
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective S-gem-difluoroallylation of unprotected peptides with 3,3-difluoroallyl sulfonium salts†
Jin-Xiu
Ren
a,
Minqi
Zhou
a,
Xiao-Tian
Feng
a,
Hai-Yang
Zhao
a,
Xia-Ping
Fu
a and
Xingang
Zhang
 *ab
*ab
aKey Laboratory of Fluorine and Nitrogen Chemistry and Advanced Materials, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: xgzhang@mail.sioc.ac.cn
bSchool of Chemistry and Material Sciences Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China
First published on 22nd May 2024
Abstract
Bench-stable 3,3-difluoroallyl sulfonium salts (DFASs), featuring tunable activity and their editable C-β and gem-difluoroallyl group, proved to be versatile fluoroalkylating reagents for site-selective S-gem-difluoroallylation of cysteine residues in unprotected peptides. The reaction proceeds with high efficiency under mild conditions (ambient temperature and aqueous and weak basic conditions). Various protected/unprotected peptides, especially bioactive peptides, are site-selectively S-gem-difluoroallylated. The newly added gem-difluoroallyl group and other functional groups derived from C-β of DFASs are poised for ligation with bio-functional groups through click and radical chemistry. This stepwise “doubly orthogonal” modification of peptides enables the construction of bioconjugates with enhanced complexity and functionality. This proof of principle is successfully applied to construct a peptide–saccharide–biotin chimeric bioconjugate, indicating its great potential application in medicinal chemistry and chemical biology.
Introduction
The tactical introduction of fluorine functionalities into organic molecules has emerged as one of the most powerful tools in modern drug discovery1 and advanced functional materials,2 because of the unique properties of fluorine atom(s) that often change the physiochemical and biological properties of organic molecules.3 In this context, the site-selective fluorofunctionalization of peptides has long been used to alter their acidity, basicity, hydrophobicity, geometry conformation, and bioavailability.4 Furthermore, owing to the absence of fluorine atoms in native biomolecules, such as peptides, fluorine functionalities can serve as probes to investigate the protein–ligand interaction and the instant conformational changes of peptides and proteins via19F NMR.5 As a result, there is a high demand for the site-selective fluorination of peptides in peptidomimetics (Fig. 1a).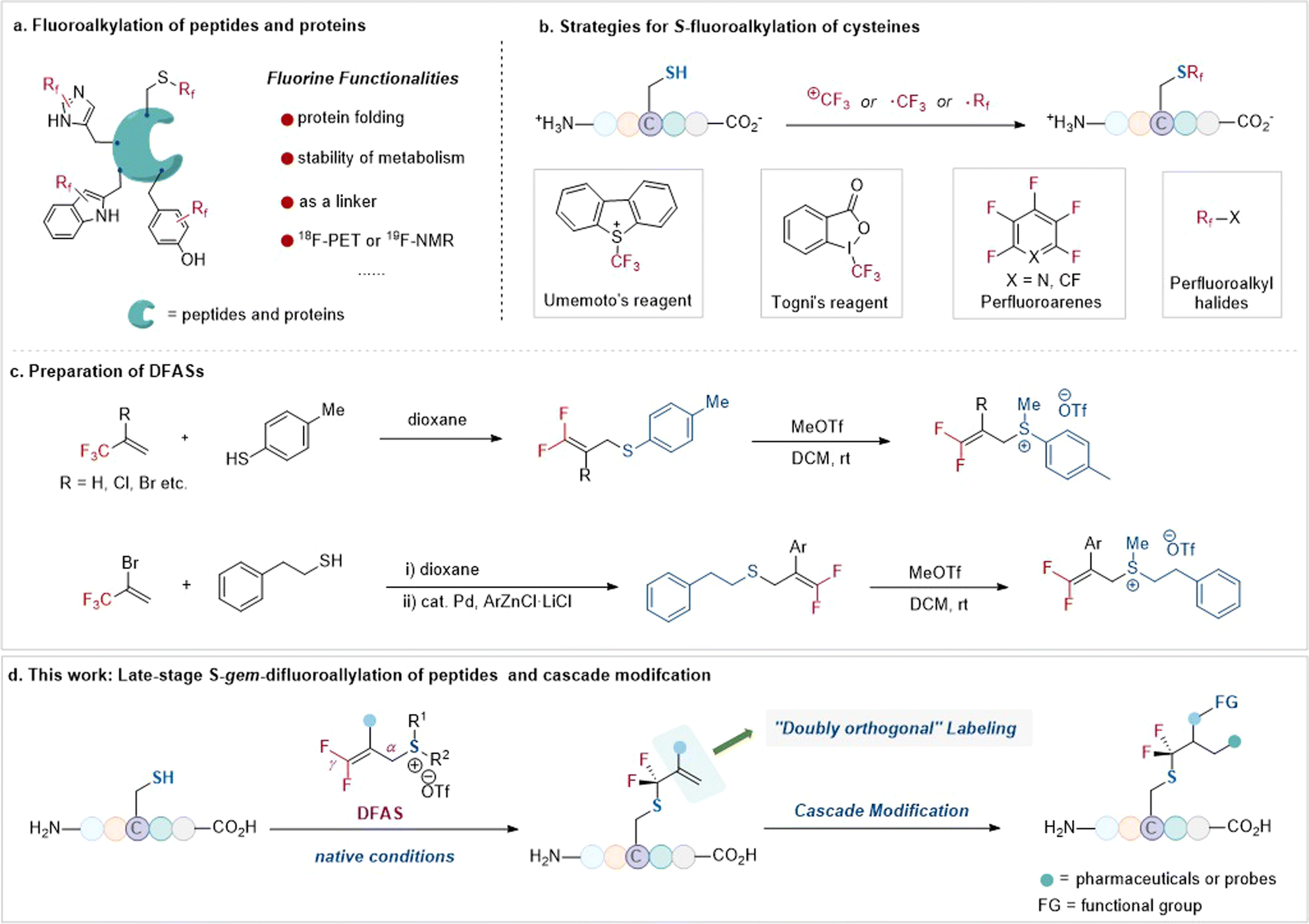 | ||
| Fig. 1 Strategies for S-fluoroalkylation of cysteines and late-stage S-gem-difluoroallylation of peptides and cascade modification (this work). | ||
Among the 20 proteinogenic amino acids, cysteine, with its sulfhydryl side chain, exhibits superior nucleophilicity compared to other amino acids, rendering it the primary choice for site-selective modification, including fluorination, with no doubt, of unprotected peptides.6 To date, the site-selective fluorination on cysteine residues has demonstrated diverse applications7–13 in chemoproteomic profiling of cysteines,718F-PET molecular imaging,8 protein–ligand interaction,9 and efficient bioconjugation.11 Nonetheless, the site-selective fluorination on cysteine residues of an unprotected peptide is severely constrained to only a few fluoroalkylating reagents, including the Togni reagent,14 Umemoto reagent,15 perfluoroarenes,10–13 and perfluoroalkyl halides compounds16 (Fig. 1b). This limitation is even more pronounced when considering fluoroalkylating reagents for other less nucleophilic amino acid residues, such as tryptophan,17 tyrosine,18 histidine, and dehydroalanine.19 Consequently, the restricted fluorination toolbox with limited fluorine space, perfluoroalkyl groups mostly, for peptides is insufficient to explore the unique “fluorine effect” in peptidomimetics.
