
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric three-component Tsuji–Trost allylation reaction enabled by chiral aldehyde/palladium combined catalysis†
Jian-Hua
Liu
a,
Wei
Wen
 a,
Zhu-Lian
Wu
a,
Tian
Cai
a,
Yan-Min
Huang
*b and
Qi-Xiang
Guo
*a
a,
Zhu-Lian
Wu
a,
Tian
Cai
a,
Yan-Min
Huang
*b and
Qi-Xiang
Guo
*a
aKey Laboratory of Applied Chemistry of Chongqing Municipality, Chongqing Key Laboratory of Soft-Matter Material Chemistry and Function Manufacturing, School of Chemistry and Chemical Engineering, Southwest University, Chongqing, 400715, China. E-mail: qxguo@swu.edu.cn
bGuangxi Key Laboratory of Natural Polymer Chemistry and Physics, Nanning Normal University, Nanning, 530001, China. E-mail: huangyanmin828@gxtc.edu.com
First published on 3rd June 2024
Abstract
Despite the long-standing exploration of the catalytic asymmetric Tsuji–Trost allylation reaction since the mid-20th century, most reported instances have adhered to a two-component approach. Here, we present a remarkably efficient three-component asymmetric allylation reaction enabled by the collaborative action of chiral aldehyde and palladium. A diverse array of NH2-unprotected amino acid esters, aryl or alkenyl iodides, and allyl alcohol esters exhibit robust participation in this reaction, resulting in the synthesis of structurally diverse non-proteinogenic α-amino acid esters with favorable experimental outcomes. Mechanistic investigations reveal the dominance of the allylation/Heck coupling cascade in reactions involving electron-rich aryl iodides, while the Heck coupling/allylation cascade emerges as the dominant pathway in reactions involving electron-deficient aryl iodides. This chiral aldehyde/palladium combining catalytic system precisely governs the chemoselectivity of C-allylation and N-allylation, the regioselectivity of linear and branched allylation, and the enantioselectivity of C-allylation products.
Introduction
The catalytic enantioselective Tsuji–Trost allylation stands as one of the most invaluable approaches leading to optically active allylic compounds.1 Since its inaugural disclosure,2 extensive efforts have been dedicated to the development of innovative catalytic systems and the advent of new reactions.3 Consequently, this fundamental reaction has found widespread application in the construction of chiral molecules through the establishment of chemical bonds such as C–C, C–N, C–O, and so forth,4 as well as in natural product synthesis (Fig. 1a).5 Typically, many reactions have been employed to form a single chemical bond through a two-component reaction pathway. To engender the formation of multiple chemical bonds in a unified process, the substrates must be judiciously tailored by integrating multiple reactive sites within a single molecule.6 In this regard, the incorporation of two leaving groups in a single reactant to facilitate consecutive allylation reactions has been the most extensively investigated strategy.7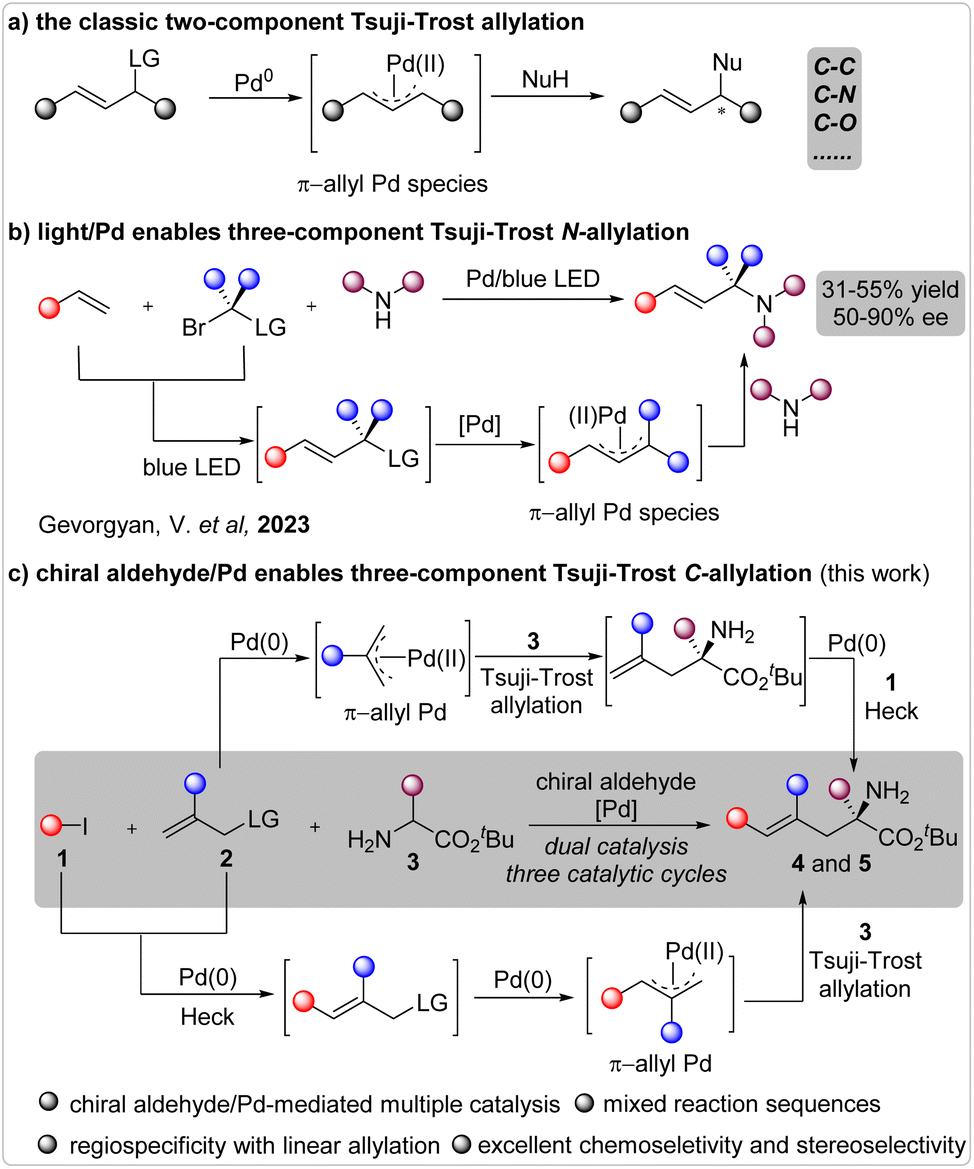 | ||
