.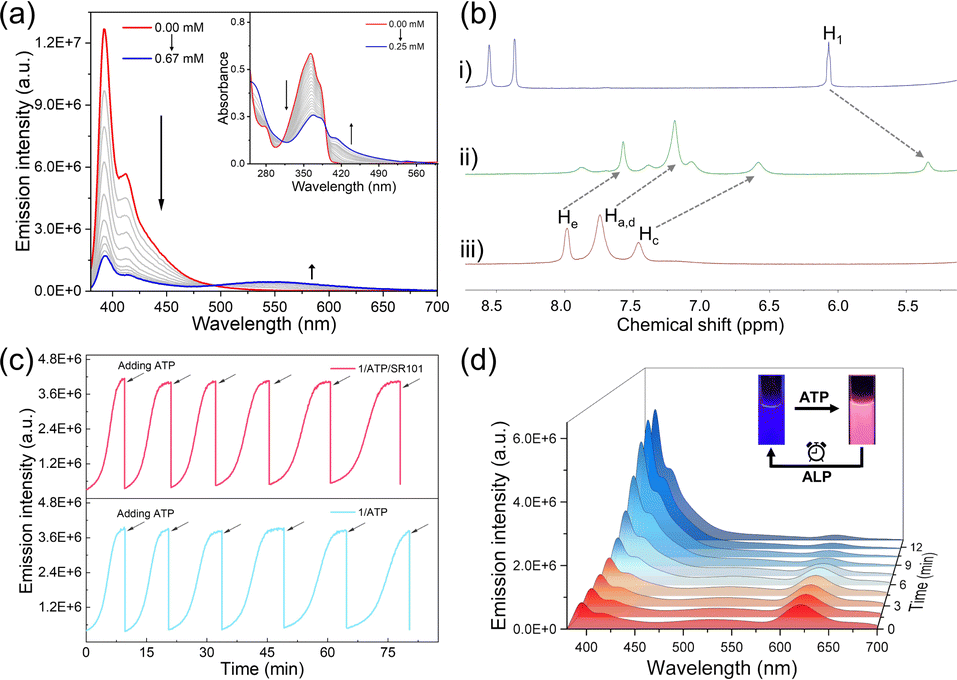 | ||
| Fig. 4 ALP mediated spontaneous fluorochromism in co-assembly 1/ATP. (a) Fluorescence and inset of UV-vis absorbance spectrum variations upon gradual addition of ATP into the aqueous solution of 1 (1.5 × 10−5 mol L−1). λex = 365 nm. (b) Partial 1H NMR (400 MHz, D2O, 298 K) spectra of (i) ATP, (ii) 1/ATP and (iii) 1. [1] = [ATP] = 1.5 × 10−2 mol L−1. (c) Time-dependent fluorescence intensity at 393 nm upon several repetitive additions of ATP into 1 and 1/SR101 in the presence of 1.0 U mL−1 of ALP. Arrows represent the (re)fuelling points with the addition of ATP ([1] = 1.5 × 10−5 mol L−1, [ATP] = 4.5 × 10−5 mol L−1, [SR101] = 3.0 × 10−7 mol L−1). (d) Fluorescence spectrum variations of 1/ATP/SR101 versus hydrolyzed time in the presence of 1.0 U mL−1 of ALP in aqueous solution. λex = 365 nm. Inset: photographs of 1/ATP/SR101 before and after hydrolysis under 365 nm excitation. | ||
Then, the ability of enzyme-induced disassembly was investigated by in situ decomposition of ATP. ALP was chosen as the enzymatic cleavage reagent, because of its ability to hydrolyze ATP to adenosine and monophosphate under ambient conditions.57,58 Since the hydrolysis products are unable to efficiently induce the formation of the co-assemblies, 1/ATP should gradually disassemble with the hydrolysis of ATP. Indeed, when ALP is present in the solution of 1/ATP, the emission intensity of 1/ATP gradually decreased, accompanied by a gradual increase in monomeric emission of 1 (ESI Fig. S22a†), indicating that ALP induced the disassembly of 1/ATP and the release of free monomers. At the same time, the emission color of the solution recovered to the original deep blue (ESI Fig. S22a,† inset). Moreover, when the amount of ALP was increased from 0.6 U mL−1 to 1.0 U mL−1 and 1.8 U mL−1, the recovery time was significantly shortened from 18.0 min to 15.1 min and 6.3 min (ESI Fig. S23†). These results undoubtedly prove that the hydrolysis reaction of ATP directly reflects the disassembly process. When ATP was re-added to the solution, the deep-blue fluorescence immediately changed to cyan, accompanied by a decrease in monomer fluorescence and the appearance of assembly emission (ESI Fig. S24†). This phenomenon indicates that the co-assembly 1/ATP was re-generated. Intriguingly, the blue fluorescence signal increased spontaneously over time, which can be attributed to the ALP already present in the solution system. Afterward, ATP was reintroduced, and the same trend as in the previous cycle occurred. As fresh batches of ATP were repeatedly added, the emission signal could be switched reversibly over six cycles (Fig. 4c bottom). However, there was a gradual decrease in the rate of hydrolysis with increasing number of cycles, which may be due to the effect of hydrolysis products (such as adenosine diphosphate) on the ATP hydrolysis process (ESI Fig. S25†).57