To bridge the gap, we envisioned that the bench-stable 3,3-difluoroallyl sulfonium salts (DFASs) (Fig. 1c)20,21 could be beneficial for site-selective fluoroalkylation of peptides, because such a type of fluoroalkylating reagent features a flexible molecular scaffold to be readily edited, satisfying varieties of demands for peptide research: (1) the electrophilicity of DFAS could be tuned by the substituents on the sulfur atom, allowing for highly chemical selectivity toward cysteine without affecting other nucleophilic residues or enabling potential proximity-driven reactivity; (2) the β-C of DFASs could be readily modified with bioorthogonal functional groups, such as alkynyl, azide, or useful probes (e.g., dyes, biomarkers); (3) upon reacting with a peptide, the newly added gem-difluoroallyl group is a good handle for successive “doubly orthogonal” modifications (Fig. 1d).
Results and discussion
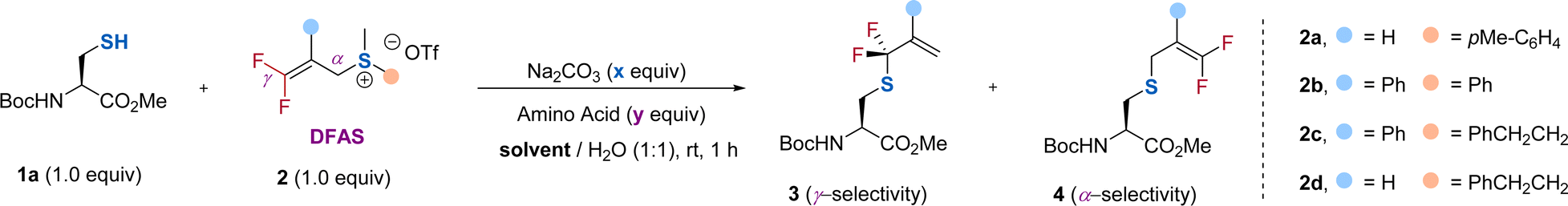
To test our hypothesis, we chose protected cysteine 1a and DFAS 2a as the model substrates (Table 1). As depicted in Fig. 1c, DFASs could be easily prepared from inexpensive and bulk chemical feedstock 3,3,3-trifluoropropene (TFP) or 2-bromo-3,3,3-trifluoroprene through a two or three-step procedure (for details, see ESI†).20,21 A total conversion of 1a was observed when the reaction was carried out with 1a (1.0 equiv.) and 2a (1.0 equiv.) in the presence of Na2CO3 (1.0 equiv.) in DMSO/H2O (1/1, v/v) at room temperature for 1 h (entry 1). Although moderate regioselectivity (3a/4a = γ/α = 3.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was obtained, this positive result encouraged us to improve the regioselectivity. We envisioned that the introduction of an aryl group at the β-position of 2a would increase the γ-selectivity, due to the ability of the aromatic ring to activate the gem-difluoroalkene, thus enabling attack of the thiol group on cysteine to the carbon–carbon double bond easier. An improved γ-selectivity (γ/α = 10:1) along with a 79% yield of 3b was obtained when 2b was used (entry 2). Replacing the aryl group on the sulfur atom with an alkyl group could exclusively provide 3b without observation of the side product 4b (entry 3). However, the absence of the aryl group at the β-position did not affect the regioselectivity and reaction efficiency (entry 4), demonstrating the critical role of the substituent on the sulfur atom. We ascribed this observation to the decreasing leaving ability of the sulfonium salt by using an alkyl group on the sulfur atom, which provides a driving force to produce the γ-selectivity through an SN2′ pathway. Considering that the introduction of an additional aryl group at the β-position of DFAS could improve the structural diversity for the cascade modification of the peptides, we used 2c for further optimization. Further optimization of the reaction conditions showed that 0.5 equiv. of Na2CO3 was enough to promote the reaction (entry 5), and DMF is the optimal co-solvent, providing 3b in almost quantitative yield (95%) (entry 6, for the examination of other solvents, see Table S3†). The competitive experiments between 1a and other amino acids bearing a nucleophilic side chain with 2c exhibited superior S-selective gem-difluoroallylation (entries 7–14). In particular, tyrosine and lysine did not interfere with the reaction (entries 7 and 12), providing an opportunity for late-stage S-gem-difluoroallylation of cysteine residues in peptides and proteins. We also found that the reaction is very fast and could completely afford 3b within 4 min, in sharp contrast to the non-fluorinated sulfonium salt 2c′, which provided the corresponding nonfluorinated product in only 14% yield (Fig. 2), thus demonstrating the unique fluorine effect on the reaction.