| Fig. 1 Catalytic asymmetric α-hydrocarbylation reaction of amino acid esters with halohydrocarbons. | ||
The catalytic enantioselective multiple-component reaction represents an exceptional approach for effecting the simultaneous construction of multiple chemical bonds in a unified fashion, and has been extensively explored in many types of transformations.8 However, the corresponding multiple-component Tsuji–Trost allylation reaction has scarcely been investigated, partly owing to the exigency of concurrently controlling the entire reaction sequence, chemoselectivity, regioselectivity, and enantioselectivity.9 Remarkably, Gevorgeyan et al. recently unveiled a sophisticated three-component N-allylation reaction facilitated by photoinduced palladium catalysis (Fig. 1b).10 Several enantioselective instances were realized with moderate yields and moderate-to-good enantioselectivities. Thus, the development of a novel methodology to achieve a highly efficient Tsuji–Trost allylation reaction involving three or more reactants remains a pressing imperative to further enrich the domain of this seminal reaction.
The chiral aldehyde/palladium combined catalysis11 has been well documented in the asymmetric α-functionalization of NH2-unprotected amino acid esters.12 However, all of these reactions have occurred via a two-component pathway, and the advancement of multiple-component reactions will significantly propel the progression of chiral aldehyde catalysis.13 Drawing from our investigations into the catalytic asymmetric Tsuji–Trost allylation12a and benzylation12c reactions, we envisaged that a reaction involving a halide, a terminal allylic alcohol ester, and an NH2-unprotected amino acid ester could occur under the influence of a chiral aldehyde/palladium combined catalytic system (Fig. 1c). However, significant challenges lie ahead. On one hand, this three-component reaction comprises three catalytic cycles, involving two distinct palladium-mediated catalytic cycles for the Heck coupling and allylation processes, as well as one chiral aldehyde-mediated catalytic cycle for the generation of active nucleophiles from amino acid esters. Additionally, it encompasses two potential reaction sequences, including the Tsuji–Trost allylation/Heck coupling or the Heck coupling/Tsuji–Trost allylation. These intricate circumstances posed a challenge in identifying a privileged catalytic system. On the other hand, precise control over the chemoselectivity of N-allylation and C-allylation, regioselectivity of linear and branched C-allylation, and enantioselectivity of the C-allylation product is imperative. Here, we present our endeavors in exploring the asymmetric three-component Tsuji–Trost allylation using chiral aldehyde/palladium combined catalysis.