Given that 1/ATP possesses an emission band centered at 540 nm similar to that of 1 in the self-assembly state (Fig. 2a), we hypothesized that 1/ATP could be used to construct a FRET system with the above mentioned dyes in water. It was true that when 5 mol% of SR101 was added to 1/ATP, the original 540 nm fluorescence intensity declined, accompanied by an enhancement of the emission band of SR101 (Fig. 4d). The solution emission color also changed significantly (Fig. 4d, inset). 1/ATP exhibited a decrease in fluorescence lifetime after the addition of SR101, further confirming the occurrence of the FRET process from 1/ATP to SR101 (ESI Fig. S26d†). Similar to 1/ATP, the co-assembly 1/ATP/SR101 underwent a disassembly process over time in the presence of ALP, with a spontaneous enhancement of the emission signal of monomer 1 and a reduction of the emission intensity of SR101 (Fig. 4d). The signals of 1/ATP/SR101 could be switched six times with the continuous addition of ATP (Fig. 4c top). For the other dyes such as EY, PB and RhB, similar FRET behaviors were realized (ESI Fig. S22b–d, S26a–c and S27†). Thus, an enzyme-mediated FRET assembled system with spontaneous fluorochromism was successfully established.
Spontaneously recoverable SDM
Inspired by the above time-dependent FRET behaviors of the co-assembled fluorochromism system, we envisioned that it could be used for constructing time-gated logic devices, which is the basis for realizing programming and erasing of signals in dynamic memory cells. The logic system defined four different components of 1, ATP, SR101 and ALP as inputs, with the emission intensity of SR101 at 617 nm as output 1 and the intensity of monomer 1 at 393 nm as output 2. For the output, the fluorescence intensity of 1/ATP/SR101 above 9.0 × 105 was indicated as “1” whereas the one below as “0”.59–61 The weights of the two outputs were set to 0.7 and 0.3, respectively. On this basis, a four-input logic gate was designed, in which multiple logic operations can be performed (Fig. 5a and ESI Table S9†). By combining different components as inputs, it is possible to construct various logic gates ranging from complex ones to a basic AND logic gate (ESI Fig. S28†). Furthermore, time-dependent outputs were successfully achieved in the designed logic gates, which could be clearly seen in the truth table and fluorescence intensity changes: over time, output 1 changed to output 2 (Fig. 5b, ESI Fig. S29, Tables S10 and S11†).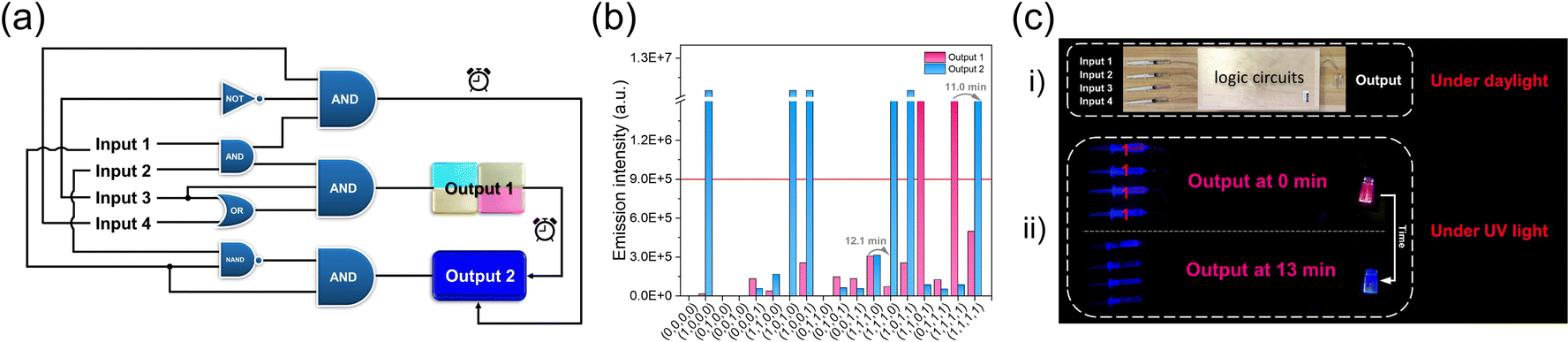 | ||