:1) was obtained, this positive result encouraged us to improve the regioselectivity. We envisioned that the introduction of an aryl group at the β-position of 2a would increase the γ-selectivity, due to the ability of the aromatic ring to activate the gem-difluoroalkene, thus enabling attack of the thiol group on cysteine to the carbon–carbon double bond easier. An improved γ-selectivity (γ/α = 10:1) along with a 79% yield of 3b was obtained when 2b was used (entry 2). Replacing the aryl group on the sulfur atom with an alkyl group could exclusively provide 3b without observation of the side product 4b (entry 3). However, the absence of the aryl group at the β-position did not affect the regioselectivity and reaction efficiency (entry 4), demonstrating the critical role of the substituent on the sulfur atom. We ascribed this observation to the decreasing leaving ability of the sulfonium salt by using an alkyl group on the sulfur atom, which provides a driving force to produce the γ-selectivity through an SN2′ pathway. Considering that the introduction of an additional aryl group at the β-position of DFAS could improve the structural diversity for the cascade modification of the peptides, we used 2c for further optimization. Further optimization of the reaction conditions showed that 0.5 equiv. of Na2CO3 was enough to promote the reaction (entry 5), and DMF is the optimal co-solvent, providing 3b in almost quantitative yield (95%) (entry 6, for the examination of other solvents, see Table S3†). The competitive experiments between 1a and other amino acids bearing a nucleophilic side chain with 2c exhibited superior S-selective gem-difluoroallylation (entries 7–14). In particular, tyrosine and lysine did not interfere with the reaction (entries 7 and 12), providing an opportunity for late-stage S-gem-difluoroallylation of cysteine residues in peptides and proteins. We also found that the reaction is very fast and could completely afford 3b within 4 min, in sharp contrast to the non-fluorinated sulfonium salt 2c′, which provided the corresponding nonfluorinated product in only 14% yield (Fig. 2), thus demonstrating the unique fluorine effect on the reaction.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 2, x | AA (y) | Solvent | 3 and 4 | |
| 3/4 yieldb (%) | γ/α | ||||
| a Reaction conditions (unless otherwise specified): 1a (0.1 mmol, 1.0 equiv.), 2 (0.5–1.5 equiv.), solvent (1 mL), and H2O (1 mL), rt, 1 h. b Determined by 19F NMR using fluorobenzene as an internal standard. | |||||
| 1 | 2a, 1.0 | — (0) | DMSO | 3a/4a = 77/23 | 3.3:1 |
| 2 | 2b, 1.0 | — (0) | DMSO | 3b/4b = 79/8 | 10:1 |
| 3 | 2c, 1.0 | — (0) | DMSO | 3b/4b = 92/— | >99:1 |
| 4 | 2d, 1.0 | — (0) | DMSO | 3a/4a = 93/— | >99:1 |
| 5 | 2c, 0.5 | — (0) | DMSO | 3b/4b = 90/— | >99:1 |
| 6 | 2c, 0.5 | — (0) | DMF | 3b/4b = 95/— | >99:1 |
| 7 | 2c, 0.5 | Boc-Tyr-OMe (1.0) | DMF | 3b/4b = 99/— | >99:1 |
| 8 | 2c, 0.5 | Boc-His-OMe (1.0) | DMF | 3b/4b = 99/— | >99:1 |
| 9 | 2c, 0.5 | Boc-Trp-OMe (1.0) | DMF | 3b/4b = 90/— | >99:1 |
| 10 | 2c, 0.5 | Boc-Ser-OMe (1.0) | DMF | 3b/4b = 95/— | >99:1 |