Results and discussion
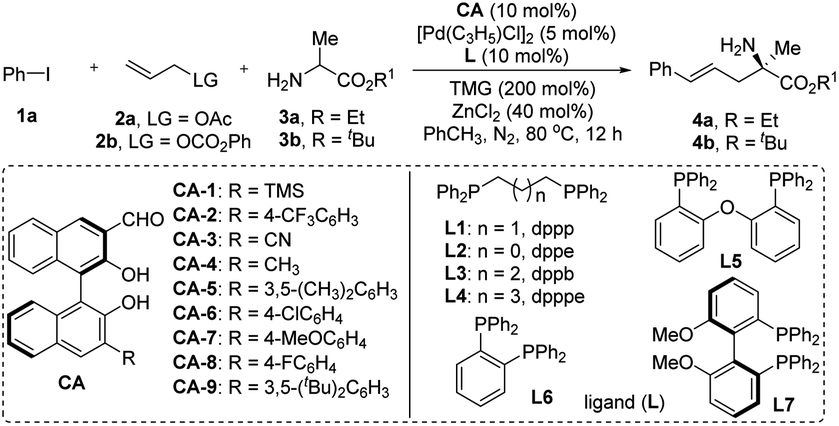
Our investigation commenced with assessing the reaction of iodobenzene 1a, allyl acetate 2a, and ethyl alaninate 3a, facilitated by the application of a combined catalytic system derived from chiral aldehyde CA-1, palladium [Pd(C3H5)Cl]2, and the Lewis acid ZnCl2. The base 1,1,3,3-tetramethylguanidine (TMG) was included to aid the deprotonation process. As anticipated, the desired product 4a was obtained in a 45% yield and 71% ee (Table 1, entry 1). Subsequently, the reaction conditions were systematically optimized. Firstly, various substituted chiral aldehydes were utilized as catalysts, with the outcomes demonstrating that CA-1 was the best choice in term of the enantioselectivity (Table 1, entries 1–9). Replacing L1 by other achiral ligands did not lead to an enhancement in the experimental findings (Table 1, entries 14). Lewis acid screening revealed that ZnBr2 marginally improved the yield of 4a (Table 1, entry 15). Thereafter, a variety of achiral and chiral ligands were re-examined, with results indicating that the chiral ligand L7 was the most suitable, harmonizing well with the chiral aldehyde CA-1 (Table 1, entry 16). To further refine the experimental outcomes, several additives were introduced to the reaction system. Of these, LiOTf exhibited a beneficial effect, yielding product 4a in 61% yield and 88% ee (Table 1, entry 17). Modest enhancements in yield were achieved by reducing the reaction temperature and increasing the equivalents of the Lewis acid ZnBr2 and the base TMG (Table 1, entries 19–21). The leaving group (LG) of reactant 2 and the alkoxy group of amino acid ester 3 exerted a discernible influence on the experimental results. Notably, utilizing 2b and 3b as reactants, product 4b was achieved in a 71% yield and 96% ee (Table 1, entry 23). Eventually, the additives were reassessed and the equivalents of 2b were meticulously adjusted. With LiBF4 as the additive and 1.8 equivalents of 2b, the reaction yielded 4b in 81% yield and 96% ee (Table 1, entry 25). Fueled by these exceptional outcomes, the optimal reaction conditions were established and employed for subsequent substrate scope investigations.|
|
||||
|---|---|---|---|---|
| Entry | CA | L | Yieldb (%) | eec (%) |
a Reaction conditions: 1a (0.2 mmol), 2 (0.3 mmol), 3 (0.3 mmol), CA (0.02 mmol), L (0.02 mmol), [Pd(C3H5)Cl]2 (0.01 mmol), ZnCl2 (0.08 mmol), TMG (0.4 mmol), in PhCH3 (0.5 mL) at 80 °C.
b Isolated yield.
c Determined by chiral HPLC.
d ZnBr2 instead of ZnCl2.
e With ent-CA-1.
f LiOTf as additive.
g At 70 °C.
h With 100 mol% ZnBr2.
i With 220 mol% TMG.
j With 2b.
k With 3b.
l With LiBF4 as additive.
m
1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2b:3b = 1.8:1:1.5. NR = no reaction, ND = not determined. :2b:3b = 1.8:1:1.5. NR = no reaction, ND = not determined.
|
||||
| 1 | CA-1 | L1 | 45 | 71 |
| 2 | CA-2 | L1 | 29 | 50 |
| 3 | CA-3 | L1 | 46 | 20 |
| 4 | CA-4 | L1 | 49 | 65 |
| 5 | CA-5 | L1 | 31 | 58 |
| 6 | CA-6 | L1 | 32 | 65 |
| 7 | CA-7 | L1 | 47 | 60 |
| 8 | CA-8 | L1 | 51 | 64 |
| 9 | CA-9 | L1 | 43 | 58 |
| 10 | CA-1 | L2 | 52 | 67 |
| 11 | CA-1 | L3 | 25 | 76 |
| 12 | CA-1 | L4 | NR | ND |
| 13 | CA-1 | L5 | 26 | 65 |
| 14 | CA-1 | L6 | NR | ND |
| 15d | CA-1 | L1 | 55 | 70 |
| 16d | CA-1 | L7 | 30 | 80 |
| 17d,e | CA-1 | L7 | 32 | 16 |
| 18d,f | CA-1 | L7 | 61 | 88 |
| 19d,f,g | CA-1 | L7 | 56 | 92 |
| 20f,g,h | CA-1 | L7 | 63 | 90 |
| 21f,g,h,i | CA-1 | L7 | 67 | 89 |
| 22f,g,h,i,j | CA-1 | L7 | 69 | 90 |
| 23f,g,h,i,j,k | CA-1 | L7 | 71 | 96 |
| 24g,h,i,j,k,l | CA-1 | L7 | 78 | 97 |
| 25g,h,i,l,m | CA-1 | L7 | 81 | 97 |
Following the establishment of the optimal reaction conditions, we proceeded to investigate the substrate scopes of this reaction. Firstly, an array of aryl iodides were employed as reactants, with the results indicating the substantial impact of steric effects of substituents on the aryl ring on both yield and enantioselectivity. Particularly, under the optimal reaction conditions, the reaction of ortho-methyl iodobenzene, 2b and 3b gave product 4c in 31% yield and 92% ee. As for ortho-fluoro iodobenzene, while product 4d was obtained in a 58% yield, the enantioselectivity (78% ee) markedly decreased. Conversely, other phenyl iodides bearing a single substituent at the corresponding meta- or para-position of the phenyl ring demonstrated successful reactivity with reactants 2b and 3b, yielding products 4e–4t in commendable yields (59–75%) and exceptional enantioselectivities (91–96% ee). Similar encouraging outcomes were observed with phenyl iodides bearing two substituents at their phenyl rings (Fig. 2, 4u–4aa). Additionally, aryl iodides bearing aryls other than phenyl were examined, and both 2-iodonaphthalene and 5-iodo-1-tosyl-1H-indole effectively participated in this reaction, yielding products 4ab and 4ac in favorable yields and excellent enantioselectivities. Furthermore, alkenyl iodides also displayed noteworthy reactivity in this reaction. All four alkenyl iodides employed in this study exhibited smooth reactivity with 2b and 3b, yielding the desired products 4ad–4ag in satisfactory yields and excellent enantioselectivities. However, aryl iodides bearing strong electron-withdrawing groups were not suitable reactants for this reaction (Fig. 2, 4ah–4al).