| Fig. 5 Time-gated logic gates. (a) Scheme of four-input and two-output logic gates. (b) Fluorescence intensity with different input combinations in the SR101 four-input logic gate system. The “false/true” threshold is the red line. (c) Time-gated logic device. (i) The logic gate device under daylight. (ii) The logic gate device under 365 nm excitation using (1, 1, 1, 1) input at 0 min and 13 min. | ||
Next, based on the design, we successfully produced a time-gated logic device (Fig. 5c). In this device, different syringes at the left end served as inputs, which contained aqueous solution with different assembly components (Fig. 5c, i). The input was connected through a flexible tube into the logic circuits constructed in the box to perform the logic operations. The mixed solution was allowed to flow into the sample bottle at the right end, serving as the output. For example, the four input ports simultaneously injected 1, ATP, SR101 and ALP into the logic circuits as (1, 1, 1, 1). After performing logic operations, output 1 of red-light emission was observed (Fig. 5c, ii). As time passed, the solution emission gradually changed to deep blue, as output 2 (Fig. 5c, ii). This phenomenon was also realized for other dye doped co-assembled systems, but with initial output states of different emission colors (ESI Fig. S28†). These results suggest that it holds great potential for the construction of complex logic circuits.
Based on the above discussion, our time-gated logic system is considered to be ideally suited for use as a spontaneously recoverable SDM device. Therefore, we constructed a storage matrix based on the AND logic gate, in which output 1 is defined as “1” and output 2 as “0”. For each data point, we employed a photoresistor to convert the fluorescent signal into electrical signals with a final series connection to a power supply (Fig. 6a). Specifically, the data points with ATP added showed a low voltage, while those without ATP showed a high voltage (Fig. 6b). Then, the hydrolysis caused by ALP brought the data points back to the initial state and enabled the data points to be recovered (Fig. 6b). In the initial state, every data point contained the 1/SR101/ALP solution and output “0” (Fig. 6b). When ATP was added at different data points to bring the matrix into the programmed state, the change in fluorescence made a difference in voltage (Vout). Next, the signals were converted into binary code, and the stored information was obtained as “029” (Fig. 6d). When the addition of ATP stopped and it was hydrolyzed by ALP, the matrix can be programmed again by reintroducing ATP, and the stored information “010” was obtained (Fig. 6e). This process could be repeated several times, as shown in the time-dependent voltage plot (Fig. 6f). All the FRET systems with different dyes show the same phenomenon (ESI Fig. S30–S32†).