| 11 | 2c, 0.5 | Pro-OMe (1.0) | DMF | 3b/4b = 96/— | >99:1 |
| 12 | 2c, 1.5 | Boc-Lys (1.0) | DMF | 3b/4b = 95/— | >99:1 |
| 13 | 2c, 1.5 | Boc-Met (1.0) | DMF | 3b/4b = 90/— | >99:1 |
| 14 | 2c, 1.5 | Boc-Gln (1.0) | DMF | 3b/4b = 99/— | >99:1 |
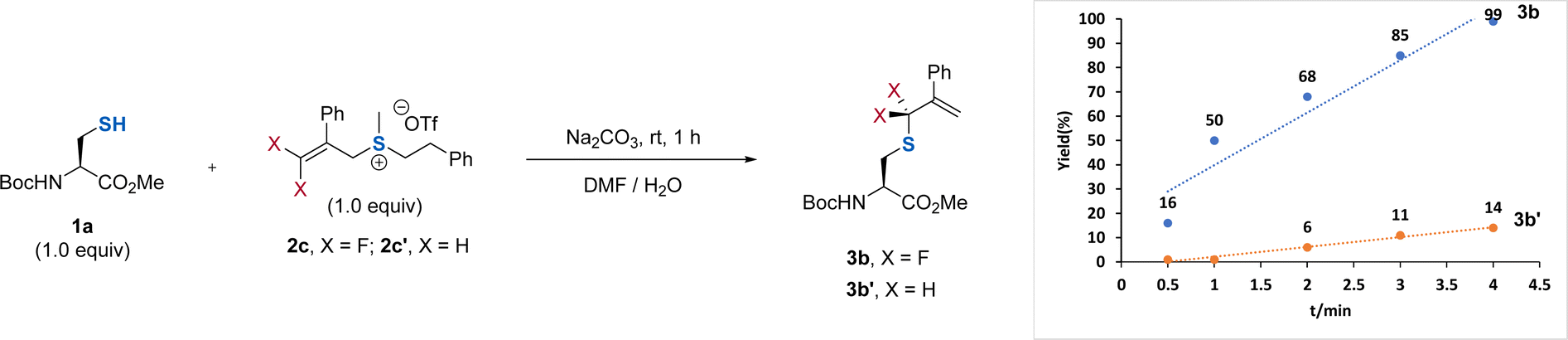 | ||
| Fig. 2 Kinetic studies of the reaction of 1a with 2c or 2c′. | ||
With the optimized conditions in hand, we examined the reaction of 2c with a variety of cysteine-containing dipeptides (Scheme 1a). The N-terminal and C-terminal of the peptides were protected for easy separation of the resulting gem-difluoroallylated products. Generally, high efficiency and excellent γ-selectivity (γ/α > 99:1) were observed. The reaction exhibited excellent functional group tolerance. The amino acid residues bearing an unprotected nucleophilic side chain, such as tyrosine (3c), serine (3d), histidine (3e), tryptophan (3f), and glutamine (3g), did not affect the reaction, without observation of the gem-difluoroallylation side products resulting from their nucleophilic side chains. Additionally, protected lysine (3h), glutamic acid (3i), proline (3j), methionine (3k), valine (3l), phenylalanine (3m), and glycine (3n) were all compatible with the reaction. Notably, unprotected glutathione (GSH), one of the most important natural antioxidants, underwent the S-selective gem-difluoroallylation smoothly (γ/α > 99:1) (3o). In contrast, the protected GSH led to a slightly lower yield when the reaction was carried out on a one mmol scale (3p). The reaction was not restricted to 2c, as DFASs 2 bearing different substituents at the β-position proceeded the reaction efficiently with excellent regioselectivity (γ/α > 99:1) (Scheme 1b). For instance, the electronic nature of the aryl substituent did not affect the regioselectivity (γ/α > 99:1) (3q, 3r), even for the modification of free GSH (3w–3y). However, a higher yield was obtained for the aryl group bearing an electron-withdrawing group (3r) because of its more substantial inducing effect to activate the alkene. Additionally, the introduction of bioorthogonal groups, such as alkyne (3s) and azide (3t), also provided good opportunities for cascade modification of the peptides. Replacing the aryl group at the β-position of DFAS with bromide furnished the corresponding product efficiently (3u). Notably, an alkyl group could also be introduced into the alkene with E-configuration as the major product (3v), demonstrating the generality of the protocol. This modification may also provide the potential to be used as a lipidation analog.22
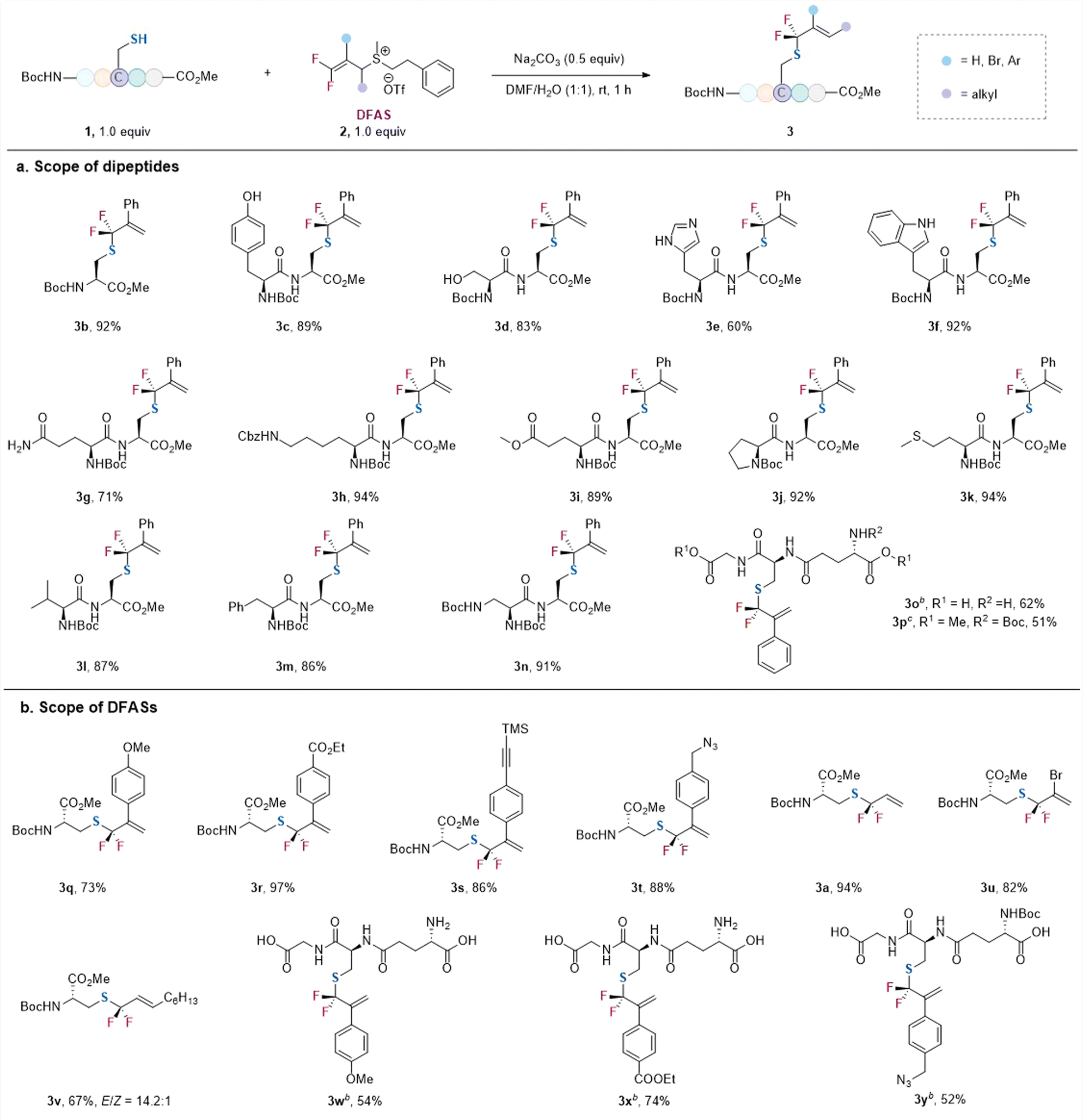 | ||
| Scheme 1 S-gem-Difluoroallylation of cysteine-containing dipeptides and tripeptides with DFASs 2.a aReaction conditions: 1 (0.2 mmol, 1.0 equiv.), 2 (1.0 equiv.), Na2CO3 (0.5 equiv.), H2O (2 mL), DMF (2 mL), rt, 1 h. b2.5 equiv. Na2CO3 was added. cThe reaction was carried out on a 1 mmol scale. All reported yields are isolated yields. | ||