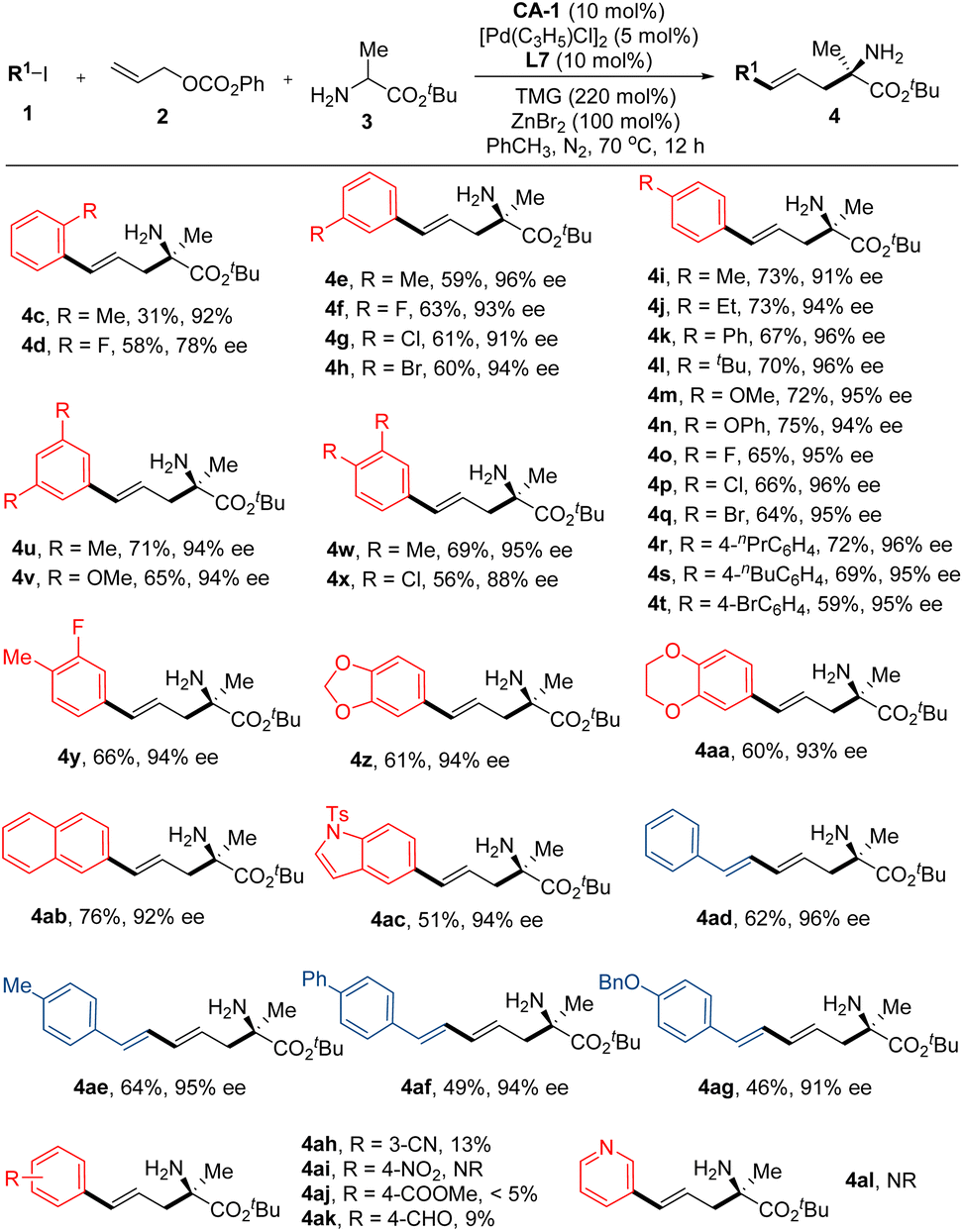 | ||
| Fig. 2 Substrate scopes of the aryl- or alkenyl-substituted iodides. | ||
Subsequently, we delved into the substrate scope of amino acid derivatives (Fig. 3). The results revealed that amino acid esters bearing alkyl groups proved to be favorable reaction partners for reactants 1 and 2a, yielding products 5a–5d in noteworthy yields and enantioselectivities. Furthermore, amino acid esters derived from phenylglycines, phenylalanines, and homophenylalanine were subjected to testing, all yielding comparable experimental results to those observed with alkyl-substituted ones (Fig. 3, 5e–5l). Other amino acid esters bearing functional groups such as ether and ester also demonstrated commendable reactivity in this reaction, yielding the corresponding products 5m–5o with favorable experimental outcomes. Although several substituted allyl alcohol esters were examined, they displayed limited reactivity in this reaction (Fig. 3, 5p–5q). This observation can be attributed to the increased steric effects upon the introduction of a substituent to the molecular skeleton of 2b.