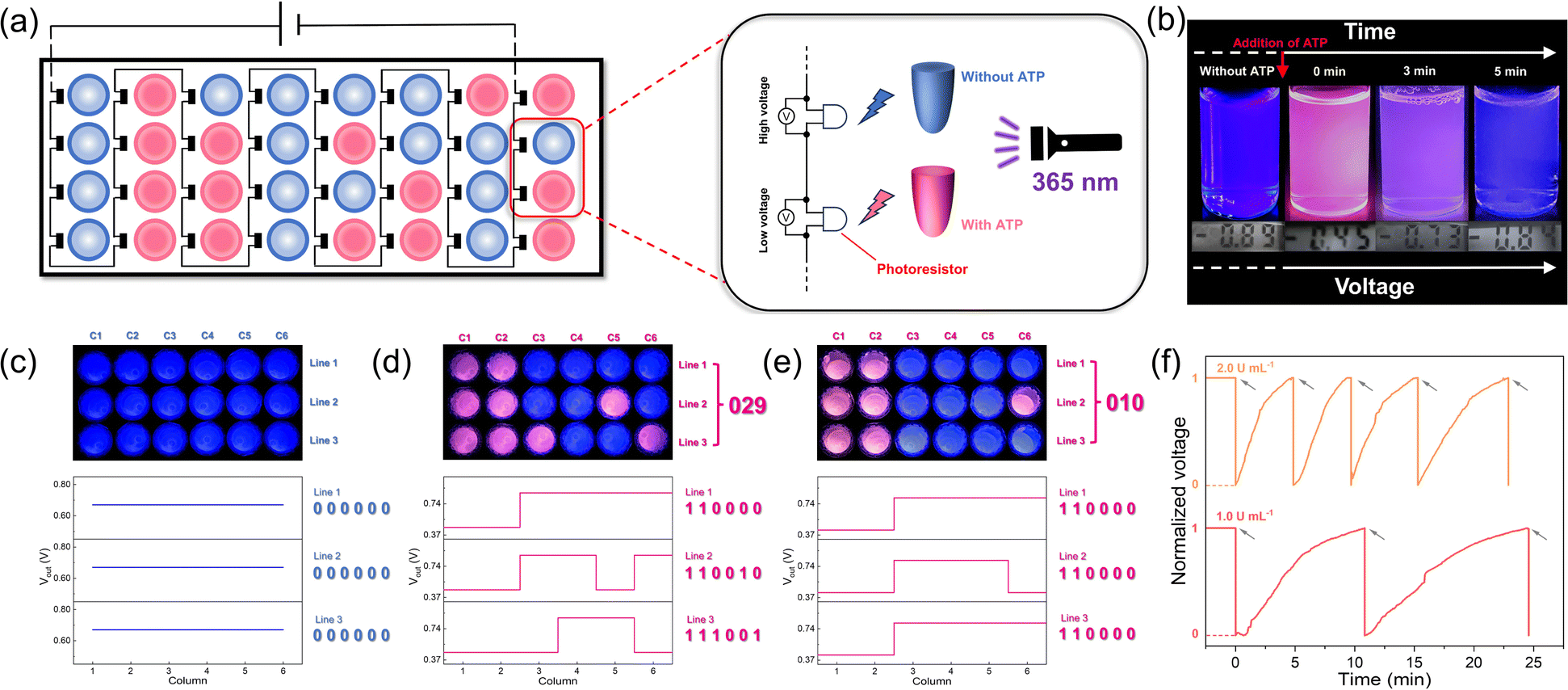 | ||
| Fig. 6 Programmable supramolecular dynamic memory devices. (a) Illustration of the designed supramolecular dynamic memory device. (b) Voltage and fluorescence photographs without ATP addition and during hydrolysis after ATP addition. (c–e) Storage matrix under 365 nm excitation and the corresponding voltages for each data point. The voltages can be converted into binary code to get the information. (f) Time-dependent normalized voltage intensity of a single data point upon several repetitive additions of ATP in the presence of 1.0 U mL−1 and 2.0 U mL−1 of ALP, respectively. Arrows represent the (re)fuelling points with the addition of ATP. The concentration of 1, ATP and SR101 is set at 1.5 × 10−5 mol L−1, 4.5 × 10−5 mol L−1 and 7.5 × 10−7 mol L−1, respectively. | ||
Moreover, the memory time could be easily modified. As mentioned above, the concentration of ALP is positively correlated with the rate of ATP hydrolysis. When the concentration of ALP increased, more program–erase cycles could be achieved in the same amount of time, thus reducing the memory time (Fig. 6f). In addition, by combining different dye systems, we have successfully achieved higher level memory, which will greatly improve storage density (ESI Fig. S33†). Due to the wide emission band of the assembly, it is possible to undergo the FRET process with diverse dyes, potentially achieving octal or even hexadecimal values. Here, each data point is equivalent to a DRAM cell in which the readout could be programmed and erased upon addition and hydrolysis of ATP.