Encouraged by the excellent performance of DFASs in achieving regio/chemo-selective S-gem-difluoroallylation of cysteine-containing dipeptides and tripeptides, particularly unprotected GSH, a wide range of unprotected cysteine-containing bioactive oligopeptides were examined, providing the corresponding S-gem-difluoroallylated peptides in moderate to good yields without observation of γ-selective allylation side products (γ/α > 99:1) (Scheme 2). Cyclic peptide RGD, the most common peptide motif responsible for cell adhesion to the extracellular matrix,23 underwent the S-selective gem-difluoroallylation smoothly (6a, 6b). Notably, the introduced azide moiety provides an excellent opportunity for downstream biorthogonal conjugation to achieve new interesting bioactive molecules or probes (6b). Acyclic peptides from pentapeptide to tridecapeptide were suitable substrates (6c–6f). Notably, nonapeptide bearing the nucleophilic side chain-containing amino acid residues, including lysine, histidine, tyrosine, and tryptophan, was applicable to the reaction (6d), thus demonstrating the generality of the current protocol. The Tyr-amyloid P component (6e) and β-amyloid peptide fragment (6f) proceeded smoothly. The low isolated yield of 6c is because of its decomposition upon purification. However, its 19F NMR yield was high (72%). We also examined disulfide-bridged bioactive peptides oxytocin (6g, 6h), the “best-understood neuropeptide”,24 and eptifibatide (6i), a glycoprotein inhibitor. Treatment of these cyclic peptides with tris(2-carboxyethyl)phosphine (TCEP)25 to break their disulfide bonds, followed by the reaction of DFAS with the freshly generated thiol group, furnished the S-gem-difluoroallylated efficiently (6g–6i), featuring the synthetic simplicity and high efficiency without removing the excess TCEP of this two-step, one-pot procedure. The excellent compatibility of DFAS with TCEP would significantly expand its applicability in site-selective fluorination of peptides. Most importantly, the newly formed two gem-difluoroallyl handles would enhance the chemical space to create more structurally diversified structures of medicinal and chemicobiological interest. Moreover, this cysteine-selective gem-difluoroallylation has proven effective at low concentrations, as low as 50 μM, thereby providing a promising tool for modifying proteins (Table S7†).
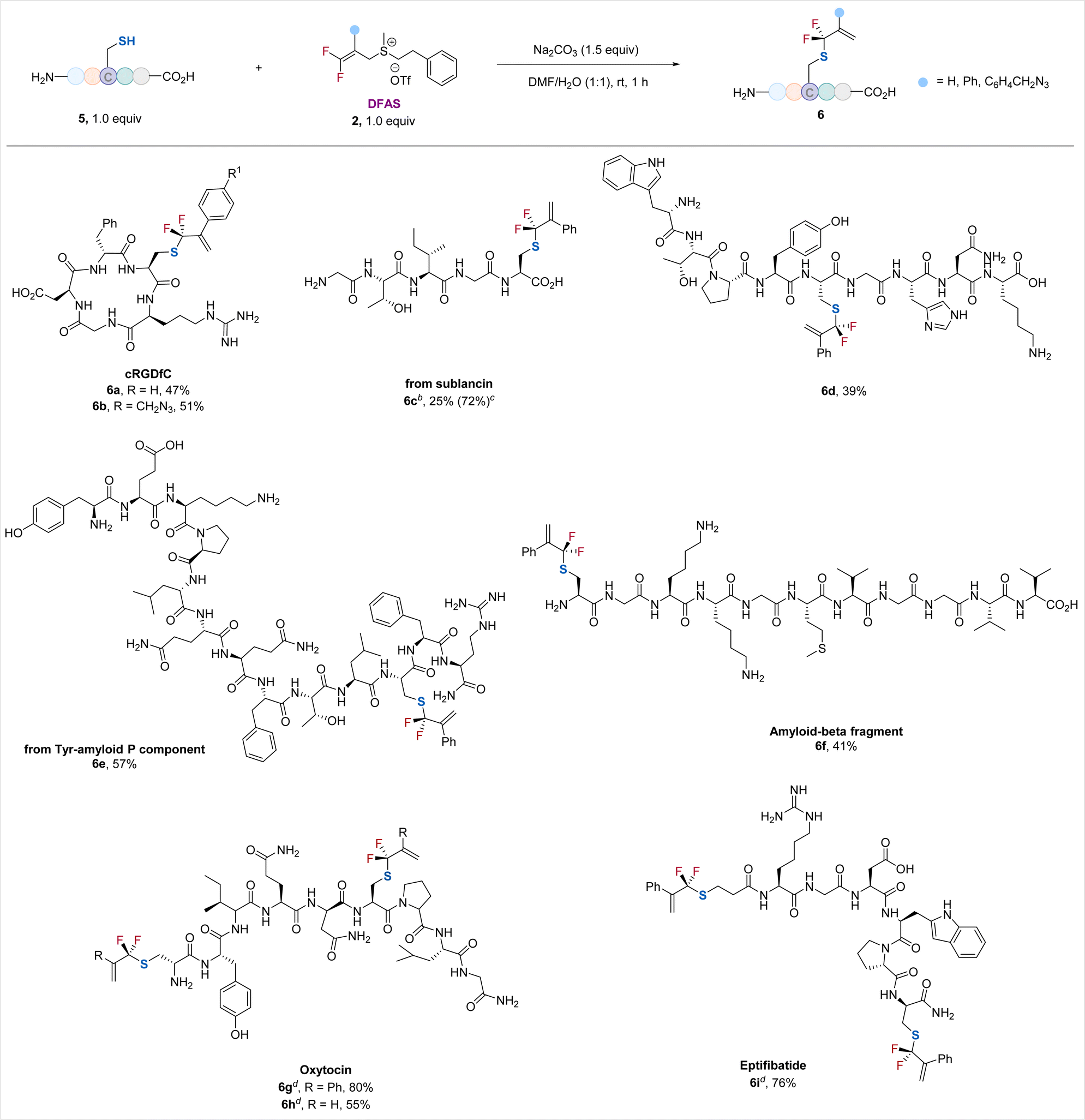 | ||
| Scheme 2 Late-stage S-gem-difluoroallylation of unprotected cysteine-containing peptides with DFASs 2.a aReaction conditions: 5 (0.02 mmol, 1.0 equiv.), 2 (1.0 equiv.), Na2CO3 (1.5 equiv.), H2O (1 mL), DMF (1 mL), rt, 1 h. bThe reaction was carried out on a 0.04 mmol scale. c19F NMR yield. d(i) 5 (0.02 mmol, 1.0 equiv.), TCEP·HCl (1.5 equiv.), Na2CO3 (3.0 equiv.), H2O (1 mL), DMF (1 mL), rt, 1 h; (ii) 2 (2.0 equiv.), rt, 1 h. | ||