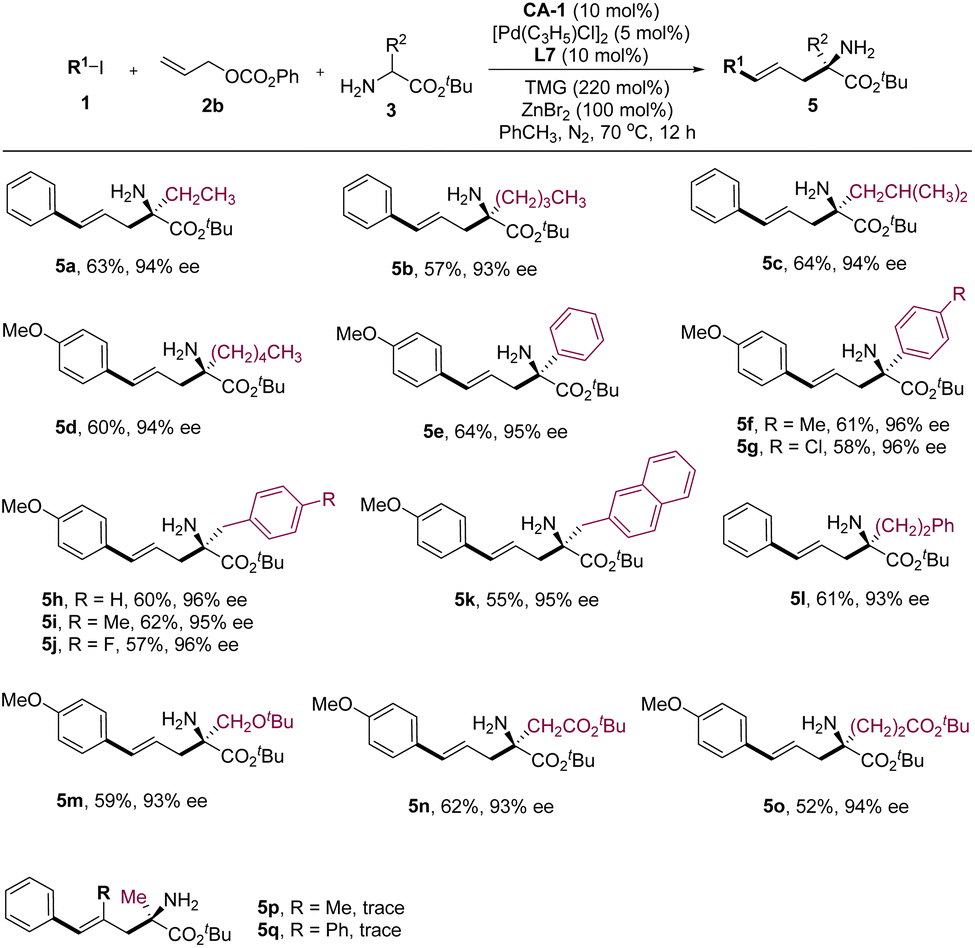 | ||
| Fig. 3 Substrate scopes of the amino acid esters. | ||
Two potential reaction sequences, the Heck/allylation and the allylation/Heck cascades, were encompassed in this reaction. To discern the dominant one, several control experiments were conducted under the optimal reaction conditions (Fig. 4a). The experimental results from the reaction of 2b and 3b were used for comparison, wherein the allylation intermediate 3a′ was produced in 79% yield and 97% ee. Assuming that this reaction proceeded via the formation of allylation intermediate 3a′, a comparable enantioselectivity should have been achieved. For the model reaction, product 4b was generated with 97% ee. However, only 88% ee was attained when 1,2-dichloro-4-iodobenzene 1b was utilized as the reactant. These findings suggested that the iodobenzene reacted with 2b and 3b through the potential allylation/Heck cascade, while the 1,2-dichloro-4-iodobenzene 1b followed a different reaction pathway.