Conclusions
In summary, we have successfully established a spontaneously recoverable SDM system derived from the enzyme-controlled co-assembled system with time-dependent FRET features. Benefitting from the promoted exciton migration (4.48 × 1015 L mol−1 s−1), the FRET ability of the self-assembly 1/SR101 is satisfactory and extends to the co-assembly 1/ATP/SR101. The emission signals of 1/ATP/SR101 can be finely switched on a time scale, by temporally regulating the consumption of ATP using its hydrolytic enzyme ALP. Based on the time-dependent co-assembled FRET system, spontaneously recoverable SDM devices are successfully constructed and enable the memory of dynamic information. In contrast to conventional dynamic memory, SDM can be observed through both optical and electrical modes that are interrelated, making it suitable for a wider range of application scenarios. Besides, SDM devices are simple, inexpensive, and feature benign tunability thanks to the most extensive supramolecular structure library available. This work not only represents a universal approach for integrating the functional programmability of supramolecular materials into memory devices, but opens up a new avenue towards high performance computational systems.Data availability
Detailed synthetic procedures, analytical data, and computational methods are provided in the ESI.†Author contributions
X. W. performed the experiments and analyzed the data under the supervision of Z. G. and W. T. X. W., Z. G. and W. T. co-wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22371230, 22071197, 22001213, and 22022107), the Postdoctoral Science Foundation of China (2023M732855 and 2022TQ0258), the Xi'an Association for Science and Technology Youth Talent Support Program (959202313031) and the Shaanxi Fundamental Science Research Project for Chemistry & Biology (22JHQ020). We would like to thank the Analytical & Testing Centre of Northwestern Polytechnical University for TEM and SEM tests.Notes and references
- Q.-D. Ling, D.-J. Liaw, C. Zhu, D. Chan, E.-T. Kang and K.-G. Neoh, Prog. Polym. Sci., 2008, 33, 917–978 CrossRef CAS.
- L. Yu, B. Chen, Z. Li, Q. Huang, K. He, Y. Su, Z. Han, Y. Zhou, X. Zhu, D. Yan and R. Dong, Chem. Soc. Rev., 2023, 52, 1529–1548 RSC.
- K. N. Lin, K. Volkel, J. M. Tuck and A. J. Keung, Nat. Commun., 2020, 11, 2981 CrossRef CAS PubMed.
- Y. Sun, X. Le, S. Zhou and T. Chen, Adv. Mater., 2022, 34, 2201262 CrossRef CAS PubMed.
- H. Lian, X. Cheng, H. Hao, J. Han, M.-T. Lau, Z. Li, Z. Zhou, Q. Dong and W.-Y. Wong, Chem. Soc. Rev., 2022, 51, 1926–1982 RSC.
- S. Gao, X. Yi, J. Shang, G. Liu and R.-W. Li, Chem. Soc. Rev., 2019, 48, 1531–1565 RSC.
- Q. Liu, S. Gao, L. Xu, W. Yue, C. Zhang, H. Kan, Y. Li and G. Shen, Chem. Soc. Rev., 2022, 51, 3341–3379 RSC.
- R. H. Dennard, Nat. Electron., 2018, 1, 372 CrossRef.
- J. D. Meindl, Q. Chen and J. A. Davis, Science, 2001, 293, 2044–2049 CrossRef CAS PubMed.
- Y. Jiang, E. Parsonnet, A. Qualls, W. Zhao, S. Susarla, D. Pesquera, A. Dasgupta, M. Acharya, H. Zhang, T. Gosavi, C.-C. Lin, D. E. Nikonov, H. Li, I. A. Young, R. Ramesh and L. W. Martin, Nat. Mater., 2022, 21, 779–785 CrossRef CAS PubMed.
- P. Schofield, A. Bradicich, R. M. Gurrola, Y. Zhang, T. D. Brown, M. Pharr, P. J. Shamberger and S. Banerjee, Adv. Mater., 2023, 35, 2205294 CrossRef CAS PubMed.
- B. Zhou, Z. Qi and D. Yan, Angew. Chem., Int. Ed., 2022, 61, e202208735 CrossRef CAS PubMed.
- F. Yang, S. Ye, W. Dong, D. Zheng, Y. Xia, C. Yi, J. Tao, C. Sun, L. Zhang, L. Wang, Q. Chen, Y. Wang and Z. Nie, Adv. Mater., 2021, 33, 2100325 CrossRef CAS PubMed.
- J. S. Lindsey and D. F. Bocian, Acc. Chem. Res., 2011, 44, 638–650 CrossRef CAS.
- Y. Han, C. Nickle, Z. Zhang, H. P. A. G. Astier, T. J. Duffin, D. Qi, Z. Wang, E. del Barco, D. Thompson and C. A. Nijhuis, Nat. Mater., 2020, 19, 843–848 CrossRef CAS.