To demonstrate the synthetic utility of the protocol, we explored the potential to construct more intricate bioconjugates by utilizing the newly added bioorthogonal groups. As shown in Scheme 3a, azide-containing RGD 6b could be easily ligated with a dansyl fluorophore or an anti-tumor drug, camptothecin, through a ring-strain promoted click reaction26 with the carbon–carbon double bond intact. The newly formed gem-difluoroallyl group can also be a good synthetic handle for bioconjugation via a radical pathway (Scheme 3b). For example, gem-difluoroallylated GSH 3p underwent radical addition smoothly when treated with readily available biotin-derived redox-active ester27 in the presence of Hantzsch ester (HE) under blue light irradiation. Similarly, the bioactive molecule lithocholic acid derivative (10b) and carbohydrate (10c) could also be easily introduced into the peptide. Encouraged by the bioorthogonal properties between the azide and alkenes, we turned our attention to the sequential “doubly orthogonal” peptide modification (Scheme 3c). Modified GSH 3y, featuring an azide group, initially underwent click reaction28 with biotin derivative 7c efficiently. Subsequently, treatment of the “single orthogonal” modified peptide 11 with monosaccharide-derived redox-active ester under irradiation led to the “doubly orthogonal” modified peptide 12 with a synthetically reasonable yield. This flexible “doubly orthogonal” peptide modification enables the convenient construction of bioconjugates with multiple functional elements, thus meeting various demands of peptidomimetics research.
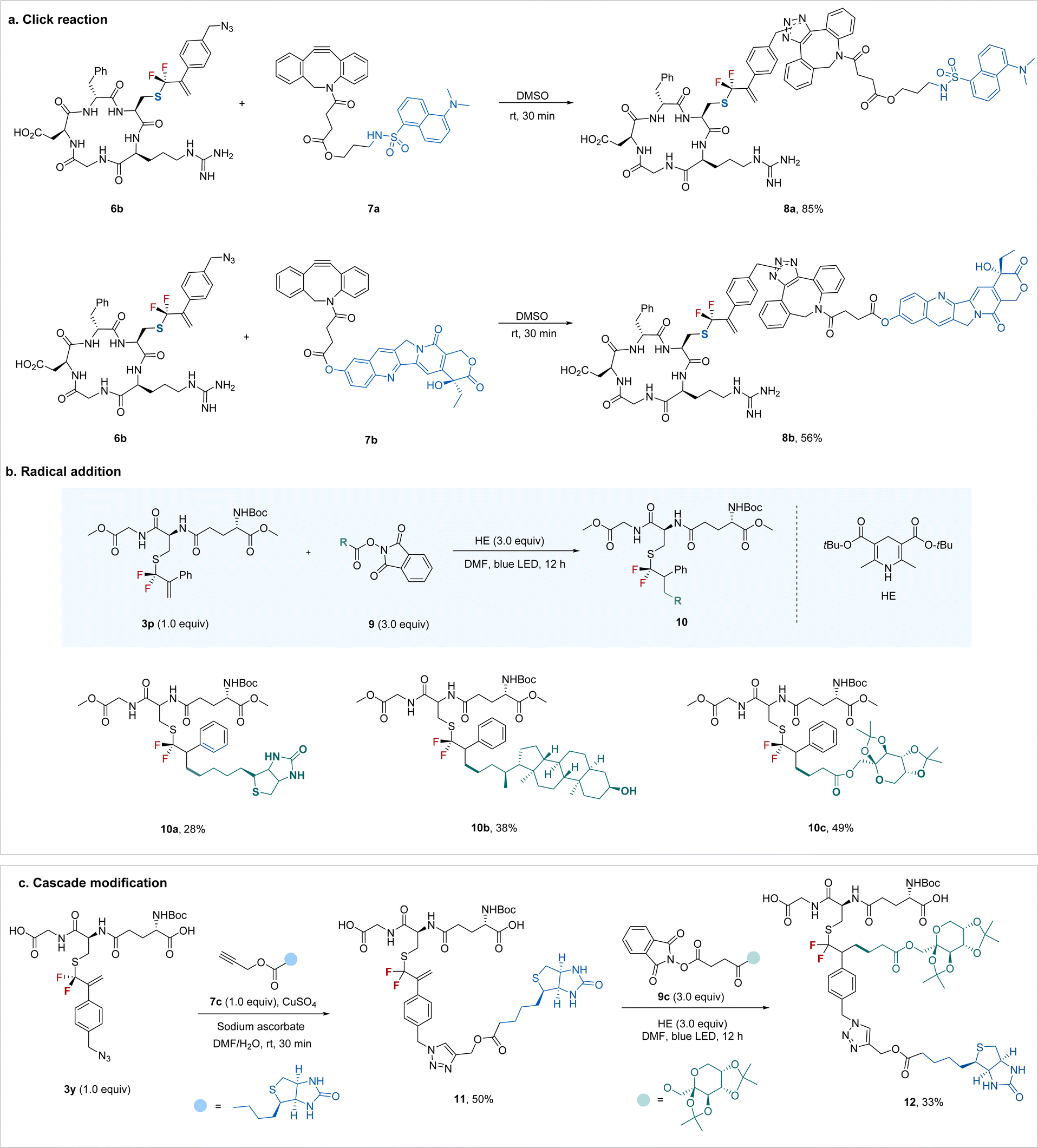 | ||
| Scheme 3 Synthetic applications of the gem-difluoroallylated peptides and “doubly orthogonal” modification. | ||
Conclusions
In conclusion, we have developed a highly chemo- and regio-selective method for late-stage gem-difluroallylation of cysteine residues in unprotected peptides with versatile DFASs. The protocol features high biocompatibility, mild reaction conditions, and synthetic simplicity. Particularly, the tunable activity of DFAS, as well as the biorthogonal property of the newly added functionalities and the gem-difluoroallyl group, allows for stepwise “doubly orthogonal” modification of peptides to meet various demands in peptidomimetics research. This proof of principle has been successfully applied to a wide range of oligopeptides, including bioactive peptides, such as GSH, RGD, and β-amyloid peptide fragment, showing great potential to explore the unique fluorine effect of the CF2 group in medicinal chemistry and chemical biology, leading to the discovery of new bioactive molecules.Data availability
All experimental data, procedures for data analysis, and pertinent data sets are provided in the ESI.†Author contributions
X. Z. and J.-X. R. conceived and designed the experiments. X. Z. directed the project. J.-X. R. performed the experiments. M. Z. and X.-T. F. prepared some starting materials. J.-X. R., F.-P. F., and H.-Y. Z. analyzed the data of ESI.† X. Z. wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the National Natural Science Foundation of China (22193072, 21931013, and 22301307), the National Key R&D Program of China (2021YFF0701700), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0590000), and the Science and Technology Commission of Shanghai Municipality (22JC1403500, 21XD1404400).Notes and references