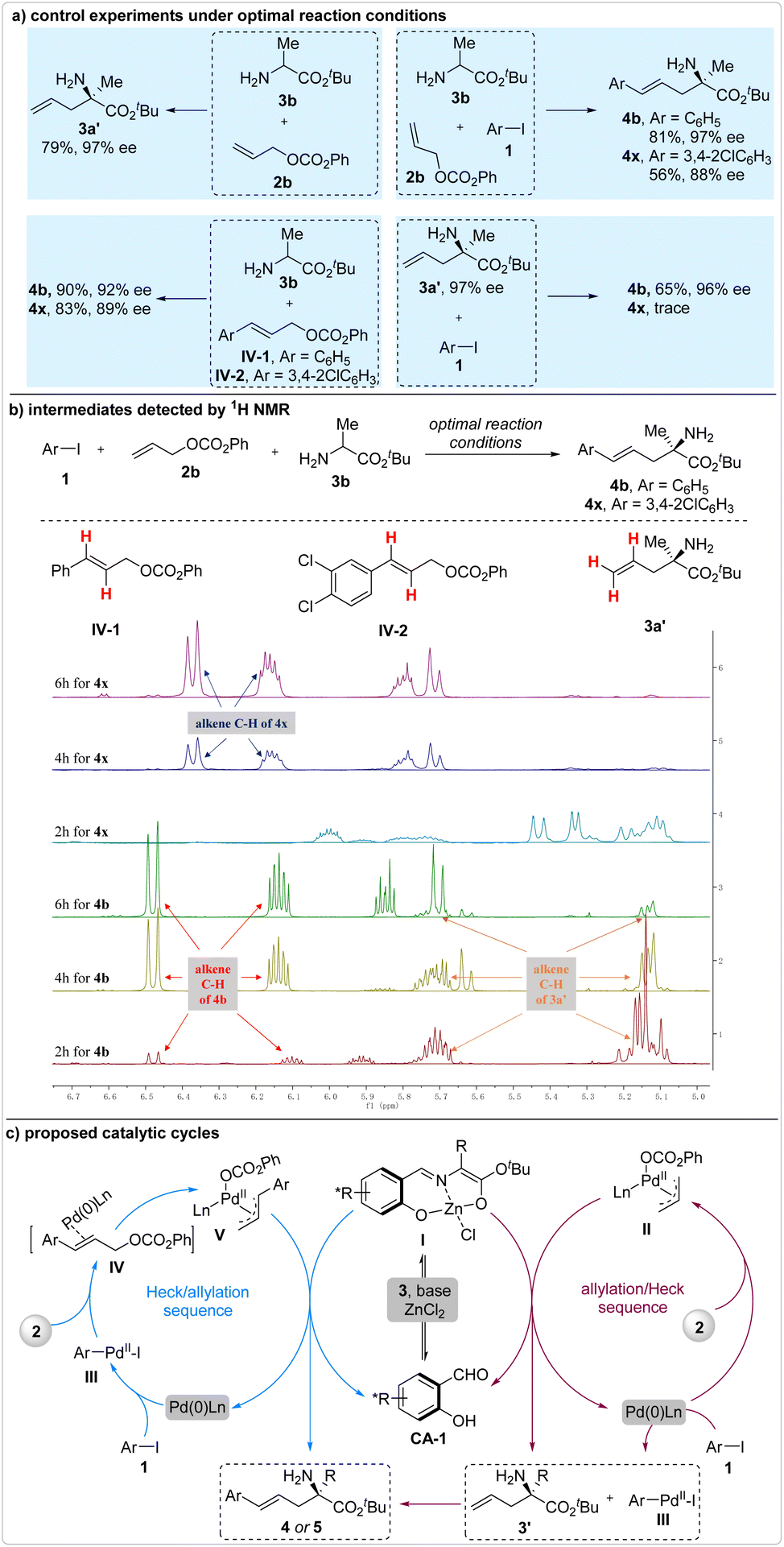 | ||
| Fig. 4 Reaction mechanism investigation. | ||
Subsequently, two additional control experiments were conducted under the optimal reaction conditions. The reaction of 3b and the potential Heck coupling intermediate IV-1 produced product 4b with a lower enantioselectivity of 92% ee, as compared to that achieved in the corresponding three-component reaction (97% ee). This outcome indicated that the iodobenzene-involved three-component reaction may not proceed via the formation of Heck coupling intermediate IV-1. Conversely, the reaction of 3b and IV-2 yielded product 4x with a comparable enantioselectivity (89% ee vs. 88% ee), suggesting that IV-2 was the likely intermediate in the 1,2-dichloro-4-iodobenzene-involved three-component reaction. Furthermore, the reaction of allylation intermediate 3a′ and iodobenzene 1a yielded product 4b with a 96% ee, while the reaction of 3a′ and 1,2-dichloro-4-iodobenzene 1b failed to proceed under the optimal reaction conditions. These results provided additional evidence that iodobenzene and 1,2-dichloro-4-iodobenzene participate in this reaction by forming different intermediates. The divergence in reaction pathways observed in the electron-deficient iodobenzene-involved reactions can be attributed to the heightened reaction rate of the associated Heck coupling.
In order to gain definitive evidence that elucidates the true reaction mechanism, we diligently monitored the entire reaction process using 1H NMR (Fig. 4b). For the reaction involving iodobenzene 1a, allyl alcohol ester 2b, and amino acid ester 3b, the allylation intermediate 3a′ was clearly observed after 2 hours. As the reaction progressed, the ratio of allylation intermediate gradually decreased, indicating that this iodobenzene-involved three-component reaction likely proceeded through the allylation/Heck coupling cascade. In contrast, the allylation intermediate 3a′ was not detected in the 1,2-dichloro-4-iodobenzene-involved three-component reaction, suggesting that the Heck/allylation cascade was the probable reaction pathway. However, neither of the Heck coupling intermediates IV could be detected. One of the most plausible reasons for this is that these intermediates existed as alkene–Pd complexes and promptly converted into the corresponding π-allyl palladium species upon completion of the Heck coupling reaction.
Based on these findings, a plausible reaction mechanism involving mixed reaction sequences has been proposed (Fig. 4c). In the case of the Heck/allylation cascade reaction, the aryl iodides 1 undergo oxidative addition to form intermediate III, which promptly transforms into the π-allyl palladium species V. Upon being attacked by the active enolate I, enantioselective generation of the corresponding product 4 or 5 occurs. On the other hand, in the allylation/Heck coupling cascade, nucleophilic attack occurs between the enolate I and the π-allyl palladium species II, yielding the allylation intermediate 3a′ enantioselectively. Subsequently, 3a′ reacts with III to produce the desired products 4 or 5. Generally, the Heck/allylation process predominates in the three-component reactions involving electron-deficient aryl iodides, whereas the allylation/Heck cascade emerges as the dominant pathway when an electron-rich aryl iodide is employed as the reactant.
Conclusions
In summary, a highly efficient asymmetric three-component Tsuji–Trost allylation reaction is accomplished through the collaborative catalysis of chiral aldehyde/palladium. Various substituted aryl and alkenyl iodides, along with amino acid esters, serve as excellent substrates in this reaction, yielding the corresponding non-proteinogenic α,α-disubstituted α-amino acid derivatives in favorable yields and exceptional enantioselectivities. Mechanistic investigations reveal that the electronic properties of the iodide reactants govern the reaction pathway, leading to either a Heck coupling/allylation sequence or an allylation/Heck coupling cascade.Data availability
All data supporting the findings of this study are available within the article and its ESI file.†Author contributions
H. Y. M. W. W. and G. Q. X. conceived this project. L. J. H. carried out the experiments. W. Z. L. and C. T. performed the HRMS analysis. G. Q. X. wrote the manuscript. All authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from NSFC (22371233, 22071199), and the Innovation Research 2035 Pilot Plan of Southwest University (SWU-XDZD22011).Notes and references
- (a) J. Tsuji, Tetrahedron, 2015, 71, 6330–6348 CrossRef CAS; (b) B. M. Trost, Tetrahedron, 2015, 71, 5708–5733 CrossRef CAS PubMed; (c) J. D. Weaver, A. Recio III, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846–1913 CrossRef CAS PubMed; (d) B. M. Trost, M. R. Machacek and A. P. Aponick, Acc. Chem. Res., 2006, 39, 747–760 CrossRef CAS PubMed; (e) B. M. Trost and D. L. VanVranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS PubMed; (f) F. Tian, J. Zhang, W. Yang and W. Deng, Chinese J. Org. Chem., 2020, 40, 3262–3278 CrossRef CAS; (g) X. Ma, J. Yu, Z. Wang, Y. Zhang and Q. Zhou, Chinese J. Org. Chem., 2020, 40, 2669–2677 CrossRef CAS.
- (a) J. Tsuji, H. Takahashi and M. Morikawa, Tetrahedron Lett., 1965, 49, 4387–4388 CrossRef; (b) B. M. Trost and T. J. Fullerton, J. Am. Chem. Soc., 1973, 95, 292–294 CrossRef CAS.
- (a) B. M. Trost, Org. Process Res. Dev., 2012, 16, 185–194 CrossRef CAS PubMed; (b) B. Sundararaju, M. Achard and C. Bruneau, Chem. Soc. Rev., 2012, 41, 4467–4483 RSC; (c) S. Noreen, F. Z. Ameer, A. Sajjad, S. Irum, I. Ali and F. Sadia, Curr. Org. Chem., 2019, 23, 1168–1213 CrossRef CAS; (d) G. Wu, J.-R. Wu, Y. Huang and Y.-W. Yang, Chem.–Asian J., 2021, 16, 1864–1877 CrossRef CAS PubMed.
- (a) L. Mohammadkhani and M. M. Heravi, Chem. Rec., 2021, 21, 29–68 CrossRef CAS PubMed; (b) Y. Liu, J. Oble, A. Pradal and G. Poli, Eur. J. Inorg. Chem., 2020, 942–961 CrossRef; (c) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2944 CrossRef CAS PubMed.
- (a) B. M. Trost, R. Madsen and S. D. Guile, J. Am. Chem. Soc., 2000, 122, 5947–5956 CrossRef CAS; (b) B. M. Trost and C. A. Kalnmals, Chem.–Eur. J., 2020, 26, 1906–1921 CrossRef CAS PubMed.
- S. K. Nanda, Chem. Commun., 2023, 59, 11298–11319 RSC.
- (a) B. M. Trost and C. B. Lee, J. Am. Chem. Soc., 2001, 123, 3671–3686 CrossRef CAS PubMed; (b) B. M. Trost and C. B. Lee, J. Am. Chem. Soc., 2001, 123, 3687–3696 CrossRef CAS PubMed; (c) B. M. Trost, J. T. Masters and A. C. Burns, Angew. Chem., Int. Ed., 2013, 52, 2260–2264 CrossRef CAS PubMed; (d) X.-H. Huo, L. Zhao, Y.-C. Luo, Y. Wu, Y.-W. Sun, G. L. Li, G. Tatiana, J.-C. Zhang, Y. Ye and W.-B. Zhang, CCS Chem., 2022, 4, 1720–1731 CrossRef CAS; (e) G.-L. Li, L. Zhao, Y.-C. Luo, Y.-B. Peng, K. Xu, X.-H. Huo and W.-B. Zhang, Chem.–Eur. J., 2022, 28, e2022002 Search PubMed.
- (a) M. Syamala, Org. Prep. Proced. Int., 2009, 41, 1–68 CrossRef CAS; (b) M. Syamala, Org. Prep. Proced. Int., 2005, 37, 103–171 CrossRef CAS.
- (a) D. Zhu, Z. Liao, Y. R. Chi, T. P. Goncalves, K. W. Huaang and J. S. Zhou, Angew. Chem., Int. Ed., 2020, 59, 5341–5345 CrossRef CAS PubMed; (b) Z. H. Kang, W. Chang, X. Tian, X. Fu, W. Zhao, X. Xu, Y. Liang and W. Hu, J. Am. Chem. Soc., 2021, 143, 20818–20827 CrossRef CAS PubMed.
- N. Kvasovs, J. Fang, F. Kliuev and V. Gevorgyan, J. Am. Chem. Soc., 2023, 145, 18497–18505 CrossRef CAS PubMed.
- (a) M. Kamlar, M. Urban and J. Veselý, Chem. Rec., 2023, 23, e202200284 CrossRef CAS PubMed; (b) D.-F. Chen and L.-Z. Gong, J. Am. Chem. Soc., 2022, 144, 2415–2437 CrossRef CAS PubMed; (c) D.-F. Chen, Z.-Y. Han, X.-L. Zhou and L.-Z. Gong, Acc. Chem. Res., 2014, 47, 2365–2377 CrossRef CAS PubMed; (d) Z. Shao and H. Zhang, Chem. Soc. Rev., 2009, 38, 2745–2755 RSC; (e) G. Chen, Y. Deng, L. Gong, A. Mi, X. Cui, Y. Jiang, M. C. K. Choi and A. S. C. Chan, Tetrahedron: Asymmetry, 2001, 12, 1567–1571 CrossRef CAS; (f) M. Nakoji, T. Kanayama, T. Okino and Y. Takemoto, Org. Lett., 2001, 3, 3329–3331 CrossRef CAS PubMed; (g) M. Á. Fernández-Ibañez, B. Maciá, D. A. Alonso and I. M. Pastor, Molecules, 2013, 18, 10108–10121 CrossRef PubMed; (h) C. Zhong and X. Shi, Eur. J. Org Chem., 2010, 2010, 2999–3025 CrossRef; (i) Z. Du and Z. Shao, Chem. Soc. Rev., 2013, 42, 1337–1378 RSC.
- (a) L. Chen, M.-J. Luo, F. Zhu, W. Wen and Q.-X. Guo, J. Am. Chem. Soc., 2019, 141, 5159–5164 CrossRef CAS PubMed; (b) F. Zhu, Q.-W. Shen, W.-Z. Wang, Z.-L. Wu, T. Cai, W. Wen and Q.-X. Guo, Org. Lett., 2021, 23, 1463–1467 CrossRef CAS PubMed; (c) J.-H. Liu, W. Wen, J. Liao, Q.-W. Shen, Y. Lin, Z.-L. Wu, T. Cai and Q.-X. Guo, Nat. Commun., 2022, 13, 2509 CrossRef CAS PubMed; (d) J.-H. Liu, Q. Zhou, Y. Lin, Z.-L. Wu, T. Cai, W. Wen, Y.-M. Huang and Q.-X. Guo, ACS Catal., 2023, 13, 6013–6022 CrossRef CAS; (e) Q. Zhou, Z.-W. Yin, Z.-L. Wu, T. Cai, W. Wen, Y.-M. Huang and Q.-X. Guo, Org. Lett., 2023, 25, 5790–5794 CrossRef CAS PubMed; (f) Q.-W. Shen, W. Wen and Q.-X. Guo, Org. Lett., 2023, 25, 3163–3167 CrossRef CAS PubMed; (g) H. Zhang, W. Wen, Z.-X. Lu, Z.-L. Wu, T. Cai and Q.-X. Guo, Org. Lett., 2024, 26, 153–159 CrossRef CAS PubMed.
- (a) Q. Wang, Q. Gu and S. L. You, Angew. Chem., Int. Ed., 2019, 58, 6818–6825 CrossRef CAS PubMed; (b) J. F. Chen, Y. E. Liu, X. Gong, L. M. Shi and B. G. Zhao, Chin. J. Chem., 2019, 37, 103–112 CrossRef CAS; (c) W. Wen and Q.-X. Guo, Synthesis, 2023, 55, 719–732 CrossRef CAS; (d) W. Wen and Q.-X. Guo, Acc. Chem. Res., 2024, 57, 776–794 CrossRef CAS PubMed; (e) X. Xiao and B. G. Zhao, Acc. Chem. Res., 2023, 56, 1097–1117 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02594f |
| This journal is © The Royal Society of Chemistry 2024 |