- M. Lanza, A. Sebastian, W. D. Lu, M. Le Gallo, M.-F. Chang, D. Akinwande, F. M. Puglisi, H. N. Alshareef, M. Liu and J. B. Roldan, Science, 2022, 376, 1066 CrossRef.
- L. Liu, J. Han, L. Xu, J. Zhou, C. Zhao, S. Ding, H. Shi, M. Xiao, L. Ding, Z. Ma, C. Jin, Z. Zhang and L.-M. Peng, Science, 2020, 368, 850–856 CrossRef CAS.
- Y. Lin, Y. Cao, S. Ding, P. Zhang, L. Xu, C. Liu, Q. Hu, C. Jin, L.-M. Peng and Z. Zhang, Nat. Electron., 2023, 6, 506–515 CrossRef CAS.
- Z. Yang, B. Li, J.-J. Wang, X.-D. Wang, M. Xu, H. Tong, X. Cheng, L. Lu, C. Jia, M. Xu, X. Miao, W. Zhang and E. Ma, Adv. Sci., 2022, 9, 2103478 CrossRef PubMed.
- J. Shen, W. Song, K. Ren, Z. Song, P. Zhou and M. Zhu, Adv. Mater., 2023, 35, 2208065 CrossRef CAS PubMed.
- K.-X. Teng, L.-Y. Niu and Q.-Z. Yang, J. Am. Chem. Soc., 2023, 145, 4081–4087 CrossRef CAS PubMed.
- T. Schlossarek, V. Stepanenko, F. Beuerle and F. Würthner, Angew. Chem., Int. Ed., 2022, 61, e2022114 CrossRef PubMed.
- D. Zhang, K.-X. Teng, L. Zhao, L.-Y. Niu and Q.-Z. Yang, Adv. Mater., 2023, 35, 2209789 CrossRef CAS PubMed.
- L. Zhang, H.-X. Wang, S. Li and M. Liu, Chem. Soc. Rev., 2020, 49, 9095–9120 RSC.
- J. Li, J. Wang, H. Li, N. Song, D. Wang and B.-Z. Tang, Chem. Soc. Rev., 2020, 49, 1144–1172 RSC.
- Z. Gao, S. Qiu, F. Yan, S. Zhang, F. Wang and W. Tian, Chem. Sci., 2021, 12, 10041–10047 RSC.
- X. Lin, G. Ouyang and M. Liu, ACS Appl. Mater. Interfaces, 2023, 15, 19741–19749 CrossRef CAS PubMed.
- H.-Y. Lin, Y.-T. Wang, X. Shi, H.-B. Yang and L. Xu, Chem. Soc. Rev., 2023, 52, 1129–1154 RSC.
- H. Shigemitsu, T. Fujisaku, W. Tanaka, R. Kubota, S. Minami, K. Urayama and I. Hamachi, Nat. Nanotechnol., 2018, 13, 165–172 CrossRef CAS PubMed.
- Z. Gao, Y. Han and F. Wang, Nat. Commun., 2018, 9, 3977 CrossRef PubMed.
- A. J. P. Teunissen, C. Pérez-Medina, A. Meijerink and W. J. M. Mulder, Chem. Soc. Rev., 2019, 47, 7027–7044 RSC.
- S. Garain, B. C. Garain, M. Eswaramoorthy, S. K. Pati and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 19720–19724 CrossRef CAS PubMed.
- W.-J. Li, X.-Q. Wang, D.-Y. Zhang, Y.-X. Hu, W.-T. Xu, L. Xu, W. Wang and H.-B. Yang, Angew. Chem., Int. Ed., 2021, 60, 18761–18768 CrossRef CAS PubMed.
- V. García-López, F. Chen, L. G. Nilewski, G. Duret, A. Aliyan, A. B. Kolomeisky, J. T. Robinson, G. Wang, R. Pal and J. M. Tour, Nature, 2017, 548, 567–572 CrossRef PubMed.
- E. Kopperger, J. List, S. Madhira, F. Rothfischer, D. C. Lamb and F. C. Simmel, Science, 2018, 359, 296–301 CrossRef CAS.
- F. Lancia, A. Ryabchun and N. Katsonis, Nat. Rev. Chem, 2019, 3, 536–551 CrossRef CAS.