- (a) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed; (b) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed; (c) C. Zhang, K. Yan, C. Fu, H. Peng, C. J. Hawker and A. K. Whittaker, Chem. Rev., 2022, 122, 167–208 CrossRef CAS PubMed; (d) Z. Q. Zhang, Y. Q. Sang, C. Q. Wang, P. Dai, X. S. Xue, J. P. Piper, Z. H. Peng, J. A. Ma, F. G. Zhang and J. Wu, J. Am. Chem. Soc., 2022, 144, 14288–14296 CrossRef CAS PubMed.
- J. Lv and Y. Cheng, Chem. Soc. Rev., 2021, 50, 5435–5546 RSC.
- (a) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed; (b) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed; (d) N. A. Mean-well, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- (a) E. S. Eberhardt, N. Panasik and R. T. Raines, J. Am. Chem. Soc., 1996, 118, 12261–12266 CrossRef CAS PubMed; (b) S. K. Holmgren, K. M. Taylor, L. E. Bretscher and R. T. Raines, Nature, 1998, 392, 666–667 CrossRef CAS PubMed; (c) S. K. Holmgren, L. E. Bretscher, K. M. Taylor and R. T. Raines, Chem. Biol., 1999, 6, 63–70 CrossRef CAS PubMed; (d) A. M. Remete, M. Nonn, S. Fustero, F. Fülöp and L. Kiss, Tetrahedron, 2018, 74, 6367–6418 CrossRef CAS; (e) P. K. Mykhailiuk, J. Org. Chem., 2022, 87, 6961–7005 CrossRef CAS PubMed; (f) L. F. Awad and M. S. Ayoup, Beilstein J. Org. Chem., 2020, 16, 1022–1050 CrossRef CAS PubMed.
- (a) M. Cheng, B. Zhang, W. Cui and M. L. Gross, Angew. Chem., Int. Ed., 2017, 56, 14007–14010 CrossRef CAS PubMed; (b) L. Fojtík, J. Fiala, P. Pompach, J. Chmelík, V. Matoušek, P. Beier, Z. Kukačka and P. Novák, J. Am. Chem. Soc., 2021, 143, 20670–20679 CrossRef PubMed; (c) D. Seebach, H. Widmer, S. Capone, R. Ernst, T. Bremi, I. Kieltsch, A. Togni, D. Monna, D. Langenegger and D. Hoyer, Helv. Chim. Acta, 2009, 92, 2577–2586 CrossRef CAS; (d) L. K. Charkoudian, C. W. Liu, S. Capone, S. Kapur, D. E. Cane, A. Togni, D. Seebach and C. Khosla, Protein Sci., 2011, 20, 1244–1255 CrossRef CAS PubMed; (e) G. Wang, Z. T. Zhang, B. Jiang, X. Zhang, C. Li and M. Liu, Anal. Bioanal. Chem., 2014, 406, 2279 CrossRef CAS PubMed; (f) E. N. G. Marsh, Acc. Chem. Res., 2014, 47, 2878–2886 CrossRef CAS PubMed; (g) A. A. Berger, J. Völler, N. Budisa and B. Koksch, Acc. Chem. Res., 2017, 50, 2093–2103 CrossRef CAS PubMed.
- (a) M.-Q. Zhou, Z. Feng and X.-G. Zhang, Chem. Commun., 2023, 59, 1434–1448 RSC; (b) J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem.–Asian J., 2009, 4, 630–640 CrossRef CAS PubMed; (c) S. B. Gunnoo and A. Madder, ChemBioChem, 2016, 17, 529–553 CrossRef CAS PubMed; (d) F.-J. Chen, M.-M. Zhang, V. Nobile and J.-M. Gao, Chem.–Eur. J., 2022, 28, e202200058 CrossRef CAS PubMed; (e) C. E. Stieger, Y. Park, M. A. R. de Geus, D. Kim, C. Huhn, J. S. Slenczka, P. Ochtrop, J. M. Müchler, R. D. Süssmuth, J. Broichhagen, M.-H. Baik and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2022, 61, e202205348 CrossRef CAS PubMed; (f) J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666–1676 RSC; (g) P. M. S. D. Cal, G. J. L. Bernardes and P. M. P. Gois, Angew. Chem., Int. Ed., 2014, 53, 10585–10587 CrossRef CAS PubMed.
- D. Abegg, M. Tomanik, N. Qiu, D. Pechalrieu, A. Shuster, B. Herzon and A. Adibekian, J. Am. Chem. Soc., 2021, 143, 20332–20342 CrossRef CAS PubMed.
- S. Verhoog, C. W. Kee, Y. L. Wang, T. Khotavivattana, T. C. Wilson, V. Kersemans, S. Smart, M. Tredwell, B. G. Davis and V. Gouverneur, J. Am. Chem. Soc., 2018, 140, 1572–1575 CrossRef CAS PubMed.
- (a) E. N. G. Marsh and Y. Suzuki, ACS Chem. Biol., 2014, 9, 1242–1250 CrossRef CAS PubMed; (b) H. Duewel, E. Daub, V. Robinson and J. F Honek, Biochemistry, 1997, 36, 3404–3416 CrossRef CAS PubMed.
- C. Zhang, M. Welborn, T. Y. Zhu, N. J. Yang, M. S. Santos, T. V. Voorhis and B. L. Pentelute, Nat. Chem., 2016, 8, 120–128 CrossRef CAS PubMed.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu and Y.-S. Lin, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- C. Zhang, A. M. Spokoyny, Y. Zou, M. D. Simon and B. L. Pentelute, Angew. Chem., Int. Ed., 2013, 52, 14001–14005 CrossRef CAS PubMed.
- C. Zhang, P. Dai, A. M. Spokoyny and B. L. Pentelute, Org. Lett., 2014, 16, 3652–3655 CrossRef CAS PubMed.
- (a) I. Kieltsch, P. Eisenberger and A. Togni, Angew. Chem., Int. Ed., 2007, 46, 754–757 CrossRef CAS PubMed; (b) S. Capone, I. Kieltsch, O. Flögel, G. Lelais, A. Togni and D. Seebach, Helv. Chim. Acta, 2008, 91, 2035–2056 CrossRef CAS.
- H. Jia, A. P. Häring, F. Berger, L. Zhang and T. Ritter, J. Am. Chem. Soc., 2021, 143, 7623–7628 CrossRef CAS PubMed.
- C. Bottecchia, X.-J. Wei, K. P. L. Kuijpers, V. Hessel and T. Noël, J. Org. Chem., 2016, 81, 7301–7307 CrossRef CAS PubMed.
- (a) M. Imiołek, G. Karunanithy, W.-L. Ng, A. J. Baldwin, V. Gouverneur and B. G. Davis, J. Am. Chem. Soc., 2018, 140, 1568–1571 CrossRef PubMed; (b) M. Imiołek, P. G. Isenegger, W.-L. Ng, A. Khan, V. Gouverneur and B. G. Davis, ACS Cent. Sci., 2021, 7, 145–155 CrossRef PubMed.
- C. W. Kee, O. Tack, F. Guibbal, T. C. Wilson, P. G. Isenegger, M. Imiołek, S. Verhoog, M. Tilby, G. Boscutti, S. Ashworth, J. Chupin, R. Kashani, A. W. J. Poh, J. K. Sosabowski, S. Macholl, C. Plisson, B. Cornelissen, M. C. Willis, J. Passchier, B. G. Davis and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 1180–1185 CrossRef CAS PubMed.
- H. Wright, B. J. Bower, J. M. Chalker, G. J. L. Bernardes, R. Wiewiora, W.-L. Ng, R. Raj, S. Faulkner, M. R. J. Vallée, A. Phanumartwiwath, O. D. Coleman, M.-L. Thézénas, M. Khan, S. R. G. Galan, L. Lercher, M. W. Schombs, S. Gerstberger, M. E. Palm-Espling, A. J. Baldwin, B. M. Kessler, T. D. W. Claridge, S. Mohammed and B. G. Davis, Science, 2016, 354, 597 CrossRef PubMed.
- X.-T. Feng, J.-X. Ren, X. Gao, Q. Q. Min and X.-G. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202210103 CrossRef CAS PubMed.
- X. Gao, X.-X. Ren, W. Deng and X.-G. Zhang, Chin. J. Chem., 2023, 41, 3521–3527 CrossRef CAS.
- H. Jiang, X. Y. Zhang, X. Chen, P. Aramsangtienchai, Z. Tong and H. N. Lin, Chem. Rev., 2018, 118, 919–988 CrossRef CAS PubMed.
- R. Hatley, S. Macdonald, R. Slack, J. Le, S. Ludbrook and P. Lukey, Angew. Chem., Int. Ed., 2018, 57, 3298–3321 CrossRef CAS PubMed.
- (a) B. Jurek and I. D. Neumann, Physiol. Rev., 2018, 98, 1805–1908 CrossRef CAS PubMed; (b) M. Muttenthaler, Å. Andersson, I. Vetter, R. Menon, M. Busnelli, L. Ragnarsson, C. Bergmayr, S. Arrowsmith, J. R. Deuis, H. S. Chiu, N. J. Palpant, M. O'Brien, T. J. Smith, S. Wray, I. D. Neumann, C. W. Gruber, R. J. Lewis and P. F. Alewood, Sci. Signaling, 2017, 10, eaan3398 CrossRef PubMed.
- S. N. Mthembu, A. Sharma, F. Albericio and B. G. de la Torre, ChemBioChem, 2020, 21, 1947–1954 CrossRef CAS PubMed.
- (a) N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed; (b) J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- (a) S. Murarka, Adv. Synth. Catal., 2018, 360, 1735–1753 CrossRef CAS; (b) C. Zheng, G.-Z. Wang and R. Shang, Adv. Synth. Catal., 2019, 361, 4500–4505 CrossRef CAS.
- (a) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed; (b) V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef CAS PubMed; (c) A. K. Agrahari, P. Bose, M. K. Jaiswal, S. Rajkhowa, A. S. Singh, S. Hotha, N. Mishra and V. K. Tiwari, Chem. Rev., 2021, 121, 7638–7956 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, and analytical data for all new compounds. See DOI: https://doi.org/10.1039/d4sc02681k |
| This journal is © The Royal Society of Chemistry 2024 |