- E. Fuentes, M. Gerth, J. A. Berrocal, C. Matera, P. Gorostiza, I. K. Voets, S. Pujals and L. Albertazzi, J. Am. Chem. Soc., 2020, 142, 10069–10078 CrossRef CAS PubMed.
- Z. Gao, F. Yan, L. Shi, Y. Han, S. Qiu, J. Zhang, F. Wang, S. Wu and W. Tian, Chem. Sci., 2022, 13, 7892–7899 RSC.
- F. Xu, L. Pfeifer, S. Crespi, F. K.-C. Leung, M. C. A. Stuart, S. J. Wezenberg and B. L. Feringa, J. Am. Chem. Soc., 2021, 143, 5990–5997 CrossRef CAS PubMed.
- S. Takahashi and S. Yagai, J. Am. Chem. Soc., 2022, 144, 13374–13383 CrossRef CAS PubMed.
- F. Xu, K. W. Hershey, R. J. Holmes and T. R. Hoye, J. Am. Chem. Soc., 2016, 138, 12739–12742 CrossRef CAS PubMed.
- P. A. Korevaar, C. Schaefer, T. F. A. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482–13491 CrossRef CAS PubMed.
- Z. Chen, A. Lohr, C. R. Saha-Möllera and F. Würthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC.
- Z. Gao, L. Shi, F. Yan, Y. Han, W. Yuan and W. Tian, Angew. Chem., Int. Ed., 2023, 62, e2023022 Search PubMed.
- A. V. Sorokin, I. I. Filimonova, R. S. Grynyov, G. Ya. Guralchuk, S. L. Yefimova and Y. V. Malyukin, J. Phys. Chem. C, 2010, 114, 1299–1305 CrossRef CAS.
- C. Giansante, C. Schäfer, G. Raffy and A. D. Guerzo, J. Phys. Chem. C, 2012, 116, 21706–21716 CrossRef CAS.
- H.-Q. Peng, Y.-Z. Chen, Y. Zhao, Q.-Z. Yang, L.-Z. Wu, C.-H. Tung, L.-P. Zhang and Q.-X. Tong, Angew. Chem., Int. Ed., 2012, 51, 2088–2092 CrossRef CAS PubMed.
- Y. Liu, J. Jin, H. Deng, K. Li, Y. Zheng, C. Yu and Y. Zhou, Angew. Chem., Int. Ed., 2016, 55, 7952–7957 CrossRef CAS PubMed.
- G. Chadha, Q.-Z. Yang and Y. Zhao, Chem. Commun., 2015, 51, 12939–12942 RSC.
- Z. Li, Y. Han and F. Wang, Nat. Commun., 2019, 10, 3735 CrossRef PubMed.
- A. Del Guerzo, A. G. L. Olive, J. Reichwagen, H. Hopf and J.-P. Desvergne, J. Am. Chem. Soc., 2005, 127, 17984–17985 CrossRef CAS PubMed.
- S. Banerjee, R. K. Das, P. Terech, A. de Geyer, C. Aymonier, A. Loppinet-Serani, G. Raffy, U. Maitra, A. Del Guerzo and J.-P. Desvergne, J. Mater. Chem. C, 2013, 1, 3305–3316 RSC.
- K.-T. Wong and D. M. Bassani, NPG Asia Mater., 2014, 6, e116 CrossRef CAS.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- C. Kulkarni, R. Munirathinam and S. J. George, Chem.–Eur. J., 2013, 19, 11270–11278 CrossRef CAS PubMed.
- Z. Gao, Z. Li, Z.-C. Gao and F. Wang, Nanoscale, 2018, 10, 14005–14011 RSC.
- L. Zhang, R. Buchet and G. Azzar, Biophys. J., 2004, 86, 3873–3881 CrossRef CAS PubMed.
- A. Mishra, S. Dhiman and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 2740–2756 CrossRef CAS PubMed.
- M. Tian, Z. Wang, X. Yuan, H. Zhang, Z. Liu and Y. Liu, Adv. Funct. Mater., 2023, 33, 2300779 CrossRef CAS.
- X. He, Z. Li, M. Chen and N. Ma, Angew. Chem., Int. Ed., 2014, 53, 14447–14450 CrossRef CAS PubMed.
- M. Massey, I. L. Medintz, M. G. Ancona and W. R. Algar, ACS Sens., 2017, 2, 1205–1214 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02756f |
| ‡ X. Wang and Z. Gao contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |